

ABSTRACT
Vascular endothelial growth factor (VEGF), a key angiogenic and permeability factor, plays an important role in new blood vessel formation. However, abnormal VEGF-induced VEGFR2 signaling leads to hyperpermeability. We have shown previously that Rap1, best known for promoting cell adhesion and vessel stability, is a critical regulator of VEGFR2-mediated angiogenic and shear-stress EC responses. To determine the role of Rap1 role in endothelial barrier dynamics, we examined vascular permeability in EC-specific Rap1A- and Rap1B-knockout mice, cell–cell junction remodeling and EC monolayer resistivity in Rap1-deficient ECs under basal, inflammatory or elevated VEGF conditions. Deletion of either Rap1 isoform impaired de novo adherens junction (AJ) formation and recovery from LPS-induced barrier disruption in vivo. However, only Rap1A deficiency increased permeability in ECs and lung vessels. Interestingly, Rap1B deficiency attenuated VEGF-induced permeability in vivo and AJ remodeling in vitro. Therefore, only Rap1A is required for the maintenance of normal vascular integrity. Importantly, Rap1B is the primary isoform essential for normal VEGF-induced EC barrier dissolution. Deletion of either Rap1 isoform protected against hyper permeability in the STZ-induced diabetes model, suggesting clinical implications for targeting Rap1 in pathologies with VEGF-induced hyperpermeability.
KEY WORDS: Vascular permeability, Endothelial barrier function, VEGF, Adherens junctions, LPS
Summary: Rap1 has been defined as an adhesion-promoting molecule but Rap1B is shown to promote adherens junction disassembly, whereas Rap1A is required for junction maintenance; both isoforms promote junction re-formation following injury.
INTRODUCTION
The endothelial cell layer lining blood vessels constitutes a barrier that restricts the movement of water, proteins and blood cells between the intravascular and interstitial compartments. Exacerbated permeability associated with pathological conditions such as inflammation or diabetes leads to vascular leakage, edema and organ failure (Dejana et al., 2009; Nagy et al., 2008). While maintenance of the endothelial barrier (EB) is critical for end organ function (Rodrigues and Granger, 2015), dynamic regulation of the EB is required for angiogenesis during development and for wound healing after birth. In particular, vascular endothelial growth factor (VEGF), a major angiogenic factor first identified as a permeabilizing factor (Senger et al., 1983), plays a key role in regulating physiological vascular permeability during vessel formation.
The EB is maintained by adherens junctions (AJs), tight junctions and their interactions with the underlying actomyosin cytoskeleton. At the core of AJs are homophilic, trans interactions of transmembrane receptor vascular endothelial (VE)-cadherin (Harris and Nelson, 2010). The cytoplasmic tail of VE-cadherin is connected to the actin cytoskeleton via a number of proteins, including β-catenin, and is involved in transmitting signals that affect both the VE-cadherin adhesive function and outside-in signaling, both of which are crucial for vessel formation (Carmeliet et al., 1999). These functions of VE-cadherin are mediated by several signaling partners (Dejana and Giampietro, 2012), including the small GTPase Rap1, which has been shown to promote AJ assembly (Cullere et al., 2005; Glading et al., 2007; Kooistra et al., 2005; Wittchen et al., 2005) and attenuate thrombin-induced junction dissolution (Birukova et al., 2008). In isolated endothelial cells (ECs), active, GTP-bound Rap1 promotes the EB via direct enforcement of VE-cadherin adhesion and junction integrity via Krit-1/CCM1 (Béraud-Dufour et al., 2007; Chrzanowska-Wodnicka, 2013; Glading et al., 2007), and indirectly via dynamic regulation of the actomyosin cytoskeleton and Rho-dependent tension (Ando et al., 2013; Chrzanowska-Wodnicka, 2017; Noda et al., 2010; Post et al., 2013, 2015; Tawa et al., 2010; Wilson et al., 2013). AJ disassembly, in turn, is triggered by signaling initiated by vascular permeability factors. Among them, VEGF plays a key role, as VEGF-induced activation of VEGF receptor 2 (VEGFR2) on ECs triggers several signaling pathways that modulate VE-cadherin adhesive properties, leading to junction dissolution. These pathways include activation of Src family kinases (Eliceiri et al., 1999; Zachary and Gliki, 2001), and converge on phosphorylation and internalization of VE-cadherin and its dissociation from the actin cytoskeleton (Gavard, 2009), altering adhesive properties of VE-cadherin (Esser et al., 1998) and leading to AJ dissolution and increased permeability (Carmeliet, 2003; Lin et al., 2003). Inhibition of VE-cadherin phosphorylation blocks angiogenesis (Lin et al., 2003).
While most studies, particularly in vitro, define the role of Rap1 as a positive regulator of cell–cell adhesion, in vivo studies suggest that Rap1 regulation of the EB is more complex. Two closely related isoforms of Rap1: Rap1A and Rap1B, with identical effector domains and overall 95% homology but divergent C-terminus involved in membrane attachment, are expressed in different proportions in most mammalian cells, with Rap1A preferentially localized to cell–cell junctions in cultured ECs (Wittchen et al., 2011). While EC-specific deletion of either isoform does not lead to obvious vascular defects (Chrzanowska-Wodnicka et al., 2015), EC-specific double knockout of Rap1A and Rap1B isoforms (Rap1-ECKO) leads to lethality around E13.5 (Chrzanowska-Wodnicka et al., 2015), with hemorrhage and engorgement of perineural vessels, and bleeding as a likely cause of death, suggesting a redundant function of Rap1A and Rap1B in vascular barrier formation. Interestingly, however, EC-specific deletion of both Rap1 isoforms after birth did not interfere with normal cell–cell junction formation or lead to a gross vascular defect (Lakshmikanthan et al., 2015). This suggests that Rap1 may have different functions at different stages of vessel formation and maintenance. Furthermore, our recent findings implicate Rap1 as an important positive regulator of VEGFR2 signaling. In response to VEGF, Rap1 promotes VEGFR2 activation in ECs via integrin αvβ3 (Lakshmikanthan et al., 2011) and is required for VEGF-dependent angiogenesis (Carmona et al., 2009; Chrzanowska-Wodnicka et al., 2008; Lakshmikanthan et al., 2011). More recently, we demonstrated that Rap1 is required for shear stress-induced VEGFR2 transactivation, leading to nitric oxide (NO) release and endothelial homoeostasis (Lakshmikanthan et al., 2015). These findings suggested that Rap1 might also act as a positive regulator of VEGF-induced permeability.
In this study, we took advantage of EC-specific Rap1A-knockout (Rap1A-ECKO) and EC-specific Rap1B-knockout (Rap1B-ECKO) mice, to address, for the first time to our knowledge, the function of the two Rap1 isoforms in EB maintenance, de novo formation after barrier disruption, and in particular, VEGF-induced remodeling. We examined the effect of Rap1A and Rap1B deficiency on AJs in vitro. Interestingly, we found that the two isoforms play a similar, redundant function promoting de novo junction formation. However, once the EB is established, there is an isoform-specific requirement for EB maintenance. Lastly, because our study suggested a novel role of Rap1 in promoting VEGF-induced EB disassembly, we examined the effect of endothelial Rap1 deletion on pathological hyper-permeability associated with early diabetes. Our study suggests a novel role of two Rap1 isoforms in preventing pathological hyperpermeability.
RESULTS
Endothelial Rap1A promotes vessel barrier maintenance in lungs in vivo
First, to determine the functional significance of each Rap1 isoform in EB maintenance in vivo, we examined the effect of endothelial-specific deletion of Rap1A or Rap1B on basal permeability following perfusion with Evan's Blue. The lung is often used as a model organ because of the prevalence of continuous endothelium, in which cell–cell junctions are a primary determinant of permeability (Tse and Stan, 2010). We found that while basal permeability was unaltered in lungs from Rap1B-ECKO mice, it was slightly but significantly increased in lungs from Rap1A-ECKO mice (Fig. 1A). Interestingly, permeability was also increased in lungs of mice missing all but one Rap1b allele, but not in mice missing all but one Rap1a allele (Fig. S1), suggesting that heterozygous expression of Rap1A is sufficient to maintain a normal endothelial barrier. Because Tie2-Cre is also active in BM-derived cells, we examined basal permeability in the Rap1A-ECKO engrafted with WT bone marrow (WT–Rap1A-ECKO chimeras) in order to attribute the phenotype to ECs. We found that basal vascular permeability in chimeric Rap1A-ECKO mice engrafted with WT marrow was still elevated, indicating that Rap1a deficiency in the endothelium was responsible for decreased barrier function (Fig. 1B). We observed a similar increase in lung permeability in endothelial-restricted Rap1A (Rap1AiΔEC)-knockout but not Rap1B (Rap1BiΔEC)-knockout mice (Fig. 1C). Interestingly, we did not detect increased vascular permeability in other organs examined (Fig. 1C). Because Rap1B is the predominant Rap1 isoform in mouse endothelium (Lakshmikanthan et al., 2011) and Rap1A-ECKO mice have a higher total Rap1 content than Rap1B-ECKO mice, this suggested that Rap1A specifically is required for maintenance of basal permeability. These findings suggested that the elevated basal permeability in Rap1A-ECKO mice is not due to a quantitative difference in total Rap1 content but is rather due to the specific role of Rap1A in the maintenance of the EB in the continuous endothelium in lungs.
Fig. 1.

Rap1A is required for protection of endothelial barrier function. (A) Increased basal permeability to Evans Blue-labeled BSA in Rap1A-ECKO (Tie2-Cre+/0; rap1af/frap1b+/+) but not in Rap1B-ECKO (Tie2-Cre+/0; rap1a+/+rap1bf/f) lungs compared with Tie2-Cre0/0 controls in vivo. Evans Blue dye concentration in perfused lung tissue was determined spectrophotometrically at 640 nm following dye extraction with formamide. Illustration below graph shows Rap1 isoform content (filled circle) as an approximation of total Rap1 content in presented strains, based on immunoblots of EC lysates (bottom). (B) Elevated lung vessel permeability in Rap1A-ECKO is endothelium autonomous; elevated vascular permeability in Tie2-Cre+/0; rap1af/frap1b+/+ mice grafted with control bone marrow. Shown are actual values and mean; error bars are s.e.m. (C) Increased basal permeability to Evans Blue-labeled BSA in Rap1AiΔEC [Cdh5(PAC)-CreERT2+/0; rap1af/frap1b+/+] but not in Rap1BiΔEC [Cdh5(PAC)-CreERT2+/0; rap1a+/+rap1bf/f] lungs compared with Cdh5(PAC)-CreERT20/0 controls.
Rap1A is required for maintenance of adherens junction organization in vitro
To gain insight into the role of Rap1A in EB maintenance, we utilized primary mouse lung endothelial cells (MLECs) from Rap1-ECKO and WT mice to examine the effect of Rap1 isoform depletion on the architecture of EC junctions (Fig. 2), as alteration of AJ architecture correlates with increased endothelial permeability (Dejana et al., 2009). MLECs from Rap1-ECKO mice provide a good model of Rap1 deficiency, but also, being of mostly microvascular origin, provide a more physiologically relevant model of microvessel permeability than commonly studied human umbilical vein endothelial cells (HUVECs). Because surface expression of VE-cadherin was unchanged in ECs from Rap1-deficient mice, compared with expression in Cre-negative controls (Fig. S2), we examined the morphology of adherens junctions (AJs) in MLECs from these mice. VE-cadherin and β-catenin immunofluorescence was examined in EC monolayers by confocal microscopy. We found that in WT and Rap1B-deficient (ΔRap1B) ECs, the AJ complex proteins VE-cadherin and β-catenin localized predominantly to the plasma membrane, where they formed a continuous smooth line, consistent with the presence of mature AJs (Fig. 2). In contrast, in Rap1A-deficient (ΔRap1A) EC monolayers, VE-cadherin and β-catenin formed fragmented, disorganized AJs with short clusters perpendicular to the plasma membrane, typical of immature AJs (Fig. 2). To quantify this effect, we plotted histograms of β-catenin fluorescence intensities along a line perpendicular to the contacts, as previously described (Smutny et al., 2010). The shape of fitted histograms for both WT and ΔRap1B ECs was very similar and formed a sharp, narrow fluorescence peak corresponding to a narrow localized β-catenin distribution (Fig. 2). However, the fluorescence peak of the ΔRap1A EC histogram was much wider, and area under the curve was larger, consistent with a wider, more disorganized β-catenin distribution. Therefore, whereas Rap1B is redundant, Rap1A is required for maintenance of steady-state endothelial AJ organization in vitro, a finding consistent with a preferred localization of Rap1A to the plasma membrane (Wittchen et al., 2011).
Fig. 2.

Rap1A deficiency leads to disorganized AJs. (A) Confocal images of control, ΔRap1A or ΔRap1B primary mouse lung ECs (MLECs) grown to confluence and stained for AJ proteins, as indicated. Dispersed VE-cadherin and β-catenin staining is visible in ΔRap1A ECs, but not in ΔRap1B ECs. The same β-catenin-stained control and ΔRap1A MLECs are also shown in Fig. 5A. Scale bar: 20 μm. (B) Fluorescence intensity distribution across cell–cell junctions was measured in three independent images with 10 randomly selected contacts per experimental condition. After correcting for background fluorescence, data were nonlinear Lorentzian curve-fitted to each profile using Prism5, as previously described (Smutny et al., 2010) and area under the curve was calculated (bottom). (C) Representative immunoblots of total Rap1 content in Rap1-deficient ECs; actin is shown as loading control. Control and ΔRap1B MLECs were isolated from control (Tie2-Cre0/0; rap1a+/+rap1bf/f) or Rap1B-ECKO (Tie2 Cre+/0; rap1a+/+rap1bf/f) P6-9 lungs. ΔRap1A ECs were obtained by knocking down Rap1A in control cells using ‘pooled’ siRap1A.
Rap1A and Rap1B promote barrier re-formation following LPS-induced disruption
Next, to investigate the role of both Rap1 isoforms in the re-formation of EB function in vivo following its disruption, we measured lung vascular permeability in the LPS-induced inflammation model, in which increased endothelial permeability occurs as a result of loss of cell–cell contacts (Mehta and Malik, 2006). We found that endothelial deletion of either Rap1A or Rap1B led to increased vascular permeability, compared with Cre-negative controls (Fig. 3A). Perturbed barrier function, increased leakiness and edema are major factors that contribute to organ damage in sepsis (Ait-Oufella et al., 2010; Ince et al., 2016). To determine the functional consequences of increased vascular permeability on organ function, we examined mouse survival in the LPS-induced sepsis model. We found that deletion of either isoform led to increased lethality in LPS-induced sepsis (Fig. 3B). These findings suggest that deficiency of either Rap1A or Rap1B leads to a decreased ability to reform an EB, with significant consequences on recovery following the inflammatory insult.
Fig. 3.

Both Rap1 isoforms are required for junction reformation. (A,B) Increased vascular permeability and mortality in Rap1A-ECKO and Rap1B-ECKO mice under LPS-induced inflammatory conditions. (A) Increased vascular permeability to Evans Blue-labeled BSA in Rap1A-ECKO and Rap1B-ECKO mice 5 h after retro-orbital injection of lipopolysaccharides (LPS) from Escherichia coli 055:B5 (30 mg/kg). Evans Blue dye concentration was determined as in Fig. 1. Shown are average values, normalized, as indicated; error bars are s.e.m. (n≥12). (B) Kaplan–Meier survival curves of mice following 30 mg/kg LPS IP injection (n=14). (C,D) Both Rap1 isoforms, and in particular Rap1B, are required for de novo AJ formation. (C) Delayed VE-cadherin fluorescence recovery in AJs of Rap1-deficient ECs following AJ disruption by Ca2+ switch. Confluent MLECs (‘steady state’) were treated with 4 mM EGTA for 15 min at 37°C to disrupt endothelial cell–cell contacts (‘disassembly’). MLECs were allowed to reform AJs by further incubation in 1.8 mM Ca2+-containing culture medium for 2 h (‘early re-formation’) and 6 h (‘late re-formation’). Representative confocal images are shown. Insert in the top row shows DAPI nuclear staining. Scale bar: 20 μm. (D) The intensity of VE-cadherin fluorescence in AJs was quantified in 10 boxed areas/view/experimental condition, as shown in the example image on the left. Graph indicates significantly decreased fluorescence intensity in early AJ in Rap1-deficient MLECs (n=3; error bars are s.e.m.). (E) Rap1 deletion leads to attenuated VE-cadherin homophilic binding. Time course of adhesion of calcein-AM-loaded ECs onto Fc-VE-cadherin-coated plates protein as a measure of the ability of ECs to form AJs. Fc-IgG-coated wells serve as negative controls (error bars are s.e.m.; n≥3).
Barrier re-formation in vitro
Increased vascular permeability followed by an increased death rate of Rap1-deficient mice in response to LPS suggests that endothelial barrier re-formation following injury is impaired in Rap1-ECKO mice (Ait-Oufella et al., 2010). Re-establishment of VE-cadherin homophilic binding is essential for barrier re-formation after disruption (Vestweber et al., 2009). To determine whether defective VE-cadherin-based junction formation in ECs underlies the increased permeability in ΔRap1 ECs, we examined the effect of deleting each isoform on the kinetics of AJ re-formation in a ‘calcium switch’ method (Martinez-Palomo et al., 1980). To this end, WT, ΔRap1A and ΔRap1B ECs were incubated in the presence of the calcium chelator EDTA, which leads to the disruption of the Ca2+-dependent VE-cadherin junction. They were then incubated with regular Ca2+-containing culture medium to enable synchronized re-formation of AJs (Fig. 3C,D, Fig. S3). We found that in all tested conditions, after initial EDTA-induced loss of cell–cell contacts, VE-cadherin AJs partially re-formed 2 h after the re-addition of regular medium (early re-formation) and further strengthened at a late re-formation stage, 6 h after a return to regular medium (Fig. 3C). However, junction re-formation was slower in ΔRap1B ECs, as indicated by lower VE-cadherin fluorescence intensity in the early re-formation stage (Fig. 3D). At the same stage, deletion of Rap1A alone led to morphologically distinct junctions than in control or ΔRap1B ECs, as observed under basal conditions (Fig. 2A), but did not result in quantitative differences in VE-cadherin fluorescence intensity. These findings suggest that Rap1, and in particular the Rap1B isoform is required for establishment of VE-cadherin homophilic binding. To further examine the role of Rap1 in the ability of VE-cadherin to form homophilic bonds, we examined Rap1A- and Rap1B-dependent EC adhesion to substrates coated with Fc-VE-cadherin recombinant protein (Potter et al., 2005). We found that adhesion of Rap1A KO ECs versus WT controls was significantly decreased only at later time points (60 and 120 min), whereas adhesion of Rap1B KO ECs was significantly decreased at all time points (Fig. 3E). Moreover, Rap1B deletion led to a greater impairment of adhesion at all time points examined. These data show that following disruption of the EB, both Rap1 isoforms promote re-establishment of VE-cadherin homophilic binding and junction re-formation, and deficiency of either isoform delays AJ re-formation. A functional assessment of endothelial monolayer permeability also indicated a trend towards initial faster recovery of the barrier in Rap1A-deficient (or siRap1A) ECs; however, the delay was not statistically significant in Rap1B-deficient (or siRap1B) cells (Fig. S4A).
Rap1 in VEGF-induced permeability in vivo
The studies above demonstrate that both Rap1 isoforms promote de novo AJ formation and EB establishment. Our previous studies identified Rap1 as an important regulator of VEGF–VEGFR2 signaling, acting both downstream (Chrzanowska-Wodnicka et al., 2008) and upstream from VEGFR2 to promote angiogenesis (Lakshmikanthan et al., 2011) and endothelial function (Lakshmikanthan et al., 2015). VEGF is the critical factor responsible for new blood vessel formation but is also a potent permeabilizing factor, which, under normal conditions, enables the EC junction dissolution that is required for the formation of new vessels (Olsson et al., 2006). This raises an interesting and so far unrecognized possibility that Rap1 may be involved in the physiological response to VEGF and promote dissociation of EC junctions. To examine the role of Rap1 isoforms in VEGF-induced vascular permeability in vivo, we subjected Rap1A-ECKO and Rap1B-ECKO mice to the Miles assay, in which VEGF-induced extravasation of IP-injected Evans Blue is measured in skin vessels. As expected, we found that the VEGF caused an increase in dye leakage not only in control mice but also in Rap1A-ECKO mice (Fig. 4). Interestingly, VEGF treatment did not lead to elevated dye leakage compared with carrier alone (BSA) in Rap1B-ECKO mice (Fig. 4). This suggests that Rap1B is required for VEGF-induced skin microvessel permeability, whereas Rap1A is dispensable for that process.
Fig. 4.
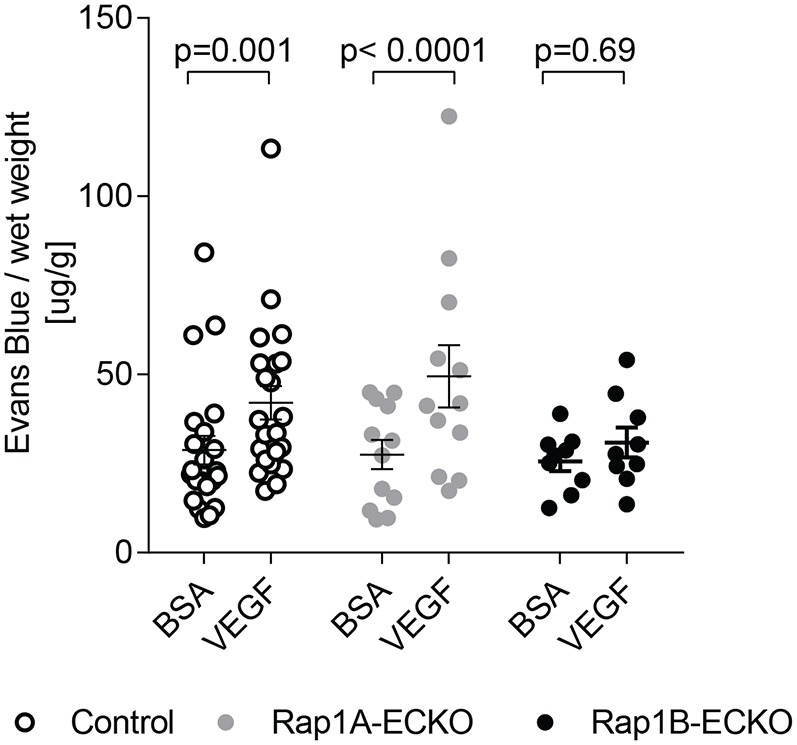
Rap1B-ECKO mice are resistant to VEGF-induced vascular permeability. The Miles skin permeability assay was performed in anesthetized mice retro-orbitally injected with 1 mg Evans Blue dye and then subcutaneously with 10 μl VEGF (0.013 mg/kg body weight in 0.1% BSA in PBS) or 0.1% BSA in PBS alone (BSA) into each side of the abdomen. After 15 min, Evans Blue dye concentration in the tissue was determined as described in Fig. 1. Shown are actual values and means, normalized to wet skin weight; error bars are s.e.m.; P-values are indicated.
Rap1B promotes VEGF-induced AJ remodeling and EC permeability
The above in vivo studies suggested that Rap1B might play a role in the dynamic regulation of EC junctions in response to VEGF stimulation. To address this hypothesis, we examined the effect of Rap1 isoform deficiency on VEGF-induced AJ remodeling in cultured ECs by using confocal microscopy to examine the kinetics of junctional VE-cadherin and β-catenin immunofluorescence intensity following VEGF treatment (Fig. 5, Fig. S5). In WT ECs, the intensity of VE-cadherin and β-catenin staining was decreased 30 min after VEGF treatment (Fig. 5B), consistent with VEGF-induced AJ disassembly (Fig. 5A). AJ immunofluorescence intensity and cell–cell junctions were re-established after 45 min of VEGF treatment (Fig. 5). While similar dynamics of β-catenin fluorescence intensity was observed in ΔRap1A ECs, AJ fluorescence intensity remained unchanged in ΔRap1B ECs following VEGF treatment (Fig. 5A), demonstrating the resistance of AJs to VEGF remodeling in Rap1B-deficient ECs.
Fig. 5.

Rap1b deficiency impairs the ability of VEGF to remodel AJs. (A) Confocal images of β-catenin staining in ΔRap1A, ΔRap1B or control confluent MLECs, serum-starved for 6 h as in Fig. 2 (control and ΔRap1A MLECs are reproduced from Fig. 2A) and treated for indicated time with 50 ng/ml VEGF. Scale bar: 20 μm. (B) AJ fluorescence intensity distribution and calculation of area under the curve at corresponding time points of VEGF treatment, measured as described in Fig. 2. After 30 min of VEGF treatment, AJs are dispersed in WT and ΔRap1A ECs, but not in ΔRap1B ECs (A, middle row), in which fluorescence intensity is unchanged (B, middle graph). Representative images of n=3 independent experiments are shown.
To examine the role of Rap1 isoforms in dynamic changes in endothelial barrier function in response to VEGF treatment, we employed electric cell substrate impedance sensing (ECIS), an in vitro system for quantitative measurement of cell adhesive behavior (Szulcek et al., 2014). By measuring impedance over a range of frequencies and applying a mathematical model, this approach allows differentiation between junctional impedance (cell–cell contact tightness) and impedance resulting from cell–substrate adhesion (Giaever and Keese, 1991; Lo et al., 1995). EC monolayers with intact Rap1 or ECs depleted of either Rap1 isoform were subjected to VEGF and the resistivity of cell–cell contacts to the current flow, Rb, was measured. As expected, VEGF induced a transient drop in Rb, representing transient increased permeability in WT cells (Fig. 6A). The response to VEGF was further increased in Rap1A-deficient ECs, consistent with decreased stability of AJs and, therefore, increased susceptibility to VEGF-induced disruption. In contrast, the response to VEGF was consistently and significantly reduced in Rap1B-deficient cells, representative of a resistance to VEGF-induced permeability in these cells. This result is consistent with decreased VEGF-induced AJ remodeling (Fig. 5).
Fig. 6.

Rap1B is required for VEGF-induced EC responses and signaling: Src activation and VE-cadherin phosphorylation. (A,B) Rap1B-deficient EC monolayers are resistant to VEGF-induced permeability. VEGF treatment (A, arrow) induces a rapid, reversible drop in resistivity in siControl ECs and siRap1A ECs, but a reduced response in siRap1B ECs. EC barrier function was measured using ECIS in early passage HMVECs transfected with 50 nM ‘pooled siRNAs’ for 24 h, seeded onto 5 µg/ml fibronectin-coated 8W10E+ ECIS array, allowed to reach confluence and treated with VEGF (100 ng/ml). (B) Rap1A overexpression rescues the loss of response to VEGF-induced barrier dissolution in siRap1B ECs. 24 h after siRap1B knockdown, ECs were transduced with lentiviral particles expressing WT-Rap1A (Myc-tagged) at 5 MOI and cultured for an additional 36 h prior to measuring barrier function by ECIS. Right panel shows Rap1B knockdown and Myc/Rap1A overexpression confirmed by western blotting of lysates from EC in parallel samples. Plotted are average Rb values obtained in a representative experiment (of >3 performed) using multiple frequency/time (MFT) setting from triplicate wells per condition, modeled as previously described (Szulcek et al., 2014) and normalized to baseline. Values are averages of triplicate wells per condition, error bars are s.d. At the time point of maximum barrier dissolution, mean values of experimental samples were compared with mean values of controls. Arrow indicates VEGF treatment. (C) VEGF-induced VE-cadherin and Src tyrosine 416 phosphorylation was examined in HMVECs transfected with control, scrambled siRNA or Rap1A siRNA or Rap1B siRNA. Representative immunoblots of phosphorylated and total protein expression are shown, together with actin as loading control. Note that the Src pY416 panel is from a longer exposure of the same blot to reveal protein. (D) Quantification of fold change in VEGF-induced phosphorylation of signaling molecules in Rap1 isoform-deficient and control ECs. Rap1B, but not Rap1A deficiency leads to decreased phosphorylation of Src and VE-cadherin. Note that the kinetics of respective phosphorylation events implicates non-linearity in their response to VEGF stimulation. Values are means±s.e.m. (n≥3).
The differential effects of the two Rap1 isoforms on VEGF-induced EB dynamics suggested differential isoform function, with Rap1B but not Rap1A required for VEGF-induced permeability. To further investigate whether Rap1B is the only isoform mediating VEGF-induced permeability, we examined the ability of exogenous Rap1A to rescue the defective VEGF response in Rap1B-deficient ECs. We found that overexpression of (siRap1a-resistant) Rap1A in siRap1B ECs restored the permeability response to VEGF, similar to that in siControl ECs (Fig. 6B). Therefore, when overexpressed, Rap1A can also promote the EC response to VEGF.
Rap1B promotes VEGF signaling to VE-cadherin phosphorylation
VEGF-induced destabilization of cell–cell junctions at the molecular level is mediated by signaling leading to VE-cadherin phosphorylation (Esser et al., 1998; Monaghan-Benson and Burridge, 2009). Specifically, VEGF-induced Src-mediated VE-cadherin tyrosine phosphorylation leads to junction disassembly (Eliceiri et al., 1999). Decreased AJ dynamics and defective EB modulation in response to VEGF treatment in siRap1B ECs (Fig. 6) suggested that Rap1B might be involved in VEGF-induced VE-cadherin phosphorylation. We found, as expected, that VEGF treatment led to a rapid increase in phosphotyrosine 685 in control ECs (Wallez et al., 2007) (Fig. 6C). While depletion of Rap1A did not affect that response, depletion of Rap1B led to attenuated VEGF-induced VE-cadherin phosphorylation (Fig. 6D). To examine whether defective signaling from VEGF to VE-cadherin contributes to decreased VE-cadherin phosphorylation, we examined VEGF-induced Src tyrosine 416 phosphorylation corresponding to the Src activation site. Interestingly, we found that whereas in WT and siRap1A ECs, VEGF induced rapid Src tyrosine 416 phosphorylation, such induction was not present in siRap1B ECs, consistent with decreased Src activation. Therefore, Rap1B promotes signaling from VEGFR2, causing phosphorylation of VE-cadherin, which leads to dissolution of VE-cadherin junctions (Fig. 6).
Whereas a normal permeability response to VEGF is required for physiological angiogenesis, and its loss may lead to defective development or wound healing, an aberrant response to VEGF, such as under pathological conditions of elevated VEGF, can result in edema and tissue damage. In particular, in early diabetes, blood–retinal barrier (BRB) breakdown leads to excessive leakage, macular edema and vision loss. These events are largely initiated and propagated by elevated VEGF (Qaum et al., 2001). Because our above results indicate that Rap1 is required for VEGF-induced permeability, we postulated that Rap1 deficiency might offer protection against aberrant VEGF-induced permeability in early diabetes. To test this hypothesis, we examined the effect of endothelial-specific Rap1 isoform deficiency on retinal vascular permeability in the streptozotocin-induced diabetes model (Du et al., 2002). Three weeks following induction of diabetes, retinal vascular permeability was examined in Rap1A-ECKO, Rap1B-ECKO and control diabetic mice. Endothelial deletion of either Rap1A or Rap1B (Fig. 7A) conferred a protective effect. This indicates that both Rap1 isoforms are required for STZ-induced permeability and suggests that inhibition of Rap1 function offers protection against pathological VEGF permeability.
Fig. 7.

Synergistic role of Rap1 isoforms in VEGFR2 signaling. Rap1 deficiency protects against increased retinal vascular permeability in the STZ-induced diabetes model. (A) Representative retinal flat-mounts from diabetic control, Rap1A-ECKO and Rap1B-ECKO mice injected with fluorescent dextran. Leakage is shown as hyperfluorescence extravasating from vessels. Scale bar: 50 μm. Quantification of fluorescent leakage, obtained from a single confocal slice, demonstrates reduced leakage in diabetic Rap1-ECKO mice compared with diabetic control mice (n≥8). (B) Proposed role of Rap1 isoforms in VEGF-mediated modulation of EC barrier function; a model built on current and published results. Rap1B, acting downstream from VEGFR2, is the predominant isoform responsible for VEGF-induced AJ dissociation and endothelial permeability, while both Rap1 isoforms promote full VEGFR2 activation and as such, participate in full response to VEGF. Rap1A is the main Rap1 isoform involved in structural maintenance of AJs, and its depletion may increase AJ susceptibility to VEGF-induced dissolution.
DISCUSSION
Rap1 signaling has been implicated in increased cell adhesion and is associated with enhanced EB function in ECs, with the roles of two closely related Rap1 isoforms, Rap1A and Rap1B, not clearly delineated in that process. Our in vivo studies in EC-specific Rap1-knockout mice, showing the overlapping, but essential roles of both isoforms in development but their redundancy after the formation of the EB, suggested differential significance of Rap1 in vessel integrity establishment during development (Chrzanowska-Wodnicka et al., 2015) and after birth (Lakshmikanthan et al., 2015). Importantly, our studies underscored the importance of Rap1 in promoting VEGFR2 signaling (Chrzanowska-Wodnicka et al., 2008; Lakshmikanthan et al., 2011, 2015). These findings prompted us to investigate the in vivo role of Rap1A and Rap1B in EB maintenance, re-formation after disruption and, importantly, VEGF-mediated modulation of EB function. The seminal findings of this paper are that both Rap1 isoforms are involved in de novo junction formation, which is delayed in ECs lacking either isoform because of decreased VE-cadherin binding and resulting in increased permeability and increased organ damage and mortality in vivo under conditions of barrier disruption such as during inflammation (LPS study). Importantly, our study demonstrates that once the EB is established, only the Rap1A isoform is required for maintenance of normal AJ architecture and EB function specifically in the lungs, while Rap1B is redundant for these functions. Significantly, however, in response to VEGF stimulation, Rap1B, but not Rap1A, is required for normal AJ disassembly and EB dissociation. Therefore, our study identifies a novel role of Rap1 as a factor required for cell–cell junction disassembly, extending the understanding of the role of Rap1 in dynamic regulation of cell–cell adhesions and EB function.
The specific role of each Rap1 isoform in regulation of endothelial permeability has not been extensively studied. However, in cultured ECs, Rap1A was shown to preferentially localize to cell–cell junctions, and its depletion led to a weakening of the EB (Wittchen et al., 2011). Here, we demonstrate a functional importance of the Rap1B, but not Rap1A isoform, in VEGF-mediated EB dissolution (Fig. 7B). This differential role of Rap1 isoforms in regulation of the VEGF response may be explained by the differential participation of the two isoforms in Rap1-VEGFR2 feed-forward signaling. We have previously shown that both Rap1 isoforms act upstream of VEGFR2, promoting its activation (Lakshmikanthan et al., 2011, 2015); however, only Rap1B is activated downstream of VEGFR2 as there is no detectable active Rap1 in Rap1B-knockout ECs (Chrzanowska-Wodnicka et al., 2008). Therefore, it is conceivable that depletion of Rap1B removes the feed-forward mechanism required for activation and downstream signaling from VEGFR2, leading to the phosphorylation of VE-cadherin and AJ dissolution. Alternatively, depletion of Rap1A, which is less abundantly expressed, may have a quantitatively lesser effect on VEGFR2 signaling; thus, when overexpressed, Rap1A is able to rescue the deficient response to VEGF treatment in Rap1B-knockout cells. Conversely, deletion of Rap1A, by compromising AJ stability, may increase susceptibility to VEGF-induced dissolution (Fig. 6, Fig. 7B). Furthermore, it is likely that in addition to decreasing linear signaling from VEGFR2 to Src, deletion of Rap1B may perturb additional VEGF-mediated AJ dissolution pathways (Pannekoek et al., 2014). Molecular details of the complex signaling by Rap1 to VEGFR2, involving other receptors (Lakshmikanthan et al., 2011), remain to be fully elucidated in future studies.
Our results demonstrating that both Rap1 isoforms have a similar role in promoting de novo AJ formation are consistent with existing in vitro models implicating Rap1 in promoting de novo junction formation (Chrzanowska-Wodnicka, 2017). Importantly, these results may help to explain the physiological and pathological consequences of endothelial Rap1 deficiency in vivo (Birukova et al., 2015; Chrzanowska-Wodnicka et al., 2015; Lakshmikanthan et al., 2015). We show that deletion of either isoform, and in particular Rap1B, delays AJ formation and EB establishment but does not prevent it. Consistently, knockout of a single Rap1 isoform in ECs does not have a major impact on vessel integrity during vessel development, whereas deletion of both isoforms leads to vessel leakiness, hemorrhage and death by embryonic day 13.5 (Chrzanowska-Wodnicka et al., 2015). In contrast, after AJs are formed and the EB is established, neither Rap1 isoform is absolutely required for the maintenance of the EB under physiological conditions; consistently, inducible deletion of both Rap1 isoforms after birth does not grossly affect cell–cell junction organization or vascular permeability (Lakshmikanthan et al., 2015) (Fig. 1). However, the ability of both Rap1 isoforms to promote re-formation of EC junctions, although redundant during development, becomes essential under pathological conditions. Specifically, both Rap1 isoforms are involved in EC junction re-formation and promote maintenance of the EB during LPS-induced inflammation, as endothelial knockout of each isoform leads to increased permeability, organ damage and death (Fig. 3). Previously, Rap1A has been implicated in barrier re-formation in inflammation (Birukova et al., 2015); our study shows that both isoforms play a key role in EB repair, and both are required for the prevention of organ damage in sepsis. A normal permeability response to VEGF is required for angiogenesis and any defect may interfere with normal development or wound healing; however, an aberrant response to VEGF, such as under pathological conditions of elevated VEGF levels, leads to chronic vascular hyperpermeability (Nagy et al., 2008) resulting in leaky vessels, edema and tissue damage associated with multiple diseases (Weis and Cheresh, 2005). Normalization of vessels by targeting VEGF signaling is a validated anti-angiogenesis therapy for tumor metastasis and retinopathy (Ferrara et al., 2007). Because our results show that inhibition of Rap1B signaling reduces VEGF-induced permeability, this suggested that inhibiting Rap1B function might offer protection against pathological VEGF permeability. Indeed, we found that endothelial deletion of Rap1B protects against excessive vascular leakage in early diabetes, associated with excessive VEGF signaling (Qaum et al., 2001). Interestingly, we found that deletion of Rap1A was also protective under these conditions (Fig. 7A), supporting a synergistic role of both isoforms in promoting signaling from VEGFR2 (Fig. 7B; Lakshmikanthan et al., 2011). These findings underscore context-specific, dynamic regulation of both Rap1 isoforms with potential clinical implications for specific targeting of Rap1B for the treatment of VEGF pathologies and inhibition of pathological hyper permeability.
MATERIALS AND METHODS
Reagents
Unless otherwise indicated, chemicals were purchased from Sigma-Aldrich. Human microvascular lung ECs (HMVECs) were purchased from LONZA. RNAiMAX was from Life Technologies. Antibodies against Rap1 (rabbit polyclonal; #4938; 1:1000), Rap1B (clone 36E1 rabbit mAb; #2326; 1:1000) and phospho-c-Src-pY416 (rabbit polyclonal; #2101generated against a synthetic phosphopeptide corresponding to residues surrounding Tyr419 of human Src; 1:1000) were from Cell Signaling. Antibodies against actin (clone I-19 goat polyclonal; #sc-1616; 1:1000), VE-cadherin (C-19 goat polyclonal; #sc-6458; 1:1000; F-8 mouse mAb; #sc-9989; 1:1000) were from Santa Cruz Biotechnology. Antibody against phospho-VE-cadherin pY685 (rabbit polyclonal; #ab119785; 1:1000) was from Abcam. Antibodies against phospho-VE-cadherin pY658 (rabbit polyclonal; #44-1144G and 44-1145G; 1:1000) were from Thermo Fisher and were used as previously described (Adam et al., 2010; Benn et al., 2016; Wessel et al., 2014). All HRP-conjugated secondary antibodies were from Jackson ImmunoResearch.
Genetic mouse models
All mouse procedures were performed according to approved Medical College of Wisconsin Institutional Animal Use and Care Committee protocol 00001206. Generation of endothelial lineage-restricted Rap1-knockout mice (Tie2-Cre+/0; Rap1 f/fRap1b+/+, Rap1A-ECKO, and Tie2-Cre+/0; Rap1a+/+Rap1 f/f, Rap1B-ECKO) has been previously described (Lakshmikanthan et al., 2011). Inducible endothelial-restricted (Rap1iΔEC) mice were obtained by crossing mice containing floxed Rap1a or Rap1b alleles (Rap1af/fRap1b+/+ or Rap1a+/+Rap1bf/f mice) (Pan et al., 2008) with Cdh5(PAC)-CreERT2+/0 mice and inducing Cre activity with tamoxifen injections, as previously described (Lakshmikanthan et al., 2015; Pitulescu et al.). Three- to twelve-month-old male and female mice were used for experiments. Mice were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) cocktail.
In vivo permeability assays
Basal vascular permeability to Evans Blue-labeled BSA was determined in lungs, heart, kidney and liver, as previously described (Lakshmikanthan et al., 2015). Lipopolysaccharide (LPS)-induced permeability was induced by intraperitoneal injection of Escherichia coli O55:B5 at 30 mg/kg, and lung vascular permeability to Evans Blue-labeled BSA was determined 5 h post-LPS injection. Survival in the LPS-induced sepsis model was assessed for 7 days following intraperitoneal injection of a predetermined EC50 dose of LPS (30 mg/kg). Post-LPS challenge, mice were monitored every 4–6 h and critically ill animals were euthanized. Skin microvascular permeability to VEGF was determined using the Miles assay (Miles and Miles, 1952). Briefly, following retro-orbital injection with 0.5% Evans Blue with 4% BSA in PBS, anesthetized mice were injected subcutaneously with 10 µl VEGF (0.013 mg/kg body weight in 0.1% BSA in PBS), or with 10 µl of 0.1% BSA into each side of the abdomen. After 15 min, mice were perfused with 4% paraformaldehyde in PBS and skin was dissected. Concentration of Evans Blue dye was determined spectrophotometrically at 640 nm following dye extraction with formamide and the amount of Evans Blue was calculated from a standard curve.
Cell culture, transfection and Ca2+ switch experiments
For immunofluorescence studies primary mouse lung endothelial cells (MLECs) isolated from Tie2-Cre-negative mice (Control ECs), or Tie2 Cre-negative mice and transfected with siRap1A (ΔRap1A ECs), Tie2-Cre+/0; Rap1a+/+Rap1f/f or Tie2-Cre+/0; rap1a+/+rap1f/+ mice and transfected with siRap1B (ΔRap1B ECs), were cultured in VascuLife EnGS Endothelial Medium Complete Kit, LL-0002, as previously described (Sobczak et al., 2010). Primary human microvascular lung ECs (HMVECs; LONZA) were used as an established model to study endothelial permeability by ECIS and VE-cadherin signaling in response to VEGF; Rap1 deficiency was obtained by EC transfection with 50 nM Rap1a or Rap1b siGENOME siRNA pool (Dharmacon) or with scrambled siRNA using Opti-MEM medium and RNA iMAX reagent (Life Technologies) for 6 h and cultured for additional 36 h in EC-complete medium, until cells reached 85% confluency.
For Ca2+ switch experiments, following siRNA knockdown, confluent MLECs were exposed to 4 mM EGTA for 15 min, followed by incubation with fresh culture medium containing 1.8 mM Ca2+ for 6 h at 37°C, as previously described (Martinez-Palomo et al., 1980).
Western blotting
Following siRNA knockdown, 85% confluent HMVEC cultures were serum-starved for 10 h in basal EC medium prior to VEGF-A (40 ng/ml) induction. Following treatment, as described, ECs were rapidly washed once with ice-cold PBS and lysed with 1× RIPA buffer plus protease/phosphatase inhibitor cocktail. Cell lysates were clarified by centrifugation at 13,800 g for 10 min at 4°C. Supernatants were flash frozen with liquid nitrogen and stored at −80°C until further analysis. Post BCA assay, normalized total lysates were resolved on 4–12% gradient gel and blotted using specific antibodies, as indicated. Signal intensity was determined by densitometry of X-ray film, values obtained for phosphoproteins were normalized to actin in the same sample and fold induction of phosphorylation over the basal phosphorylation level was calculated.
Immunofluorescence, confocal microscopy and image analysis
MLECs were plated on fibronectin-coated coverslips (2.5 µg/ml in PBS), allowed to reach confluence, usually 24–48 h. Following treatment, as specified, cells were fixed in 3.7% formaldehyde in PBS, permeabilized with 0.5% Triton X-100 in PBS on ice, blocked with 1% BSA in PBS or 5% horse serum (Atlanta Biologicals) and stained with anti-VE-cadherin (BD Pharmingen; #555289; 1:50) and monoclonal anti β-catenin (BD Pharmingen; #610153; 1:50) antibodies, followed by appropriate secondary antibodies (Jackson ImmunoResearch; 1:75) or Hoechst nuclear stain (1:1000 in PBS). Images were acquired using confocal Olympus FluoView FV1000 MPE scanning microscope and analyzed with FV10-ASW 2.0 software. In all experiments, laser power was maintained at the same level for all experimental conditions per each fluorophore and fluorescence intensity was not altered between samples during quantification.
AJ morphology was assessed by quantifying β-catenin fluorescence distribution at cell–cell contacts, performed as in Smutny et al. (2010). Briefly, independent images with 10 randomly selected contacts were used for quantification at every time point. A 16-μm-long line was drawn centered on, and perpendicular to, every contact, and pixel intensities along the selected line were recorded. Data for each profile were then imported into Prism 5 and intensities were corrected for background fluorescence by subtracting a constant background value. A nonlinear Lorentzian curve was fitted to each profile using Prism5 (n=30). For analysis of junction re-formation, the intensity of VE-cadherin fluorescence in AJs was quantified at 2 h (‘early re-formation’) and 6 h (‘late re-formation’) after Ca2+ switch in 10 boxed areas per view, per experimental condition (n=3).
VE-cadherin-dependent adhesion
Recombinant Human VE-Cadherin Fc Chimera His-tag Protein, CF (R&D Systems; #938-VC-050; 100 µg/ml), IgG (non-specific binding control) or fibronectin (a positive control), at 5 µg/ml were coated onto a 96-well plate format overnight at 4°C. The following day, wells were washed with PBS, blocked with 3% BSA for 2 h at RT and washed with PBS. Briefly, control or Rap1A-KO or Rap1B-KO MLECs (passage 3) were stained with 12.5 µM calcein-AM in 0.1% BSA/PBS for 30 min at room temperature and washed once with 0.1% BSA/PBS. Calcein-AM-labeled ECs were seeded at 30,000 cells/well density for the indicated times and fluorescence at 515 nm measured at indicated time points (total cell fluorescence) using a Vallac VICTOR fluorescent plate reader (PerkinElmer, Vallac, Waltham, MA). Non-adherent ECs were removed by washing with 0.1% BSA/PBS and fluorescence was measured again (adherent cell fluorescence). VE-Cadherin homophilic adhesion was calculated as the adherent cell fluorescence to total cell fluorescence ratio. Shown are average values from three experiments performed in triplicate.
Electrical cell impedance sensing (ECIS)
Following siRap1/control knockdown, HMVECs were seeded onto fibronectin (5 µg/ml)-coated 8W10E+ ECIS array (Applied Biophysics, MA) and allowed to reach confluence overnight. Baseline barrier was recorded and established for at least 60 min using multiple frequency/time (MFT) setting and Rb was modeled as previously described (Szulcek et al., 2014). VEGF-induced permeability was measured following 100 ng/ml VEGF-A treatment. Fresh media was added after junction re-formation, ∼30 min after VEGF-A treatment. For the gain-of-function (GOF) experiment, siRNA-transfected-HMVECs were transduced with 6 μg/ml polybrene (hexadimethrine bromide) lentiviral particles expressing WT Rap1a (Myc-Rap1a) at a multiplicity of infection (MOI) of 5 and ECs were cultured as described above prior to analysis VEGF-A-induced permeability. Following the experiment, Rap1 expression was confirmed by anti-Myc western blot analysis.
STZ diabetes model and retinal permeability assay
All animal studies were performed under a protocol approved by the Institutional Animal Care and Use Committee and in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Adult Rap1A-ECKO and Rap1B-ECKO mice and Cre-negative control mice were made diabetic using multiple doses of streptozotocin (STZ) as previously described (Dominguez et al., 2016). Diabetes was confirmed by the measurement of blood glucose in excess of 250 mg/dl on two separate days. After 3 weeks of diabetes, mice underwent assessment of retinal vascular permeability (Cai et al., 2011). Mice received a tail vein injection of FITC-labeled albumin (0.5 mg in 50 µl vehicle). After 2 h, the mice were euthanized and eyes enucleated. Retinas were removed, flat-mounted and kept on glass slides with Fluoromount mounting medium (Sigma-Aldrich). Images were acquired using an Olympus FluoView 1000 scanning laser confocal microscope and at least five different view areas were selected to collect images for each sample. All images were generated using identical settings within each experiment. Fluorescence intensity, from a single confocal slide, was determined using ImageJ software (http://rsb.info.nih.gov).
Statistical analysis
For in vivo permeability studies, the calculated amount of extravasated Evans dye was normalized to body weight and EB/weight measurements were analyzed. For studies involving more than two groups, one- or two-way ANOVA was used, as appropriate, with Holm–Šidák (Figs 1 and 5) or Dunnett's (Figs 3 and 7) adjustment for multiple comparisons. For analysis of ECIS data (Fig. 6A,B), at the time point of maximum barrier dissolution, mean values of siRap1A or siRap1B samples were compared with mean values of controls. For analysis of VEGF-A signaling data within each experiment, fold change in VE-cadherin phosphorylation induction between Rap1-isoform-deficient and control values were calculated for each condition. Mean values and s.e.m. are shown. Statistical significance of group differences was determined using unpaired two-tailed Student's t-test.
Supplementary Material
Acknowledgements
We thank Ms Jennifer Idsvoog and Dr Yongwei Zheng for excellent technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.L., M.S., M.B.G., M.C.-W.; Methodology: S.L., M.S., S.L.C., L.S., M.B.G.; Formal analysis: S.L., M.S., S.L.C., L.S., M.B.G., M.C.-W.; Investigation: S.L., M.S., S.L.C.; Resources: M.B.G., M.C.-W.; Data curation: S.L., M.S., S.L.C., L.S.; Writing - original draft: M.S., M.C.-W.; Writing - review & editing: S.L., M.B.G., M.C.-W.; Visualization: S.L., S.L.C., L.S.; Supervision: M.B.G., M.C.-W.; Project administration: M.C.-W.; Funding acquisition: M.B.G., M.C.-W.
Funding
This work was supported by American Heart Association grant 0950118G (to M.C.-W.), the National Institutes of Health grants (HL111583 to M.C.-W. and EY07739, EY12601 and DK-09-0730 to M.B.G.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.207605.supplemental
References
- Adam A. P., Sharenko A. L., Pumiglia K. and Vincent P. A. (2010). Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J. Biol. Chem. 285, 7045-7055. 10.1074/jbc.M109.079277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Oufella H., Maury E., Lehoux S., Guidet B. and Offenstadt G. (2010). The endothelium: Physiological functions and role in microcirculatory failure during severe sepsis. Intensive Care Med. 36, 1286-1298. 10.1007/s00134-010-1893-6 [DOI] [PubMed] [Google Scholar]
- Ando K., Fukuhara S., Moriya T., Obara Y., Nakahata N. and Mochizuki N. (2013). Rap1 potentiates endothelial cell junctions by spatially controlling myosin I activity and actin organization. J. Cell Biol. 202, 901-916. 10.1083/jcb.201301115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benn A., Bredow C., Casanova I., Vukičević S. and Knaus P. (2016). VE-cadherin facilitates BMP-induced endothelial cell permeability and signaling. J. Cell Sci. 129, 206-218. 10.1242/jcs.179960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béraud-Dufour S., Gautier R., Albiges-Rizo C., Chardin P. and Faurobert E. (2007). Krit 1 interactions with microtubules and membranes are regulated by Rap1 and integrin cytoplasmic domain associated protein-1. Febs J. 274, 5518-5532. 10.1111/j.1742-4658.2007.06068.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova A. A., Zagranichnaya T., Alekseeva E., Bokoch G. M. and Birukov K. G. (2008). Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protection. J. Cell. Physiol. 215, 715-724. 10.1002/jcp.21354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova A. A., Meng F., Tian Y., Meliton A., Sarich N., Quilliam L. A. and Birukov K. G. (2015). Prostacyclin post-treatment improves LPS-induced acute lung injury and endothelial barrier recovery via Rap1. Biochim Biophys Acta. 1852, 778-791. 10.1016/j.bbadis.2014.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J., Wu L., Qi X., Li Calzi S., Caballero S., Shaw L., Ruan Q., Grant M. B. and Boulton M. E. (2011). PEDF regulates vascular permeability by a γ-secretase-mediated pathway. PLoS ONE 6, e21164 10.1371/journal.pone.0021164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. (2003). Angiogenesis in health and disease. Nat. Med. 9, 653-660. 10.1038/nm0603-653 [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Lampugnani M.-G., Moons L., Breviario F., Compernolle V., Bono F., Balconi G., Spagnuolo R., Oosthuyse B. and Dewerchin M. (1999). Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98, 147-157. 10.1016/S0092-8674(00)81010-7 [DOI] [PubMed] [Google Scholar]
- Carmona G., Gottig S., Orlandi A., Scheele J., Bauerle T., Jugold M., Kiessling F., Henschler R., Zeiher A. M., Dimmeler S. et al. (2009). Role of the small GTPase Rap1 for integrin activity regulation in endothelial cells and angiogenesis. Blood 113, 488-497. 10.1182/blood-2008-02-138438 [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M. (2013). Distinct functions for Rap1 signaling in vascular morphogenesis and dysfunction. Exp. Cell Res. 319, 2350-2359. 10.1016/j.yexcr.2013.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M. (2017). Rap1 in endothelial biology. Curr. Opin Hematol. 24, 248-255. 10.1097/MOH.0000000000000332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M., Kraus A. E., Gale D., White G. C. II and Vansluys J. (2008). Defective angiogenesis, endothelial migration, proliferation, and MAPK signaling in Rap1b-deficient mice. Blood 111, 2647-2656. 10.1182/blood-2007-08-109710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M., White G. C., Quilliam L. A. and Whitehead K. J. (2015). Small GTPase Rap1 is essential for mouse development and formation of functional vasculature. PLoS ONE 10, e0145689 10.1371/journal.pone.0145689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullere X., Shaw S. K., Andersson L., Hirahashi J., Luscinskas F. W. and Mayadas T. N. (2005). Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 105, 1950-1955. 10.1182/blood-2004-05-1987 [DOI] [PubMed] [Google Scholar]
- Dejana E. and Giampietro C. (2012). Vascular endothelial-cadherin and vascular stability. Curr. Opin Hematol. 19, 218-223. 10.1097/MOH.0b013e3283523e1c [DOI] [PubMed] [Google Scholar]
- Dejana E., Tournier-Lasserve E. and Weinstein B. M. (2009). The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev. Cell 16, 209-221. 10.1016/j.devcel.2009.01.004 [DOI] [PubMed] [Google Scholar]
- Dominguez J. M. II, Hu P., Caballero S., Moldovan L., Verma A., Oudit G. Y., Li Q. and Grant M. B. (2016). Adeno-associated virus overexpression of angiotensin-converting enzyme-2 reverses diabetic retinopathy in type 1 diabetes in mice. Am. J. Pathol. 186, 1688-1700. 10.1016/j.ajpath.2016.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y., Smith M. A., Miller C. M. and Kern T. S. (2002). Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J. Neurochem. 80, 771-779. 10.1046/j.0022-3042.2001.00737.x [DOI] [PubMed] [Google Scholar]
- Eliceiri B. P., Paul R., Schwartzberg P. L., Hood J. D., Leng J. and Cheresh D. A. (1999). Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 4, 915-924. 10.1016/S1097-2765(00)80221-X [DOI] [PubMed] [Google Scholar]
- Esser S., Lampugnani M., Corada M., Dejana E. and Risau W. (1998). Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 111, 1853-1865. [DOI] [PubMed] [Google Scholar]
- Ferrara N., Mass R. D., Campa C. and Kim R. (2007). Targeting VEGF-A to treat cancer and age-related macular degeneration. Annu. Rev. Med. 58, 491-504. 10.1146/annurev.med.58.061705.145635 [DOI] [PubMed] [Google Scholar]
- Gavard J. (2009). Breaking the VE-cadherin bonds. FEBS Lett. 583, 1-6. 10.1016/j.febslet.2008.11.032 [DOI] [PubMed] [Google Scholar]
- Giaever I. and Keese C. R. (1991). Micromotion of mammalian cells measured electrically. Proc. Natl. Acad. Sci. U.S.A. 88, 7896-7900. 10.1073/pnas.88.17.7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glading A., Han J., Stockton R. A. and Ginsberg M. H. (2007). KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 179, 247-254. 10.1083/jcb.200705175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris E. S. and Nelson W. J. (2010). VE-cadherin: at the front, center, and sides of endothelial cell organization and function. Curr. Opin. Cell Biol. 22, 651-658. 10.1016/j.ceb.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince C., Mayeux P. R., Nguyen T., Gomez H., Kellum J. A., Ospina-Tascón G. A., Hernandez G., Murray P. and De Backer D. (2016). The endothelium in sepsis. Shock 45, 259-270. 10.1097/SHK.0000000000000473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra M. R. H., Corada M., Dejana E. and Bos J. L. (2005). Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 579, 4966-4972. 10.1016/j.febslet.2005.07.080 [DOI] [PubMed] [Google Scholar]
- Lakshmikanthan S., Sobczak M., Chun C., Henschel A., Dargatz J., Ramchandran R. and Chrzanowska-Wodnicka M. (2011). Rap1 promotes VEGFR2 activation and angiogenesis by a mechanism involving integrin alphavbeta(3). Blood 118, 2015-2026. 10.1182/blood-2011-04-349282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmikanthan S., Zheng X., Nishijima Y., Sobczak M., Szabo A., Vasquez-Vivar J., Zhang D. X. and Chrzanowska-Wodnicka M. (2015). Rap1 promotes endothelial mechanosensing complex formation, NO release and normal endothelial function. EMBO Rep. 16, 628-637. 10.15252/embr.201439846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M.-T., Yen M. L., Lin C. Y. and Kuo M. L. (2003). Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of Src-dependent vascular endothelial cadherin tyrosine phosphorylation. Mol. Pharmacol. 64, 1029-1036. 10.1124/mol.64.5.1029 [DOI] [PubMed] [Google Scholar]
- Lo C. M., Keese C. R. and Giaever I. (1995). Impedance analysis of MDCK cells measured by electric cell-substrate impedance sensing. Biophys. J. 69, 2800-2807. 10.1016/S0006-3495(95)80153-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Palomo A., Meza I., Beaty G. and Cereijido M. (1980). Experimental modulation of occluding junctions in a cultured transporting epithelium. J. Cell Biol. 87, 736-745. 10.1083/jcb.87.3.736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D. and Malik A. B. (2006). Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279-367. 10.1152/physrev.00012.2005 [DOI] [PubMed] [Google Scholar]
- Miles A. A. and Miles E. M. (1952). Vascular reactions to histamine, histamine-liberator and leukotaxine in the skin of guinea-pigs. J. Physiol. 118, 228-257. 10.1113/jphysiol.1952.sp004789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan-Benson E. and Burridge K. (2009). The regulation of vascular endothelial growth factor-induced microvascular permeability requires rac and reactive oxygen species. J. Biol. Chem. 284, 25602-25611. 10.1074/jbc.M109.009894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy J. A., Benjamin L., Zeng H. Y., Dvorak A. M. and Dvorak H. F. (2008). Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 11, 109-119. 10.1007/s10456-008-9099-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda K., Zhang J., Fukuhara S., Kunimoto S., Yoshimura M. and Mochizuki N. (2010). Vascular endothelial-cadherin stabilizes at cell-cell junctions by anchoring to circumferential actin bundles through α- and β-catenins in cyclic AMP-Epac-Rap1 signal-activated endothelial cells. Mol. Biol. Cell 21, 584-596. 10.1091/mbc.E09-07-0580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson A.-K., Dimberg A., Kreuger J. and Claesson-Welsh L. (2006). VEGF receptor signalling - in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359-371. 10.1038/nrm1911 [DOI] [PubMed] [Google Scholar]
- Pan B.-X., Vautier F., Ito W., Bolshakov V. Y. and Morozov A. (2008). Enhanced cortico-amygdala efficacy and suppressed fear in absence of Rap1. J. Neurosci. 28, 2089-2098. 10.1523/JNEUROSCI.5156-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannekoek W. J., Post A. and Bos J. L. (2014). Rap1 signaling in endothelial barrier control. Cell Adhesion and Migration 8, 100-107. 10.4161/cam.27352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu M. E., Schmidt I., Benedito R. and Adams R. H. (2010). Inducible gene targeting in the neonatal vasculature and analysis of retinal angiogenesis in mice. Nat. Protoc. 5, 1518-1534. 10.1038/nprot.2010.113 [DOI] [PubMed] [Google Scholar]
- Post A., Pannekoek W.-J., Ross S. H., Verlaan I., Brouwer P. M. and Bos J. L. (2013). Rasip1 mediates Rap1 regulation of Rho in endothelial barrier function through ArhGAP29. Proc. Natl. Acad. Sci. USA 110, 11427-11432. 10.1073/pnas.1306595110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post A., Pannekoek W. J., Ponsioen B., Vliem M. J. and Bos J. L. (2015). Rap1 spatially controls ArhGAP29 to inhibit Rho signaling during endothelial barrier regulation. Mol. Cell. Biol. 35, 2495-2502. 10.1128/MCB.01453-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter M. D., Barbero S. and Cheresh D. A. (2005). Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and β-catenin and maintains the cellular mesenchymal state. J. Biol. Chem. 280, 31906-31912. 10.1074/jbc.M505568200 [DOI] [PubMed] [Google Scholar]
- Qaum T., Xu Q., Joussen A. M., Clemens M. W., Qin W., Miyamoto K., Hassessian H., Wiegand S. J., Rudge J., Yancopoulos G. D. et al. (2001). VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest. Ophthalmol. Vis. Sci. 42, 2408-2413. [PubMed] [Google Scholar]
- Rodrigues S. F. and Granger D. N. (2015). Blood cells and endothelial barrier function. Tissue Barriers 3, e978720 10.4161/21688370.2014.978720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senger D., Galli S., Dvorak A., Perruzzi C., Harvey V. and Dvorak H. (1983). Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219, 983-985. 10.1126/science.6823562 [DOI] [PubMed] [Google Scholar]
- Smutny M., Cox H. L., Leerberg J. M., Kovacs E. M., Conti M. A., Ferguson C., Hamilton N. A., Parton R. G., Adelstein R. S. and Yap A. S. (2010). Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat. Cell Biol. 12, 696-702. 10.1038/ncb2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak M., Dargatz J. and Chrzanowska-Wodnicka M. (2010). Isolation and culture of pulmonary endothelial cells from neonatal mice. J. Vis. Exp. 46, 2316. 10.3791/2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulcek R., Bogaard H. J. and van Nieuw Amerongen G. P. (2014). Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. J. Vis. Exp. 85, 51300. 10.3791/51300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawa H., Rikitake Y., Takahashi M., Amano H., Miyata M., Satomi-Kobayashi S., Kinugasa M., Nagamatsu Y., Majima T., Ogita H. et al. (2010). Role of afadin in vascular endothelial growth factor- and sphingosine 1-phosphate-induced angiogenesis. Circ. Res. 106, 1731-1742. 10.1161/CIRCRESAHA.110.216747 [DOI] [PubMed] [Google Scholar]
- Tse D. and Stan R. V. (2010). Morphological heterogeneity of endothelium. Semin. Thromb. Hemost. 36, 236-245. 10.1055/s-0030-1253447 [DOI] [PubMed] [Google Scholar]
- Vestweber D., Winderlich M., Cagna G. and Nottebaum A. F. (2009). Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 19, 8-15. 10.1016/j.tcb.2008.10.001 [DOI] [PubMed] [Google Scholar]
- Wallez Y., Cand F., Cruzalegui F., Wernstedt C., Souchelnytskyi S., Vilgrain I. and Huber P. (2007). Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: Identification of tyrosine 685 as the unique target site. Oncogene 26, 1067-1077. 10.1038/sj.onc.1209855 [DOI] [PubMed] [Google Scholar]
- Weis S. M. and Cheresh D. A. (2005). Pathophysiological consequences of VEGF-induced vascular permeability. Nature 437, 497-504. 10.1038/nature03987 [DOI] [PubMed] [Google Scholar]
- Wessel F., Winderlich M., Holm M., Frye M., Rivera-Galdos R., Vockel M., Linnepe R., Ipe U., Stadtmann A., Zarbock A. et al. (2014). Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 15, 223-230. 10.1038/ni.2824 [DOI] [PubMed] [Google Scholar]
- Wilson C. W., Parker L. H., Hall C. J., Smyczek T., Mak J., Crow A., Posthuma G., Mazière A. D., Sagolla M., Chalouni C. et al. (2013). RASIP1 regulates vertebrate vascular endothelial junction stability through EPAC1-RAP1 signaling. Blood 122, 3678-3690. 10.1182/blood-2013-02-483156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittchen E. S., Worthylake R. A., Kelly P., Casey P. J., Quilliam L. A. and Burridge K. (2005). Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J. Biol. Chem. 280, 11675-11682. 10.1074/jbc.M412595200 [DOI] [PubMed] [Google Scholar]
- Wittchen E. S., Aghajanian A. and Burridge K. (2011). Isoform-specific differences between Rap1A and Rap1B GTPases in the formation of endothelial cell junctions. Small GTPases 2, 65-76. 10.4161/sgtp.2.2.15735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary I. and Gliki G. (2001). Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc. Res. 49, 568-581. 10.1016/S0008-6363(00)00268-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.