

Abstract
Background
O6-methylguanine-DNA-methyltransferase (MGMT) promoter methylation status is a predictive biomarker in glioblastoma. We investigated whether this marker furthermore defines a molecularly distinct tumor subtype with clinically different outcome.
Methods
We analyzed copy number variation (CNV) and methylation profiles of 1095 primary and 92 progressive isocitrate dehydrogenase wildtype glioblastomas, including paired samples from 49 patients. DNA mutation data from 182 glioblastoma samples of The Cancer Genome Atlas (TCGA) and RNA expression from 107 TCGA and 55 Chinese Glioma Genome Atlas samples were analyzed.
Results
Among untreated glioblastomas, MGMT promoter methylated (mMGMT) and unmethylated (uMGMT) tumors did not show different CNV or specific gene mutations, but a higher mutation count in mMGMT tumors. We identified 3 methylation clusters. Cluster 1 showed the highest average methylation and was enriched for mMGMT tumors. Seventeen genes including gastrulation brain homeobox 2 (GBX2) were found to be hypermethylated and downregulated on the mRNA level in mMGMT tumors. In progressive glioblastomas, platelet derived growth factor receptor alpha (PDGFRA) and GLI2 amplifications were enriched in mMGMT tumors. Methylated MGMT tumors gain PDGFRA amplification of PDGFRA, whereas uMGMT tumors with amplified PDGFRA frequently lose this amplification upon progression. Glioblastoma patients surviving <6 months and with mMGMT harbored less frequent epidermal growth factor receptor (EGFR) amplifications, more frequent TP53 mutations, and a higher tumor necrosis factor–nuclear factor-kappaB (TNF-NFκB) pathway activation compared with patients surviving >12 months.
Conclusions
MGMT promoter methylation status does not define a molecularly distinct glioblastoma subpopulation among untreated tumors. Progressive mMGMT glioblastomas and mMGMT tumors of patients with short survival tend to have more unfavorable molecular profiles.
Keywords: glioblastoma biomarker, NFκB, O6-methylguanine-DNA-methyltransferase (MGMT), PDGFRA, TERT
Importance of the study
This study compares more than 1200 glioblastomas from 3 sources for differences in methylation, CNV, mutation, and RNA expression according to their MGMT promoter methylation status. There was an uneven distribution of MGMT promoter methylation in 3 defined methylation clusters, despite otherwise similar molecular profiles. The study is of major relevance for upcoming clinical trials that will stratify and include patients with newly diagnosed glioblastoma according to their MGMT promoter methylation status and frequently withhold temozolomide in the experimental study arms. Differential analysis of primary and progressive tumors revealed differences upon progression, including activation of the TNF-NFκB pathway and CNV changes in mMGMT tumors that are distinct from uMGMT tumors.
O6-methylguanine-DNA-methyltransferase (MGMT) promoter methylation status has been consistently identified and used as a predictive biomarker for response to alkylating chemotherapy in patients with glioblastoma.1–3 MGMT is a protein that repairs damage induced by alkylating chemotherapies, including temozolomide. Methylated MGMT (mMGMT) promoter leads to reduced MGMT expression.4 Especially patients with wildtype isocitrate dehydrogenase 1/2 genes (IDH1/2) and an unmethylated MGMT (uMGMT) promoter show minimal response to chemotherapy, with a median survival of just over one year when treated with radiation and chemo after maximal safe resection.5
The concept of trials with replacement of temozolomide in the experimental arm in favor of a combination of an experimental systemic therapy with radiotherapy has been successfully established.6–9 Preclinical data do not suggest an impact of MGMT on radiotherapy, but other treatments have not been systematically evaluated.10 The German Neuro-Oncology Society NOA-04 trial provided a clinical basis for MGMT promoter methylation status being predictive also in IDH wildtype anaplastic gliomas,11,12 as did NOA-08 for elderly patients with glioblastoma.13
In the present study, we aimed to evaluate the molecular differences between glioblastomas with a methylated or unmethylated MGMT promoter in different datasets.
Methods
Glioblastoma Patient Cohorts
As of August 2, 2016, we screened the Heidelberg 450k methylation array database. We identified 1028 glioblastoma samples at diagnosis and 66 glioblastoma samples at tumor relapse (see Supplementary Methods) with IDH wildtype. MGMT promoter methylation status was determined by the algorithm of Bady et al.14 Samples were classified as “unsure” and excluded from the analysis if the confidence interval of the prediction included the cutoff value of 0.358. Patients provided informed consent concerning the use of their tissue samples for research purposes. The study was approved by the local ethics committee (#206/2005). Of the 1028 primary tumors, 143 with clinical trial grade follow-up data and treatment according to the present standard of care were accessible for survival analysis.
Illumina 450k Array Platform Analysis
The Illumina Infinium HumanMethylation450 (450k) array was used to obtain the DNA methylation status at 482421 cytosine-phosphate-guanine (CpG) sites at the Genomics and Proteomics Core Facility of the German Cancer Research Center (DKFZ) (see Supplementary Methods).15
Copy Number Variation Analysis
Copy numbers of single genes were assessed from 450k array data (details in the Supplement).
The Cancer Genome Atlas Data Analysis
Data from The Cancer Genome Atlas (TCGA) of 261 glioblastoma specimens at diagnosis were downloaded from the cBio portal (www.cbioportal.org)16,17 and from Firebrowse (Broad institute, http://firebrowse.org)18 on September 12, 2016 (Supplementary Table S1). Only tumor samples with IDH1/2 wildtype and defined MGMT promoter methylation status were used for analysis. Clinical data, copy number variations (CNVs), and mutation and expression subtype according to the classification of Verhaak et al19 were downloaded from the cBio portal. Methylation cluster types of Ceccarelli et al20 and Sturm et al21 were obtained from the original paper. RNA sequencing (RNAseq) V3 normalized data were downloaded from Firebrowse and available for 107 samples meeting the above-mentioned criteria. Data from 450k array methylation were downloaded from the data portal of TCGA (https://portal.gdc.cancer.gov/) and available for 67 of the above-mentioned tumor samples.
Chinese Glioma Genome Atlas Data Analysis
Messenger RNA microarray data from the Chinese Glioma Genome Atlas (CGGA) of diffuse gliomas22 including clinical information were downloaded from the CGGA website (http://www.cgga.org.cn) on October 25, 2016. Tumors with IDH mutations were excluded. For analysis of short and long surviving patients, only samples of patients with survival times <6 months and >12 months of altogether 55 patients were used (Supplementary Table S1).
Messenger RNA Data and Ingenuity Pathway Analysis
RNAseq data from TCGA samples were downloaded from firebrowse.org as described above. We only used samples with available RNAseq data and IDH wildtype as well as determined MGMT promoter methylation status (n = 107) (details in the Supplement).
Gene Set Enrichment Analysis
GSEA was performed with RNAseq data from TCGA samples and microarray data from CGGA samples downloaded as described above from firebrowse.org and cgga.org.cn (see Supplement).
Cluster Analysis
Consensus clustering using k-means was performed with the 10000 most variable methylation positions of the indicated dataset. Clusters were calculated with the R extension package “ConsensusClusterPlus”23 version 1.38.0. Euclidean distance was used for distance measure. The maximum number of evaluated clusters was k = 8.
Statistical Analysis and Graphics
Univariate survival analyses were performed using the Kaplan–Meier estimator and the log-rank test using Sigmaplot 12.5 software (Systat Software). Fisher’s exact test was used for significance testing for experiments with a 2 × 2 matrix. Values of P < 0.05 were considered significant and asterisked. Multiple testing was corrected using the Benjamini–Hochberg procedure. Circos plots were generated using R version 3.3.2 with the extension package “OmicCircos.”24 Heatmaps and all further graphics were generated using the extension packages “gplots” and “ggplot2.”
Results
Differences in the Molecular Profile of mMGMT and uMGMT Tumors at Diagnosis
There were 435/1028 (42%) mMGMT and 593 (58%) uMGMT samples. Patients with mMGMT promoter in the Heidelberg cohort had a better outcome (P = 0.001; Fig. 1A). In the cohort from TCGA, survival analysis of patients who received chemotherapy showed a better outcome in the mMGMT tumor subgroup, but no difference in patients not receiving chemotherapy (P = 0.008, P = 0.001, and P = 0.94, respectively; Fig. 1A, B).
Fig. 1.

Copy number variations in primary glioblastoma according to MGMT promoter methylation. (A) Left panel: overall survival of 143 glioblastoma patients with IDH1 wildtype tumors according to MGMT promoter methylation. Right panel: survival analysis of patients with tumors from the dataset from TCGA regardless of the treatment. (B) Left panel: survival analysis of tumors of the TCGA glioblastoma database not receiving chemotherapy according to MGMT promoter methylation. Right panel: survival analysis of the dataset from TCGA treated with chemotherapy. (C) Graphical illustration of the percentage of CNV in the mMGMT and uMGMT groups. (D) Relative fractions of DNA copy number variations detected in the database from TCGA according to MGMT promoter methylation. Shown are the means ± standard deviation. (E) Average fraction of mutations detected in the dataset from TCGA according to MGMT promoter methylation. Shown are the means ± standard deviation.
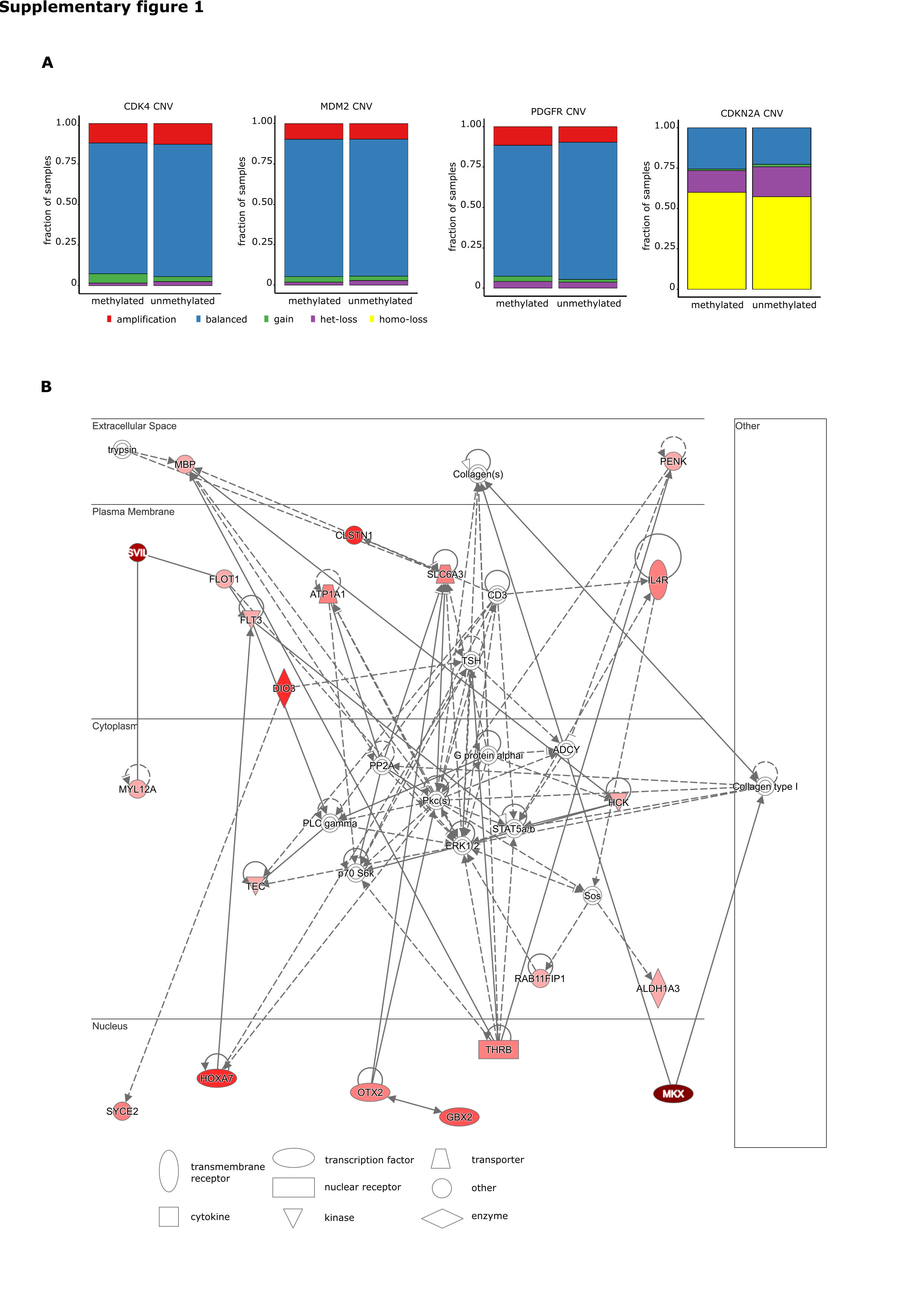
No significant differences in cyclin dependent kinase 4 (CDK4), mouse double minute human homolog (MDM2), cyclin dependent kinase inhibitor 2A (CDKN2A), and platelet derived growth factor alpha (PDGFRA) were found when stratifying for MGMT promoter methylation status (Supplementary Figure S1A).
There were no differences between mMGMT and uMGMT tumors in the general DNA copy number profile (Fig. 1C). The percentage of copy number altered genomes between mMGMT and uMGMT tumors did not differ in the dataset from TCGA (Fig. 1D).
In TCGA,25 a higher overall number of mutations was reported for mMGMT tumors. The absolute number of mutations was higher in mMGMT tumors in the current TCGA cohort (46 vs 38, P < 0.001; Fig. 1E). None of the mutation frequencies of any single gene differed significantly between mMGMT and uMGMT tumors, but there was a trend toward a higher frequency of phosphatase and tensin homolog (PTEN) mutations in the mMGMT group (31.4% vs 18.5%, P = 0.071). Telomerase reverse transcriptase (TERT) promoter mutations and BRAF mutations were analyzed in a large dataset by Arita el al.26 After exclusion of IDH mutant and lower-grade tumors, no differences in TERT promoter mutations and BRAF mutation frequencies were found according to MGMT promoter methylation (TERT 57.9% vs 57.8% and BRAF 2.8% vs 2.5%, for mMGMT and uMGMT, respectively).
After excluding CpGs in proximity to the MGMT gene, we found 2024 differentially methylated positions (DMPs) with an adjusted P-value ≤0.001 and 438 differentially methylated regions (DMRs) between mMGMT and uMGMT tumors in our 450k array datasets from TCGA (Fig. 3A). Filtering by including only CpGs within the promoter region of a gene identified 814 DMPs in 419 different genes (Supplementary Table S2). All but 3 of these positions were hypermethylated in mMGMT tumors. GSEA of differentially methylated positions and regions revealed that the top molecular functions of genes in hypermethylated regions in mMGMT tumors involve transcription regulation and DNA binding (Supplementary Table S3). Applying a more stringent gene selection, we included genes with at least 3 differentially methylated CpGs in one DMR. Ingenuity Pathway Analysis (IPA) of the 87 genes matching these criteria found the highest scoring network associated with the functions cell death and survival (Supplementary Figure S1B).
Fig. 3.

IDH wildtype glioblastoma cluster into 3 different clusters. (A) Heatmaps of 2024 positions that are differentially methylated between mMGMT and uMGMT tumors in the dataset of primary tumors, excluding MGMT related and intergenomic positions. (B) Consensus clustering using k-means of all samples of the primary tumor dataset. Shown is the result for k = 3. (C) Plot showing the relative change in area under the cumulative distribution function (CDF) curve in the k-means unsupervised clustering of primary glioblastoma. (D) CDF plot with examples from k = 2 to k = 8. Three clusters were chosen as the optimal number. (E) Distribution of mMGMT and uMGMT primary tumors into the clusters 1, 2, and 3. (F) Comparison of the cluster assignments with the clusters from Ceccarelli et al20 and Sturm et al21 for 65 glioblastoma samples of the database from TCGA. (G) Mean methylation beta-values of the 2024 differentially methylated CpGs, (H) relative TERT expression, and (I) TNF-NFκB score according to methylation clusters. The lower and the upper hinges correspond to the first and third quartiles. The upper and lower ends of the whiskers correspond to the 1.5× interquartile range from the hinge.
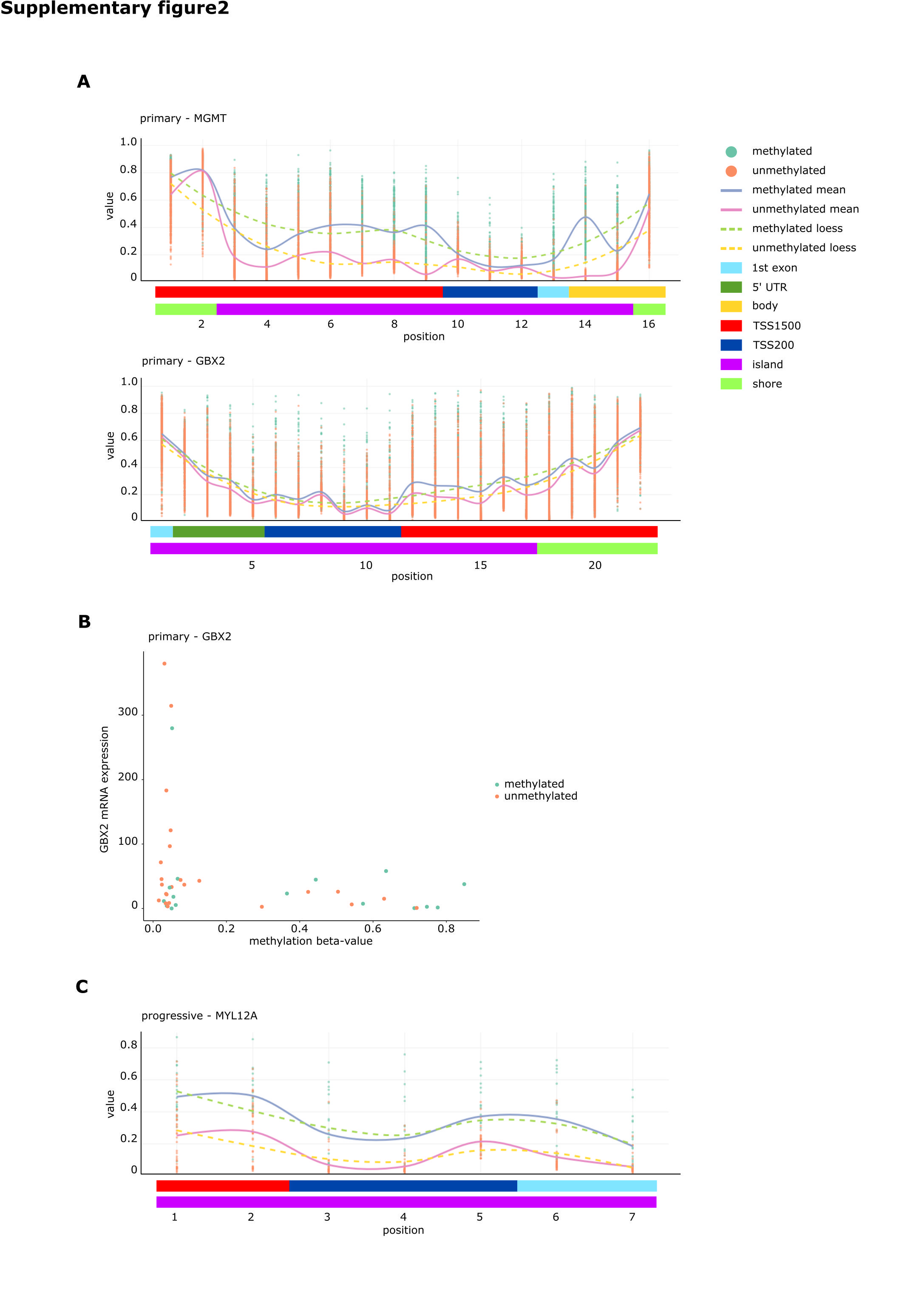
Gastrulation brain homeobox 2 (GBX2), a transcription factor known to stimulate proliferation of prostate cancer cells,27 was hypermethylated at 5 CpGs in the transcription start site of the methylation dataset (adjusted P-values = 3.7 × 10−6 to 6.5 × 10−7; Supplementary Figure S2A) in the mMGMT group and furthermore more than 2-fold downregulated on the RNA level in the mMGMT group in the dataset from TCGA, suggesting transcriptional repression through promoter methylation in mMGMT tumors (Fig. 2). Direct comparison in 40 samples from TCGA with methylation and RNAseq data revealed relevant GBX2 expression only in samples with low methylation of GBX2, and these tumors were predominantly uMGMT (Supplementary Figure S2B). In addition to GBX2, 16 genes were hypermethylated at a minimum of 3 CpGs in the combined methylation analysis and downregulated in the RNAseq dataset of TCGA (Fig. 2, Supplementary Table S2).
Fig. 2.

Differential methylation in primary glioblastoma according to MGMT promoter methylation. From outside to inside of the circle: First circle: hypermethylated genes in mMGMT tumors according to their chromosome position. Genes that were additionally downregulated in the RNAseq TCGA dataset are labeled in black, others in light red. Second circle: all DMPs within a promoter region of a gene. Third circle: area of each DMR between mMGMT and uMGMT glioblastomas. Fourth circle: all DMPs, including MGMT related on chromosome 10. Center: GBX2 mRNA expression in the dataset from TCGA in mMGMT and uMGMT glioblastomas.
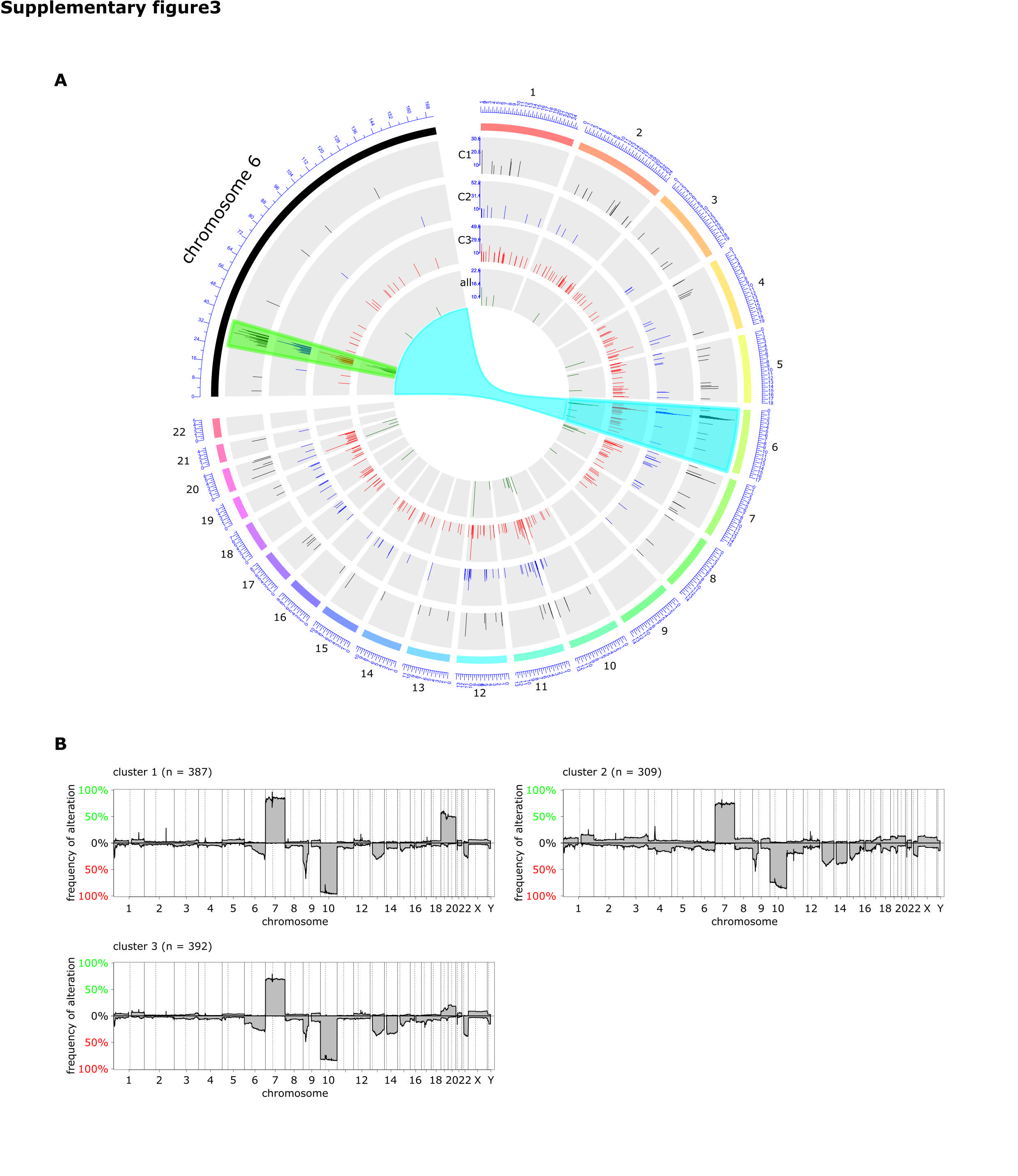
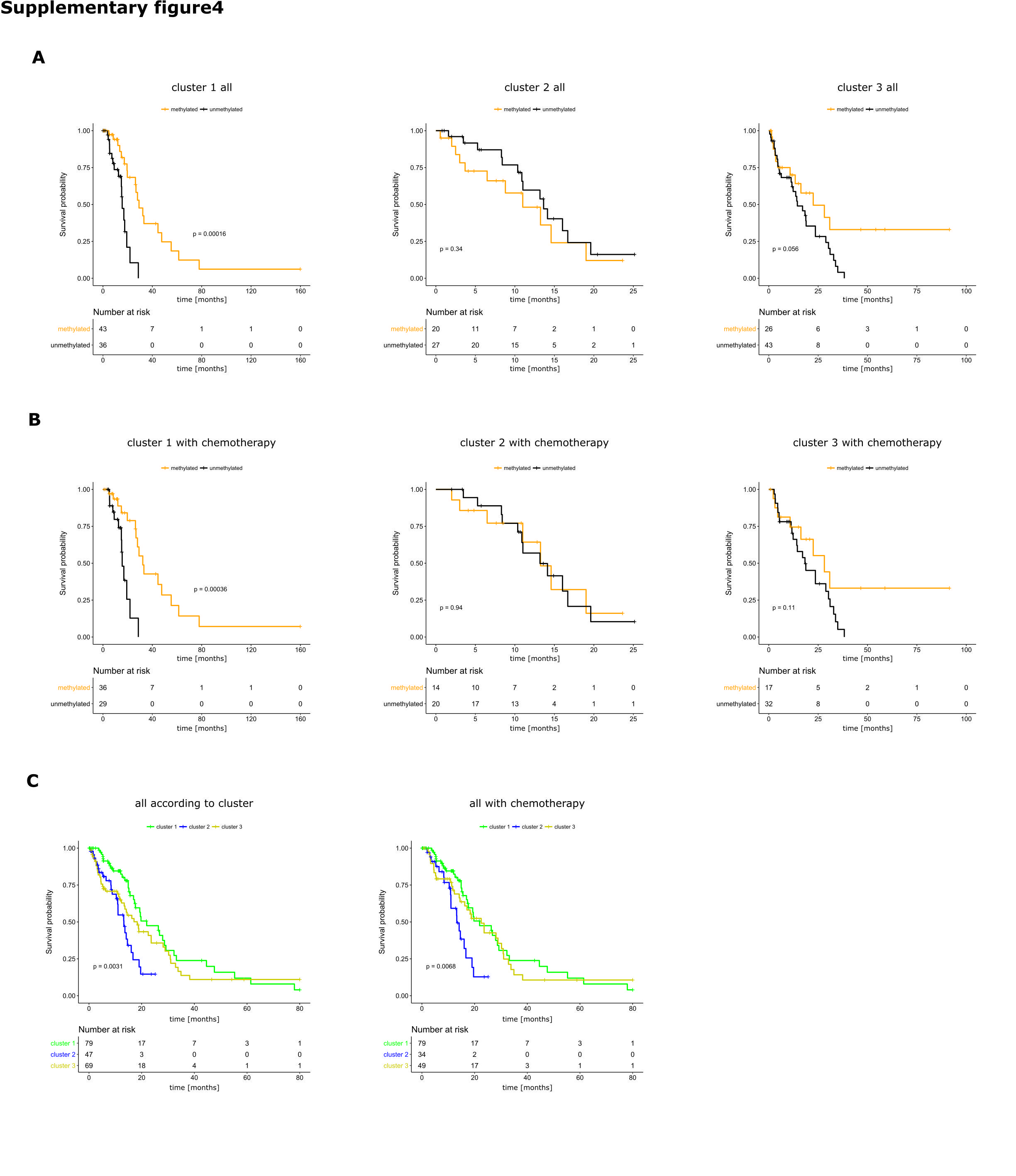
Consensus clustering using k-means of all samples revealed 3 different subgroups (Fig. 3A–D). Methylated MGMT tumors were overrepresented in cluster 1 (44% vs 31%, P = 0.0001) and underrepresented in cluster 3 (28% vs 40%, P = 0.0001; Fig. 3E). Comparison of tumors from TCGA included in the analyses by Ceccarelli et al20 and Sturm et al21 revealed that cluster 1 contains mainly the “LGm4” tumors of Ceccarelli et al20 and the “RTKII classic” tumors of the Sturm et al classification21 (Fig. 3F). Tumor samples in cluster 1 showed higher mean methylation beta-values of differentially methylated CpGs between mMGMT and uMGMT tumors than tumors in clusters 2 and 3 (P = 2 × 10−92 and P = 2 × 10−186; Fig. 3G). A hotspot of DMRs on chromosome 6p21-6p22 is described in the Supplement. Age at diagnosis was similar between the 3 clusters (cluster 1: 65.5 y, cluster 2: 62.7 y, cluster 3: 61.7 y; P-values: cluster 1 vs 2: 0.09, cluster 1 vs 3: 0.05, cluster 2 vs 3: 0.65). The survival difference between patients with mMGMT and uMGMT tumors was largest in patients with tumors of cluster 1 (P < 0.001 for cluster 1; Supplementary Figure S4A), and no significant survival difference according to MGMT promoter methylation was found for clusters 2 and 3. Chemotherapy was given more often in patients of cluster 1, but survival curves were similar when including only patients who received chemotherapy (Supplementary Figure S4B). Overall, patients of cluster 2 showed worse survival (Supplementary Figure S4C). Of note, Nguyen et al28 suggested a survival benefit of patients with mMGMT only in TERT promoter mutated tumors. As TERT promoter mutation increases TERT expression, we analyzed the tumors in the different clusters with available RNAseq data. Tumors of cluster 1 showed considerably higher TERT expression levels than tumors of clusters 2 and 3 (P-values: cluster 1 vs 2: 0.021, cluster 1 vs 3: 0.010; Fig. 3H). Conversely, the tumor necrosis factor (TNF)–nuclear factor-kappaB (NFκB) score was highly enriched in tumors of cluster 3 (P-values: cluster 1 vs 3: 0.007, cluster 2 vs 3: 0.002; Fig. 3I).
MGMT showed the lowest P-value for downregulation of all recorded genes in the dataset from TCGA (P = 3.9 × 10–13, fold change 0.38 for methylated vs unmethylated). Applying P-value for difference of P < 0.05 and a fold change of >2 by RNAseq analysis, there were 175 differentially regulated genes.
Analysis of Paired Tumor Samples
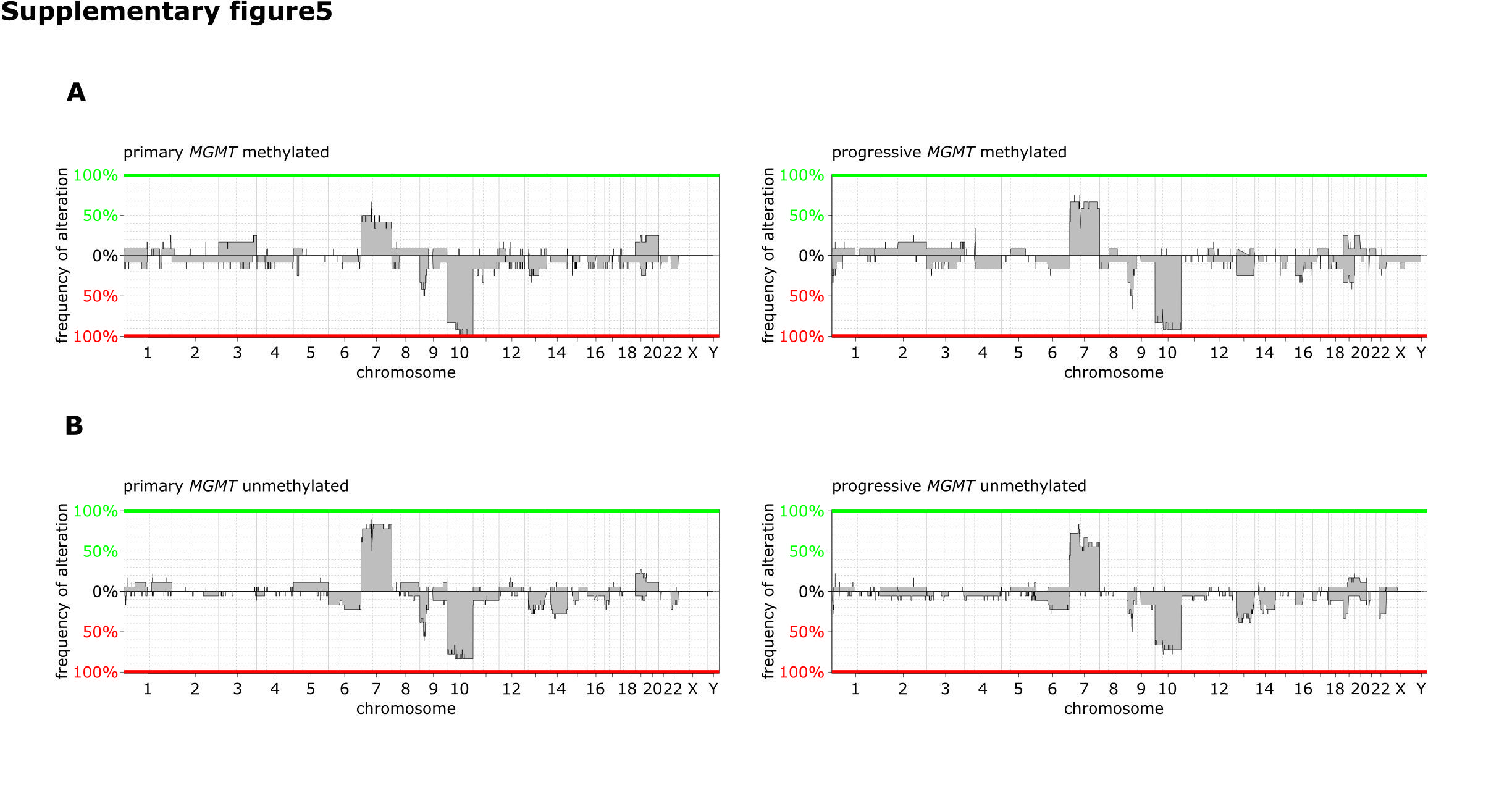
Twenty (48%) of paired tumor samples showed MGMT promoter methylation, whereas 29 (52%) did not (Fig. 4A). Only samples with the same MGMT promoter methylation status at diagnosis and progression were included for paired analysis. Chemoradiotherapy was received by 42/49 (86%) patients, 3 patients received chemotherapy alone, and 4 received radiotherapy alone (Supplementary Table S4). Three of the 20 (15%) mMGMT tumors acquired PDGFRA amplification or gain at progression, whereas none of the uMGMT tumors did. Instead, 3 progressive uMGMT tumors lost their PDGFRA amplification (Fig. 4A, B). The uMGMT tumor 19 is shown (Fig. 4C upper panel) as an example of an acquired loss of PDGFRA and an acquired gain of CDKN2A gene dosage at progression that occurred frequently only in uMGMT tumors (21% vs 0%, P = 0.031; Fig. 4A, B). Loss of the CDKN2A deletion in uMGMT tumor 16 was accompanied by acquired CDK4 amplification upon progression (Fig. 4C middle panel). Methylated MGMT tumor 15 is an example of loss of CDKN2A upon progression paralleled by loss of preexisting CDK4 and MDM2 amplifications (Fig. 4C lower panel). No major differences were observed in the global CNV profiles of paired tumor samples according to MGMT promoter methylation status (Supplementary Figure S5A, B).
Fig. 4.

CNV changes in pairs of primary and progressive glioblastoma. (A) CNV of the genes CDK4, CDKN2A, MDM2, and PDGFRA in a set of 49 paired glioblastoma samples at primary diagnosis and progression. MGMT promoter methylation is indicated in the first row. (B) Shift in gene dosages of CDK4, CDKN2A, MDM2, and PDGFRA in paired samples of primary and progressive glioblastoma according to MGMT promoter methylation. (C) Examples of copy number profiles of 3 paired primary and progressive glioblastoma samples. Color code: PDGFRA = red, CDKN2A = yellow, CDK4 = blue, MDM2 = green.
The Frequency of PDGFRA, CDK4, and MDM2 Amplification in Progressive Glioblastomas Differs According to MGMT Promoter Methylation
In 85 samples of progressive glioblastoma (including 66 samples from the Heidelberg database, 13 samples from TCGA patients, and 6 samples from Wang et al29), CNV of CDK4, MDM2, PDGFRA, and CDKN2A was assessed (Fig. 5A). Of these tumors, 32 (38%) were MGMT promoter methylated and 53 (62%) were unmethylated. Amplification or gain of the PDGFRA gene was found in 22% of the mMGMT tumors and 7.5% of uMGMT samples among progressive tumors (P = 0.092; Fig. 5B right panel). Furthermore, amplification or gain of CDK4 and MDM2 was found more frequently in progressive uMGMT tumors compared with mMGMT tumors (3.1% vs 18.9%, P = 0.046 and 0% vs 13.2%, P = 0.042, respectively; Fig. 5B left and middle panels). When compared with newly diagnosed glioblastomas, the frequency of CDK4 or MDM2 amplifications was significantly lower in mMGMT tumors in the progressive setting but was comparable in uMGMT tumors (Supplementary Figure S6). In progressive tumors with 450k array data available, we analyzed a set of 19 frequently altered genes in glioblastoma. Besides the differences in CDK4, MDM2, and PDGFRA amplifications described above, we found a gain or amplification of the C19MC microRNA cluster and the GLI2 gene more often in mMGMT compared with uMGMT tumors (54% vs 22% and 14% vs 0%, respectively). A higher percentage of myeloblastosis (MYB) deletions was detected in uMGMT tumors (36% vs 9%; Fig. 5C). The higher rate of C19MC amplifications in the mMGMT group was accompanied by a trend toward a higher amplification rate of the whole chromosome 19 (Fig. 5D).
Fig. 5.

Molecular differences between uMGMT and mMGMT progressive glioblastomas. (A) CNV of CDK4, MDM2, PDGFRA, and CDKN2A in the cohort of the progressive tumors, grouped according to MGMT promoter methylation. (B) Comparison of CDK4, MDM2, and PDGFRA copy number variations between mMGMT and uMGMT tumors in the progressive situation. (C) Heatmap of CNVs of a set of 19 frequently altered genes in the progressive tumor collective. High frequencies of alterations are shown in red, low frequencies in blue. MGMT promoter methylation is indicated in the first row. (D) Copy number variation profiles in mMGMT and uMGMT tumors in the progressive situation.
Principal component analysis of progressive tumors in the 450k methylation array dataset did not separate tumors according to MGMT status (Supplementary Figure S7A). Applying the same clustering algorithm as we did for primary tumors, we identified 3 different methylation groups. Methylated MGMT tumors were overrepresented in cluster 1 (57% vs 20%, P = 0.005) and underrepresented in cluster 3 (10% vs 43%, P = 0.009; Supplementary Figure S7B–E).
We found 346 DMPs and 182 DMRs between progressive mMGMT and uMGMT tumors (Supplementary Figure S7F). After filtering for positions within the promoter region of a gene, we identified 196 positions in 152 genes that were differentially regulated between both groups. These positions were all hypermethylated in mMGMT tumors. GSEA did not show significantly enriched molecular functions in both groups.
Twenty-one of the 152 genes (14%) had at least one DMP in the dataset of primary tumors. Of these, within the promoter region of the myosin light chain (MYL12A), which may be involved in DNA damage repair,30 we identified 5 positions that were hypermethylated in progressive mMGMT tumors (Supplementary Figure S2C). Three of exactly these 5 positions were already found to be hypermethylated in the dataset of primary tumors.
Comparison of Patients with Short and Long Survival Revealed Differentially Regulated Pathways
We created groups within the dataset from TCGA containing samples of patients who survived for either less than 6 months (mMGMT: n = 31, uMGMT: n = 34) or more than 12 months (mMGMT: n = 43, uMGMT: n = 44) after primary diagnosis. Within both the mMGMT and uMGMT groups, patients with a short survival time less frequently received alkylating chemotherapy (57% vs 88% in the methylated group and 47% vs 86% in the unmethylated group) and were older at diagnosis (67.9 y vs 56.8 y in the methylated and 65.7 y vs 60.0 y in the unmethylated group; Supplementary Table S5). Amplification of the epidermal growth factor receptor gene (EGFR) was less frequent in tumors with a methylated MGMT promoter in patients with death within 6 months after initial diagnosis compared with those who lived longer than 12 months or had an unmethylated MGMT promoter or both (29% vs 50%, P = 0.0048; Fig. 6A). This corresponded to a reduced mRNA expression of EGFR in uMGMT patients with short survival in 2 independent datasets of the cohort from TCGA derived from RNAseq and microarray data.
Fig. 6.

Comparison between glioblastomas of patients with different survival times (<6 mo vs >12 mo) according to MGMT promoter methylation status. (A) Copy number variations of CDK4, EGFR, and PDGFRA according to MGMT promoter methylation status and survival groups (<6 mo vs >12 mo) of glioblastoma patients in the cohort of TCGA. (B) Mutations of EGFR and TP53 according to MGMT promoter methylation status and survival groups of TCGA glioblastoma patients. (C) GSEA (left) and IPA (right) comparing results obtained for mMGMT tumors of patients with survival times <6 months vs >12 months. Prediction for upregulation is indicated in orange. (D) IPA of patients with survival times <6 months vs >12 months. Enriched differentially regulated functions are visualized and the topics cellular movement, cell survival, signaling, and proliferation as well as infectious disease are highlighted in red. (E) GSEA (left) and IPA (right) comparing results obtained for uMGMT tumors of patients with survival times <6 months vs >12 months. Prediction for upregulation is indicated in orange, for downregulation in blue.
TP53 mutations were common in MGMT promoter unmethylated tumors and in methylated tumors with a short survival but were significantly less common in mMGMT tumors and a survival time of more than 1 year compared with the group of patients with short survival of less than 6 months (30% vs 4.5%, P = 0.041; Fig. 6B). Like the lower amplification rate of EGFR, there was a trend toward a lower EGFR mutation frequency in the mMGMT patients with short survival.
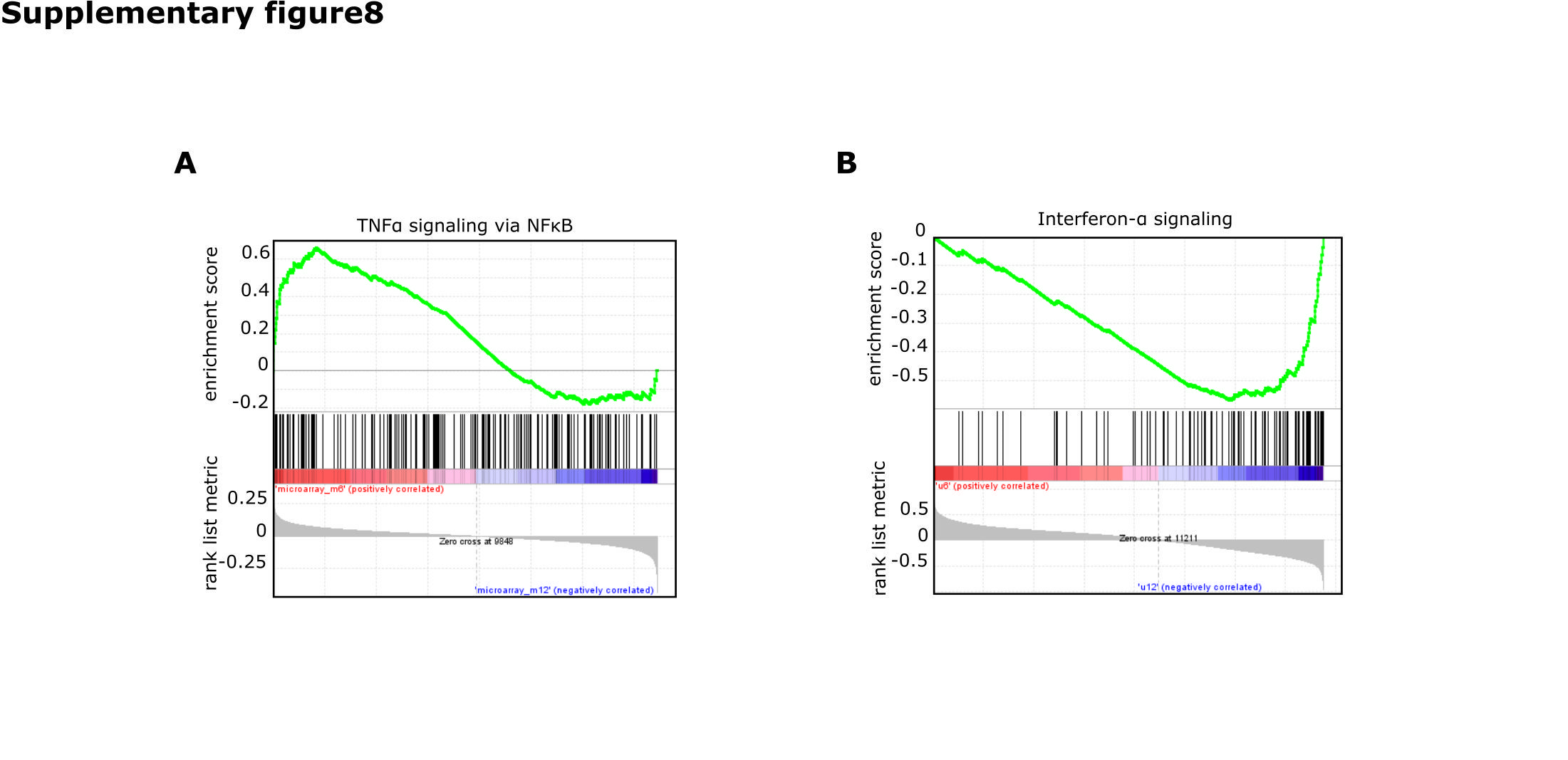
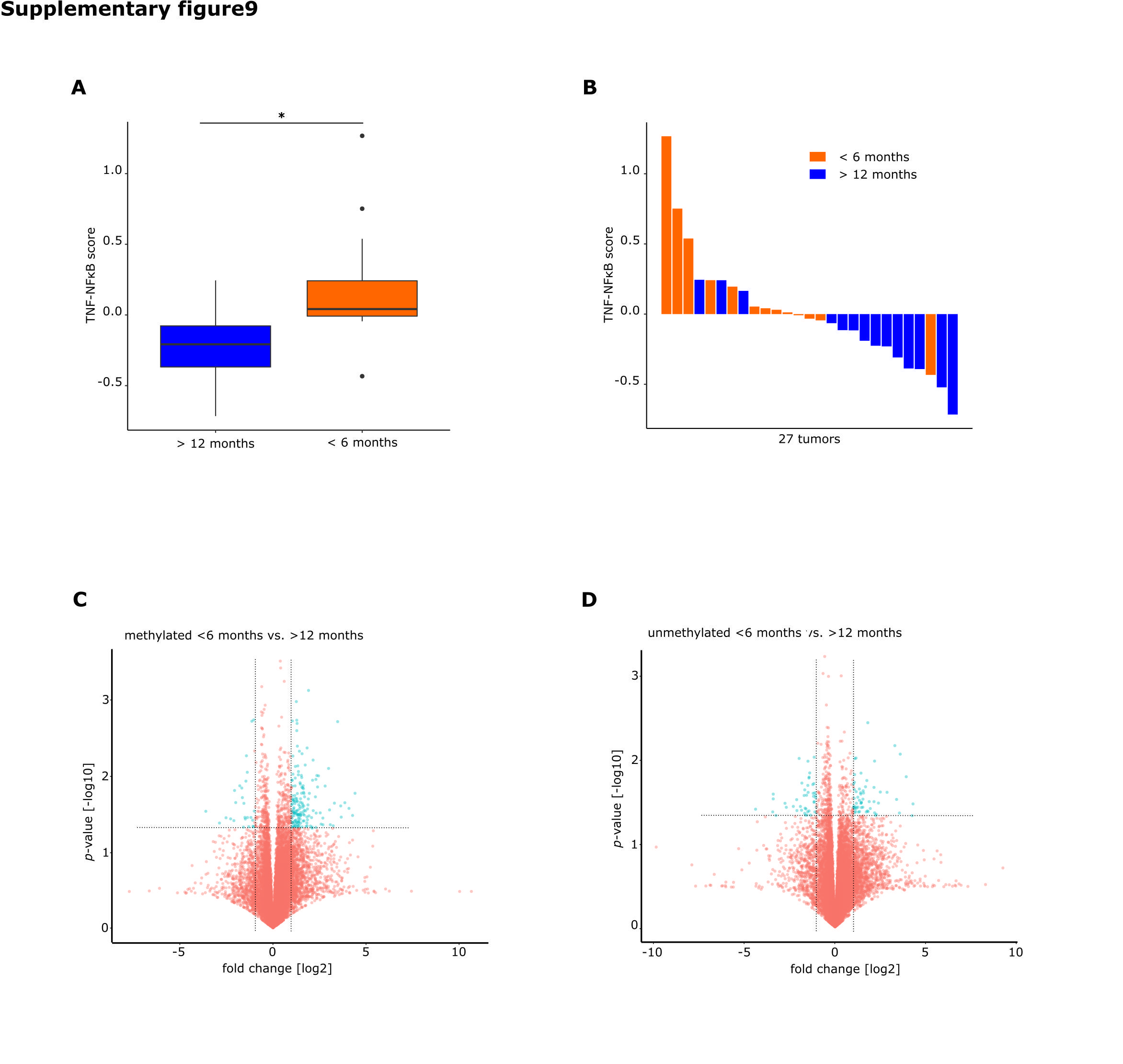
RNAseq data of tumors of the groups was available for 50 patients. IPA of differentially regulated genes on RNA level (Fig. 6C, Supplementary Table S6) revealed a strong prediction of upregulation of TNF-α and the NFκB complex in tumors from patients with short survival with mMGMT compared with patients with longer survival. To confirm this finding, we conducted a GSEA with a set of genes for signaling of TNF-α through NFκB provided by the Broad Institute. This gene set was highly enriched in patients with mMGMT tumors and short survival (Fig. 6C, Supplementary Table S7). Conclusively, cellular movement and proliferation were enhanced in short surviving mMGMT tumors in IPA (Fig. 6D). Further evidence comes from the CGGA cohort, where in mMGMT tumors signaling of TNF-α through NFκB was enriched in a patient cohort with worse prognosis.31 Exceptionally high TNF-NFκB scores were seen in 3/13 (23%) tumors of patients surviving <6 months. Nine of 13 (62%) tumors of the <6 months group and 3/14 (21%) tumors of the >12 months group had scores >0 for NFκB target genes (Supplementary Figure S9A, B). Additionally, the same pathway was enriched in the dataset of tumors from the collective of TCGA with available microarray data (Supplementary Figure S8A).
Signaling through interferon (INF)-α was the only pathway strongly activated in the group of tumors from patients with longer survival (Fig. 6E, Supplementary Table S6) translating into a decreased function “infectious disease” (Fig. 6D), which has been confirmed by GSEA and CGGA, as response to INF-α was the top enriched in patients with high MGMT gene expression and survival times of more than 12 months (Supplementary Figure S8B). Signal transducer and activator of transcription 3 (STAT3) was predicted to be upregulated in both mMGMT and uMGMT tumors of patients with short survival compared with patients with longer survival times (Fig. 6C, E).
Discussion
Most of the IDH mutant glioblastomas are MGMT promoter methylated.32 In this study, genetic, epigenetic, and transcriptional differences between MGMT methylated and unmethylated IDH wildtype glioblastomas have been explored.
CNV and mutations do not differ between newly diagnosed mMGMT and uMGMT tumors. Survival data of patients from the cohort from TCGA who received sole radiotherapy showed no difference in overall survival of mMGMT and uMGMT patients, making it likely that minor changes in the molecular profile do not affect the clinical course of the disease in the absence of temozolomide.
Methylation analysis with consensus clustering of more than 1000 glioblastoma samples revealed 3 different methylation clusters sharing similarities with published work.20,21 The allocation into 3 clusters is relatively stable between different datasets. We found that mMGMT tumors were enriched in cluster 1 and uMGMT tumors in cluster 3. Cluster 1 shows higher average methylation values, explaining why almost all differentially methylated CpGs between mMGMT and uMGMT tumors are hypermethylated in mMGMT tumors. There are no distinct methylation patterns of mMGMT and uMGMT, but MGMT promoter methylation is unevenly distributed within 3 main methylation clusters, with a higher chance of having a methylated MGMT promoter in the cluster with the highest average methylation. The survival advantage of MGMT promoter methylation was only present in the more favorable cluster 1, supporting that the prognostic impact of MGMT is dependent on the global methylation profile. Tumors of cluster 1 express higher levels of TERT, suggesting that TERT promoter mutations are more frequently in that cluster where survival was strongly associated with MGMT promoter methylation. Patients with tumors of clusters 2 and 3 have low TERT expression and do not show a survival benefit with mMGMT, confirming mMGMT is only a survival advantage in high TERT expressing tumors.28 Furthermore, TERT expression/promoter mutations might be associated with specific epigenetic subgroups with high methylation levels and the enriched high TNF-NFκB scores in cluster 3 may partly explain the chemoresistance in this cluster. However, these findings should be validated in further studies.
A hypermutated genotype in some MGMT promoter methylated tumors upon progression had been published.8 We specifically analyzed CNV and found PDGFRA predominantly amplified in MGMT promoter methylated progressive tumors. In paired samples, we found that uMGMT tumors lose PDGFRA amplifications upon progression, whereas mMGMT tumors tend to gain amplifications of PDGFRA. This might also be a resistance mechanism to therapy, especially in mMGMT tumors that are more vulnerable to chemotherapy with temozolomide.
The frequency of CDK4 and MDM2 amplifications tended to be higher in progressive MGMT promoter unmethylated tumors, and in such cases preexisting CDKN2A losses may change upon progression to a balanced state, which is consistent with the finding that CDKN2A loss and CDK4 amplification rarely occur in parallel regardless of MGMT promoter methylation status.33 These specific changes cannot be explained by the function of the MGMT protein and are likely to be a response of the tumor to different efficacy of the alkylating chemotherapy. However, the CNV differences of PDGFRA, CDK4, and MDM2 are based on a limited tumor dataset and should therefore be viewed as preliminary. Robust statistical validation from a larger number of samples will be required.
In our comparison of patient groups with shorter versus longer survival we found differences in specific CNV as well as mutation frequencies and different gene expressions according to MGMT promoter methylation. However, a potential bias could be due to the lower number of patients receiving alkylating chemotherapy in both the methylated and unmethylated groups of patients who survived for less than 6 months compared with the respective groups of patients living longer than 12 months.
The predicted activation of the TNF-NFκB pathway in the group of short surviving mMGMT patients is supported by previous preclinical data showing upregulation of MGMT through high NFκB signaling and therefore increased chemoresistance,34 although this may not be uniformly dependent on the level of promoter inactivity, possibly contributing to the short survival times in patients with otherwise favorable MGMT promoter methylated tumors. Of note, it is more likely that glioblastomas of patients with longer survival show a downregulation of TNF-NFκB signaling, as we also found higher NFκB signaling in unmethylated tumors not selected for survival compared with all methylated tumors and no difference when comparing tumors from patients with short survival and MGMT promoter methylated tumors against all patients with unmethylated glioblastomas.
Likewise, INF signaling through INF-α/β can sensitize MGMT promoter unmethylated glioblastomas to temozolomide.35,36 There is an upregulation of the INF-α pathway in uMGMT tumors and long survival compared with both patients with unmethylated tumors and short survival as well as patients with methylated tumors and long survival, suggesting that patients who survive relatively longer with the unfavorable uMGMT might have this survival benefit from increased INF-α signaling and therefore increased chemotherapy sensitivity. This is of particular importance because most of the patients in the group with longer survival despite having tumors with uMGMT received temozolomide. The data are supported by a dataset of the CGGA, although caution is required because of the low sample size. The predicted upregulation observed in our study supports the recently proposed therapeutic inhibition of STAT3 in glioblastoma patients,37 at least for patients with glioblastomas lacking MGMT promoter methylation.
In conclusion, MGMT promoter methylation status serves as a biomarker for temozolomide therapy and does not determine an otherwise molecularly distinct glioblastoma subpopulation in the newly diagnosed setting. The chance to find a molecular target for trial inclusion is not per se worse in the MGMT unmethylated population. Further, there is a clear signal from these data to demand a new tissue analysis for every trial at recurrence. Lastly, the data provide evidence for the TNF-NFκB pathway to be revisited in future trials. Temozolomide might be safely omitted in patients with MGMT promoter unmethylated glioblastomas in clinical trials, especially when meaningful molecular markers predicting the response to experimental therapies are found by predictive molecular analyses.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This research was funded from the Heidelberg Center for Personalized Oncology, project number K25K.
Conflict of interest statement. G.T. served on advisory boards for Bristol Myers Squibb, received travel grants from Medac, and received a research grant from Roche Diagnostics. G.R. received research grants from Roche and Merck as well as honoraria for advisory boards and lectures from Amgen, Celldex, and Medac.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1. Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–1850. [DOI] [PubMed] [Google Scholar]
- 2. Louis DN, Perry A, Reifenberger G et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 3. Wick W, Weller M, van den Bent M et al. MGMT testing–the challenges for biomarker-based glioma treatment. Nat Rev Neurol. 2014;10(7):372–385. [DOI] [PubMed] [Google Scholar]
- 4. Hegi ME, Diserens AC, Gorlia T et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
- 5. Stupp R, Mason WP, van den Bent MJ et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 6. Wick W, Steinbach JP, Platten M et al. Enzastaurin before and concomitant with radiation therapy, followed by enzastaurin maintenance therapy, in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation. Neuro Oncol. 2013;15(10):1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Herrlinger U, Schäfer N, Steinbach JP et al. Bevacizumab plus irinotecan versus temozolomide in newly diagnosed O6-methylguanine-DNA methyltransferase nonmethylated glioblastoma: The Randomized GLARIUS Trial. J Clin Oncol. 2016;34(14):1611–1619. [DOI] [PubMed] [Google Scholar]
- 8. Wick W, Gorlia T, Bady P et al. Phase II study of radiotherapy and temsirolimus versus radiochemotherapy with temozolomide in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation (EORTC 26082). Clin Cancer Res. 2016;22(19):4797–4806. [DOI] [PubMed] [Google Scholar]
- 9. Nabors LB, Fink KL, Mikkelsen T et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 2015;17(5):708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hermisson M, Klumpp A, Wick W et al. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J Neurochem. 2006;96(3):766–776. [DOI] [PubMed] [Google Scholar]
- 11. Wick W, Roth P, Hartmann C et al. ; Neurooncology Working Group (NOA) of the German Cancer Society Long-term analysis of the NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with PCV or temozolomide. Neuro Oncol. 2016;18(11):1529–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wick W, Hartmann C, Engel C et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol. 2009;27(35):5874–5880. [DOI] [PubMed] [Google Scholar]
- 13. Wick W, Platten M, Meisner C et al. ; NOA-08 Study Group of Neuro-oncology Working Group (NOA) of German Cancer Society Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13(7):707–715. [DOI] [PubMed] [Google Scholar]
- 14. Bady P, Sciuscio D, Diserens AC et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol. 2012;124(4):547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Morris TJ, Butcher LM, Feber A et al. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics. 2014;30(3):428–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cerami E, Gao J, Dogrusoz U et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao J, Aksoy BA, Dogrusoz U et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Center BITGDA. Analysis-ready standardized TCGA data from Broad GDAC Firehose 2016_01_28 run Broad Institute of MIT and Harvard; Dataset. http://doi.org/10.7908/C11G0KM9. 2016. [Google Scholar]
- 19. Verhaak RG, Hoadley KA, Purdom E et al. ; Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ceccarelli M, Barthel FP, Malta TM et al. ; TCGA Research Network Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sturm D, Witt H, Hovestadt V et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–437. [DOI] [PubMed] [Google Scholar]
- 22. Sun Y, Zhang W, Chen D et al. A glioma classification scheme based on coexpression modules of EGFR and PDGFRA. Proc Natl Acad Sci U S A. 2014;111(9):3538–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu Y, Yan C, Hsu CH et al. OmicCircos: a simple-to-use R package for the circular visualization of multidimensional omics data. Cancer Inform. 2014;13:13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arita H, Yamasaki K, Matsushita Y et al. A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol Commun. 2016;4(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao AC, Lou W, Isaacs JT. Enhanced GBX2 expression stimulates growth of human prostate cancer cells via transcriptional up-regulation of the interleukin 6 gene. Clin Cancer Res. 2000;6(2):493–497. [PubMed] [Google Scholar]
- 28. Nguyen HN, Lie A, Li T et al. Human TERT promoter mutation enables survival advantage from MGMT promoter methylation in IDH1 wild-type primary glioblastoma treated by standard chemoradiotherapy. Neuro Oncol. 2017;19(3):394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang J, Cazzato E, Ladewig E et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48(7):768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hopker K, Hagmann H, Khurshid S et al. AATF/Che-1 acts as a phosphorylation-dependent molecular modulator to repress p53-driven apoptosis. EMBO J. 2012;31(20):3961–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang W, Zhang L, Wang Z et al. A three-gene signature for prognosis in patients with MGMT promoter-methylated glioblastoma. Oncotarget. 2016;7(43):69991–69999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Molenaar RJ, Verbaan D, Lamba S et al. The combination of IDH1 mutations and MGMT methylation status predicts survival in glioblastoma better than either IDH1 or MGMT alone. Neuro Oncol. 2014;16(9):1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmidt EE, Ichimura K, Reifenberger G, Collins VP. CDKN2 (p16/MTS1) gene deletion or CDK4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994;54(24):6321–6324. [PubMed] [Google Scholar]
- 34. Lavon I, Fuchs D, Zrihan D et al. Novel mechanism whereby nuclear factor kappaB mediates DNA damage repair through regulation of O(6)-methylguanine-DNA-methyltransferase. Cancer Res. 2007;67(18): 8952–8959. [DOI] [PubMed] [Google Scholar]
- 35. Shen D, Guo CC, Wang J et al. Interferon-α/β enhances temozolomide activity against MGMT-positive glioma stem-like cells. Oncol Rep. 2015;34(5):2715–2721. [DOI] [PubMed] [Google Scholar]
- 36. Motomura K, Natsume A, Kishida Y et al. Benefits of interferon-β and temozolomide combination therapy for newly diagnosed primary glioblastoma with the unmethylated MGMT promoter: a multicenter study. Cancer. 2011;117(8):1721–1730. [DOI] [PubMed] [Google Scholar]
- 37. Han TJ, Cho BJ, Choi EJ et al. Inhibition of STAT3 enhances the radiosensitizing effect of temozolomide in glioblastoma cells in vitro and in vivo. J Neurooncol. 2016;130(1):89–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.