

ABSTRACT
Genetic factors underlying the human limb abnormality congenital talipes equinovarus (‘clubfoot’) remain incompletely understood. The spontaneous autosomal recessive mouse ‘peroneal muscular atrophy’ mutant (PMA) is a faithful morphological model of human clubfoot. In PMA mice, the dorsal (peroneal) branches of the sciatic nerves are absent. In this study, the primary developmental defect was identified as a reduced growth of sciatic nerve lateral motor column (LMC) neurons leading to failure to project to dorsal (peroneal) lower limb muscle blocks. The pma mutation was mapped and a candidate gene encoding LIM-domain kinase 1 (Limk1) identified, which is upregulated in mutant lateral LMC motor neurons. Genetic and molecular analyses showed that the mutation acts in the EphA4–Limk1–Cfl1/cofilin–actin pathway to modulate growth cone extension/collapse. In the chicken, both experimental upregulation of Limk1 by electroporation and pharmacological inhibition of actin turnover led to defects in hindlimb spinal motor neuron growth and pathfinding, and mimicked the clubfoot phenotype. The data support a neuromuscular aetiology for clubfoot and provide a mechanistic framework to understand clubfoot in humans.
KEY WORDS: Limk1, Axon guidance, Clubfoot, Limb development, Chicken
Highlighted Article: The mutation in the PMA mouse model of human clubfoot was mapped and a candidate gene, Limk1, identified that was shown to cause sciatic nerve and limb abnormalities when overexpressed.
INTRODUCTION
Congenital talipes equinovarus (CTEV, also known as ‘clubfoot’) is a human lower limb developmental defect with a worldwide prevalence of 1-3 cases for every 1000 live births, making it one of the most common pediatric orthopedic conditions (Cartlidge, 1984; Dobbs et al., 2000). Clubfoot is characterised by the inward rotation and downward flexion of the foot, which persists after birth and, unless corrected, causes permanent disability. However, our understanding of the aetiological and genetic factors underlying clubfoot is incomplete. Most clubfoot births, where the infant has unilateral or bilateral CTEV but no other clinical problems, are largely unexplained. Most patients respond well to serial casting (Ponseti manipulation) followed by Achilles tenotomy in infancy but a significant proportion (10-15%) experience relapse and most post-clubfoot individuals experience some degree of leg fatigue (Ippolito et al., 2009).
Several hypotheses have been put forward to explain clubfoot aetiology. Although intuitively clubfoot may be assumed to arise from a skeletal patterning defect, there is in fact little evidence for this and the primary defect appears to be hindlimb developmental arrest from around day 44 of gestation (Miedzybrodzka, 2003). The hindlimb must rotate during late embryogenesis, such that the sole of the foot, which lies initially in the plane of the body axis, faces down, with toes lying in the horizontal plane. Clubfoot results when this rotation fails to complete, and it has been hypothesised that a musculoskeletal inter-relationship is required for limb rotation (Isaacs et al., 1977; Stewart, 1951; Bechtol and Mossman, 1950; Dittrich, 1930; Ponseti and Campos, 1972). Several lines of clinical evidence show that weakness, inactivity, or absence of calf muscles can be associated with clubfoot (Flynn et al., 2007; Ohno et al., 1986), and there is persistent loss of muscle density in the calves of individuals with clubfoot (Duce et al., 2013).
Twin studies and population genetics have provided strong evidence for a genetic basis of clubfoot (Miedzybrodzka, 2003). Complex segregation analyses suggest that the most likely inheritance pattern is a single gene of major effect but low penetrance, with either dominant or recessive inheritance, operating against a polygenic background (Wang et al., 1988; de Andrade et al., 1988; Rebbeck et al., 1993; Chapman et al., 2000). However, the identity of the major gene(s) involved remains elusive. Mutations in the genes encoding the hindlimb transcription factors PITX1 and TBX4 have been shown to lead to a reduction in lower limb musculature and classic clubfoot phenotypes in humans and mice, although these may also be associated with syndromic long bone growth defects (Gurnett et al., 2008; Alvarado et al., 2010, 2011; Lu et al., 2012; Peterson et al., 2014). Approximately 5% of human cases of familial isolated clubfoot are associated with microduplications of TBX4 (Dobbs and Gurnett, 2013). There are other mouse models [e.g. ephrin receptor EphA4 knockout mice exhibit clubfoot as part of a syndrome of motor and cognitive abnormalities (Helmbacher et al., 2000)] but none of these has led to the discovery of other major human causative genes, and most cases of clubfoot in humans are idiopathic (i.e. with no known cause).
A spontaneous mouse mutant, the peroneal muscular atrophy (PMA) mouse, shows an inherited, hindlimb-restricted, bilateral clubfoot-like phenotype at birth that is comparable to the human pathology (Nonaka et al., 1986; Duce et al., 2010) (Fig. 1). The mutation is autosomal recessive with high penetrance, and has been previously mapped to mouse chromosome 5 (Katoh et al., 2003). However, no candidate gene has been identified and at least 67 genes are contained within the 4.8 Mb mapped region. Adult pma/pma mice have anterior-lateral regional defects of the hindlimbs only, including absence of the peroneal nerve and atrophy of the anterior-lateral calf muscle compartment (Nonaka et al., 1986; Ashby et al., 1993). It is hypothesised that, because the outer (dorsal and lateral) calf muscles are atrophied but the inner (ventral and posterior) are normal, the foot remains in a CTEV-like position during development. It is not known why the peroneal branch of the sciatic nerve fails in PMA mice. Normally, the motor neurons that contribute to the sciatic nerve project from the lateral motor columns (LMCs) of the ventral neural tube. Those that will contribute to dorsal limb nerves project from the lateral LMC (lLMC), whereas those that will project ventrally originate in the medial region of the LMC (mLMC) (Landmesser, 1978). Both populations project through the ventral roots of the lumbar neural tube and coalesce at the lumbosacral plexus before entering the hindlimb (Wang and Scott, 2008; Jessell, 2000). Here, they branch into the ventral limb, becoming the precursors of the tibial and sural nerves, or into the dorsal limb, becoming the peroneal nerve.
Fig. 1.
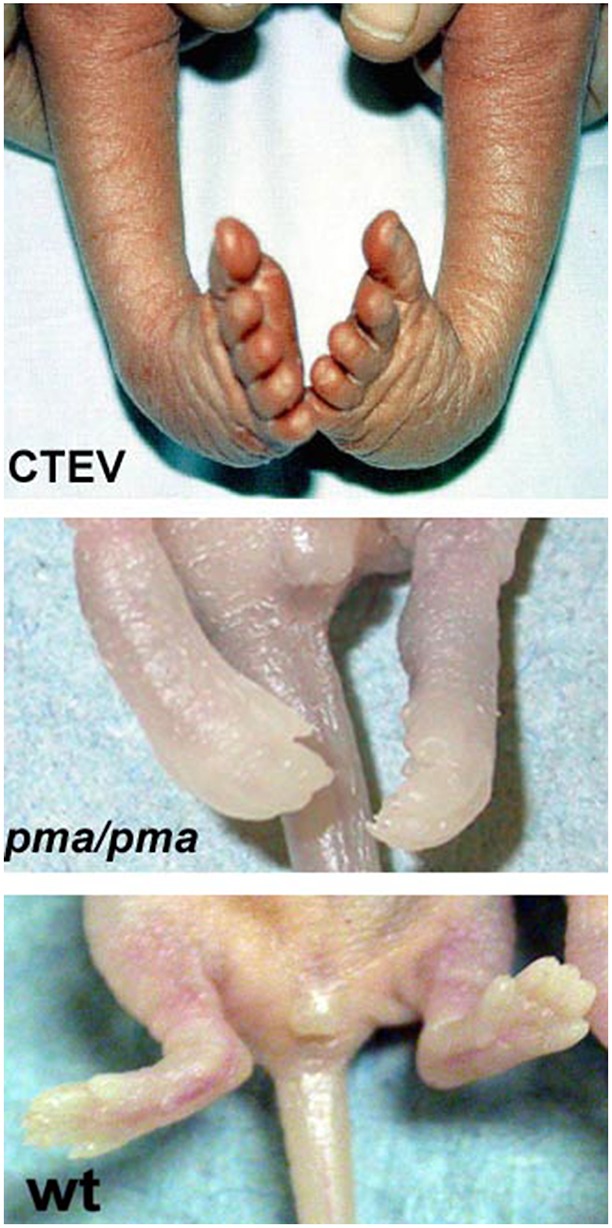
Clubfoot phenotype in pma/pma mice. Human newborn with congenital talipes equinovarus (CTEV, commonly known as clubfoot) (top) compared with newborn pma/pma mouse (middle) and newborn wild-type mouse (bottom).
The PMA mouse is potentially a clinically relevant model of clubfoot, but the genetic basis of the phenotype has not been described, and the primary defect, whether developmental or degenerative, is poorly understood. This work explores the aetiology and genetics of clubfoot using the PMA animal model. By genetic mapping, sequencing and gene expression analysis, we identify Limk1 as a candidate gene underlying the pma mutation and show, using mouse and chicken models, that overexpression of this gene delays neuronal growth and drives aberrant neuron guidance, resulting in muscular defects, failure of foot rotation, and clubfoot.
RESULTS
Peroneal nerve development is aborted in pma/pma embryos
It has been shown previously that the common peroneal nerve is absent in adult pma/pma mice (Nonaka et al., 1986). This is the dorsal branch of the sciatic nerve that innervates the anterior-dorsal ‘peroneal’ calf muscles, which are atrophied in the PMA mouse and in patients with clubfoot (Duce et al., 2010, 2013). Immunohistochemistry on tissue sections and whole-mount preparations at embryonic day (E)16.5 confirmed the complete absence of motor innervation in the dorsal-anterior muscles of the lower leg of pma/pma homozygotes (the tibialis anterior, the extensor digitorum longus and the peroneus longus), with normal innervation of ventral calf muscles (Fig. 2). Immunohistochemical analysis showed that the anterior-dorsal muscles were normally vascularised in pma/pma homozygotes (Fig. S1). The data confirm that the clubfoot phenotype in PMA mice is associated with neural rather than vascular failure. To define the earliest developmental stage at which a defect is observed, homozygous pma/pma mouse embryos were examined histologically at E10.5-E16.5. It was found that, at E11.5, the sciatic nerve had entered the hindlimb of wild-type mouse homozygotes and the beginnings of branching were visible, but in stage-matched pma/pma homozygote embryos, the nerve had neither entered the limb nor started to branch (Fig. 3A,B). At E12.5, wild-type sciatic nerves had forked into clearly demarcated, fasciculated dorsal peroneal and ventral tibial trunks (Fig. 3C,C′). The E12.5 pma/pma littermates had a morphologically normal tibial trunk and no obvious peroneal nerve, suggesting failure of normal branching. However, whole-mount immunohistochemistry showed a dorsal projection of a small number of defasciculated axons projecting dorsally in E12.5 pma/pma embryos that may represent an abortive peroneal nerve (Fig. 3D,D′). The fate of these axons is unknown, because no peroneal innervation was detectable at later stages, as described above (Fig. 3E,F). The peroneal nerve was confirmed by dissection to be absent postnatally in pma/pma mice, consistent with the observations of Nonaka et al. (1986) (Fig. S2).
Fig. 2.
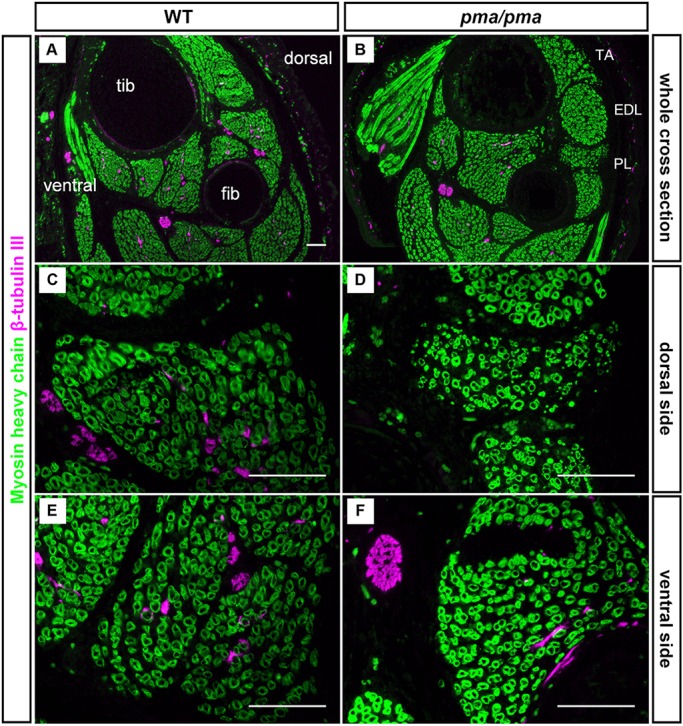
Absence of innervation of dorsal (peroneal) calf muscles in pma/pma homozygotes. Immunohistochemistry of cross-sections of stage-matched E16.5 wild-type (WT; left) and pma/pma homozygotes (right) using antibodies against myosin heavy chain to label muscle blocks (green) and β-III-tubulin to label nerves (red). (A,B) Low-magnification images with ventral muscles to the left and dorsal muscles to the right. (C,D) Higher-magnification images of dorsal muscle blocks showing innervation of wild-type dorsal muscles (C) but not of dorsal muscles in pma/pma homozygotes (D). (E,F) Magnification of ventral muscles showing innervation of both wild-type (E) and pma/pma (F) mice EDL, extensor digitorum longus; fib, fibula; PL, peroneus longus; TA, tibialis anterior; tib, tibia. Scale bars: 25 µm.
Fig. 3.
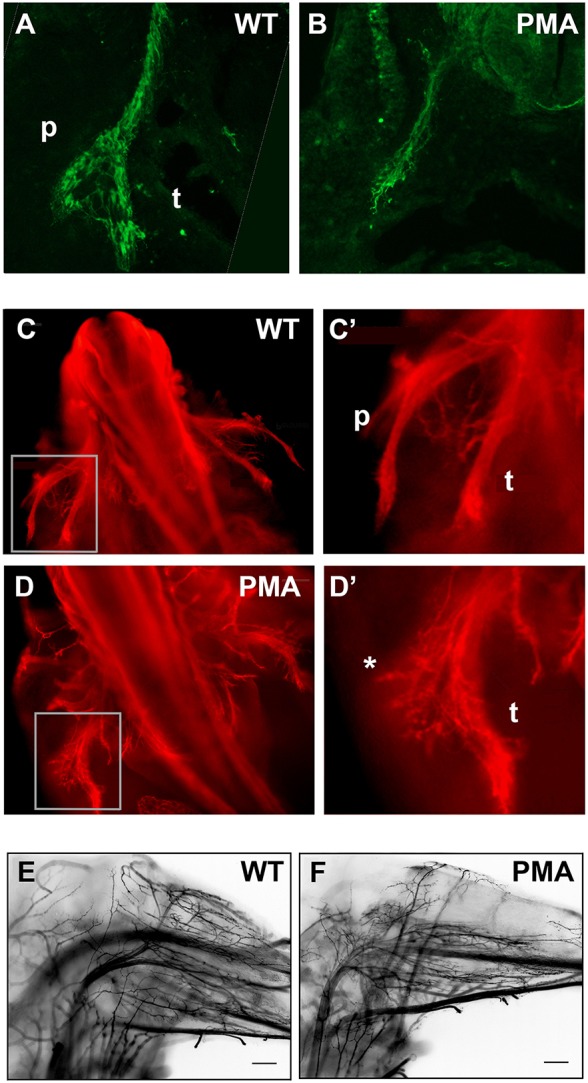
Retardation of nerve growth and abortive innervation of dorsal muscles in PMA mice. (A,B) β-III-tubulin immunohistochemistry (green) in transverse sections of stage-matched E11.75 wild-type (WT; A) and pma/pma mice (B). The wild-type sciatic nerve has projected further than that in the pma/pma mice and, unlike the PMA nerve, started to branch into discrete dorsal (peroneal, p) and ventral (tibial/sural, t) components. (C,D) Whole-mount β-III-tubulin immunohistochemistry (red) on wild-type (C,C′) and pma/pma (D,D′) embryos. C′ and D′ show magnifications of the boxed areas over the left-hand-side nerves in C and D, respectively. The peroneal (p) and tibial/sural (t) components are labelled. In pma/pma embryos, the tibial/sural branch is grossly normal, but only a few defasciculated axons are observable (asterisk) in place of the peroneal nerve, presenting a feather-like appearance. (E,F) Whole-mount β-III-tubulin immunohistochemistry on lower hindlimbs of E16.5 wild-type (left) and pma/pma embryos (right). Dorsal is to top: the dorsal muscles of the pma/pma foetuses are completely aneural, suggesting that the putative peroneal axons noted at E12.5 have not survived. Scale bars: 50 µm.
We previously observed degeneration of the peroneal muscles in adult pma/pma hindlimbs (Duce et al., 2010). To determine whether this was secondary to the loss of innervating nerves, we scrutinised early muscle development in pma/pma limbs. Abnormal myotube structures were observed specifically in myosin-positive developing dorsal muscles only at E16.5, with an approximate twofold increase in apoptosis (Fig. 4). These results confirmed at a molecular level the previous observations of regional muscular defects in PMA mice by our and other groups (Nonaka et al., 1986; Ashby et al., 1993; Duce et al., 2010) and showed that muscle failure occurred subsequent to failure of innervation.
Fig. 4.
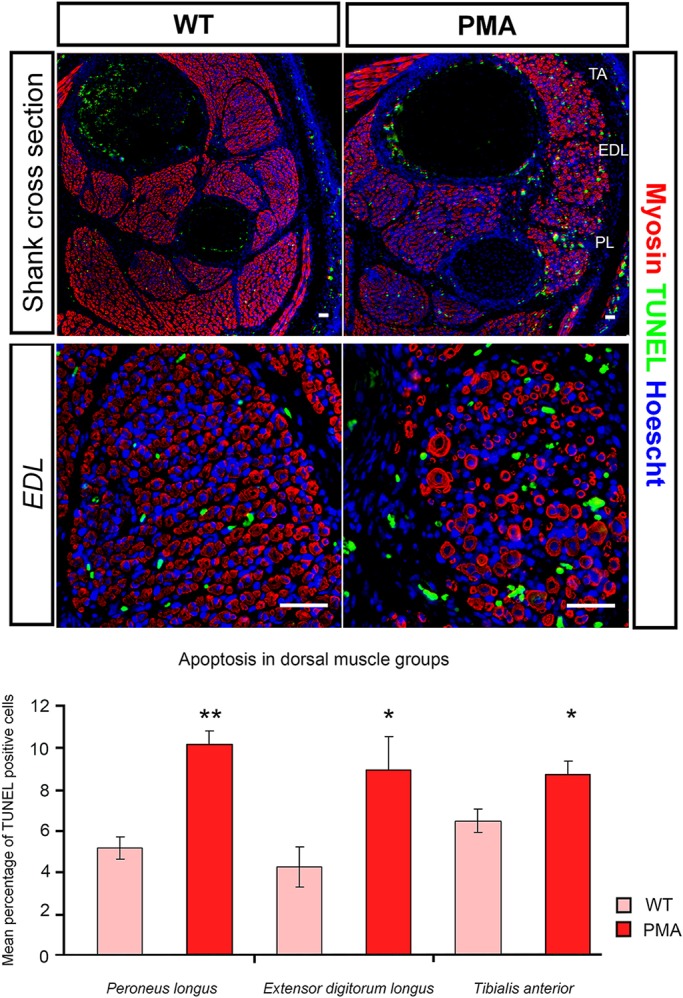
Increased apoptosis in dorsal muscle blocks of the pma hindlimb. Top: TUNEL labelling (green) to visualise apoptotic cells in cross-sections of E16.5 wild-type (WT; left) and pma/pma foetuses (right), combined with immunohistochemistry for myosin heavy chain (red) and Hoechst nuclear stain (blue). Higher magnification of the one dorsal muscle, the extensor digitorum longus, is shown. Bottom: Although apoptosis occurs in all muscles, the percentage of TUNEL-positive cells was significantly greater in the three major dorsal muscle blocks of pma/pma foetuses than in wild-type controls (n=8 for both groups). *P<0.05; **P<0.01. Error bars represent s.e.m. Scale bars: 50 µm.
Molecular specification is normal but axon growth is reduced and apoptosis increased in PMA motor neurons
To delineate whether the eventual absence of the peroneal nerve in PMA mice resulted from either the death of putative dorsal-projecting LMC neurons or their misdirection, we measured the cross-sectional area of the sciatic nerve in adult pma/pma and wild-type mice of equal weight. The mean area of the pma/pma nerve (98.98±0.43×103 µm2; n=5) was significantly reduced compared with controls (111.41±1.37×103 µm2; n=9; t-test, P=0.014), suggesting that motor neuron death occurred. Although terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) labelling at E11.5-E14.5 revealed no gross abnormal apoptotic events (Fig. 5A), by E16.5 the number of lumbar LMC nuclei was reduced in pma/pma homozygotes compared with wild-type stage-matched controls (Fig. 5B). Sacral LMC neurons were not affected. Patterns of proliferation were investigated by bromodeoxyuridine (BrdU) labelling and immunohistochemistry and were found to be normal in pma/pma embryos at E11.5 (Fig. S3), suggesting dorsal motor neuron death at E14.5-E16.5, secondary to failure to target the dorsal muscle blocks.
Fig. 5.
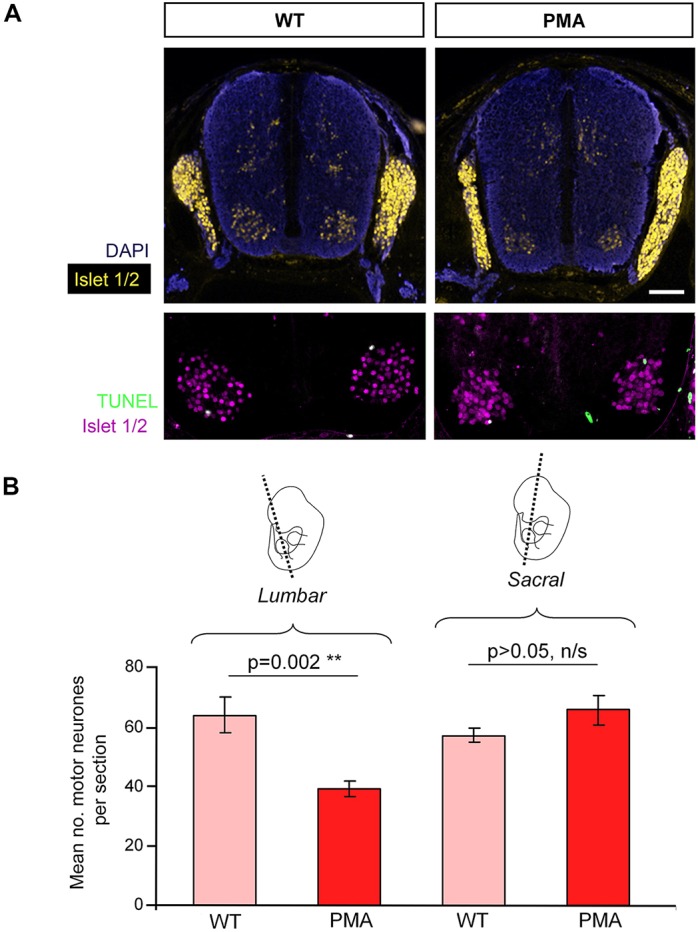
Apoptosis and motor neuron survival in lumbar neural tube of pma/pma homozygotes. (A) Immunohistochemistry on cross-sections of lumbar neural tube of E14.5 wild-type (WT; left) and pma/pma (right) foetuses. Islet 1 staining (yellow/magenta) is evident in the dorsal root ganglia and more weakly so in the lateral motor columns of both genotypes. There are rare apoptotic events in both genotypes (TUNEL labelling, green) but quantification was not possible as so few apoptotic cells were detected. (B) Quantification of motor neuron numbers in the neural tube of E16.5 wild-type and pma/pma foetuses. There are significantly fewer surviving motor neurons in pma/pma foetuses specifically at the level of the hindlimb (lumbar) but not more posteriorly (sacral). Error bars represent s.e.m. n/s, not significant. Scale bar: 50 µm.
Sciatic motor neurons of the hindlimb are derived from the LMC of the developing lumbar neural tube. The dorsal (peroneal) branch of the sciatic nerve is derived from neurons of the lateral component of the LMC (lLMC), which are characterised by expression of genes encoding marker LIM homeodomain proteins Lim1 (also known as Lhx1) and Islet 2 (Isl2), and the ventral (tibial/sural) branch is derived from the medial LMC neurons (mLMC) expressing Islet 1 and 2 (reviewed by Jessell, 2000). Lim1-deficient lLMCs project illicitly along the ventral pathway (Kania et al., 2000). Dorsoventral patterning of the neural tube and specification of the lLMC and mLMC were investigated in pma/pma mice. No defects of patterning were found: the dorsal and mediolateral markers Pax3 and Pax6, respectively, were localised normally from E11.5 to E14.5 (Fig. 6A). The molecular identity of the LMC populations was found to be maintained in pma/pma embryos: Lim1-positive lLMC neurons that should form the peroneal nerve were present as normal (Fig. 6B). These data suggest that any loss of peroneal neurons in the pma/pma sciatic nerve is not a primary failure of patterning or cell proliferation at E11.5-E12.5, but is secondary to the cellular changes that prevent normal patterns of axonal projection.
Fig. 6.

Normal neural tube patterning but reduced extension of motor neurons in the pma/pma hindlimb. (A) Immunohistochemistry on cross-sections of the neural tube at E11.5 in pma/pma embryos and stage-matched wild-type (WT) controls. Pax6 most strongly labels medial-ventral progenitors at the level of the motor neurons. Pax3 is localised to more-dorsal progenitors. Both genes are expressed normally in PMA mice and there is no evidence of dorsalisation of the neural tube or loss of motor neuron identity. (B) Immunohistochemistry for the lLMC marker Lim1 (green) and the mLMC marker Islet1 (magenta) at E12.5 in wild-type and pma/pma stage-matched embryos. Lim1-positive neurons are observable in the pma/pma lLMC, indicating that peroneal-fated neurons are correctly specified. DAPI nuclear counterstain is shown separately. There is some encroachment of Islet1-positive cells into the lLMC, but green Lim1-positive cells are discernible within the region of encroachment. (C-E) LMC axon growth in wild-type and pma/pma cultures. Double immunohistochemistry for β-III-tubulin (C; magenta) and Lim1 (D; green) in a LMC culture from a wild-type embryo. Time-lapse analysis of axon extension in motor neuron cultures (E) showed significantly reduced growth in pma/pma cultures. (See also Fig. S5.) *P<0.05. Error bars represent s.e.m. Scale bars: 50 µm.
No defects in genes required for normal dorsoventral patterning of the hindlimb mesenchyme were detected in E11.5-E12.5 pma/pma embryos by western blot or qPCR (Fig. S4). To investigate whether the motor neurons of the PMA mouse were inherently defective, E11.5 lLMCs were dissected from stage-matched pma/pma and wild-type embryos and cultured in identical media for 72 h. Immunocytochemical analysis showed projecting axons with β-III-tubulin and cell body localisation of Lim1, confirming their lLMC identity (Fig. 6C,D). Time-lapse measurement of growth cone position showed that pma/pma axons projected at approximately half the speed (2.09 µm/100 min; n=50) of wild-type axons (4.03 µm/100 min; n=97) (Fig. 6E, Fig. S5). These data confirm an autonomous defect of pma/pma motor neurons.
The data suggest an autonomous qualitative axon growth defect of motor neurons in pma/pma embryos, such that limb innervation is delayed. The reasons why this leads to failure of the peroneal branch but not of the tibial/sural are discussed below, but it is postulated that dorsal-specified axons miss their permissive time window for successful projection into the limb. It was concluded that the pma mutation has arisen in a gene required for normal extension and ultimate survival of lumbar motor neuron axons.
Genetic mapping of the pma mutation
The pma mutation was previously mapped to a 4.8 Mb region of chromosome 5 (Katoh et al., 2003). This region contains 67 genes with no standout clubfoot candidate. Genomic PCR was performed for 20 genes spanning the candidate region in pma/pma animals and all genes were found to be present, suggesting no large deletion (Fig. S6). We repeated the mapping using a higher density of microsatellites across the candidate region (full details described in supplementary Materials and Methods). The results are presented in Table 1. There were 12 potentially informative crossovers identified, which located the mutation to 2.5 Mb bounded by D5Mit166 (Chr5:133146598-133146706 bp) and D5Mit60 (Chr5:135715435-135715564 bp) (Fig. 7). This smaller candidate region contained 39 genes.
Table 1.
Microsatellite mapping of pma mutation in recombinant mice

Fig. 7.

Genetic mapping of the pma mutation. The region of chromosome 5 linked to the pma mutation by Katoh et al. (2003) is represented by the green horizontal bar, with the location of all genes listed below. Positions of microsatellites (μSat) used for genetic mapping are shown by black bars. The region eliminated from the candidate area by this study is shown by orange bars (see Table 1), with additional orange bars showing the minimum area defined by Katoh et al. (2003). The 3.04 Mb region sequenced in crossover animals, PMA and BALB/c founders is indicated by the blue bar. The red bar and pink shaded area represent the region within the crossovers identified by sequencing, and delineate a 0.86 Mb region most likely to encompass the pma mutation, containing 13 genes.
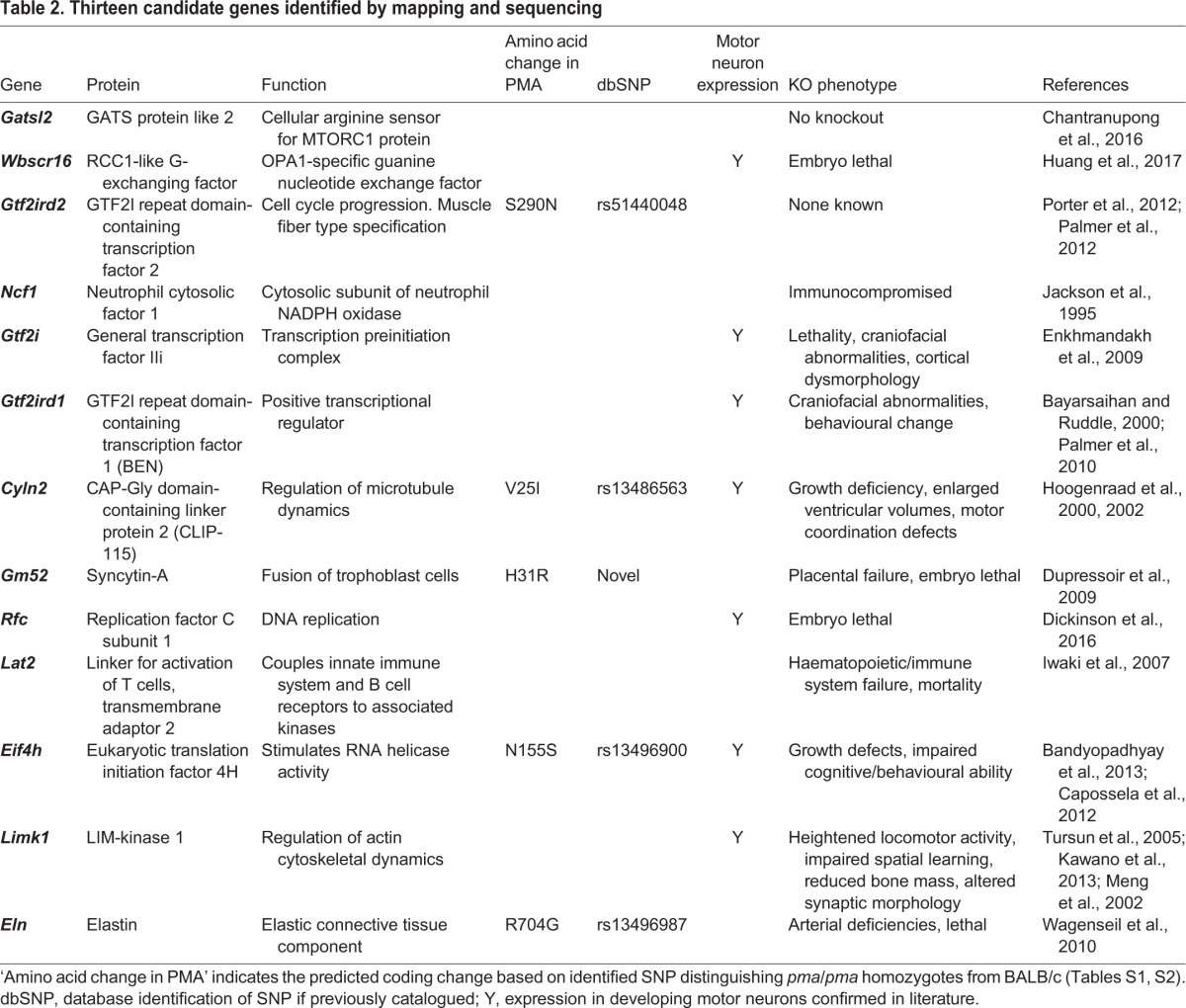
To further define the candidate region, targeted next-generation resequencing was performed on two recombinant mice (one with crossover distal to the mutation, and one with crossover proximal to the mutation), three parental pma/pma and three parental BALB/c mice. A 3.04 Mb stretch encompassing the entire candidate region between D5Mit166 and D5Mit60 was obtained from all mice. The full data set with details of alleles, location, sequencing depth and reproducibility, and inferred sites of crossovers is presented in Table S1. This analysis defined crossovers at approximate positions Chr5: 134514245 and Chr5: 134483438, representing a 0.89 Mb candidate region containing 13 genes: Gatsl2; Wbscr16 (Rcc1l); Gtf2ird2; Ncf1; Gtf2i; Gtf2ird1; Cyln2 (Clip2); Lat2; Gm52 (Syna); Rfc2; Eif4h; Limk1 and Eln (Fig. 7). Within this 0.89 Mb candidate region, nearly 4075 SNPs and small insertions or deletions (indels) were identified that were unambiguously homozygous for the pma allele in all clubfoot crossover animals and not found in BALB/c mice, any one of which could be the ‘pma mutation’. Restricting the analysis only to predicted nonsynonymous SNPs and indels within annotated genes identified 23 single nucleotide polymorphisms (SNPs) that were predicted to cause changes in either the amino acid sequence of the gene product (five SNPs – Table 2) or the untranslated region (UTR) of the processed mRNA (18 SNPs, all located in 3′ UTR) (Table S2). Nearly all these changes were known SNPs and could be identified as existing in the homozygous state in at least one of the C57BL/6, A/J, AKR/J, FVB/NJ and/or CAST/EiJ inbred strains without causing a clubfoot phenotype. The only novel nonsynonymous coding mutation was found in the Gm52 gene (Table 2). This gene encodes an envelope glycoprotein (now known as syncytin-A), which is a placenta-specific product required for normal architecture of the syncytiotrophoblast-containing labyrinth (Dupressoir et al., 2009). Gm52 knockout mice die in utero with placental failure, indicating that this gene is a weak functional candidate for the pma phenotype. These data suggest that the pma phenotype is not associated with an amino acid-coding mutation.
Table 2.
Thirteen candidate genes identified by mapping and sequencing
In addition, a microRNA, mmu-miR590-5p, is localised to this region (accession number MIMAT0004895), nesting within Eif4h (Fig. S7). No mutation was present within the mature 22 base pair miR sequence.
The possibility of gene amplification or deletion was directly tested. Genomic DNA was isolated from pma homozygotes and wild-type controls, and real-time PCR was performed for five primer pairs involving Cyln2, Gtf2ird1, Limk1 (two primer sets) and Pde6b, a gene 20 Mb outside the candidate region. No change in copy number in PMA mice was detected with any primer combination (Fig. S8).
The data suggested the pma mutation is not a null mutation (causing total loss of protein product or activity) in any of the genes in the candidate region. In addition to the lack of novel, potentially pathogenic coding mutations identified by sequencing, published knockout mice exist for ten of the genes, none of which produces a clubfoot phenotype or any other relevant motor neuron defect (Table 2). Furthermore, the pma candidate region is syntenic to the region of human chromosome 7 that is heterozygously deleted in patients with Williams–Beuren syndrome (Francke, 1999), a morphological and neurodevelopmental disorder associated with developmental delay, mental retardation and behavioural abnormalities. Williams–Beuren syndrome is not associated with clubfoot (Morris, 1999). Therefore, it was hypothesised that the problem was one of misexpression of one or more genes resulting from a regulatory mutation (i.e. one of the several thousand SNPs and small indels affecting an important enhancer or promoter sequence). This was investigated further, as detailed below.
Overexpression of Limk1 in pma homozygotes
Of the 13 genes within the mapped region, Limk1, Cyln2 and Eif4h were the best candidates to underlie the motor neuron defects in the PMA mouse on account of known or inferred function in central nervous system (CNS) axon guidance. Limk1 encodes LIM-domain kinase 1, an enzyme that phosphorylates and inactivates the actin-depolymerising protein cofilin (Yang et al., 1998). Limk1 activity modulates growth cone extension/retraction decisions and acts in a biochemical pathway that is downstream of EphA4, which was previously shown to be mutated in a clubfoot mouse model (Helmbacher et al., 2000). Cyln2 encodes the CAP-GLY domain-containing linker protein 2 (also known as CLIP-115), which acts as a microtubule-associate linking protein in the CNS (Hoogenraad et al., 1998, 2000). Cyln2 was shown to be strongly expressed in the developing motor neurons of the sciatic nerve (Fig. S9). Eif4h encodes a neurally expressed translation initiation factor responsible for modulating normal neuronal number and complexity in the CNS (Capossela et al., 2012).
Expression of Limk1, Eifh4 and Cyln2 was investigated in the PMA mouse by qPCR on cDNA from limbs and neural tubes of E11.5 pma/pma embryos and stage-matched wild types (Fig. 8A). No change in Cyln2 or EIf4h expression was detected, but the data suggested variable but significant four- to sevenfold upregulation of Limk1 in both the neural tube (P=0.014) and hindlimb (P=0.027) of pma/pma homozygotes.
Fig. 8.

Upregulation of Lim-kinase 1 (Limk1) in pma/pma mice. (A) qPCR data of the top-three candidate ‘clubfoot’ genes in the PMA mouse: Cyln2, Eif4h and Limk1. All data are means from three independent replicates, comparing E11.5 pma/pma homozygotes with C57BL/6 controls. cDNA was prepared separately from hindlimbs and neural tube of both genotypes. The error bars represent the 95% confidence interval from the statistical randomisation method from the REST software. Limk1 transcripts were significantly over-represented in both hindlimbs and the neural tube of pma/pma homozygotes. *P<0.05. (B) Western blot of E11.5 neural tube and hindlimbs of wild-type (WT) and pma/pma embryos. Limk1 and p-Limk1 were present at higher levels in pma/pma homozygotes. Total levels of cofilin (the main substrate of Limk1) were not affected but levels of phosphorylated cofilin increased, as would be expected with upregulation of Limk1. Cyln2 was not affected by pma genotype. β-actin was used as the loading control. (C) Immunohistochemistry for Limk1 (brown) in the E11.5 neural tube of wild-type and pma/pma embryos. Limk1 is present at higher levels in PMA mice, especially in the medial and lateral LMC. (D) Immunohistochemistry for p-Limk1 in dorsal (peroneal) and ventral (tibial) branches of the sciatic nerve at E12.5. p-Limk1 is present at higher levels in the developing peroneal nerve. See also Fig. S11.
The putative upregulation of Limk1 was confirmed by immunohistochemistry and western blot on protein extracts of E11.5 neural tube and hindlimbs. Normalised to β-actin, Limk1 and active (phosphorylated) p-Limk1 were localised at higher levels in pma/pma neural tubes and hindlimbs compared with stage-matched wild types (Fig. 8B). Furthermore, phosphorylation of the major substrate of Limk1, the actin-depolymerising protein, cofilin, was shown to be higher in pma/pma embryos, whereas total cofilin levels were unchanged. Cyln2 levels were also unchanged in pma/pma homozygotes. This suggests that the observed increase in Limk1 leads to phosphorylation and inactivation of cofilin, which in turn could impact the dynamics of actin cytoskeleton remodelling in the growth cones of the sciatic nerve.
We next wanted to understand the Limk1 profile throughout progression of clubfoot in the pma/pma mice. Repeating the western blots at E11.5-E16.5 showed that the upregulation of Limk1 expression was transient. Limk1 levels were increased in pma/pma mice at E11-E12.5, at the time when the peroneal nerve fails to form and enter the hindlimb, but were reduced to wild-type levels by E16.5 (Fig. S10).
To determine whether the upregulation of Limk1 detected by western blot indicated a genuine per-cell overexpression or misexpression of the gene in ectopic locations, immunohistochemistry was performed. In E11.5 wild types, at the level of the lumbar neural tube, Limk1 protein was observed to be present at higher levels in the neural tube, in particular the lLMC, of pma/pma homozygotes compared with controls (Fig. 8C). In pma/pma embryos processed in parallel with the wild types, there was a general upregulation of the gene, but protein levels were very high in the lLMC in the neurons that form the peroneal branch of the sciatic nerve.
Limk1 protein was found to be differentially localised in the peroneal and tibial branches of the wild-type sciatic nerve at E12.5. Immunohistochemistry showed that, in wild-type embryos, higher levels of p-LIMK1 were present in the dorsal (peroneal) branch of the sciatic nerve compared with the ventral (tibial) branch (Fig. 8D, Fig. S11).
Sciatic nerve plexus defects caused by jasplakinolide exposure mimic Limk1 overactivation
Regulatory mutations appear to have an important role in human pathology, particularly in hindlimb deformities (VanderMeer and Ahituv, 2011). Our data suggest that the pma mutation is in a cis-regulatory element of the Limk1 gene, increasing its expression.
The predicted consequence of increased Limk1 activity is increased phosphorylation (hence inactivation) of cofilin, leading to inhibition of actin treadmilling in the extending growth cone. This effect can be mimicked pharmacologically using jasplakinolide, a cell-permeable peptide that has been shown to inhibit actin depolymerisation in vivo (Bubb et al., 2000; Rosso et al., 2004). For practical purposes, the pharmacological manipulation was performed in chicken embryos: we have recently shown that disruption of neuromuscular function in the hindlimb of chicken embryos causes a clubfoot-like phenotype (N.V., E. Kilby, C.N., S.B., Z.M. and J.M.C., unpublished), establishing the chicken as a useful model of this condition. Jasplakinolide (20 µg/ml) was applied as a microsponge to the dorsal-lumbar region of Hamilton–Hamburger (HH) stage 20-21 chicken embryos in ovo, before nerve entry to the limb (approximately equivalent to E10.5 mice). This was followed by whole-mount immunohistochemistry for β-III-tubulin to identify changes in sciatic nerve projection compared with the contralateral untreated limbs and vehicle-treated controls. Axon projection defects were induced after 24 h in 11/29 jasplakinolide-treated limbs but in none of the 15 controls (Fisher exact test: P=0.0079) (Fig. 9). In particular, the rostral-most roots exiting the neural tube, which are the main contributors to the peroneal nerve, were truncated. It was concluded that peroneal nerve disruption is affected by addition of jasplakinolide, consistent with the model that Limk1 overactivation can prevent normal peroneal nerve development in vivo.
Fig. 9.
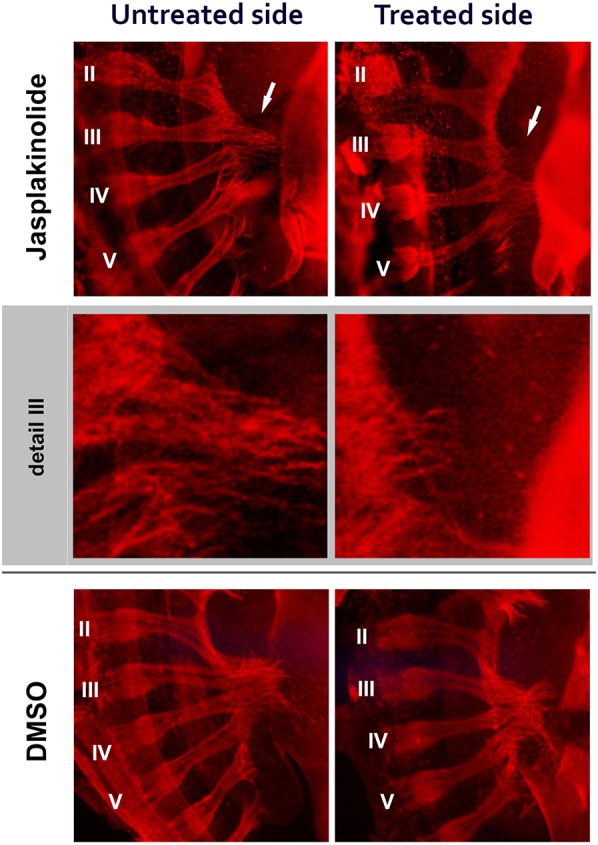
Jasplakinolide addition to the developing chicken embryo can induce sciatic nerve guidance defects in vivo. Whole-mount immunohistochemistry for β-III tubulin 16 h after application of a microsponge containing 20 µg/ml jasplakinolide (or 2% DMSO vehicle) to one side of the HH stage 22-23 chicken embryo at the level of the hindlimb. The spinal cord roots are numbered from I to VI, where root I (not shown) is associated with the anterior femoral nerve and roots III-V contribute to the posterior sciatic nerve. Root VI is only associated with the sciatic nerve later in development. Addition of jasplakinolide induced neuronal defects in the hindlimb. Top row shows jasplakinolide-treated (right) and contralateral untreated (left) sides highlighting the loss of axons on the treated side, especially from rostral-most root III, which contributes to the peroneal nerve (arrows). Shaded in grey is detail from root III. The vehicle, DMSO, was used as a control treatment and no defects were observed (bottom).
Electroporation of Limk1 in chicken neural tube disrupts sciatic nerve innervation of the hindlimb
Electroporation of a plasmid expressing Limk1 into the lumbar neural tube of stage 11 chicken embryos was performed to experimentally overexpress the gene in hindlimb motor neurons. This caused significant disruption and loss of plexus formation and nerve projection into the limb on the electroporated side of the embryo at stage 24, compared with the contralateral side (Fig. 10A). Electroporation of a control empty vector plasmid had no such effect. Cartilage preparations were made from 12 embryos that were incubated in ovo for a further 5 days. Although 9/12 were superficially normal, three showed unilateral clubfoot phenotype on the electroporated side (Fig. 10B). These data confirm that overexpression of Limk1 alone disrupts sciatic nerve formation and suggest that overexpression of Limk1 at the sensitive developmental period leads to clubfoot.
Fig. 10.
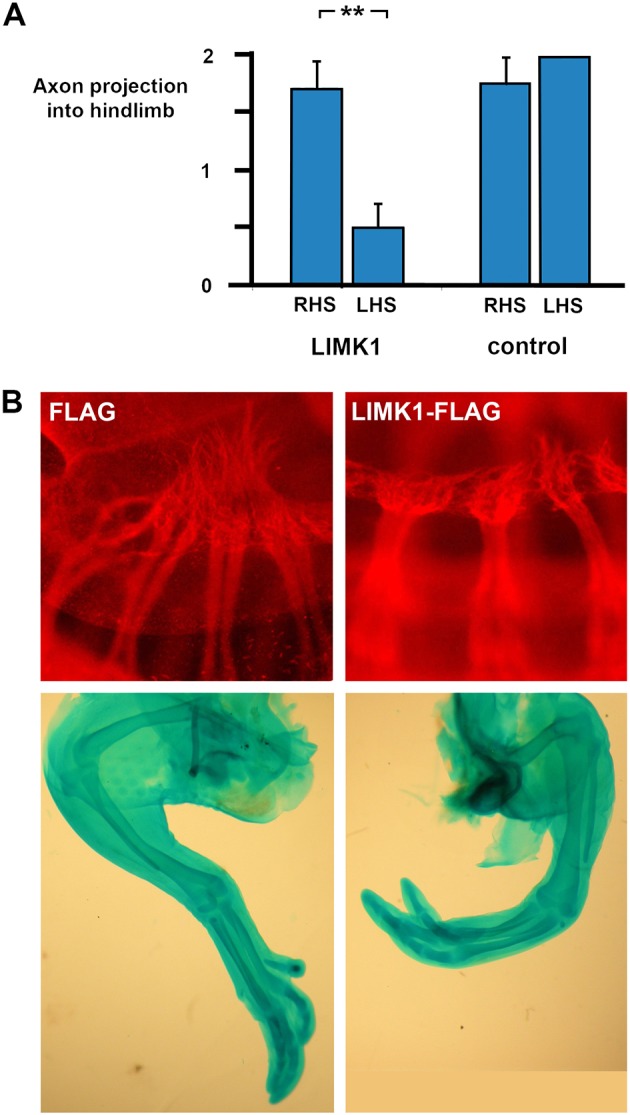
Electroporation of LIMK1 into chicken neural tube causes axon loss and clubfoot. Electroporation of plasmids expressing LIMK1 or empty vector controls into HH stage 11 chickens followed by immunohistochemistry for β-III-tubulin 72 h later or Alcian Blue cartilage staining after a further 5 days. (A) Nerve projection scored 0-2 as described in the Materials and Methods for electroporated (left-hand side, LHS) and non-electroporated contralateral sides (right-hand side, RHS) of each embryo, for LIMK1 and control vectors, and shows significant inhibition of axon growth in LIMK1-treated nerves, but not in empty-vector electroporations. LIMK1 electroporation: RHS nerve score=1.67±0.25, n=9; LHS score=0.5±0.22, n=8 (one embryo damaged cf. RHS). Control FLAG electroporation: RHS nerve score=1.75±0.25, n=4; LHS score=2±0.00, n=3. **P=0.001 (paired t-test). Error bars represent s.e.m. (B) (Top) Whole-mount β-III-tubulin immunohistochemistry (red) on chicken embryos showing normal sciatic plexus formation and axon projection (score 2) after an empty ‘FLAG’ vector transfection compared with representative failure of nerve plexus formation (score 0) after LIMK1 transfection. (Bottom) Alcian Blue cartilage preparation of (left) a control FLAG-electroporated chicken limb and (right) limb of one of the chickens (3/12) that exhibited a mild clubfoot-like phenotype after transfection with LIMK1.
Genetic interaction between EphA4 mutation and pma
EphA4−/− mice show a very high incidence of clubfoot (Helmbacher et al., 2000). Analysis of EphA4−/− E16.5 hindlimbs, prior to the failure of limb rotation that characterises clubfoot, showed that only two muscle blocks, the tibialis anterior and the extensor digitorum longus, were aneural, suggesting that the peroneus longus is of lesser consequence for clubfoot (Fig. S12).
We hypothesised that the EphA4 mutation acts on the sciatic nerve by modulating the Limk1-cofilin pathway. In support of this, increased labelling of p-cofilin was observed by immunohistochemistry in both E11.5 pma/pma and EphA4−/− mice compared with wild-type littermates, consistent with Limk1 regulating cofilin activity (Fig. 11A). To test a genetic interaction between EphA4 and pma mutations, EphA4+/− mice were bred with pma/pma homozygotes or wild-type C57BL/6 to generate littermates that were EphA4+/− pma/+, EphA4+/+ pma/+, or EphA4+/+ +/+ and EphA4+/− +/+. All progeny were scored as normal, unilateral or bilateral clubfoot at weaning on the basis of gross limb morphology, and dissections were performed to score the presence or absence of the peroneal nerve (Fig. S13). No clubfoot or nerve loss was observed in either EphA4+/+ +/+ mice (n=37 from these litters) or EphA4+/+ pma/+ (n=44). Mice that were heterozygous only for EphA4 were usually normal (19/24 mice) but 20.8% (5/24) had unilateral loss of peroneal nerve (either complete loss in four cases or retention of an extremely thin remnant in one case). By contrast, only 32% (12/37) of double heterozygote EphA4+/− pma/+ littermates were normal; 45% (17/37) were scored as unilateral clubfoot and showed loss of peroneal nerve on the affected side, whereas 21% (8/37) were bilateral clubfoot with loss or atrophy of nerves (Fig. 11B) (full data set: Table S3). The occurrence of bilateral clubfoot in the double heterozygotes had never been observed in any of the pma/+ or EphA4+/− single heterozygotes bred over the course of the project. The increased incidence of peroneal nerve loss in the double heterozygotes was significant (5/24 EphA4+/− +/+ mice versus 25/37 EphA4+/− pma/+. χ2=12.43, P=0.0004). Hence, the phenotype of the double mutation is more severe than the sum of the phenotypes of the single mutations. This is formal genetic evidence that the pma mutation lies in the same molecular pathway as EphA4. It suggests that the phenotypic occurrence of clubfoot can be associated with the cumulative effect of heterozygosity for predisposing mutations in more than one gene.
Fig. 11.
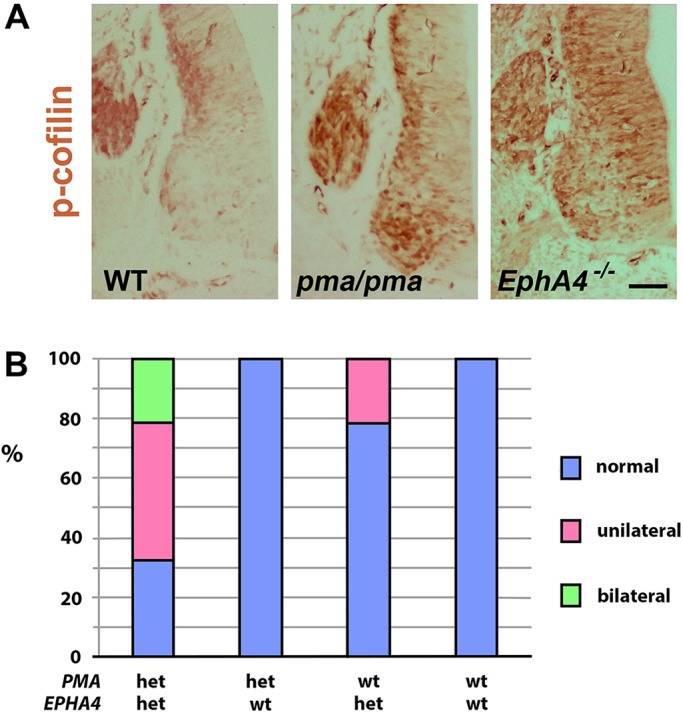
Interaction between pma and Epha4 mutations. (A) P-cofilin immunohistochemistry (brown) on transverse sections of neural tube of E11.5 wild-type (WT), pma/pma and Epha4−/− mouse embryos. Compared with wild type, p-cofilin is at higher levels in both pma/pma and Epha4 mutant mice, suggesting a shared biochemical pathway. Similar levels of protein in the dorsal root ganglia provide an internal control for staining. (B) Representation of frequency occurrence of peroneal nerve loss in postnatal day 21 mice that are compound heterozygotes for EphA4 and pma, compared with single heterozygous littermates for either gene, or littermates wild-type at both loci. See main text for details. Scale bar: 35 µm.
DISCUSSION
Clubfoot is a complex heritable abnormality with a range of described anatomical defects and no simple association with any one genetic or environmental factor (Miedzybrodzka, 2003). In this work, the spontaneous autosomal recessive mouse model, peroneal muscular atrophy (PMA), was used to understand the underlying developmental causes of clubfoot. The pma mutation was mapped, identifying several candidate genes, of which, Limk1, was upregulated in mutant mice. We showed in chickens that LIMK1 upregulation can cause sciatic nerve defects and a clubfoot phenotype. We also demonstrated proof of principle in mice that expression of the clubfoot phenotype can occur because of the cumulative effect of predisposing alleles at more than one locus.
Limk1 as the pma clubfoot gene
We have previously shown that Limk1 is dynamically expressed during embryogenesis (Lindström et al., 2011). Neural tube expression patterns of Limk1 and patterns of p-cofilin localisation described in this study are consistent with those previously reported in embryos (Phan et al., 2010). Limk1 is present in all neurons of the wild-type developing neural tube at E11.5, with higher levels in the lumbar LMC. P-cofilin is primarily localised to commissural neurons in wild-type mice, although with detectable staining in the LMC (this study). Immunohistochemical analysis confirmed the upregulation of Limk1 throughout the E11.5 neural tube in pma/pma mutants, particularly obvious in the LMC. Limk1 is a serine/threonine kinase that phosphorylates cofilin, inhibiting its actin filament depolymerisation activity; thus, overexpression of Limk1 would be predicted to inhibit actin turnover (Sarmiere and Bamburg, 2004). Consistent with this, we showed that motor neurons from the Limk1-overexpressing PMA mouse have high levels of p-cofilin and extend axons poorly, in comparison with wild type. In vivo, the most severe consequence of this would appear to be loss of the peroneal nerve. To determine empirically whether Limk1 overactivation can result in neuronal defect, Limk1 activity was mimicked in ovo pharmacologically using jasplakinolide to inhibit actin turnover. Delayed or repressed growth of the dorsal-projecting axons was observed in jasplakinolide-treated limbs, indicating that Limk1 pathway dysregulation may disrupt peroneal nerve development. These findings were further confirmed by additional electroporation of LMC in ovo to induce Limk1 overexpression during in vivo development of the chicken embryo.
Actin turnover, mediated by Limk1, the ADF/cofilin family, and slingshot proteins, is essential to growth cone motility (Endo et al., 2003; Wen et al., 2007). The importance of Limk1 for CNS neuronal morphology and function has been established (Meng et al., 2002), but the effects of changing Limk1 dosage are context dependent. Short-term overexpression of Limk1 in cultured hippocampal neurons promotes axon development through cofilin phosphorylation, but longer-term overexpression of Limk1 leads to growth cone collapse (Rosso et al., 2004). Similarly, in chicken dorsal root ganglia overexpressing Limk1, axon extension and growth cone mobility are inhibited (Endo et al., 2003). Phan et al. (2010) showed that overexpression of LIMK1 in chicken commissural neurons increases p-cofilin levels and stalls commissural growth cones, whereas lowering LIMK1 accelerates axon extension. The inhibition of commissural growth cone extension could be rescued by co-electroporation of a construct expressing a non-phosphorylatable cofilinS3A mutant. Hence, excessive stabilisation of F-actin at the growth cone either inhibits growth cone dynamics or facilitates myosin-driven growth cone retraction (Gallo et al., 2002).
The fact that Limk1 is widely expressed in CNS neurons makes it counterintuitive that the PMA mouse has only a hindlimb phenotype. Similarly, Gdnf/Ret and Hoxc10/d10 double mutants all lead specifically to peroneal nerve loss, despite the expression of these genes in many other neurons (Tarchini et al., 2005; Kramer et al., 2006). Why the peroneal nerve should be overtly susceptible to failure is not known, but we show in this study that this nerve has higher basal levels of phosphorylated (active) Limk1 than the tibial nerve, which suggests that the pma mutation raises this further to a level where nerve growth stalls. Limk1 activity should be assayed in the other models above to determine whether this is a general mechanism.
The timing of Limk1 overexpression overlaps the end of the previously described ‘waiting period’ of the sciatic nerve plexus (Wang and Scott, 2008). This waiting period is characterised by the arrest of axonal growth at the hindlimb sciatic nerve plexus, before limb innervations initiate, mediated through retinoic acid signalling. In Ret−/− mice (Kramer et al., 2006), the ventral pathway choice becomes available to dorsal axons that are unable to project dorsally, indicating that delayed axons may also be able to select the ventral pathway. Therefore, timing of limb innervation is crucial to correct patterning, and we hypothesise that reduced growth rate of pma motor neurons causes dorsal-fated axons to miss their time window for entry to the limb, resulting in atrophied peroneal muscles and clubfoot.
Limk1/EPHA4 interaction
EphA4−/− animals show a similar phenotype to the PMA mice and specifically lack peroneal nerves (Helmbacher et al., 2000). EPHA4/Ephrin-A receptor signalling is known to modulate Limk1 activity: it leads to phosphorylation of ephexin and Src kinases, which signal through small GTPase RhoA and the Rho-associated protein kinase (ROCK) to modulate the phosphorylation of Limk1 and Limk2 (Maekawa et al., 1999; Sahin et al., 2005; Sumi et al., 2001). In addition to the genetic and biochemical interaction between Limk1 and EPHA4 shown here in clubfoot mice, it has also been shown that overactivation of Src kinases redirects dorsal axons to the ventral mesenchyme in the limb (Kao et al., 2009), suggesting a potential ‘clubfoot pathway’ controlling innervation of the calf muscles.
The PMA mouse as a model of human clubfoot
Previous clinical candidate gene and genome-wide association studies have implicated several loci in the development of clubfoot (Basit and Khoshhal, 2017). These include the hindlimb-specifying genes PITX1 and TBX4 described above, members of the limb patterning HOXA and HOXD clusters (Ester et al., 2009), mutations in genes encoding pro-apoptosis caspases (Ester et al., 2009; Heck et al., 2005), and genes required for normal metabolism of environmental factors, such as folic acid and tobacco smoke (Sharp et al., 2006; Hecht et al., 2007). Collagen (COL9A1) and a number of genes encoding muscle contractile proteins may also be mutated in clubfoot, supporting the model of hindlimb rotational arrest resulting from musculoskeletal dysfunction (Liu et al., 2007; Weymouth et al., 2011). The only other gene required for actin cytoskeletal function to have been associated with clubfoot is that encoding Filamin B (Yang et al., 2016). LIMK1 has not directly been implicated in clubfoot in humans; however, a high incidence of clubfoot occurs in patients with microduplications in chromosome 7q11.23, syntenic with the pma candidate region on mouse chromosome 5. Although Williams–Beuren syndrome (deletion of 7q11.23) does not cause clubfoot, patients with duplications of the region have up to a 25% incidence of clubfoot and all patients with microduplications and clubfoot have duplicated LIMK1 (Torniero et al., 2007; Van der Aa et al., 2009). This is consistent with the link between clubfoot and increased Limk1 activity shown in this study.
Circumstantial and direct anatomical evidence suggests that most patients with clubfoot do have a peroneal nerve but many cases, often associated with a drop-toe phenotype, have a neurological origin, including peroneal nerve palsy (Song et al., 2008; Edmonds and Frick, 2009; Yoshioka et al., 2010). Peroneal nerve dysfunction is rare in clubfoot in humans (8/837 patients in the study by Yoshioka et al., 2010) and the complete peroneal nerve loss described in pma/pma mice cannot be regarded as a common cause of clubfoot. Our work outlined in this study, together with other work (N.V., E. Kilby, C.N., S.B., Z.M. and J.M.C., unpublished), is consistent with a hypothesis that muscle loss is the primary cause of clubfoot. Whether the muscle weakness is a direct failure of the muscle, or secondary to other defects, such as failure of vasculature or of nerve function, is perhaps of little consequence to the clubfoot phenotype presented, although the complex clubfoot cases with an underlying neurological basis are among those that do not respond well to normal treatment (i.e. Ponseti manipulation) (Song et al., 2008; Yoshioka et al., 2010). Therefore, identification of the genetic pathway that underlies neurological clubfoot is central to the screening and optimisation of patient care. Upregulation of LIMK1 in human tissue samples cannot be assayed by normal genomic sequencing, but the EPHA4–ephexin–SRC–small GTPase–LIMK1–cofilin pathway has many components that can be screened by simple exome sequencing. Thus, our data support a general model that predisposing mutations in multiple genes in pathways affecting (directly or indirectly) muscle development in the lower limb are causal to clubfoot. The fact that so many such genes exist may well explain why the genetics of clubfoot is so complex in humans.
MATERIALS AND METHODS
Mice and genetic mapping
Experiments were performed under licence after approval by the Aberdeen University Animal Ethical Review Committee. The pma/pma homozygotes were obtained from Professor Cheryll Tickle (University of Bath, UK) on an inbred CF1-derived background, and maintained by homozygous mating. The mice were poor breeders, so for generation of experimental animals and controls, the pma mutation was bred onto CD1 and C57BL/6 lines. Wild-type CD1 and C57BL/6 mice were used as controls. The PMA phenotype was qualitatively identical irrespective of genetic background, with full bilateral clubfoot. The data presented here are for CD1 background pma/pma mice unless stated otherwise (e.g. in the genetic mapping and experiments investigating interaction with EphA4). Embryos were stage matched according to date of gestation, crown-rump length and forelimb morphology. The pma mutation produced an idiopathic clubfoot phenotype on all pure and mixed genetic backgrounds.
Epha4+/− mice were obtained from Patrick Charnay (Ecole Normale Supérieure, Paris, France) and maintained on a C57BL/6 background.
For timed matings, the date of vaginal plug was taken as E0.5.
A preliminary pan-genomic screen for informative microsatellite alleles showed that the original pma inbred CF1-derived strain maintained by our group was genetically more distinct from BALB/c than from C57BL/6 (Table S4). Therefore, Balb/c crossing was chosen for genetic mapping. Homozygous pma/pma male mice on the original inbred CF1-derived background were mated with wild-type BALB/c females, and the F1 pma/+ progeny crossed to the pma/pma males to derive a mixed pma/pma and pma/+ B1 (F2) generation, within which it was inferred would be some informative crossovers to map the pma mutation. All mice were scored at birth as ‘clubfoot’ (inferred pma/pma) or ‘normal’ (inferred pma/+). Mice were killed and dissected to confirm the absence or hypotrophy of the peroneal nerve in clubfoot mice and presence of normal peroneal nerve in mice without clubfoot. Tail-tip DNA was isolated from all clubfoot F2 mice (also three non-clubfoot mice, two F1s and both parental strains) by proteinase K digestion (80 µg/ml at 56°C overnight) followed by ethanol precipitation (3:1 ethanol:sample, −20°C for several hours and centrifugation at 12,000 g for 20 min). Microsatellite genotyping was performed by PCR, using nine primer sets for microsatellites spanning and within the candidate region on chromosome 5 (Table S5). Fluorophore-conjugated forward primers (fluorophores 6FAM, VIC, PET or NED) were synthesised by Eurogentec. PCRs were multiplexed in groups of three using three different fluorophores conjugated to the forward primers (D5Mit218, D5Mit166 and D5Mit428 together; D5Mit282, D5Mit219 and D5Mit60; D5Mit33, D5Mit32 and D5Mit97) (Table S5). Each PCR was run for 35 cycles with an annealing temperature of 61°C and the product sizes analysed using an Applied Biosystems3130 Series Genetic Analyzer. Expected band sizes were determined using DNA from founder pma/pma homozygotes and BALB/c mice, as well as their F1 progeny. All PCRs were run on the clubfoot backcross generation mice to identify clubfoot mice that were heterozygous at any of the microsatellite loci, indicative of an informative crossover event.
EphA4 genotyping and crosses
Genomic DNA was isolated from tissue samples by proteinase K digestion using the Qiagen DNA Mini Kit according to the manufacturer's instructions, with elution in 100 µl of buffer AE. Genotyping PCR was performed on EphA4-mutant mice and littermates using primers SEK1.1 (5′-TTCTGCCACTGCTATTGGTCACGAG-3′), SEK1.2 (5′-AACTGGTTCTGAGCTCCAGAAGACC-3′) and SEK1.3 (5′-GATGGGCGCATCGTAACCGTGCATC-3′), with an annealing temperature of 65°C, for 35 cycles. The EphA4 wild-type allele produced a band of ∼200 bp from primer combination SEK1.1/1.2, and the mutant allele a ∼300 bp band from primer combination SEK1.1/1.3 (Helmbacher et al., 2000).
For study of compound heterozygotes, EphA4+/− mice were crossed to pma/pma homozygotes to produce pma/+ heterozygotes that were either EphA4+/− or EphA4+/+. All mice were scored at or before weaning superficially as ‘normal’, unilateral clubfoot, or bilateral clubfoot. For statistical analysis, to avoid misdiagnosis of gross morphology, all mice from these crosses were dissected and the peroneal nerve was scored as ‘normal’, ‘absent’, or ‘thin’ if a remnant of peroneal nerve was visible (Fig. S13). Nerve scoring was performed blind by an observer unaware of the genotype of the mice.
Gene detection and copy number variation assay
Livers were dissected from one C57BL/6 control mouse and one pma/pma homozygote, and genomic DNA was isolated from 50 mg samples using the MagMAX DNA Multi-Sample Kit (ThermoFisher Scientific) according to the manufacturer's protocol, with elution into 300 µl volume. gDNA was diluted to 5 ng/µl.
Presence or absence of genes was assayed by genomic PCR using primers for 20 genes described in Table S6.
Copy number variation was assayed by real-time PCR using the TaqMan(R) Copy Number Assay (Applied Biosystems) using manufacturer's validated assays for murine Limk1 (Mm00561325_cn and Mm00165295_cn), Clip2/Cyln2 (Mm00149430_cn), Gtf2ird1 (Mm00158525_cn) and Pde6b, a linked gene that lies outside the pma candidate region (Mm00164399_cn). All genes were multiplexed and normalised to the manufacturer's internal reference assay (RNAse P).
Histology and immunohistochemistry
Samples were fixed in 4% paraformaldehyde (PFA) in PBS overnight, washed several times in PBS and dehydrated through a series of ethanol and xylene washes for embedding in paraplast wax embedding medium (Sigma). Wax sections (7 µm) were then cut. For staining, the samples were dewaxed by immersing the slides in histoclear solution (National Diagnostics) and then rehydrated through an ethanol series. Antigen retrieval was performed by boiling slides, 4×5 min, in 0.01 M sodium citrate, pH 6. The samples were blocked in PBS, 4% normal serum, 0.3% bovine serum albumin (BSA) for 1 h. Primary antibody was added and diluted in blocking buffer at 4°C overnight. The primary antibodies used were: anti-β-III-tubulin (T2200, Sigma; 1:1000); anti-PAX6 [PAX6c, Developmental Studies Hybridoma Bank, University of Iowa (DSHB); 1:400]; anti-Cyln1/Clip2 (ab80976, Abcam; 1:200); anti-myosin heavy chain (MF20, DSHB; 1:20); anti-myogenin (F5D-c, DSHB; 1:200); anti-BrdU (ab8955, Abcam; 1:200); anti-Limk1 (611749, BD Transduction Laboratories; 1:200); anti-phospho-LIMK1 (ab38508, Abcam; 1:200); anti-cofilin (ab47401, Abcam; 1:200); anti-phospho-cofilin (sc-21867, Santa Cruz; 1:400); anti-EPHA4 (AF641, R&D Systems; 1:200); anti-EPHB4 (sc-5536, Santa Cruz; 1:100); anti-frizzled 9 (AF2440, R&D Systems; 1:100); anti-islet 1 (sc-23590, Santa Cruz; 1:400); anti-islet 2 (sc-66454, Santa Cruz; 1:400); anti-Pax3 (PAX3-s, DSHB; 1:40); anti-Lmx1b (sc-21231, Santa Cruz; 1:100); anti-Lim1 (4F2, DSHB; 1:20); and anti-neurofilament-associated antigen (3910-s, DSHB; 1:40). No-primary-antibody controls were performed for each antibody. After washing with PBS, fluorescent secondary antibodies were used 1:250 in blocking buffer for 3 h at room temperature as follows (all Molecular Probes, Invitrogen): Alexa594-conjugated chicken anti-rabbit (A21442); Alexa594-conjugated donkey anti-rabbit (A21207); Alexa594-conjugated goat anti-mouse IgG2a (A21135); Alexa594-conjugated goat anti-mouse IgG1 (A21125); Alexa568-conjugated donkey anti-mouse (A10037); Alexa488-conjugated rabbit anti-goat (A11078); Alexa488-conjugated goat anti-mouse IgG2b (A21141); Alexa488-conjugated goat anti-mouse IgG1 (A21121); Alexa488-conjugated donkey anti-rabbit (A21206); and Alexa350-conjugated goat anti-rat (A21093). After PBS washes, the samples were mounted in Vectashield with DAPI (H-1200, Vector Laboratories) to counterstain nuclei.
For immunohistochemistry with a 3,3′-diaminobenzidine (DAB) endpoint, the samples were treated as described above but, before the antigen retrieval, the slides were immersed in 3% H2O2 (Sigma-Aldrich) in PBS for 15 min and then washed in PBS for 5 min. The secondary antibodies were biotin-conjugated rabbit anti-mouse (E0354; DakoCytomation) and biotin-conjugated goat anti-rabbit (B7389; Sigma). After PBS washes, the samples were treated with avidin/biotin complex (ABC) reagent (‘R.T.U. Vectastain Kit’; Vector Laboratories) and then incubated in 830 µg/ml DAB (Sigma-Aldrich), 20 mM Tris pH 7.4, 0.005% w/v H2O2. The DAB reaction was stopped by immersing the samples in running water. The samples were counterstained in Haematoxylin.
Measurement of nerve diameters was performed after dissection and fixation of the sciatic nerve in adult mice approximately midway along the femur. Nerves were fixed in 4% PFA, dehydrated and embedded vertically in wax. Transverse sections (7 µm thick) were cut, rehydrated and stained with Toluidine Blue. Sections were imaged at ×200 on a scale-calibrated Nikon Eclipse 400 light microscope. The diameters of tibial, sural and, if present, peroneal branches of the nerve were measured in at least two places on each of three sections, and the mean total area calculated in µm2.
Whole-mount antibody staining (immunohistochemistry)
Mouse or chicken embryos were fixed in Dent's fixative (1 part DMSO: 4 parts methanol) for 24 h at 4°C with rocking to permeabilise the tissue. Embryos were bleached with Dent's bleach (1 part H2O2: 2 parts Dent's fixative) for 24 h at 4°C with rocking. Embryos were rinsed five times with methanol and post-fixed in Dent's fixative for at least 24 h. Embryos were washed three times with 1×PBS for 1 h, with rocking. Primary antibody, anti-β-III-tubulin (TuJ1, Sigma), was applied at a dilution of 1:1000 in blocking solution (75% 1×PBS, 20% DMSO, 5% donkey serum). β-III-tubulin was used because it is highly expressed in differentiating neurons. Embryos were incubated overnight at room temperature. Embryos were rinsed three times with 1×PBS and then washed five times with 1×PBS for 1 h. Secondary antibody, Alexa 594-donkey anti-rabbit (Molecular Probes), was applied at 1:300 in blocking buffer. Embryos were incubated overnight at room temperature and in the dark.
Embryos were rinsed three times with PBS, then washed with PBS, 5×1 h, 1:1 PBS:methanol, 5 min, and methanol, 3× for 20 min. Following the third methanol wash, half of methanol was removed and replaced with BABB (1 part benzyl alcohol: 2 parts benzyl benzoate) for 5 min. This solution was replaced with 100% BABB and kept at 4°C in the dark. All analyses of the whole-mount IHC results were carried out on a Nikon fluorescence dissecting microscope.
In vivo chicken work
Fertilised White Leghorn eggs (Henry Stewart & Co., Louth, Lincolnshire, UK) were stored at 14°C until needed. To allow embryonic development, eggs were incubated in humidified incubator at 38°C for the required amount of time to reach the desired developmental stage.
Jasplakinolide exposure
A 1 mg/ml solution of jasplakinolide (J4580, Sigma) was prepared in DMSO and diluted to 20 µg/ml with PBS. The eggs were windowed and the stage of the embryo was visually confirmed. Microsponges (1 mm×1 mm) soaked in 20 µg/ml jasplakinolide were placed on the dorsal top of the lumbar and hindlimb regions of HH stage 22 embryos (Fig. S14). Sponges soaked in 2% DMSO were used as vehicle controls. The eggs were closed with adhesive tape and incubated at 38°C for 16 h. Embryos were fixed using Dent's fixative and processed for whole-mount β-III-tubulin immunohistochemistry as described above.
Electroporation
pWZL_Neo_Myr_Flag_LIMK1, expressing full-length Flag-tagged human LIMK1, and control pWZL_Neo_Myr_Flag plasmids were obtained from Addgene (plasmids #20512 and #15300, respectively, deposited by Jean Zhao) and prepared to a concentration of 5 mg/ml using Qiagen Endonuclease-free DNA Maxi Kit. In ovo electroporation was performed as described previously (Odani et al., 2008; Nakamura and Funahashi, 2001). LIMK1-Flag DNA or Flag DNA mixed with Fast Green (dye tracer) was injected into the lumbar region of the neural tube of HH stage 11 (E1.5) chicken embryos in windowed eggs (Hamburger and Hamilton, 1951). Platinum-coated electrodes spaced at 4 mm were placed on both sides of the injection site, parallel to the long axis of the embryo. The anode was placed on the left of the neural tube and the cathode was placed on the right of the neural tube. Five square pulses (25 volts, 50 ms/s) were applied by a CUY21 electroporator (Nepagene) and the eggs resealed. Surviving embryos were analysed 48-72 h later for whole-mount β-III-tubulin immunohistochemistry, or after a further 5 days for cartilage preparation. Nerve growth was blind-scored independently by two workers, for LIMK1 and control-electroporated embryos, with ‘0’ representing abnormal plexus formation and/or complete failure of axon to project towards the limb, ‘2’ representing normal projection into the limb (Fig. 10), and ‘1’ representing complete or partial failure of projection from at least one lumbar segment.
Alcian Blue cartilage staining
Chicken embryos were fixed overnight in 5% trichloroacetic acid (TCA; Sigma) at 4°C. The next day, the samples were incubated in 0.1% Alcian Blue (Sigma) in 70% ethanol for 8 h and de-stained overnight in 1% HCl in 70% ethanol at 4°C. The embryos were washed 3×1 h in 100% ethanol and then cleared and stored in methyl salicylate (Sigma).
TUNEL
Analysis of apoptosis was performed on tissue sections using the In situ Cell Death Detection Kit (fluorescein) (11684795910, Roche Diagnostics) according to the manufacturer's instructions, with permeabilisation using Proteinase K according to instructions. Apoptosis of muscle blocks was quantified manually on transverse sections of the mid-calf of pma/pma homozygotes and wild-type controls. Each of eight limbs was treated as an independent data point. Four sections were counted per limb, and used to count a mean and standard error of % apoptotic cells for each muscle block in wild types and mutants.
BrdU analysis
Timed pregnant mice were given a single intraperitoneal injection of 10 mg/ml BrdU to label all cells in S phase, and killed 1 h later. Embryos were fixed in 4% PFA, wax embedded and sectioned for anti-BrdU immunohistochemistry using antibodies and conditions as described above.
Motor neuron cultures
E11.5 pma/pma and wild-type embryos were stage matched on basis of crown-rump length. The lumbar neural tube was microdissected. The neural tube was further dissected by removing the dorsal region, allowing for purification of the ventral spinal cord, where the LMC neurons are located. A matrix made of bovine collagen (BD Biosciences), rat collagen (BD Biosciences) with DMEM (Gibco) and 20 µl sodium bicarbonate was prepared on a 4-well plate. DEMEM/F12 with 5% foetal bovine serum and 1% pen/strep was used as the culture medium. The dissected neural tube tissue was placed on the collagen matrix and incubated for 3 days at 37°C, 5% CO2. Fresh culture medium containing 25 mM HEPES buffer was added to the explants before initiating the time-lapse experiment. Time-lapse analysis of growing axons was performed by tracking the movement of multiple growth cones in phase-contrast over a 16 h period using a Leica Inverted microscope at 200× magnification with Volocity imaging software. Full culture conditions are provided in the supplementary Materials and Methods.
Next-generation sequencing and analysis
Genomic DNA was isolated from livers of three Balb/c, three pma/pma and two crossover mice (identified by microsatellite analysis above) using DNAzol (ThermoFisher Scientific) according to the manufacturer's instructions. Capture and sequencing of the 2.5 Mb Chr 5p23 region was performed on a contract basis by The GenePool (Next Generation Sequencing and Bioinformatics Platform at the University of Edinburgh, UK). In brief, Illumina sequencing libraries were prepared from each parental strain mouse and the two crossovers. The eArray web-based application (Agilent) was used to design custom DNA baits to capture candidate region sequences. The SureSelect system (Agilent) was used for sequence capture. Captured sequences from each sample were sequenced to at least 30× coverage using 50 bp-end reads on the Illumina platform (GAITx) in-house at The GenePool. Raw sequences were returned to the University of Aberdeen as .gz compressed text files for analysis.
BALB/c sequence reads were aligned to BALB/c reference genome mm9 using bwa (Li and Durbin, 2009), discrepancies identified and a new BALB/c reference generated. The PMA mice and crossovers were then aligned against the new reference and all SNPs and indels identified. Reads were re-aligned around indels and the base quality score recalibrated using GATK (McKenna et al., 2010). Duplicates were marked using Picard (http://broadinstitute.github.io/picard/). SNPs and indels were annotated as exon, intron, untranslated or intergenic sequences. Variant analysis was performed with Samtools 0.1.14 (Li et al., 2009). Annotation of variants was performed with Seqgene v 2.3 (Deng, 2011). SNPs and indels with variant and mapping quality >20 and present in all replicate samples were marked as potentially significant.
Quantitative PCR
For quantitative PCR (qPCR) analysis, neural tubes and hind limbs were microdissected from E11.5 pma/pma and wild-type C57BL/6 mouse embryos. Total RNA was isolated using the peqGOLD total RNA kit (Peqlab) according to the manufacturer's instructions. cDNA was synthesised by reverse transcription using SuperScript II Reverse Transcriptase kit (Life Technologies) using poly-T primer (Promega). Primers used to amplify genes in the pma candidate region were manufactured by Sigma-Aldrich and were as follows: Limk1 forward, 5′-GCTACTTTGTTGCACCTGGAG-3′; Limk1 reverse, 5′-CACACAGGCAACTCGCTTC-3′; Eif4h forward, 5′-TCAGGAAAGGTGGACCTGAT-3′; Eif4h reverse, 5′-TCCCATCCACCTCTAGATTCTC-3′; Cyln2 forward, 5′-CGGTTCTGCCAACGGTATT-3′; Cyln2 reverse, 5′-GCGGTCAGTGTTTGTCCTC-3′. qPCR was performed using the Roche Universal Probe system. For each sample, 1 μl of a probe, 0.2 μg cDNA, 10 μl Sensimix (Bioline) and 4 μl DEPC-treated water were combined in each well of a 384-well plate. All reactions were run on a Roche Lightcycler 480 (Roche) and denatured at 95°C for 15 min, followed by 50 cycles of 15 s at 95°C and 30 s at 60°C. All samples were in triplicate and normalised to Gapdh in the same run. Statistical analysis of changes in gene expression was performed using the REST statistical package.
Western blot
For western blotting, proteins were extracted from embryonic neural tube and hindlimbs in 5% SDS, 1:100 protease and phosphatase inhibitor cocktails (P2714 and P5726, Sigma-Aldrich) and stored at −20°C. SDS-PAGE was performed in 12% polyacrylamide gels with 10% SDS and proteins were blotted onto nitrocellulose. Membranes were blocked in Tris-buffered saline (TBS) with 0.3% Tween-20 and 10% skimmed milk, and incubated in primary antibody as above, diluted in 2% BSA, 0.05% Tween-20, and TBS overnight at 4°C and washed. Horseradish peroxidase-conjugated species-specific secondary antibody was applied for 1 h before washing in TBS and 0.05% Tween-20, with detection using the enhanced chemiluminescence (ECL) kit (Amersham). Peroxidase-conjugated anti-β-actin (A3854, Sigma) was used as an internal loading control. Secondary antibodies were peroxidase-conjugated anti-rabbit (#7074, Cell Signaling Technology) and peroxidase-conjugated rabbit anti-mouse (A9044, Sigma).
Statistics
The randomisation method of the Relative Expression Software Tool (REST) software was used for qPCR analysis. All error propagation calculations followed the equation:
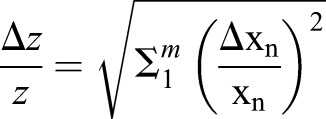 |
where z is the mean calculated value, Δz is the calculated standard error of the mean (s.e.m.), x is the experimental value, Δx is the experimental s.e.m., and m is the categories considered.
χ2 tests were performed online at https://www.graphpad.com. Fisher exact tests were performed online at http://www.socscistatistics.com/tests/fisher/Default2.aspx.
Supplementary Material
Acknowledgements
We thank Professors Cheryll Tickle and Françoise Helmbacher for discussion and reagents. We thank staff at the Aberdeen Medical Research Facility for specialist technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.M.C.; Methodology: J.M.C., N.O.L., C.N., K.W., C.M., S.D., Z.M., N.V.; Software: C.M.; Formal analysis: J.M.C., N.O.L., C.N., K.W., C.M., M.N., N.V.; Investigation: J.M.C., N.O.L., C.N., K.W., R.H.C., Z.K.R., A.M.F., I.M., K.O., M.N., S.B., S.D.; Resources: J.M.C.; Data curation: J.M.C.; Writing - original draft: J.M.C., C.N.; Writing - review & editing: N.O.L., C.N., K.W., C.M., R.H.C., Z.K.R., A.M.F., I.M., K.O., M.N., S.B., S.D., Z.M., N.V.; Visualization: J.M.C.; Supervision: J.M.C., Z.M., N.V.; Project administration: J.M.C., S.B., S.D., Z.M., N.V.; Funding acquisition: J.M.C., S.B., Z.M., N.V.
Funding
This study was funded by Medical Research Council project grant reference G0800901 (grant ID: 87379). C.N. was funded by the Fundação para a Ciência e a Tecnologia (Portugal). K.O. was funded by a Gurdon Scholarship from the British Society for Developmental Biology. Z.K.R. was funded by an Anatomical Society Summer Studentship. Deposited in PMC for release after 6 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.160093.supplemental
References
- Alvarado D. M., Aferol H., McCall K., Huang J. B., Techy M., Buchan J., Cady J., Gonzales P. R., Dobb M. B. and Gurnett C. A. (2010). Familial clubfoot is associated with recurrent chromosome 17q23.1q23.2 microduplications containing TBX4. Am. J. Hum. Genet. 87, 154-160. 10.1016/j.ajhg.2010.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarado D. M., McCall K., Aferol H., Silva M. J., Garbow J. R., Spees W. M., Patel T., Siegel M., Dobbs M. B. and Gurnett C. A. (2011). Pitx1 haploinsufficiency causes clubfoot in humans and a clubfoot-like phenotype in mice. Hum. Mol. Genet. 20, 3943-3952. 10.1093/hmg/ddr313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashby P. R., Pinconraymond M., Harris A., Pinon-Raymond M. and Harris A. J. (1993). Regulation of myogenesis in paralyzed muscles in the mouse mutants peroneal muscular atrophy and muscular dysgenesis. Dev. Biol. 156, 529-536. 10.1006/dbio.1993.1099 [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay U., Cotney J., Nagy M., Oh S., Leng J., Mahajan M., Mane S., Fenton W. A., Noonan J. P. and Horwich J. L. (2013). RNA-Seq profiling of spinal cord motor neurons from a presymptomatic SOD1 ALS mouse. PLoS ONE 8, e53575 10.1371/journal.pone.0053575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basit S. and Khoshhal K. I. (2017). Genetics of clubfoot; recent progress and future perspectives. Eur. J. Med. Genet., S1769-7212(17)30255-0 10.1016/j.ejmg.2017.09.006 [DOI] [PubMed] [Google Scholar]
- Bayarsaihan D. and Ruddle F. H. (2000). Isolation and characterization of BEN, a member of the TFII-I family of DNA-binding proteins containing distinct helix-loop-helix domains. Proc. Natl. Acad. Sci. USA 97, 7342-7347. 10.1073/pnas.97.13.7342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtol C. O. and Mossman H. W. (1950). Clubfoot: an embryological study of associated muscle abnormalities. J. Bone Joint Surg. Am. 32-A, 827-838. [PubMed] [Google Scholar]
- Bubb M. R., Spector I., Breyer B. B. and Fosen K. M. (2000). Effects of jasplakinolide on the kinetics of actin polymerization. J. Biol. Chem. 275, 5163-5170. 10.1074/jbc.275.7.5163 [DOI] [PubMed] [Google Scholar]
- Capossela S., Muzio L., Bertolo A., Bianchi V., Dati G., Chaabane L., Godi C., Politi L. S., Biffo S., D'Adamo P. et al. (2012). Growth defects and impaired cognitive-behavioral abilities in mice with knockout for Eif4h, a gene located in the mouse homolog of the Williams-Beuren syndrome critical region. Am. J. Pathol. 180, 1121-1135. 10.1016/j.ajpath.2011.12.008 [DOI] [PubMed] [Google Scholar]
- Cartlidge I. (1984). Observations on the epidemiology of club foot in Polynesian and Caucasian populations. J. Med. Genet. 21, 290-292. 10.1136/jmg.21.4.290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L., Scaria S. M., Saxton R. A., Gygi M. P., Shen K., Wyant G. A., Wang T., Harper J. W., Gygi S. P. and Sabatini D. M. (2016). The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell 165, 153-164. 10.1016/j.cell.2016.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman C., Stott N., Port R. and Nicol R. (2000). Genetics of club foot in Maori and Pacific people. J. Med. Genet. 37, 680-683. 10.1136/jmg.37.9.680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Andrade M., Barnholtz J. S., Amos C. I., Lochmiller C., Scott A. and Risman M. (1998). Segregation analysis of idiopathic talipes equinovarus in a Texan population. Am. J. Hum. Genet. 79, 97-102. [DOI] [PubMed] [Google Scholar]
- Deng X. (2011). SeqGene: a comprehensive software solution for mining exome- and transcriptome- sequencing data. BMC Bioinformatics 12, 267 10.1186/1471-2105-12-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson M. E., Flenniken A. M., Ji X., Teboul L., Wong M. D., White J. K., Meehan T. F., Weninger W. J., Westerberg H., Adissu H. et al. (2016). High-throughput discovery of novel developmental phenotypes. Nature 537, 508-514. 10.1038/nature19356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittrich R. (1930). Pathogenesis of congenital club-foot (Pes Equinovarus) – an anatomical study. J. Bone Joint Surg. Am. 12, 373-399. [Google Scholar]
- Dobbs M. B. and Gurnett C. A. (2013). Genetics of clubfoot. J. Pediatr. Orthop. B 21, 7-9. 10.1097/BPB.0b013e328349927c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs M. B., Morcuende J. A., Gurnett C. A. and Ponseti I. V. (2000). Treatment of idiopathic clubfoot: an historical review. Iowa Orthop. J. 20, 59-64. [PMC free article] [PubMed] [Google Scholar]
- Duce S., Madrigale L., Schmidt K., Cunningham C., Liu G., Barker S., Tennant G., Tickle C., Chudek S. and Miedzybrodzka Z. (2010). Micro-magnetic resonance imaging and embryological analysis of wild-type and pma mutant mice with clubfoot. J. Anat. 216, 108-120. 10.1111/j.1469-7580.2009.01163.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duce S. L., D'Alessandro M., Du Y., Jagpal B., Gilbert F. J., Crichton L., Barker S., Collinson J. M. and Miedzybrodzka Z. (2013). 3D MRI Analysis of the lower legs of treated idiopathic congenital talipes equinovarus (clubfoot). PLoS One 8, e54100 10.1371/journal.pone.0054100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupressoir A., Vernochet C., Bawa O., Harper F., Pierron G., Opolon P. and Heidmann T. (2009). Syncytin-A knockout mice demonstrate the critical role in placentation of a fusogenic, endogenous retrovirus-derived, envelope gene. Proc. Natl. Acad. Sci.USA 106, 12127-12132. 10.1073/pnas.0902925106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds E. W. and Frick S. L. (2009). The drop toe sign: an indicator of neurologic impairment in congenital clubfoot. Clin. Orthop. Relat. Res. 467, 1238-1242. 10.1007/s11999-008-0690-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M., Ohashi K., Sasaki Y., Goshima Y., Niwa R., Uemura T. and Mizuno K. (2003). Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. J. Neurosci. 23, 2527-2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkhmandakh B., Makeyev A. V., Erdenechimev L., Ruddle F. H., Chimge N.-O., Tussie-Luna M. I., Roy A. L. and Bayarsaihan D. (2009). Essential functions of Williams-Beuren syndrome-associated TFII-I genes in embryonic development. Proc. Natl. Acad. Sci. USA 106, 181-186. 10.1073/pnas.0811531106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester A. R., Weymouth K. S., Burt A., Wise C., Scott A. Gurnett C. A., Dobbs M. B. Blanton S. H. and Hecht J. T. (2009). Altered transmission of HOX and apoptotic SNPs identify a potential common pathway for clubfoot. Am. J. Med. Genet. A 149A, 2745-2752. 10.1002/ajmg.a.33130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn J. M., Ramirez N., Cornier A. S. and Colon Negron E. (2007). Unilateral congenital absence of the calf muscle. J. Ped. Orthop. Part B 16, 70-72. 10.1097/01.bpb.0000236233.64893.5c [DOI] [PubMed] [Google Scholar]
- Francke U. (1999). Williams-Beuren syndrome: genes and mechanisms. Hum. Mol. Genet. 8, 1947-1954. 10.1093/hmg/8.10.1947 [DOI] [PubMed] [Google Scholar]
- Gallo G., Yee H. F. Jr and Letourneau P. C. (2002). Actin turnover is required to prevent axon retraction driven by endogenous actomyosin contractility. J. Cell Biol. 158, 1219-1228. 10.1083/jcb.200204140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurnett C. A., Alaee F., Kruse L. M., Desruisseau D. M., Hecht J. T., Wise C. A., Bowcock A. M. and Dobbs M. B. (2008). Asymmetric lower-limb malformations in individuals with homeobox PITX1 gene mutation. Am. J. Hum. Genet. 83, 616-622. 10.1016/j.ajhg.2008.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V. and Hamilton H. L. (1951). A series of normal stages in the development of the chick embryo. J. Morphol. 88, 49-92. 10.1002/jmor.1050880104 [DOI] [PubMed] [Google Scholar]
- Hecht J. T., Ester A., Scott A., Wise C. A., Iovannisci D. M., Lammer E. J., Langlois P. H. and Blanton S. H. (2007). NAT2 variation and idiopathic talipes equinovarus (clubfoot). Am. J. Med. Genet. A 143A, 2285-2291. 10.1002/ajmg.a.31927 [DOI] [PubMed] [Google Scholar]
- Heck A. L., Bray M. S., Scott A., Blanton S. H. and Hecht J. T. (2005). Variation in CASP10 gene is associated with idiopathic talipes equinovarus. J. Pediatr. Orthop. 25, 598-602. 10.1097/01.bpo.0000173248.96936.90 [DOI] [PubMed] [Google Scholar]
- Helmbacher F., Schneider-Maunoury S., Topilko P., Tiret L. and Charnay P. (2000). Targeting of the EphA4 tyrosine kinase receptor affects dorsal/ventral pathfinding of limb motor axons. Development 127, 3313-3324. [DOI] [PubMed] [Google Scholar]
- Hoogenraad C. C., Eussen B. H. J., Langeveld A., van Haperen R., Winterberg S., Wouters C. H., Grosveld F., De Zeeuw C. I. and Galjart N. (1998). The murine CYLN2 gene: genomic organization, chromosome localization, and comparison to the human gene that is located within the 7q11.23 Williams syndrome critical region. Genomics 53, 348-358. 10.1006/geno.1998.5529 [DOI] [PubMed] [Google Scholar]
- Hoogenraad C. C., Ahkmanova A., Grosveld F., De Zeeuw C. I. and Galjart N. (2000). Functional analysis of CLIP-115 and its binding to microtubules. J. Cell Sci. 113, 2285-2297. [DOI] [PubMed] [Google Scholar]
- Hoogenraad C. C., Koekkoek B., Akhmanova A., Krugers H., Dortland B., Miedema M., van Alphen A., Kistler W. M., Jaegle M., Koutsourakis M. et al. (2002). Targeted mutation of Cyln2 in the Williams syndrome critical region links CLIP-115 haploinsufficiency to neurodevelopmental abnormalities in mice. Nat. Genet. 32, 116-127. 10.1038/ng954 [DOI] [PubMed] [Google Scholar]
- Huang G., Massoudi D., Muir A. M., Joshi D. C., Zhang C.-L., Chiu S. Y. and Greenspan D. S. (2017). WBSCR16 is a guanine nucleotide exchange factor important for mitochondrial fusion. Cell Rep. 20, 923-934. 10.1016/j.celrep.2017.06.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito E., De Maio F., Mancini F., Bellini D. and Orefice A. (2009). Leg muscle atrophy in idiopathic congenital clubfoot: is it primitive or acquired? J. Child. Orthop. 3, 171-178. 10.1007/s11832-009-0179-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs H., Handelsman J. E., Badenhorst M. and Pickering A. (1977). The muscles in club foot- a histological, histochemical and electron microscope study. J. Bone Joint Surg. Br. 59B, 465-472. [DOI] [PubMed] [Google Scholar]
- Iwaki S., Jensen B. M. and Gilfillan A. M. (2007). NTAL/LAB/LAT2. Int. J. Biochem. Cell Biol. 39, 868-873. 10.1016/j.biocel.2006.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. H., Gallin J. I. and Holland S. M. (1995). The p47phox mouse knock-out model of chronic granulomatous disease. J. Exp. Med. 182, 751-758. 10.1084/jem.182.3.751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell T. M. (2000). Neuronal specification in the spinal cord: Inductive signals and transcriptional codes. Nat. Rev. Genet. 1, 20-29. 10.1038/35049541 [DOI] [PubMed] [Google Scholar]
- Kania A., Johnson R. L. and Jessell T. M. (2000). Coordinate roles of LIM homeobox genes in directing the dorsoventral trajectory of motor axons in the vertebrate limb. Cell 102, 161-173. 10.1016/S0092-8674(00)00022-2 [DOI] [PubMed] [Google Scholar]
- Kao T.-J., Palmesino E. and Kania A. (2009). Src family kinases are required for limb trajectory selection by spinal motor axons. J. Neurosci. 29, 5690-5700. 10.1523/JNEUROSCI.0265-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh H., Watanabe Y., Ebukuro M., Muguruma K., Takabayashi S. and Shiroishi T. (2003). Chromosomal mapping of the peroneal muscular atrophy (pma) gene in the mouse. Exp. Anim. 52, 433-436. 10.1538/expanim.52.433 [DOI] [PubMed] [Google Scholar]
- Kawano T., Zhu M., Troiano N., Horowitz M., Bian J., Gundberg C., Kolodziejczak K. and Insogna K. (2013). LIM kinase 1 deficient mice have reduced bone mass. Bone 52, 70-82. 10.1016/j.bone.2012.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer E. R., Knott L., Su F., Dessaud E., Krull C. E., Helmbacher F. and Klein R. (2006). Cooperation between GDNF/Ret and ephrinA/EphA4 signals for motor-axon pathway selection in the limb. Neuron 50, 35-47. 10.1016/j.neuron.2006.02.020 [DOI] [PubMed] [Google Scholar]
- Landmesser L. (1978). The development of motor projection patterns in the chick hind limb. J. Physiol. 284, 391-414. 10.1113/jphysiol.1978.sp012546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754-1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G. and Durbin R.. and 1000 Genome Project Data Processing Subgroup (2009). The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078-2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström N. O., Neves C., McIntosh R., Miedzybrodzka Z., Vargesson N. and Collinson J. M. (2011). Tissue-specific characterization of Lim-kinase 1 expression during mouse embryogenesis. Gene Expr. Patterns 11, 221-232. 10.1016/j.gep.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. Y., Jin C. L., Cao D. H., Zhao N., Lin C. K. and Sun K. L. (2007). [Analysis of association between COL9A1 gene and idiopathic congenital talipes equinovarus] (in Chinese). Yi Chuan. 29, 427-432. 10.1360/yc-007-0427 [DOI] [PubMed] [Google Scholar]
- Lu W., Bacino C. A., Richards B. S., Alvarez C., VanderMeer J. E., Vella M., Ahituv N., Sikka N., Dietz F. R., Blanton S. H. et al. (2012). Studies of TBX4 and chromosome 17q23.1q23.2: an uncommon cause of nonsyndromic clubfoot. Am. J. Med. Genet. 158A, 1620-1627. 10.1002/ajmg.a.35418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K. and Narumiya S. (1999). Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895-898. 10.1126/science.285.5429.895 [DOI] [PubMed] [Google Scholar]
- McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y., Zhang Y., Tregoubov V., Janus C., Cruz L., Jackson M., Lu W. Y., MacDonald J. F., Wang J. Y., Falls D. L. et al. (2002). Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35, 121-133. [DOI] [PubMed] [Google Scholar]
- Miedzybrodzka Z. (2003). Congenital talipes equinovarus (clubfoot): a disorder of the foot but not the hand. J. Anat. 202, 37-42. 10.1046/j.1469-7580.2003.00147.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris C. A. (1999). Williams Syndrome. In GeneReviews [Internet] (ed. Pagon R. A., Adam M. P., Ardinger H. H., et al.). Seattle, WA: University of Washington, Seattle. [Google Scholar]
- Nakamura H. and Funahashi J. (2001). Introduction of DNA into chick embryos by in ovo electroporation. Methods 24, 43-48. 10.1006/meth.2001.1155 [DOI] [PubMed] [Google Scholar]
- Nonaka I., Kikuchi A., Suzuki T. and Esaki K. (1986). Hereditary peroneal muscular atrophy in the mouse: an experimental model for congenital contractures (arthrogryposis). Exp. Neurol. 91, 571-579. 10.1016/0014-4886(86)90053-1 [DOI] [PubMed] [Google Scholar]
- Odani N., Ito K. and Nakamura H. (2008). Electroporation as an efficient method of gene transfer. Dev. Growth Diff. 50, 443-448. 10.1111/j.1440-169X.2008.01037.x [DOI] [PubMed] [Google Scholar]
- Ohno K., Monji J., Sasaki T., Inoue K. and Shiono H. (1986). [Congenital absence of the anterior compartment muscle of both legs - a case report] in Japanese. Nihon Seikeigeka Gakkai Zasshi 60, 485-493. [PubMed] [Google Scholar]
- Palmer S. J., Santucci N., Widagdo J., Bontempo S. J., Taylor K. M., Tay E. S. E., Hook J., Lemckert F., Gunning P. W. and Hardeman E. C. (2010). Negative autoregulation of GTF2IRD1 in Williams-Beuren syndrome via a novel DNA binding mechanism. J. Biol. Chem. 285, 4715-4724. 10.1074/jbc.M109.086660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S. J., Taylor K. M., Santucci N., Widagdo J., Chan Y.-K. A., Yeo J.-L., Adams M., Gunning P. W. and Hardeman E. C. (2012). GTF2IRD2 from the Williams–Beuren critical region encodes a mobile-element-derived fusion protein that antagonizes the action of its related family members. J. Cell Sci. 125, 5040-5050. 10.1242/jcs.102798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J. F., Ghaloul-Gonzalez L., Madan-Khetarpal S., Hartman J., Surti U., Rajkovic A. and Yatsenko S. A. (2014). Familial microduplication of 17q23.1–q23.2 involving TBX4 is associated with congenital clubfoot and reduced penetrance in females. Am. J. Med. Genet. A 164, 364-369. 10.1002/ajmg.a.36238 [DOI] [PubMed] [Google Scholar]
- Phan K. D., Hazen V. M., Frendo M., Jia Z. and Butler S. J. (2010). The bone morphogenetic protein roof plate chemorepellant regulates the rate of commissural axonal growth. J. Neurosci. 30, 15430-15440. 10.1523/JNEUROSCI.4117-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponseti I. V. and Campos J. (1972). Observations on pathogenesis and treatment of congenital clubfoot. Clin. Orthop. Relat. Res. 84, 50-60. 10.1097/00003086-197205000-00011 [DOI] [PubMed] [Google Scholar]
- Porter M. A., Dobson-Stone C., Kwok J. B. J., Schofield P. R., Beckett W. and Tassabehji M. (2012). A role for transcription factor GTF2IRD2 in executive functions in Williams-Beuren Syndrome. PLoS One 7, e47457 10.1371/journal.pone.0047457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck T. R., Dietz F. R., Murray J. C. and Beutow K. H. (1993). A single-gene explanation for the probability of having idiopathic talipes equinovarus. Am. J. Hum. Genet. 53, 1051-1063. [PMC free article] [PubMed] [Google Scholar]
- Rosso S., Bollati F., Bisbal M., Peretti D. and Sumi T. (2004). LIMK1 regulates Golgi dynamics, traffic of Golgi-derived vesicles, and process extension in primary cultured neurons. Mol. Biol. Cell 15, 3433-3449. 10.1091/mbc.E03-05-0328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin M., Greer P. L., Lin M. Z., Poucher H., Eberhart J., Schmidt S., Wright T. M., Shamah S. M., O'Connell S., Cowan C. W. et al. (2005). Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron 46, 191-204. [DOI] [PubMed] [Google Scholar]
- Sarmiere P. D. and Bamburg C. R. (2004). Regulation of the neuronal actin cytoskeleton by ADF/cofilin. J. Neurobiol. 58, 103-117. 10.1002/neu.10267 [DOI] [PubMed] [Google Scholar]
- Sharp L., Miedzybrodzka Z., Cardy A. H., Inglis J., Madrigal L., Barker S., Chesney D., Clark C. and Maffulli N. (2006). The C677T polymorphism in the methylenetetrahydrofolate reductase gene (MTHFR), maternal use of folic acid supplements and risk factor of isolated clubfoot: a case parent triad analysis. Am. J. Epidemiol. 164, 852-861. 10.1093/aje/kwj285 [DOI] [PubMed] [Google Scholar]
- Song K. S., Kang C. H., Min B. W., Bae G. C., Cho C. H. and Lee J. H. (2008). Congenital clubfoot with concomitant peroneal nerve palsy in children. J. Pediatr. Orthop. B 17, 85-89. 10.1097/BPB.0b013e3282f548fc [DOI] [PubMed] [Google Scholar]
- Stewart S. F. (1951). Club-foot: its incidence, cause, and treatment; an anatomical-physiological study. J. Bone Joint Surg. Am. 33-A, 577-590. [PubMed] [Google Scholar]
- Sumi T., Matsumoto K. and Nakamura T. (2001). Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J. Biol. Chem. 276, 670-676. 10.1074/jbc.M007074200 [DOI] [PubMed] [Google Scholar]
- Tarchini B., Huynh T. H. N., Cox G. A. and Duboule D. (2005). HoxD cluster scanning deletions identify multiple defects leading to paralysis in the mouse mutant Ironside. Genes Dev. 19, 2862-2876. 10.1101/gad.351105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torniero C., dalla Bernardina B., Novara F., Vetro A., Ricca I., Darra F., Pramparo T., Guerrini R. and Zuffardi O. (2007). Cortical dysplasia of the left temporal lobe might explain severe expressive-language delay in patients with duplication of the Williams-Beuren locus. Eur. J. Hum. Genet. 15, 62-67. 10.1038/sj.ejhg.5201730 [DOI] [PubMed] [Google Scholar]
- Tursun B., Schlüter A., Peters M. A., Viehweger B., Ostendorff H. P., Soosairajah J., Drung A., Bossenz M., Johnsen S. A., Schweizer M. et al. (2005). The ubiquitin ligase Rnf6 regulates local Lim kinase 1 levels in axonal growth cones. Genes Dev. 19, 2307-2319. 10.1101/gad.1340605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Aa N., Rooms L., Vandeweyer G., van den Ende J., Reyniers E., Fichera M., Romano C., Delle Chiaie B., Mortier G., Menten B. et al. (2009). Fourteen new cases contribute to the characterization of the 7q11.23 microduplication syndrome. Eur. J. Med. Genet. 52, 94-100. 10.1016/j.ejmg.2009.02.006 [DOI] [PubMed] [Google Scholar]
- VanderMeer J. E. and Ahituv N. (2011). cis-regulatory mutations are a genetic cause of human limb malformations. Dev. Dyn. 240, 920-930. 10.1002/dvdy.22535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenseil J. E., Ciliberto C. H., Knutsen R. H., Levy M. A., Kovacs A. and Mecham R. P. (2010). The importance of elastin to aortic development in mice. Am. J. Physiol. Heart Circ. Physiol. 299, H257-H264. 10.1152/ajpheart.00194.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. and Scott S. A. (2008). Retinoid signaling is involved in governing the waiting period for axons in chick hindlimb. Dev. Biol. 321, 216-226. 10.1016/j.ydbio.2008.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. H., Palmer R. M. and Chung C. S. (1988). The role of major gene in clubfoot. Am. J. Hum. Genet. 42, 772-776. [PMC free article] [PubMed] [Google Scholar]
- Wen Z., Han L., Bamburg J. R., Shim S., Ming G. L. and Zheng J. Q. (2007). BMP gradients steer nerve growth cones by a balancing act of LIM kinase and Slingshot phosphatase on ADF/cofilin. J. Cell Biol. 178, 107-119. 10.1083/jcb.200703055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weymouth K. S., Blanton S. H., Bamshad M. J., Beck A. E., Alvarez C., Richards S., Gurnett C. A., Dobbs M. B., Barnes D., Mitchell L. E. et al. (2011). Variants in genes that encode muscle contractile proteins influence risk for isolated clubfoot. Am. J. Med. Genet. A 155, 2170-2179. 10.1002/ajmg.a.34167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., Higuchi O., Ohashi K., Nagata K., Wada A., Kangawa K., Nishida E. and Mizuno K. (1998). Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809-812. [DOI] [PubMed] [Google Scholar]
- Yang H., Zheng Z., Cai H., Li H., Ye X., Zhang X., Wang Z. and Fu Q. (2016). Three novel missense mutations in the filamin B gene are associated with isolated congenital talipes equinovarus. Hum. Genet. 135, 1181-1189. 10.1007/s00439-016-1701-7 [DOI] [PubMed] [Google Scholar]
- Yoshioka S., Huisman N. J. and Morcuende J. A. (2010). Peroneal nerve dysfunction in patients with complex clubfeet. Iowa Orthopaedic J. 30, 24-28. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.