

Abstract
We report the development of a method to parameterize and predict the performance of structurally flexible β-turn-containing peptide catalysts, using the atroposelective bromination of 3-arylquinazolin-4(3H)-ones as a case study. The multivariate correlations obtained for tetrapeptides of two β-turn types, type I′ pre-helical and type II′ β-hairpin, indicate that while one conformer may be associated with a more dominant contribution to the observed enantioselectivity, it is possible that multiple conformers contribute to a complex transition state ensemble.
TOC image
Many small molecule, chiral catalysts have been intentionally designed to have relatively rigid structures to preclude complications associated with conformational flexibility.1 An exception is peptidic catalysts, which can adopt a range of conformations, owing to the many rotatable bonds inherent in polypeptide frameworks. One family of peptide-based catalysts that likely adopt predictable conformations is the so-called β-turn-containing peptides, which have proven to be effective for many applications in enantioselective catalysis.2 Notably, the identification of optimal catalysts using these templates often reveals subtle variations in the peptide sequence that prove critical for enhanced reactivity and/or selectivity, yet the effects of these alterations are not well-understood. Additionally, computing these tetrapeptides presents particular challenges to the capabilities of current DFT methods, presumably due to the large size and flexibility of peptides and our presently limited proficiency to forecast and quantify weak, noncovalent interactions (NCIs), including those associated with solvent.3 Whether they be intramolecular within the catalyst scaffold or associated with bimolecular, or even higher order ensembles, the culmination of these NCIs present key energetic differences that define the enantiodetermining transition states.4 Further contributing to this challenge is the myriad of accessible conformations that such catalysts may populate, essentially all of which may be catalytically competent.5
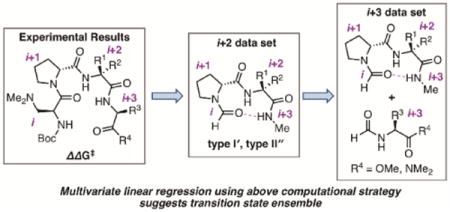
Although β-turn secondary structures can be induced by careful choice of amino acid residues (e.g., tetrapeptide catalysts used in the Miller group often incorporate proline at the i+1 position and an α,α-disubstituted amino acid in the i+2 position), many of these catalysts sample multiple distinct conformations within the β-turn classification.6 For instance, type I′ and II′ β-turns, differing mainly by the orientation of the loop amide region (blue boxes, Figure 1b),7 have been observed in the solid and solution states for several variations of 3.8 Ultimately, methods to simultaneously interrogate the flexibility and dynamics of these catalysts will facilitate an understanding of the enantioselectivity imparted in a given reaction and provide additional opportunities to predict new peptide catalyst performance.
Figure 1.
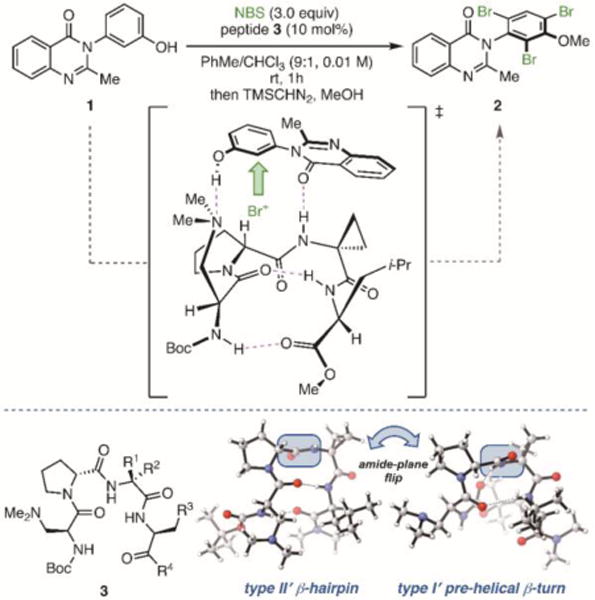
(a) Atroposelective bromination of quinazolinone 1 catalyzed by β-turn-containing peptide 3. (b) Major observed conformations of β-turn-containing peptides by NMR and X-ray crystallography.
The application of modern physical organic tools holds great promise to address the challenges associated with catalyst optimization and the mechanistic interrogation of these flexible peptides. Specifically, developing structure-function relationships within this context would provide direct and quantitative insight into the structural features of the peptides that most influence the reaction outcome. The reaction we chose to study is the atroposelective bromination of 3-arylquinazolin-4(3H)-ones (quinazolinones, e.g., 1) catalyzed by tertiary amine-containing peptide catalysts with the general structure type of 3 (Figure 1b).9 Under the optimized conditions, these catalysts have been shown to provide access to atropisomerically enriched products (e.g., 2) with high levels of enantioinduction (up to 99:1 er). Moreover, seemingly subtle structural permutations of the catalyst led to non-intuitive changes in enantioselectivity. Herein, we probe several catalytically relevant motifs of peptides 3a–m using truncated structures for which parameters are readily obtained computationally, allowing for mathematical correlations to enantioselectivity. The models derived below provide substantial insight into the structural features that make these conformationally flexible tetrapeptides effective catalysts and illustrate that the accessibility of multiple conformers is an important facet of these peptidic systems.5b
One significant difficulty envisioned at the outset of this project was how to readily describe both the functional group-rich nature of the peptide-based catalysts and their conformational dynamics.10 To design the computational platform for parameterization, recent extensive structural studies were considered. On the basis of X-ray crystallographic and NMR experiments, one key structural feature that was incorporated into our strategy from the start is the observed hydrogen bond between N–Hi+3 and Oi, as well as between the carbonyl of the substrate and N–Hi+2 (Figure 1a).8, 9, 11 Within this motif, several conformations are accessible, as the prototypical peptide catalyst 3 has been shown to adopt as many as five distinct states in the solid state, more broadly characterized as either a type II′ β-hairpin or type I′ pre-helical β-turn (Figure 1b). In solution, NMR analysis of peptide 3a was also consistent with populated states that map onto pre-helical or nonhairpin β-turn conformers.11 Given the structural flexibility, it seemed likely that both conformer families could require unique parameter sets to achieve meaningful correlations.
We also considered the original structure-enantio-selectivity relationships for the bromination reaction, which showcased that changes to the i+2 residue lead to the most significant differences in enantioselectivity, while modifications to the i+3 residue exhibit a more subtle effect in the high-selectivity regime.10 Furthermore, a linear correlation between the crystallographic main-chain angle (τ) of the i+2 position (τ(i+2)) and enantioselectivity has been identified, although τ(i+2) exhibits clustering at 111° and 117°.8,12 These experimental results suggested that initial modeling strategies should be directed at understanding how changes in the i+2 residue affect catalysis. Thus, a homologous series of peptides varying only at i+2 were synthesized and used as catalysts in the atroposelective bromination reaction to probe the effects of this residue on enantioselectivity (Figure 2).
Figure 2.
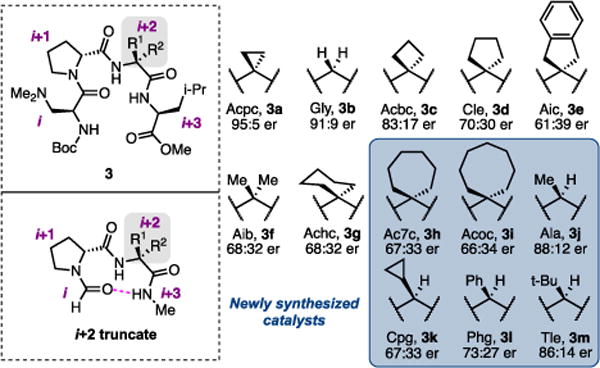
Homologous series of peptidic catalysts varying at i+2 and the truncation used computationally. See Supporting Information for abbreviation nomenclature.
At the outset of the modeling process, we removed the i+3 amino acid, replacing it with a computationally simpler N-methyl amide cap (Figure 2). Additionally, the β-dimethylaminoalanine (Dmaa) moiety was removed to further reduce the computational cost, as it is conserved within the catalyst library. Finally, the ten-membered ring hydrogen bond characteristic of β-turn peptides was maintained by locking the distance between C=Oi and H–Ni+3 in the initial computation (Figure 2).13 This constraint was then relaxed in a subsequent ground state optimization to obtain the calculated structures (M06-2X/def2tzvp).
For each i+2 variation, both type I′ and type II′ conformers were calculated so that parameters from each could be obtained, such as natural bond orbital (NBO) charges,14 torsional angles, Sterimol values,15 and infrared (IR) frequencies and intensities.16 Multivariate linear regression analysis was performed on both conformer parameter sets using a previously reported algorithm (see Supporting Information) to reveal statistically significant correlations.17 From the initial training set of nine catalysts varying only at the i+2 position, a correlation using two parameters derived from the type II′ conformer was identified (R2 = 0.88, Q2 = 0.81): the intensity of the N–Hi+3 stretch and the NBO charge on Oi+1 (Figure 3a). The intensity of the N–Hi+3 likely describes the overall catalytic importance of the C=Oi•••H–Ni+3 hydrogen bond strength, as IR intensities are correlated to a change in dipole moment.19
Figure 3.
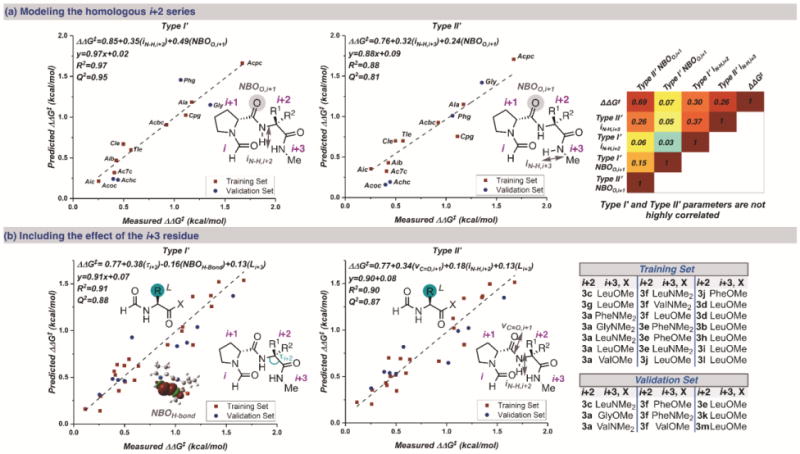
(a) Models of the homologous i+2 series for the type I′ (left) and type II′ (middle) β-turn conformers. In the correlation map (right), the text is the R2 value for the linear relationship between the parameters. (b) Models that include the effect of the i+3 residue for the type I′ (left) and type II′ (middle) conformers using the training and validation sets (right).
Six additional catalysts (3h–3m) were synthesized to test the model’s predictive capability. Ultimately, four catalysts in the series were randomly selected as external validations (Figure 3a).18 Similarly, a good correlation was obtained using the parameter set for the type I′ conformer, again using two parameters (R2=0.97, Q2=0.95): the NBO charge on Oi+1, similar to above, and a different parameter, the intensity of the N–Hi+2 stretch (Figure 3a). The N–Hi+2 intensity suggests that an inter- or intramolecular hydrogen bond with this amide is important for catalysis.9 The statistical significance of the individual models suggests that both conformers could be catalytically relevant, albeit the calculated type II′ conformers are consistently lower in ground state energy (0.9–6 kcal/mol) than the corresponding type I′ structures with the basis set that we employed. Notably, although the NBO charge of Oi+1 is included in both models, the parameter values derived from both conformers are not intercorrelated (Figure 3a). These two observations suggest that each conformer may be productive for catalysis, albeit with different substrate interactions. The importance of the electron density on Oi+1 may also allude to this oxygen playing a role in directing the initial bromination, the enantiodetermining event.20
As a next step, we sought to identify a comprehensive strategy to interrogate the entire tetrapeptide structure. To do this, we pursued a separate structural model system to describe substituents at the i+3 residue. We elected to obtain parameters for this residue, independent from the rest of the peptide, to assess a wider array of structural features with limited computational cost. This is in part due to the greater structural flexibility in the solid state observed at the i+3 position.8 Additionally, incorporating the i+3 residue into the truncated i+2 model system would require the inclusion of the Dmaa residue to maintain the second hydrogen bond between N–Hi and C=Oi+3 that defines a β-hairpin conformation, thus increasing the computational effort needed to acquire descriptors even further.21 Ultimately, as the catalytic moiety and D-proline are conserved for each of the tetrapeptides, we calculated the corresponding i+3 amino esters and amides to further account for the observed end-cap differences (Figure 1b). By including the i+3 residue separately, a combinatorial library could be generated, enabling rapid catalyst optimization for other peptide-mediated systems.
After collecting parameters for the i+3 amino esters and amides in conjunction with both parameter sets for the type I′ and type II′ conformers, multivariate linear regression was performed, which resulted in two models, respectively, with three parameters each.17 The only descriptor conserved in both equations is the Sterimol L parameter derived from the separate i+3 residue (Figure 3b). This implies that the sterics at the i+3 position have a similar effect in each case. Moreover, as before, the parameter sets between conformer types are not intercorrelated. Overall, these descriptors support our previous observations by identifying structural features that likely describe the catalysts’ conformational flexibility and abilities to bind to the substrate. In the type I′ case (R2=0.91, Q2=0.88), the unique parameters used are the τ(i+2) and the strength of the H-bond as determined through NBO analysis (Figure 3b).16c Although τ(i+2) is structural by definition, these values also correlate to other parameters including electronically based descriptors, such as the NBO of Oi+1, which was previously used to model the homologous i+2 series (Figure 3a). For the type II′ conformer (R2=0.90, Q2=0.87), the identified model uses the frequency of C=Oi+1 and the N–Hi+2 stretching intensity (Figure 3b). The use of parameters that represent analogous residue characteristics suggest that similar transition states lead to the desired brominated atropisomer 2. Overall, the models developed with the larger set of these β-turn-containing tetrapeptides are consistent with the likelihood of multiple catalytically active peptide conformations.
In summary, we have successfully developed a method to parameterize β-turn-containing peptide catalysts, which have been shown to adopt multiple conformers in solution and in the solid state. While it seems plausible that the type II′ β-turn may be a significant, and even dominant contributor to enantioselectivity, the work presented herein suggests that multiple conformers may contribute to a diffuse, complex transition state ensemble. In this same vein, it is implicit that each catalyst interrogated, inliers and outliers alike, creates a uniquely composed transition state ensemble leading to the varied levels of enantioinduction in the bromination reaction studied. These features are largely hidden in conventional mechanistic studies of enantioselective catalysts, especially those with conformational flexibility, and the results presented herein provide a strategy for the analysis and design of conformationally flexible catalysts. Furthermore, this method allows for enhanced mechanistic understanding and streamlined catalyst optimization for a broad range of peptide-catalyzed transformations.
Supplementary Material
Acknowledgments
M.S.S. and S.J.M. thank the NIH (1 R01 GM121383) for supporting this work. J.M.C. and A.J.M. acknowledge the support of the NSF Graduate Research Fellowship Program. E.A.S. acknowledges the research support of the NIH Molecular Biophysics Predoctoral Training Grant (T32 GM008283). We also thank Dr. Margaret Hilton for helpful discussions. The support and resources from the Center for High Performance Computing at the University of Utah are gratefully acknowledged.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website. Experimental and computational details, including DFT energies and geometries (PDF).
ORCID
Jennifer M. Crawford: 0000-0003-2936-4038
Elizabeth A. Stone: 0000-0002-2253-3452
Anthony J. Metrano: 0000-0002-2599-8917
Scott J. Miller: 0000-0001-7817-1318
Matthew S. Sigman: 0000-0002-5746-8830
Notes
The authors declare no competing financial interests.
References
- 1.(a) Zhou Q-L, editor. Privileged Chiral Ligands and Catalysts; Wiley-VCH Verlag GmbH & Co KGaA. 2011. [Google Scholar]; b Pfaltz A, Drury WJ. Proc Natl Acad Sci USA. 2004;101:5723–5726. doi: 10.1073/pnas.0307152101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yoon TP, Jacobsen EN. Science. 2003;299:1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 2.(a) Miller SJ. Acc Chem Res. 2004;37:601–610. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]; (b) Davie EAC, Mennen SM, Xu Y, Miller SJ. Chem Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]; (c) Wennemers H. Chem Commun. 2011;47:12036–12041. doi: 10.1039/c1cc15237h. [DOI] [PubMed] [Google Scholar]; (d) Lewandowski B, Wennemers H. Curr Opin Chem Biol. 2014;22:40–46. doi: 10.1016/j.cbpa.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 3.(a) Orlandi M, Coelho JAS, Hilton MJ, Toste FD, Sigman MS. J Am Chem Soc. 2017;139:6803–6806. doi: 10.1021/jacs.7b02311. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Neel AJ, Hilton MJ, Sigman MS, Toste FD. Nature. 2017;543:637–64.6. doi: 10.1038/nature21701. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Toste FD, Sigman MS, Miller SJ. Acc Chem Res. 2017;50:609–615. doi: 10.1021/acs.accounts.6b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walsh PJ, Kozlowski MC. Fundamentals of Asymmetric Catalysis. University Science Books; Sausalito, California: 2009. [Google Scholar]
- 5.(a) Abascal NC, Lichtor PA, Giuliano MW, Miller SJ. Chem Sci. 2014;5:4504–4511. doi: 10.1039/C4SC01440E. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schnitzer T, Wennemers H. J Am Chem Soc. 2017;139:15356–15362. doi: 10.1021/jacs.7b06194. [DOI] [PubMed] [Google Scholar]
- 6.(a) Wilmot CM, Thornton JM. J Mol Bio. 1988;203:221–232. doi: 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]; (b) Richardson JS. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 7.(a) Gunasekaran K, Gomathi L, Ramakrishnan C, Chandrasekhar J, Balaram P. J Mol Bio. 1998;284:1505–1516. doi: 10.1006/jmbi.1998.2154. [DOI] [PubMed] [Google Scholar]; (b) Hayward S. Protein Sci. 2001;10:2219–2227. doi: 10.1110/ps.23101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metrano AJ, Abascal NC, Mercado BQ, Paulson EK, Hurtley AE, Miller SJ. J Am Chem Soc. 2017;139:492–516. doi: 10.1021/jacs.6b11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Abascal NC, Miller SJ. Org Lett. 2016;18:4646–4649. doi: 10.1021/acs.orglett.6b02282. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alford JS, Abascal NC, Shugrue CR, Colvin SM, Romney DK, Miller SJ. ACS Cent Sci. 2016;2:733–739. doi: 10.1021/acscentsci.6b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diener ME, Metrano AJ, Kusano S, Miller SJ. J Am Chem Soc. 2015;137:12369–12377. doi: 10.1021/jacs.5b07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metrano AJ, Abascal NC, Mercado BQ, Paulson EK, Miller SJ. Chem Commun. 2016;52:4816–4819. doi: 10.1039/c6cc01428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou AQ, O’Hern CS, Regan L. Protein Sci. 2011;20:1166–1171. doi: 10.1002/pro.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Gardner RR, Gellman SH. J Am Chem Soc. 1995;117:10411–10412. [Google Scholar]; (b) Liang GB, Rito CJ, Gellman SH. J Am Chem Soc. 1992;114:4440–4442. [Google Scholar]
- 14.(a) Glendening ED, Reed AE, Carpenter JE, Weinhod F. NBO Version 3.1 [Google Scholar]; (b) Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735–746. [Google Scholar]; (c) Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899–926. [Google Scholar]
- 15.Verloop A. Drug Design. III Academic Press; 1976. [Google Scholar]
- 16.Milo A, Bess EN, Sigman MS. Nature. 2014;507:210–214. doi: 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]
- 17.(a) Guo JY, Minko Y, Santiago CB, Sigman MS. ACS Catal. 2017;7:4144–4151. [Google Scholar]; (b) Sigman MS, Harper KC, Bess EN, Milo A. Acc Chem Res. 2016;49:1292–1301. doi: 10.1021/acs.accounts.6b00194. [DOI] [PubMed] [Google Scholar]
- 18.Golbraikh A, Tropsha A. J Mol Graph Modell. 2002;20:269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]
- 19.Seidler P, Kongsted J, Christiansen O. J Phys Chem A. 2007;111:11205–11213. doi: 10.1021/jp070327n. [DOI] [PubMed] [Google Scholar]
- 20.(a) Denmark SE, Beutner GL. Angew Chem Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Burk MT. Proc Natl Acad Sci USA. 2010;107:20655–20660. doi: 10.1073/pnas.1005296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Haque TS, Little JC, Gellman SH. J Am Chem Soc. 1996;118:6975–6985. [Google Scholar]; (b) Karle IL, Awasthi SK, Balaram P. Proc Natl Acad Sci USA. 1996;93:8189–8193. doi: 10.1073/pnas.93.16.8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.