

Abstract
Background
Glioblastomas have been shown to rely on glycolysis as an energy source. However, recent evidence suggests that at least a subset of glioma cells with stem cell–like properties can thrive on oxidative phosphorylation. It remains unclear whether both metabolic phenotypes support tumor propagation, if they are independent, and how stable they are. The present study investigated these questions with the use of isogenic murine glioma stem cells (GSCs).
Methods
GSCs were established from tumors formed by Ink4a/Arf–null, H-RasV12-expressing glioma-initiating cells that differed in extracellular acidification potential. Metabolic characteristics of GSCs were determined by measurement of glucose, oxygen, and glutamine uptake, ATP content, and lactate production. Effects of metabolic inhibitors and changes in oxygen or nutrient availability on lactate production and tumorsphere growth were also determined.
Results
GSCs were found either to consume more glucose and produce more lactate or to consume more oxygen and maintain a higher ATP content depending on the metabolic characteristics of the tumor cells of origin. The latter, mitochondrial-type GSCs increased lactate production after treatment with the oxidative phosphorylation inhibitor oligomycin or phenformin. Exposure to hypoxia also increased lactate production and expression of glycolysis-related enzymes and metabolites in mitochondrial-type GSCs in a reversible manner.
Conclusions
Both glycolytic and mitochondrial-type energy production can sustain tumor propagation by isogenic GSCs. Whereas both phenotypes can be independent and stable, cells that rely on oxidative phosphorylation can also switch to a more glycolytic phenotype in response to metabolic stress, suggesting that plasticity is a further characteristic of GSC metabolism.
Keywords: glioma stem cell, hypoxia, metabolic heterogeneity, metabolic plasticity
Importance of the study
We found that even isogenic GSCs manifest heterogeneity of metabolic features, being able to satisfy their bioenergetic needs not only through aerobic glycolysis but also through oxidative phosphorylation. The metabolic preferences of these cells are further influenced by environmental factors such as hypoxia. Our results suggest that both metabolic phenotypes can coexist within a given tumor, and they therefore highlight the need for combinatorial metabolic targeting rather than targeting of a single energy pathway in glioblastoma.
For most types of malignant solid tumor, the quest for new, curative therapeutic approaches continues. Altered energy metabolism was recently recognized as a definitive characteristic of cancer cells,1 and drugs that target oncoenzymes or metabolites are already under development.2 As with any other therapy, the main challenge of targeted metabolic treatment will be the eradication of all cancer cells, including those equipped to survive changes in their environment and toxic interventions. Such cells, usually referred to as tumor-propagating cells, stem cells, or persisters, are defined by their ability to give rise to a number of progeny cells sufficient to sustain tumor growth, to overcome general cytotoxic therapies, and to eventually lead to tumor recurrence. They are present in many high-grade malignancies, including glioblastoma.3,4
Recent studies have begun to shed light on the metabolism of tumor-propagating cells for various tumor types. However, results are often contradictory, with either glycolysis or oxidative phosphorylation being identified as a main energy pathway for stemlike cells of the same tumor type.5 Glioma stem cells (GSCs) isolated from surgical specimens rely on glycolysis to a lesser extent than do their differentiated progeny.6 However, treatment with dichloroacetate to inhibit glycolysis and promote oxidative phosphorylation was found to reduce the viability and number of GSCs.7 Glycolytic activity was shown to be higher in the mesenchymal pool of GSCs than in proneural GSCs,8 linking metabolic characteristics to tumor subtype. Studies of the niche surrounding and protecting GSCs have also yielded conflicting results, revealing that perivascular, oxygen-rich regions9,10 as well as hypoxic regions11 can harbor GSCs. Together, these apparently discrepant findings suggest the existence of at least 2 different metabolic phenotypes for GSCs. It remains unclear, however, whether these characteristics are independent and stable or represent 2 opposite poles of a flexible phenotype.5
We have previously investigated the metabolic requirements of tumor cells during glioma initiation and found that both glycolytic and mitochondrial phenotypes are able to independently sustain the emergence of a primary tumor.12 However, malignant gliomas show a complex, branching clonal evolution,13 suggesting a dynamic remodeling in response to the environmental pressure. We therefore asked whether such selection would affect the metabolic phenotypes seen in initiating cells. Taking advantage of the fact that mouse models allow separate analysis of tumor initiation and propagation through their respective key populations, the initiating and stem cells, we have now examined whether the 2 metabolic phenotypes are also able to sustain tumor propagation and whether they are subject to adaptation in response to environmental conditions. We found that isogenic murine-induced GSCs are able to adopt either a glycolytic or mitochondrial phenotype and to stably propagate tumors in both states. Moreover, at least GSCs that rely on oxidative phosphorylation under standard culture conditions can reversibly switch between the 2 phenotypes, suggestive of metabolic plasticity.
Materials and Methods
Brain Tumor Stem Cells and Cell Culture
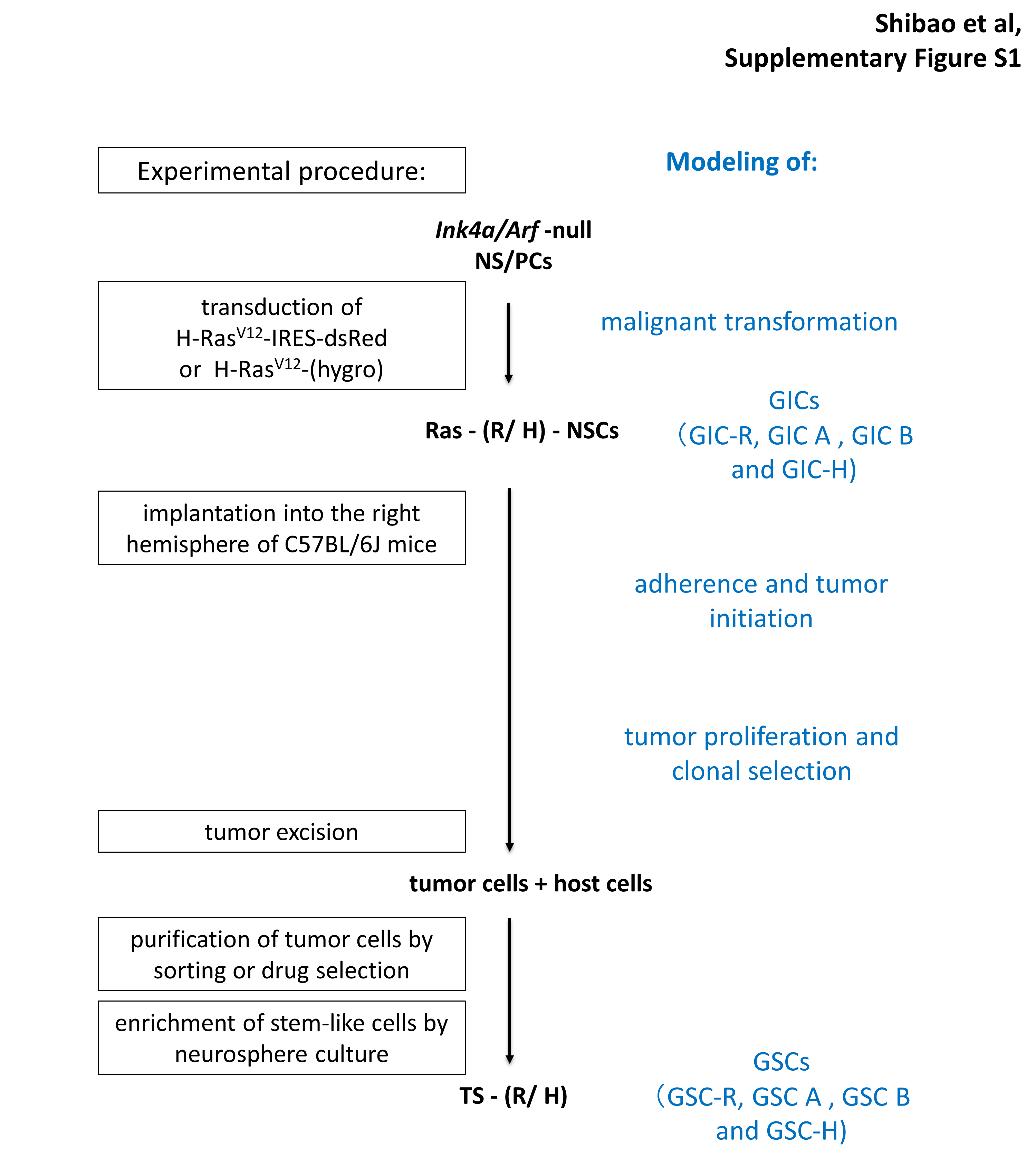
Murine Ink4a/Arf–null neural stem/progenitor cells expressing H-RasV12 and the fluorescent protein dsRed form glioma-like tumors with a 100% penetrance upon orthotopic implantation in syngeneic mice.12 These glioma-initiating cells (GICs) were termed GIC-R. GIC A and GIC B were established from GIC-R by single cell cloning.12 A second line of GICs was established by transducing Ink4a/Arf–null neural stem/progenitor cells with a construct expressing H-RasV12 and a hygromycin selection cassette and termed GIC-H. Tumors formed by orthotopic implantation of the 2 GIC lines were excised 14 days after implantation or at the onset of tumor-related symptoms. Dissociated tumor cells were purified by sorting for dsRed-positive cells or by selection with 200 µg/mL hygromycin for 14 days, and stem cells were enriched by culture in neurosphere medium (NSM) conditioned to support neurosphere growth.12 The resulting cells were termed GSC-R or GSC-H. GSC A and B are GIC A and B re-derived as described after one passage in vivo. For induction of differentiation, cells were incubated for 24 h in NSM supplemented with recombinant mouse bone morphogenetic protein 4 (BMP4) at 100 ng/mL (R&D Systems). All cells were used within 20 passages after establishment.
Orthotopic Implantation
All animal experiments were approved by the Keio University Animal Care and Use Committee. Orthotopic implantation of cells was performed as previously described.12 In brief, female C57BL/6J mice were anesthetized and placed into a stereotactic apparatus (David Kopf Instruments). A small hole was bored into the skull at 2.0 mm lateral to the bregma. Viable cells (1 × 105) in 2 μL of Hanks’ Balanced Salt Solution were injected into the right hemisphere 3 mm below the surface of the brain. Animals were monitored daily for the development of neurological deficits.
Sphere Growth Assay
Cells were manually plated in low-binding 96-well plates at a density of 100 cells (normoxia) or 1000 cells (hypoxia) per well. For one replicate, treatment groups were assayed in the same plate, with separate plates for normoxic and hypoxic conditions. Images were acquired with a Biorevo BZ9000 inverted microscope (Keyence) at 7 days after plating, and sphere area was quantified with Keyence Analysis Software. Relative sphere growth was calculated as average sphere area (treatment group)/average sphere area (control group). For analysis of sphere growth in medium without growth factors, cells were incubated in Dulbecco’s modified Eagle’s medium/F12 supplemented with B27.
Assay of Lactate and ATP
Cells were plated at a density of 5 × 105 to 1 × 106 per well in 12-well plates and cultured for 6 h (unless indicated otherwise), after which lactate concentration in the culture supernatants was measured with the use of a lactate analyzer (Lactate Pro 2, Arkray) and the ATP content of the cells was measured with the use of a CellTiter-Glo kit (Promega).
Measurement of Glucose Uptake
Glucose uptake was measured by flow cytometry with the use of the fluorescent glucose analog 2-[N-(7-nitrobenz-2-oxa-1,3-diaxol-4-yl) amino]-2-deoxyglucose (2-NBDG; Invitrogen).12 Cells were incubated in glucose-free NSM for 2 h at 37°C. After the addition of 2-NBDG to a final concentration of 0.1 mM, the cells were incubated for an additional 1 h and analyzed with an Attune cytometer (Applied Biosystems). Cells maintained on ice immediately after the addition of 2-NBDG were analyzed as controls.
Extracellular Flux Analysis
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were determined with a Seahorse XF Extracellular Flux Analyzer (Seahorse Bioscience). For evaluation of the effects of metabolic inhibitors, the concentration of each inhibitor was increased sequentially and measurements were taken for 3 time points at each concentration. Metabolic inhibitors included 2-deoxyglucose (2-DG, final concentration of 0 to 20 mM; Sigma), oligomycin (0 to 0.8 μM; Wako), and phenformin (0 to 0.4 mM; Sigma).
Immunoblot Analysis
Immunoblot analysis was performed as previously described.12 Antibodies included rabbit polyclonal antibodies to pyruvate kinase M2 (PKM2) (#3198, Cell Signaling Technology), phosphorylated PKM2 (#3827, Cell Signaling Technology), mitochondrial pyruvate dehydrogenase kinase 1 (PDK1) (ab90444, Abcam), and α-tubulin (sc-5546, Santa Cruz Biotechnology); rabbit monoclonal antibodies to lactate dehydrogenase (LDH) (ab134187, Abcam) and hexokinase 2 (HK2) (#2867, Cell Signaling Technology); and mouse monoclonal antibodies to β-actin (sc-47778, Santa Cruz Biotechnology).
Metabolome Analysis
Cells were collected by centrifugation and washed with 5% mannitol, treated with 800 μL of methanol and 550 μL of water containing internal standard 1 (Human Metabolome Technologies [HMT]), and incubated on ice for 10 min. The extracts were filtered through an Ultrafree MC-PLHCC filter unit (HMT) by centrifugation at 9100 × g, 4°C for 180 min. Concentrations of 116 metabolites were measured by HMT, according to methods previously described.14 Samples were prepared in triplicate and metabolite levels were normalized by cell number.
Statistical Analysis
All experiments were performed in triplicate unless indicated otherwise. Quantitative data are presented as means ± SD from representative experiments and were compared with the unpaired Student’s t-test (for 2 groups) or with one-way ANOVA followed by Tukey’s post-hoc test (for multiple groups). Survival curves were compared with the log-rank test. Statistical analysis was performed with GraphPad Prism. A P-value of <0.05 was considered statistically significant.
Results
Establishment and Characterization of Isogenic Mouse GSCs
To investigate the metabolic characteristics of GSCs during tumor propagation, we made use of our murine model based on Ink4a/Arf–null neural stem/progenitor cells (NS/PCs) that express the H-RasV12 oncoprotein and the fluorescent reporter dsRed12 or a hygromycin selection cassette. In this model, NS/PCs transduced with H-RasV12 and implanted into the forebrain of wild-type mice act as GICs. Moreover, cells isolated from the late stages of glioma propagation, which have interacted with the syngeneic environment and overcome different types of environmental pressure, can be sorted by fluorescence or by antibiotic selection and are subsequently cultured under stem cell culture conditions to enrich for a bona fide GSC population (Supplementary Figure S1).15 To examine the biological differences between GICs and GSCs, we first investigated the ability of these cells to expand as spheres, which is thought to reflect in vivo tumor propagation potential. Even in serum-free, growth factor–supplemented NS/PC culture medium (NSM), the growth of spheres formed by GSCs was significantly greater than that of those formed by GICs (Fig. 1A, B). When cultured in the absence of not only serum but also epidermal growth factor and basic fibroblast growth factor, the extent of sphere growth was markedly higher for GSCs than for GICs (Fig. 1A, B), suggestive of an increase in growth factor–independent expansion potential as well. This difference was most pronounced for the GIC-R/GSC-R populations, suggesting a stronger remodeling during the in vivo passage. Therefore, to investigate the metabolic characteristics of GSCs, we made use of the 2 clonal populations previously established from GIC-R, populations that differ in their extracellular acidification potential: GIC A, a population with a higher acidification capacity and overall glycolytic metabolism, and GIC B, with a higher mitochondrial reserve and greater reliance on oxidative phosphorylation.12 DsRed-positive cells sorted from tumors initiated by GIC A or GIC B cells were designated GSC A and GSC B, respectively.
Fig. 1.

Stem cell characteristics of GSC A and B populations. (A) Relative sphere growth for GIC-H and GSC-H incubated for 7 days in medium conditioned to support neurosphere growth or in medium without growth factors. *P < 0.05. (B) Relative sphere growth for GIC-R and GSC-R incubated for 7 days in medium conditioned to support neurosphere growth or in medium without growth factors. *P < 0.05. (C) Percentage sphere formation from single GSC A or GSC B cells. *P < 0.05. (D) Immunofluorescence staining (green fluorescence) for markers of undifferentiated cells (nestin), astrocytes (GFAP), or neurons (βIII-tubulin, as detected by Tuj1 antibody) in GSC A and B cells cultured in the presence of the indicated growth factors. Nuclei were counterstained with Hoechst 33342 (blue fluorescence). Scale bars, 50 μm. (E) Kaplan–Meier analysis of overall survival for wild-type mice subjected to orthotopic implantation of GSC A (n = 7) or GSC B cells (n = 6) (1 × 105). NS, not significant. (F) Hematoxylin-eosin staining and immunohistochemical staining of Ki67 of tumors formed by GSC A or B cells. The percentage of tumor cells positive for Ki67 is indicated. Scale bars, 100 μm. T, tumor; H, hemorrhage; N, necrosis.
Single cells from both GSC A and GSC B populations were able to form spheres (Fig. 1C), an ability thought to reflect in vitro self-renewal potential. Immunofluorescence analysis revealed that both types of cells were positive for the immature cell marker nestin under stem cell culture conditions and were able to differentiate into cells of the neuronal (βIII-tubulin positive) and astrocytic (glial fibrillary acidic protein [GFAP] positive) lineages on exposure to BMP4 (Fig. 1D).16 On implantation into the forebrain of wild-type mice, both GSC populations formed aggressive brain tumors with a penetrance of 100% (Fig. 1E). Histological examination revealed infiltration of the tumors into normal brain tissue, hemorrhage, and necrosis as well as a high proliferative index for tumors formed by either cell type (Fig. 1F). These results confirmed that both GSC A and GSC B cells possess stem cell characteristics, including sphere-forming ability, the ability to differentiate, and high tumorigenicity, with GSC A cells showing a slightly higher self-renewal ability in vitro.
Metabolic Characterization of GSCs
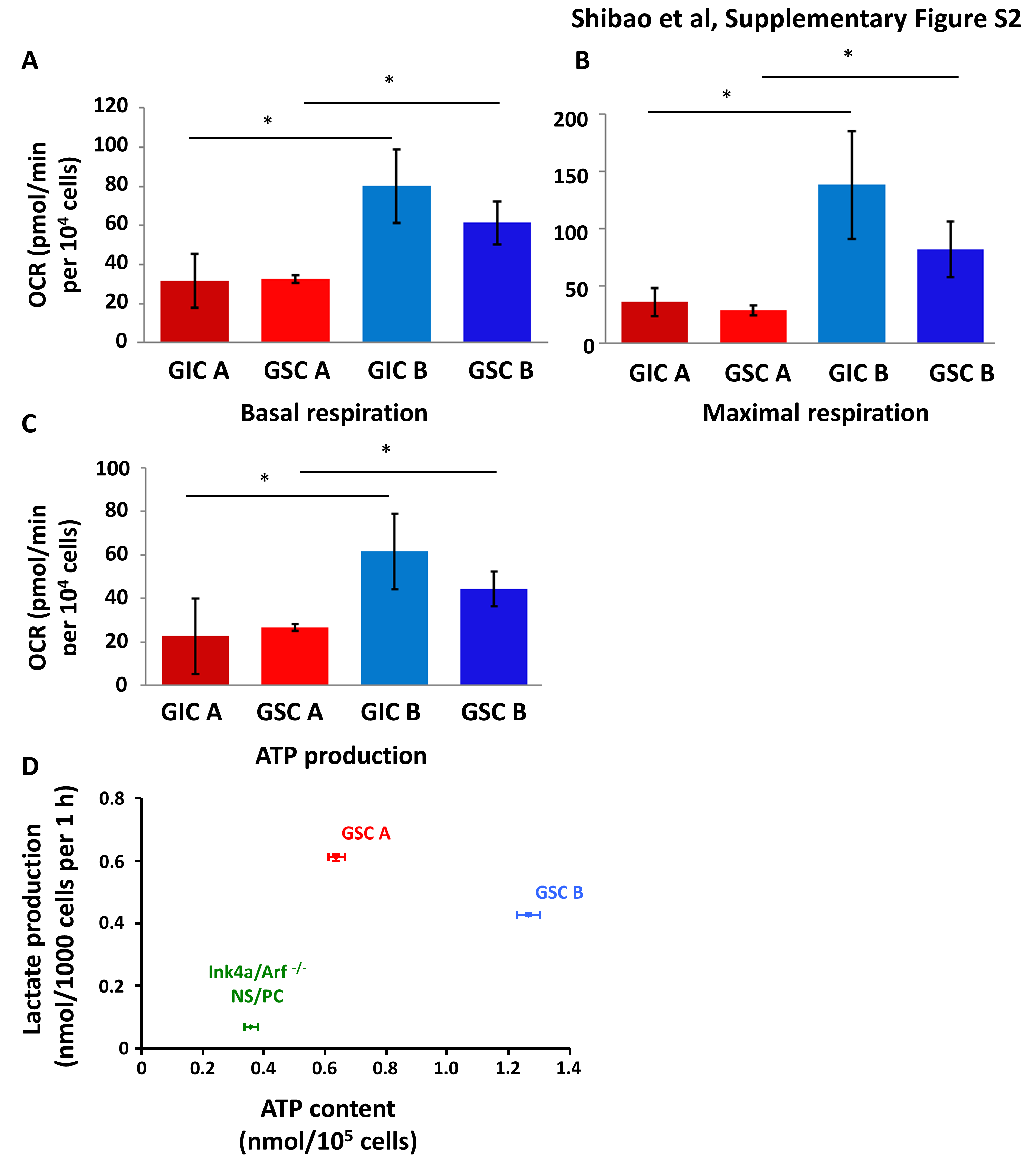
We next asked whether GSCs recapitulated the metabolic heterogeneity of GICs. GSC A cells, derived from tumors formed by glycolytic GIC A cells, showed a significantly higher uptake of the fluorescent glucose analog 2-NBDG compared with GSC B cells (Fig. 2A, B). GSC A cells also manifested a significantly higher lactate production compared with GSC B cells (Fig. 2C). In contrast, GSC B cells had higher basal respiration, maximal respiration, and ATP production, as determined by OCR measurements in extracellular flux analysis (Supplementary Figure S2A–C). Of note, the difference in these bioenergetic parameters between GSC A and GSC B cells was similar to the difference between GIC A and GIC B cells (Supplementary Figure S2A–C). GSC B cells also had a higher ATP content (Fig. 2D), consumed more glutamine (Fig. 2E), and produced more NH4+ (Fig. 2F) than did GSC A cells. Both types of GSCs showed a higher energy metabolism compared with the parental nontransformed Ink4a/Arf–null NS/PCs (Supplementary Fig. S2D).
Fig. 2.

Metabolic characteristics of GSC A and B cells. (A) Representative flow cytometric analysis of 2-NBDG uptake (solid traces), with the open traces corresponding to controls. (B) Quantification of 2-NBDG uptake as the normalized difference in mean fluorescence intensity (MFI) between experimental samples and controls. (C) Lactate production. (D) ATP content. (E) Glutamine uptake. (F) NH4+ production. (G) Immunoblot analysis of glycolysis-related enzymes in Ink4a/Arf–null NS/PCs and GSC-R cells as well as GSC A and B cells. β-Actin was examined as a loading control. *P < 0.05.
At the molecular level, the abundance of key glycolytic enzymes including HK2, PKM2 (phosphorylated and nonphosphorylated forms), PDK1, and LDH was greater in GSC A cells than in GSC B cells, a bulk population of dsRed-expressing GSCs (GSC-R), or nontransformed Ink4a/Arf–null NS/PCs (Fig. 2G), consistent with the metabolic profiles of GSC A and GSC B cells. The GSCs were thus found to recapitulate the metabolic characteristics and heterogeneity of the corresponding GICs.
Effects of Metabolic Inhibitors on GSCs
Given that targeting of energy pathways is being investigated for several refractive cancers,17,18 we next examined the effects of metabolic inhibitors on GSC A and B cells. ECAR, which is considered to be an indicator of glycolysis, was markedly attenuated in GSC A cells on exposure to the glucose analog 2-DG (Fig. 3A). OCR was not influenced by 2-DG in either cell population (Fig. 3B). In contrast, inhibition of mitochondrial ATP synthase by oligomycin19 or of complex I by phenformin20 resulted in a decrease in OCR for both clones (Fig. 3D, F), although this effect was more pronounced in GSC B cells. Oligomycin and phenformin also induced an increase in ECAR for GSC B cells (Fig. 3C, E), suggesting that GSC B cells have the potential to undergo glycolytic compensation after inhibition of oxidative phosphorylation.
Fig. 3.

Bioenergetic responses of GSC A and B cells to metabolic inhibitors. ECAR (A, C, E) and OCR (B, D, F) were measured by extracellular flux analysis for GSC A or B cells exposed to various concentrations of 2-DG (A, B), oligomycin (C, D), or phenformin (E, F). *P < 0.05 for the difference between GSC A and B cells at the indicated inhibitor concentrations.
Having determined the immediate responses of GSCs to the metabolic inhibitors 2-DG, oligomycin, and phenformin, we next examined the effects of longer-term treatment with these agents. Inhibition of glycolysis with 2-DG resulted in a significant decrease in lactate production by both GSC A and B cells, with a more pronounced decrease for GSC A (Fig. 4A) and a slight decrease in ATP content in both cells (Fig. 4B). 2-DG also inhibited the growth of spheres formed by GSC A cells to a greater extent than it did those formed by GSC B cells (Fig. 4C, Supplementary Figure S3A). In contrast, oligomycin and phenformin significantly increased lactate production for GSC B and significantly reduced the ATP content of both types of GSCs (Fig. 4D, E, G, H). Consistent with these results, inhibition of oxidative phosphorylation attenuated sphere growth to a greater extent for GSC B cells than for GSC A cells (Fig. 4F, I; Supplementary Figure S3B, C), although neither oligomycin nor phenformin completely inhibited the growth of spheres formed by GSC B cells. Together, these data suggest that inhibition of glycolysis effectively attenuates sphere growth for GSC A cells, whereas inhibition of oxidative phosphorylation attenuates that for GSC B cells to a greater extent. However, the effects of oligomycin and phenformin were also partially compensated for by increased glycolysis in GSC B cells.
Fig. 4.

Effects of metabolic inhibitors on GSC A and B cells. Lactate production by (A, D, G), ATP content of (B, E, H), and relative sphere growth for (C, F, I) GSC A or B cells exposed to the indicated concentrations of 2-DG (A–C), oligomycin (D–F), or phenformin (G–I) were measured. *P < 0.05.
Adaptation to Hypoxia and Metabolic Flexibility of GSCs
Given that inhibitors of oxidative phosphorylation induced a marked glycolytic compensation in GSC B cells, we next examined whether other conditions that limit oxygen consumption might have the same effect. Glioblastomas are known to have hypoxic regions,21 and hypoxic regions have been suggested to favor cancer stem cells22 and constitute a GSC niche.11 We therefore investigated whether hypoxia affects the metabolic characteristics of GSC B cells. Acute exposure to 1% O2 resulted in increased lactate production in both GSC A and B cells compared with that apparent under normoxic conditions, but the increase was more pronounced and started earlier in GSC B cells (Supplementary Figure S4A). ATP content was unaffected by hypoxia in GSC A cells, whereas it was slightly decreased in GSC B cells (Fig. 5A). These data suggest that GSC B cells adapt to hypoxia by switching to glycolysis within a matter of hours.
Fig. 5.

Effects of hypoxia on metabolism of GSC A and B cells. (A) ATP content of GSC A and B cells cultured for 3 h under normoxic or hypoxic conditions. (B) Lactate production by GSC A and B cells after culture for 4 days under normoxic or hypoxic conditions. (C) Real-time PCR analysis of Pkm2 and Ldha mRNA in GSC A and GSC B cells. P < 0.05. (D) Immunoblot analysis of glycolysis-related enzymes in GSC A and B cells cultured for 4 days under normoxic or hypoxic conditions. (E, F) Fold change in sphere area relative to untreated controls for GSC A or B cells in response to treatment with 5 mM 2-DG (E) or 0.1 mM phenformin (F) under normoxic or hypoxic conditions for 7 days. *P < 0.05.
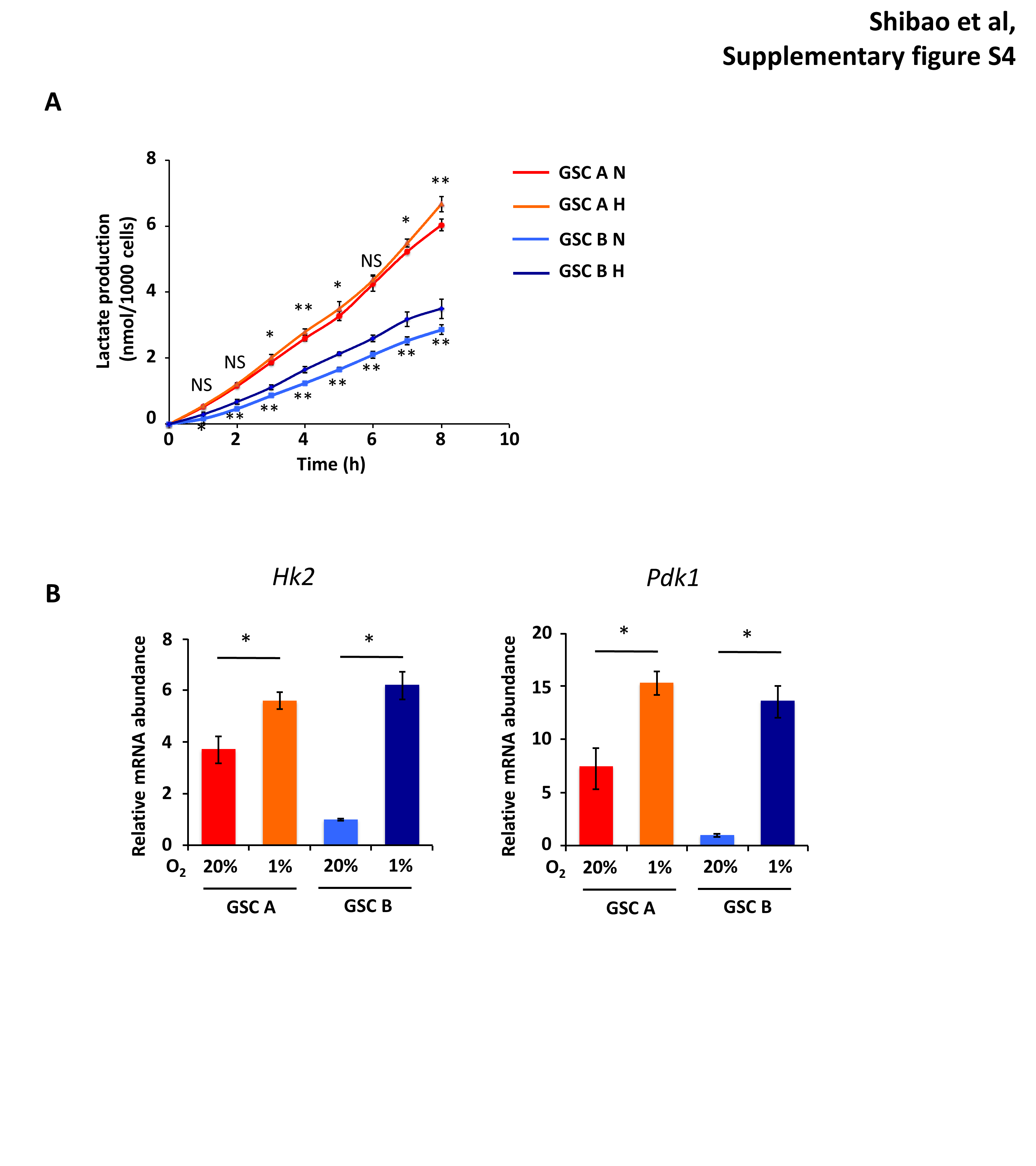
We next examined the effect of prolonged (4 days) exposure to hypoxia on GSC B cells. Whereas GSC A cells showed only a slight increase in lactate production, GSC B cells showed a pronounced increase in this parameter, achieving levels similar to those of GSC A cells (Fig. 5B). Analysis of transcript levels revealed a significant increase in abundance of Pkm2 and Ldha specific for GSC B (Fig. 5C), while Hk2 and Pdk1 were significantly increased in both clones, with a more marked increase for GSC B (Supplementary Figure S4B). Immunoblot analysis confirmed this tendency for protein levels of the glycolytic enzymes, with a pronounced upregulation of HK2, PKM2 (phosphorylated and nonphosphorylated forms), PDK1, and LDH in GSC B cells exposed to hypoxia for 4 days (Fig. 5D). While all treatments achieved statistically significant effects even in hypoxic conditions, the sensitivity of GSC B cells to metabolic inhibitors was markedly affected by hypoxia, with sphere growth being more sensitive to 2-DG (Fig. 5E) and less sensitive to phenformin (Fig. 5F). Of note, consistent with the slight increase in glycolysis in GSC A when exposed to hypoxia, sphere growth for GSC A cells also became more resistant to phenformin (Fig. 5F).
The hypoxia-induced increase in lactate production by GSC B cells was reversible, with values returning to baseline on return of the cells to the normoxic condition (Fig. 6A, Supplementary Figure S5). Hypoxia-induced upregulation of HK2, PKM2, PDK1, and LDH was also reversible (Fig. 6B). GSC A also showed a reversible change in abundance of glycolytic enzymes, which, however, did not result in a significant increase in lactic acid production (Fig. 6A, B). Analysis of the intracellular metabolites for each GSC cultured under normoxic, hypoxic, or alternating conditions, followed by principal component (PC) analysis, revealed a distinct clustering along the PC1 between GSC B and A in their basal conditions (Fig. 6C). Evaluation of factor loadings for the PC1 axis showed that several amino acids, including Thr, Val, Ile, Pro, as well as NAD+, had a high correlation with PC1 (Fig. 6C, Supplementary Table S1). Furthermore, the 2 cell types also displayed a slight separation along PC2 in their basal state. Exposure to hypoxia induced a reversible shift of GSC B toward the GSC A phenotype by the PC2 component, with a main contribution from several glycolysis and pentose phosphate pathway (PPP) intermediates (Fig. 6, Supplementary Table S1). Of note, both total adenylate and guanylate charges were similar between GSCs, irrespective of the oxygen concentration (Supplementary Fig. S5B).
Fig. 6.

Reversible adaptation of GSC B cell metabolism to hypoxia. (A) Lactate production by GSC A or B cells cultured under either normoxic (20% O2) or hypoxic (1% O2) conditions for 8 days or exposed to hypoxia for 4 days followed by normoxia for 4 days. **P < 0.01. (B) Immunoblot analysis of glycolysis-related enzymes in GSC A and B cells cultured as in (A). (C) Score plot of the principal component analysis (PCA) of intracellular metabolites for cells cultured as in (A). Percentage values represent the contribution rate of the first (PC1) and the second (PC2) principal components. Nx: normoxia, hx: hypoxia. (D) Schematic representation of metabolic heterogeneity and plasticity in GSCs. GSCgly and GSCmit indicate GSCs with predominantly glycolytic or mitochondrial-type energy metabolism, respectively.
Discussion
We have previously shown that Ink4a/Arf–null, H-RasV12-expressing murine GICs are heterogeneous with respect to their main energy source and are able to initiate tumor formation regardless of their metabolic status. In the present study, we asked whether glioma cells responsible for tumor propagation—continuous tumor growth and repopulation from a limited number of cells—are also heterogeneous and whether their metabolic features are influenced by environmental factors. GSCs established from tumors formed by cells that differ in extracellular acidification potential were found to rely predominantly on glycolysis or oxidative phosphorylation and to stably propagate tumors in each metabolic state. Furthermore, in our model, GSCs that rely on oxidative phosphorylation under homeostatic conditions were able to undergo a reversible shift in their metabolism toward a more glycolytic phenotype, suggestive of metabolic plasticity.
It might seem obvious that GSCs would recapitulate the corresponding GIC phenotype. However, whereas the terms “initiating” and “stemlike” are sometimes used interchangeably with regard to tumor cells, studies have shown that cells that initiate tumor formation (cells of origin) can differ from cells that propagate a tumor (bona fide cancer stem cells).23 Furthermore, even when these 2 types of cell are originally the same, environmental factors such as oxygen supply, nutrient availability, and the generation of reactive oxygen species might change the metabolic status of tumor stem cells.5 In addition, the presence of immune cells within a tumor can also affect tumor metabolism.24 The possibility that stem cells can ultimately adopt only 1 of the 2 types of metabolism—glycolytic or mitochondrial—would therefore not be inconsistent with these observations. Our data now provide evidence that even isogenic GSCs are heterogeneous, and while they are not completely dependent on either glycolysis or oxidative phosphorylation alone, they are able to adopt either as the predominant metabolic phenotype in an immunocompetent environment. This finding might explain apparently conflicting results regarding GSC metabolism. It also suggests the possibility that GSCs mainly dependent on glycolysis have a higher affinity for hypoxic niches, whereas GSCs that rely mainly on mitochondrial-type metabolism show a preference for perivascular regions. Further studies in this regard should also increase our understanding of GSC fate after anti-angiogenic treatments.
Our second major finding is that at least a subgroup of GSCs shows metabolic plasticity and that this plasticity affects the response to energy pathway inhibitors. Metabolic adaptation or reprogramming in cancer cells that survive treatment has been described for several tumor types.25,26 Furthermore, despite the stringent demands for oxygen in the normal brain, gliomas adapt to hypoxia and hypoxia actually promotes enrichment of GSCs.27 Our results highlight a further, less well-studied aspect of this adaptation, its reversibility (Fig. 6D). A subgroup of GSCs was found to switch reversibly between glycolysis and oxidative phosphorylation in response to changes in oxygen availability. Whereas hypoxia was the main environmental stress investigated in our study, it is plausible that adaptations to changes in nutrient availability are also dynamically regulated. The development of techniques for real-time tracking of metabolism in live tissue will likely refine our understanding of the extent of this plasticity, as well as its triggers and regulators. At present, the clinically relevant implication of the metabolic plasticity of GSCs is the possibility that both metabolic phenotypes coexist within a given tumor at any given time. This possibility suggests that caution is warranted with regard to the use of inhibitors of single energy pathways in glioblastoma as well as highlights the complexity of metabolic targeting.
In our model, the mitochondrial type of GSCs, GSC B cells, underwent a metabolic switch from oxidative phosphorylation to glycolysis after inhibition of oxidative phosphorylation, suggesting that these cells are able to adapt to metabolic stress. Indeed, the growth of spheres formed by GSC B cells could not be inhibited by >90% even at high concentrations of oligomycin or phenformin. Our results thus suggest that metabolic adaptation can result in resistance to drugs that target glycolysis or oxidative phosphorylation. One possible strategy to overcome the metabolic heterogeneity and plasticity of GSCs might be dual blockade of glycolysis and mitochondrial respiration. Indeed, in preclinical cancer models, such dual inhibition by 2-DG and metformin has been shown to be effective for suppression of tumor growth and metastasis.28 However, GSCs and progenitor cells have also been shown to tolerate concomitant inhibition of both pathways.6 We also found that mitochondrial-type GSCs showed higher intracellular concentration of several amino acids, including the aromatic amino acids phenylalanine, tryptophan, and tyrosine, as well as the branched amino acids leucine, isoleucine, and valine, compared with GSC A. This difference could be caused by differences in protein synthesis and degradation, abundance of AA transporters, or metabolizing enzymes. Given that several cancers have been shown to exhibit amino acid dependencies,29 a further investigation of the contributions of the individual amino acids to the GSC B phenotype might lead to the development of new combinatorial strategies.
Another strategy might be forced synchronization of the metabolic status of all cells and targeting of a key factor in the resulting phenotype. Mitochondrial monocarboxylate transporters (MCTs) have been shown to be important in the resistance of glycolytic tumor cells to phenformin, and genetic disruption of these transporters was found to lead to a decrease in cellular ATP content and cell death by metabolic catastrophe.30 Furthermore, MCT4 appears to be upregulated in GSCs exposed to hypoxia and to regulate the proliferation and survival of GSCs as well as the establishment and growth of xenografts formed by these cells.31 Of note, MCTs also play a key role in metabolic symbiosis, whereby lactate produced by one cell type is made use of as a fuel by another.32 Simultaneous inhibition of oxidative phosphorylation and MCTs might therefore represent a candidate strategy for combinatorial targeting.
Identification of the key regulators of metabolic plasticity in GSCs will be imperative for the development of antimetabolic strategies. In the present study, we determined several intracellular glycolysis and PPP intermediates that change significantly during the metabolic shift. While these changes likely represent the end result of this adaptation, in-depth functional studies of related enzymes could reveal potential key factors in glioma metabolic adaptation.
In conclusion, we have shown that even isogenic GSCs can be heterogeneous in their metabolic features, being able to satisfy their bioenergetic needs not only through aerobic glycolysis but also through oxidative phosphorylation. Their metabolic preferences are further influenced by environmental factors such as hypoxia. Our findings suggest that both metabolic phenotypes can coexist in a tumor, and they therefore highlight the need for combinatorial targeting, rather than targeting of individual energy pathways, in the treatment of glioblastoma.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This work was funded by the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Kakenhi grant no. 26861168 to O.S.); Project for Cancer Research and Therapeutic Evolution, Japan Agency for Medical Research and Development, AMED (to O.S.); Japan Society for the Promotion of Science (research fellowship to S.S.).
Conflict of interest statement. None.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank P. P. Pandolfi and T. Maeda for the pBabe-Hygro-H-RasV12 plasmid, I. Ishimatsu for preparing samples for histopathology, M. Sato for help in preparation of the manuscript, and the Collaborative Research Resources, School of Medicine, Keio University, for technical assistance.
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. [DOI] [PubMed] [Google Scholar]
- 2. Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10(9):671–684. [DOI] [PubMed] [Google Scholar]
- 3. Singh SK, Hawkins C, Clarke ID et al. . Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. [DOI] [PubMed] [Google Scholar]
- 4. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells—what challenges do they pose?Nat Rev Drug Discov. 2014;13(7):497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. 2016;18(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vlashi E, Lagadec C, Vergnes L et al. . Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci U S A. 2011;108(38):16062–16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michelakis ED, Sutendra G, Dromparis P et al. . Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2(31):31ra34. [DOI] [PubMed] [Google Scholar]
- 8. Mao P, Joshi K, Li J et al. . Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A. 2013;110(21):8644–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bao S, Wu Q, Sathornsumetee S et al. . Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66(16):7843–7848. [DOI] [PubMed] [Google Scholar]
- 10. Calabrese C, Poppleton H, Kocak M et al. . A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11(1):69–82. [DOI] [PubMed] [Google Scholar]
- 11. Li Z, Bao S, Wu Q et al. . Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15(6):501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saga I, Shibao S, Okubo J et al. . Integrated analysis identifies different metabolic signatures for tumor-initiating cells in a murine glioblastoma model. Neuro Oncol. 2014;16(8):1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim H, Zheng S, Amini SS et al. . Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015;25(3):316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soga T, Heiger DN. Amino acid analysis by capillary electrophoresis electrospray ionization mass spectrometry. Anal Chem. 2000;72(6):1236–1241. [DOI] [PubMed] [Google Scholar]
- 15. Sampetrean O, Saya H. Characteristics of glioma stem cells. Brain Tumor Pathol. 2013;30(4):209–214. [DOI] [PubMed] [Google Scholar]
- 16. Piccirillo SG, Reynolds BA, Zanetti N et al. . Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444(7120):761–765. [DOI] [PubMed] [Google Scholar]
- 17. Dunbar EM, Coats BS, Shroads AL et al. . Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs. 2014;32(3):452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elgogary A, Xu Q, Poore B et al. . Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci U S A. 2016;113(36):E5328–E5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roberton AM, Holloway CT, Knight IG, Beechey RB. A comparison of the effects of NNʹ-dicyclohexylcarbodi-imide, oligomycin A and aurovertin on enrgy-linked reactions in mitochondria and submitochondrial particles. Biochem J. 1968;108(3):445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 21. Rong Y, Durden DL, Van Meir EG, Brat DJ. ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65(6):529–539. [DOI] [PubMed] [Google Scholar]
- 22. Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129(3):465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim CF, Dirks PB. Cancer and stem cell biology: how tightly intertwined?Cell Stem Cell. 2008;3(2):147–150. [DOI] [PubMed] [Google Scholar]
- 24. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. 2015;43(3):435–449. [DOI] [PubMed] [Google Scholar]
- 25. Viale A, Pettazzoni P, Lyssiotis CA et al. . Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haq R, Shoag J, Andreu-Perez P et al. . Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013;23(3):302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Y, Zhou Y, Shingu T et al. . Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem. 2011;286(37):32843–32853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheong JH, Park ES, Liang J et al. . Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther. 2011;10(12):2350–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nagel R, Semenova EA, Berns A. Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep. 2016;17(11):1516–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marchiq I, Le Floch R, Roux D, Simon MP, Pouyssegur J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015;75(1):171–180. [DOI] [PubMed] [Google Scholar]
- 31. Lim KS, Lim KJ, Price AC, Orr BA, Eberhart CG, Bar EE. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene. 2014;33(35):4433–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tennant DA, Durán RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10(4):267–277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.