

ABSTRACT
Aberrations in signaling pathways that regulate tissue growth often lead to tumorigenesis. Homeodomain-interacting protein kinase (Hipk) family members are reported to have distinct and contradictory effects on cell proliferation and tissue growth. From these studies, it is clear that much remains to be learned about the roles of Hipk family protein kinases in proliferation and cell behavior. Previous work has shown that Drosophila Hipk is a potent growth regulator, thus we predicted that it could have a role in tumorigenesis. In our study of Hipk-induced phenotypes, we observed the formation of tumor-like structures in multiple cell types in larvae and adults. Furthermore, elevated Hipk in epithelial cells induces cell spreading, invasion and epithelial-to-mesenchymal transition (EMT) in the imaginal disc. Further evidence comes from cell culture studies, in which we expressed Drosophila Hipk in human breast cancer cells and showed that it enhances proliferation and migration. Past studies have shown that Hipk can promote the action of conserved pathways implicated in cancer and EMT, such as Wnt/Wingless, Hippo, Notch and JNK. We show that Hipk phenotypes are not likely to arise from activation of a single target, but rather through a cumulative effect on numerous target pathways. Most Drosophila tumor models involve mutations in multiple genes, such as the well-known RasV12 model, in which EMT and invasiveness occur after the additional loss of the tumor suppressor gene scribble. Our study reveals that elevated levels of Hipk on their own can promote both hyperproliferation and invasive cell behavior, suggesting that Hipk family members could be potent oncogenes and drivers of EMT.
KEY WORDS: Hipk, Metastasis, Tumor, Cancer
Summary: The protein kinase Hipk can promote proliferation and invasive behaviors, and can synergize with known cancer pathways, in a new Drosophila model for tumorigenesis.
INTRODUCTION
A number of evolutionarily conserved signaling pathways are used reiteratively during development to control the growth of healthy organs and tissues. Genetic aberrations in pathway components can lead to dysregulated growth signals, often resulting in uncontrolled proliferation and tumorigenesis. With time and further genetic changes, tumor cells can progress into a metastatic state by undergoing epithelial-to-mesenchymal transition (EMT), enabling cells to leave the primary tumor site and travel to other locations in the body (reviewed by Thiery et al., 2009). Many of the cellular markers and processes involved in vertebrate tumorigenesis are conserved in Drosophila, which has been used for decades to study developmental signaling pathways and has been key in revealing molecular functions of human disease and cancer-related genes (Brumby and Richardson, 2005; Gonzalez, 2013; Potter et al., 2000; Rudrapatna et al., 2012). Tissue and organ growth are often studied using the larval imaginal discs, which are epithelial sacs composed primarily of a pseudo-stratified columnar monolayer (Aldaz and Escudero, 2010). Discs undergo extensive proliferation, with subsequent patterning and differentiation to form adult structures, which requires the same key signaling pathways needed for human development and growth (Brumby and Richardson, 2005; Gonzalez, 2013; Herranz et al., 2016; Miles et al., 2011; Rudrapatna et al., 2012; Sonoshita and Cagan, 2017). Low genetic redundancy paired with powerful genetic manipulation tools make Drosophila an excellent system for the study of tumorigenesis and metastasis.
Numerous signaling pathways have been implicated in the development of tissue overgrowth and/or metastatic behavior in the fly. The majority of these studies have described tumor models that require the combination of multiple genetic aberrations in order to manifest hyperproliferation coupled with invasive behaviors. The earliest metastasis model involved activated Ras combined with loss of the tumor suppressor scribble (Pagliarini and Xu, 2003). Notch pathway activation coupled with alterations in histone epigenetic marks also led to a Drosophila tumor model (Ferres-Marco et al., 2006). Subsequent studies have identified further factors involved in both Ras- and Notch-driven tumorigenesis (Doggett et al., 2015). Other tumor studies involve Epidermal growth factor receptor (Egfr) signaling (Herranz et al., 2012) and the Sin3A histone deacetylase (HDAC) (Das et al., 2013). The Hippo pathway is a potent tumor suppressor pathway that is required to prevent hematopoietic disorders (Milton et al., 2014). Activated JAK/STAT signaling causes leukemia-like hematopoiesis defects in Drosophila (Harrison et al., 1995; Luo et al., 1997).
Homeodomain-interacting protein kinases (Hipk) are evolutionarily conserved, and vertebrates possess Hipk1-Hipk4, whereas Drosophila and Caenorhabditis elegans have only one Hipk each. Hipk family members are expressed in dynamic temporal and spatial patterns, highlighting their important roles during development (reviewed by Blaquiere and Verheyen, 2017). Hipk protein levels are highly regulated by post-translational modification and proteasomal degradation (Saul and Schmitz, 2013). Hipk family members are reported to have distinct and contradictory effects on cell proliferation and tissue growth. Overexpressing Drosophila Hipk causes tissue overgrowths in the wing, eye and legs in a dose-dependent manner (Chen and Verheyen, 2012; Lee et al., 2009a; Poon et al., 2012). In C. elegans, Hpk-1 promotes proliferation of the germline cells, and loss of hpk-1 reduces the number of proliferating cells and size of the mitotic region (Berber et al., 2013). Hipk2−/− mice have growth deficiencies, and 40% die prematurely (Chalazonitis et al., 2011; Sjölund et al., 2014; Trapasso et al., 2009). In normal human skin, Hipk2 protein expression is enriched in basal proliferating cells, whereas it is undetectable in nonproliferating cells (Iacovelli et al., 2009), and expression is reactivated when cells are stimulated to proliferate, suggesting a close link between Hipk protein function and cell proliferation. Mouse embryo fibroblasts (MEFs) from Hipk2−/− knockout mice show reduced proliferation (Trapasso et al., 2009), whereas another study claimed that such cells proliferated more than wild type (Wei et al., 2007). From these studies, it is clear that much remains to be learned about the roles of Hipk family protein kinases in proliferation and cell behavior.
Hipks regulate numerous signaling pathways required for the development of healthy tissues (Fig. S1; reviewed by Blaquiere and Verheyen, 2017). Both Drosophila and vertebrate Hipks can modulate Wnt signaling in many ways (Hikasa and Sokol, 2011; Hikasa et al., 2010; Kuwahara et al., 2014; Lee et al., 2009b; Louie et al., 2009; Shimizu et al., 2014; Swarup and Verheyen, 2011; Wu et al., 2012). Hipk proteins modulate the Hippo pathway in Drosophila, which is an essential conserved signaling pathway regulating tissue and organ growth (Chen and Verheyen, 2012; Poon et al., 2012). Yki activity requires Hipk, as hipk loss of function can suppress the effects of constitutively active Yki (YkiS168A). Hipks have also been shown to regulate Jun N-terminal kinase (JNK) signaling in numerous contexts (Hofmann et al., 2003, 2005; Huang et al., 2011; Lan et al., 2007, 2012; Rochat-Steiner et al., 2000; Song and Lee, 2003; Chen and Verheyen, 2012). Hipk is required for the full effect of JAK/STAT signaling, because loss of hipk through somatic clonal analysis causes loss of Stat92E-GFP reporter and, furthermore, loss of hipk can suppress lethality and tumor frequency in the constitutively active hopTum−L allele (Blaquiere et al., 2016 preprint).
Hipk2 is the best-characterized vertebrate Hipk family member. Studies in cell culture and cancer samples reveal conflicting results (Blaquiere and Verheyen, 2017). For example, Hipk2 acts as a tumor suppressor in the context of p53-mediated cell death after lethal DNA damage (Hofmann et al., 2013), and reduced expression of Hipk proteins is seen in several cancer types (Lavra et al., 2011; Pierantoni et al., 2002; Ricci et al., 2013; Tan et al., 2014). By contrast, Hipk2 is elevated in certain cancers, including cervical cancers, pilocytic astrocytomas and colorectal cancer cells, and in other diseases, such as thyroid follicular hyperplasia (Al-Beiti and Lu, 2008; Cheng et al., 2012; D'Orazi et al., 2006; Deshmukh et al., 2008; Jacob et al., 2009; Lavra et al., 2011; Saul and Schmitz, 2013; Yu et al., 2009). Human Hipk1 is also found at elevated levels in certain cancer cell lines and tissue samples (Kondo et al., 2003; Rey et al., 2013).
Although it is known that Drosophila Hipk is a strong inducer of tissue growth, its role in tumorigenesis is less well understood. We were intrigued to test whether Hipk contributes to this process in Drosophila, because of the genetic simplicity of this model system. Using various techniques, we provide evidence that elevation of hipk can lead to neoplasia characterized by cell invasiveness. Hipk expression drives numerous cellular changes that are hallmarks of EMT. Hipk-expressing cells can migrate through tissues, disrupt the basement membrane to exit tissues and express mesenchymal markers. Our findings are significant in that Hipk alone can promote proliferation and invasive behavior that has been previously described to arise from the perturbation of multiple pathways. We propose that Drosophila Hipk has potent oncogenic properties, and that Hipk can exert such an effect through promotion of its multiple target pathways.
RESULTS
Elevated Hipk leads to overgrowths and masses
To study the implications of elevated hipk, we used the GAL4-UAS system to overexpress Hipk in a variety of cell types. Using a combination of growth at different temperatures (which affects the potency of the GAL4 transcription factor) and copy number of UAS-transgenes, we have generated a range of Hipk overexpression phenotypes. Use of the dpp-Gal4 driver to express two copies of Hipk at 25°C caused dramatic overgrowth of eye, wing and leg imaginal discs, characterized by tissue folds and protrusions (Fig. 1A-F′). We made use of GFP labeling to mark the hipk-overexpressing cells, allowing us to visualize their behavior (Fig. 1D-F′) in comparison with wild-type cells (dpp>GFP; Fig. 1A-C′). Coexpression of hipk and GFP at 29°C (dpp>HA-hipk3M+GFP) led to overgrown wing discs (Fig. 1J). Staining for cleaved Caspase 3 (Casp3) revealed that cell death was autonomously induced within the hipk-expressing discs (Fig. 1H-H″; Fig. S2A). When we used P35 expression to block caspase-dependent cell death, cells within the Dpp domain expanded substantially and occupied almost the entire dpp>HA-hipk3M+P35+GFP discs (Fig. 1K), whereas dpp>P35 alone had no effect in this context (Fig. 1L). This implies that Hipk can induce both cell death and abundant proliferation to induce the gain-of-function phenotypes.
Fig. 1.

Hipk induces overgrowths in Drosophila imaginal discs. Control eye-antennal (A), wing (B) and leg (C) imaginal discs stained for actin to reveal tissue morphology and GFP to reveal the dpp-Gal4 expression domain. Expression of two copies of wild-type HA-hipk (HA-hipk3M and HA-hipk1M) within the dpp domain leads to overgrown eye-antennal (D), wing (E) and leg (F) imaginal discs. (G) A control wing disc pouch stained for Casp3. (H) Casp3 is autonomously induced in dpp>HA-hipk3M+GFP wing discs. (I) A control wing imaginal disc showing the dpp-GAL4 expression domain marked by GFP. (J) dpp>HA-hipk3M+GFP leads to overgrown wing discs. (K) Loss of cell death in dpp>HA-hipk3M+GFP+P35 discs worsens hipk-induced overgrowths, whereas (L) dpp>GFP+P35 appears normal. Scale bars: 50 μm.
Hipk induces melanotic masses in the hemocytes
In addition to overgrown discs, darkly pigmented stationary masses were present in both dpp>HA-hipk3M+2xGFP and dpp>HA-hipk3M+P35+GFP larvae grown at 29°C (Fig. 2B,D), whereas control larvae dpp>GFP (Fig. 2A) and dpp>P35 (Fig. 2C) displayed none. The persistence of the masses upon P35 coexpression suggests that they were not attributable to cell death. Moreover, dpp>HA-hipk3M+P35+GFP animals remained in the third larval stage for an extended period of time (beyond 10 days) and eventually died as larvae. Arrested development in tumor-ridden animals has been reported by others and is thought to be attributable to alterations in ecdysone regulation (Garelli et al., 2012; Parisi et al., 2014). We hypothesized that these tumors might arise because of expression of the dpp-Gal4 driver in larval blood cells (Ayyaz et al., 2015; Clark et al., 2011).
Fig. 2.
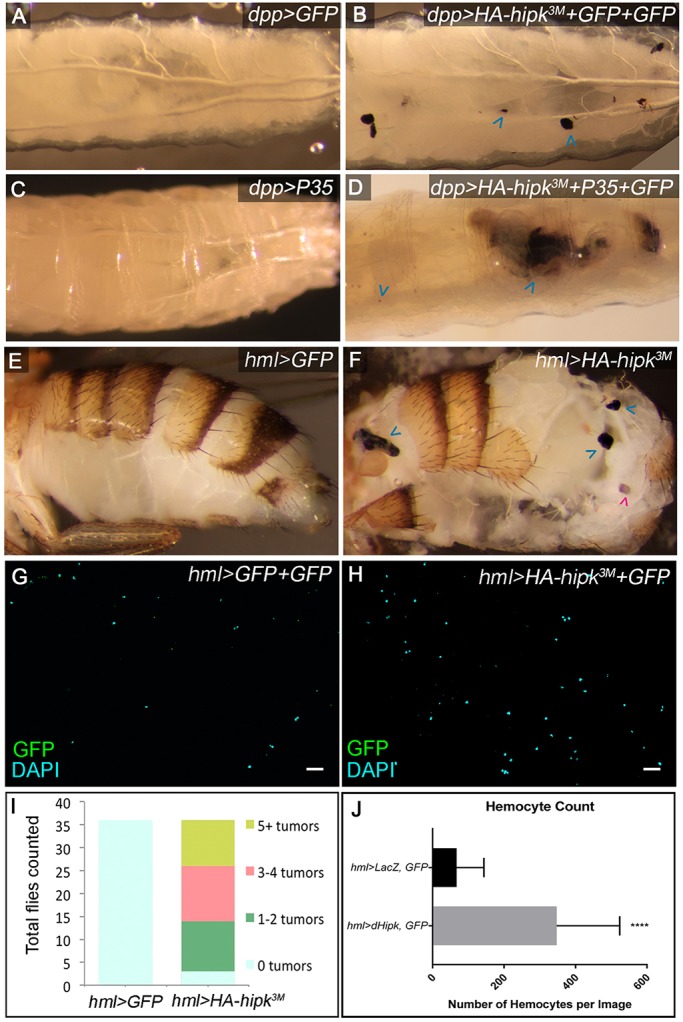
Hipk induces hemocyte-derived melanotic tumors. (A) A control dpp>GFP third instar larva. (B) Stationary melanized masses are observed in 65% of dpp>HA-hipk3M+GFP+GFP larvae (blue arrowheads; n=40). (C) dpp>P35 larvae show no tumors. (D) The masses persist when apoptotic cell death is inhibited in dpp>HA-hipk3M+P35+GFP larvae. (E) The abdomen of a control hml>GFP fly. (F) Melanized tumors are present in hml>HA-hipk3M flies (blue arrowheads). Spermathecae were not counted (magenta arrowhead). Smears of total hemolymph collected from (G) hml>2xGFP and (H) hml>HA-hipk3M+GFP third instar larvae. (I) Quantification of the number of tumors scored from dissected abdomens of flies shown in (E) and (F), n=36 for both groups. (J) Quantification of mean number of hemocytes per defined sampling area counted from genotypes in (G) and (H). Each data point represents the mean of five cell counts from one larva, hml>GFP+LacZ (n=16 samples, n=80 cell counts) and hml>HA-hipk3M+GFP (n=16 samples, n=80 cell counts), ****P<0.0001.
Melanotic tumors arise as a result of over-amplification and melanization of hemocytes, which are fly hematopoietic cells (Hanratty and Ryerse, 1981). Therefore, we next tested directly whether hipk could cause tumors when overexpressed in the circulating hemocytes and lymph gland using hemolectin-GAL4 (hml-GAL4) (Sinenko and Mathey-Prevot, 2004). Of the hml>HA-hipk3M flies, 91.7% exhibited at least one clearly visible melanotic tumor, with the average being three or four tumors (Fig. 2F,I), compared with 0% of hml>GFP flies (Fig. 2E,I). To test whether Hipk increased the number of circulating hemocytes, we isolated the total hemolymph from third instar larvae. The mean number of hemocytes in each hml>HA-hipk3M+GFP sampling area (see Materials and methods) was 348, compared with 67 per hml>GFP+GFP sampling area (Fig. 2G,H,J). These data suggest that the abdominal tumors induced by Hipk are derived from hyperproliferating hemocytes.
Hipk induces cell invasiveness
Valuable methods have been developed that allow one to assay for invasive behavior using the GAL4-UAS system (Ferres-Marco et al., 2006; Herranz et al., 2014; Pallavi et al., 2012). In the wing disc, dpp is expressed in the anterior-most cells of the anterior-posterior (A/P) boundary. Thus, in dpp>GFP discs, a sharp border of GFP-expressing and non-GFP-expressing cells is produced (Fig. 1B). In dpp>HA-hipk3M+GFP discs, multiple isolated GFP+ cells were found outside the dpp domain, suggesting that cells migrated away from their original location in the disc (Fig. 1E).
To provide further evidence of cell spreading, dpp>HA-hipk3M+GFP wing discs were co-stained for the anterior marker Cubitus interruptus (Ci) and for the posterior marker Engrailed (En) (Fig. 3A-C). In normal conditions, the A/P boundary is well defined, in which the Dpp domain (marked by GFP) is restricted within the anterior compartment (Fig. 3A,B). However, in discs with elevated hipk, GFP+ cells originating from the anterior compartment were found in the posterior domain as isolated clusters of cells (Fig. 3C′). On rare occasions, GFP+ clusters simultaneously expressed Ci and En, suggesting that these cells have either lost their ability to interpret A/P positional cues from the tissue or are in a period of fate transition (Fig. S3A). We also found that individual GFP+ cells move from the central Dpp domain towards both anterior and posterior parts of the discs (Fig. 1E).
Fig. 3.

Hipk induces cell spreading. (A,B) A dpp>GFP wing disc stained for GFP, Ci and En. (C) In dpp>HA-hipk3M+GFP discs, anterior fated cells are found within the posterior wing compartment (arrowheads). (D) An ectopic eye seen within the thorax of an ey>HA-hipk1M+HA-hipk3M fly (frequency 1%, n=100). (D′) Dissection reveals the size of the ectopic eye (arrowhead). (E) A smaller ectopic eye is seen in the abdominal region of a dpp>HA-hipk3M+GFP fly (arrowhead; frequency 2%, n=100). Scale bars: 50 μm.
Another phenotype associated with metastatic behavior in Drosophila is the migration of retinal tissue into the body of the fly (Ferres-Marco et al., 2006; Pallavi et al., 2012). When hipk was expressed in the eye disc using eyeless-GAL4 (ey-GAL4), a large cluster of pigmented retinal cells was observed in the thorax of the adult fly (Fig. 3D). The endogenous eyes were fully intact, which suggests this was not likely to have been a disc eversion defect, but rather a metastatic event where retinal tissue migrated away from the eye disc and lodged into the thorax. This phenotype also occurred in dpp>HA-hipk3M+GFP flies, in which ectopic pigmented eye cells can occasionally be observed in the abdomen (Fig. 3E), indicating that Hipk-expressing eye disc cells can migrate within the body and proliferate.
The data presented thus far suggest that elevating hipk promotes proliferation, cell migration and, possibly, metastatic behavior. To test for this, we used live imaging and witnessed cell extrusion in real time. In dpp>HA-hipk1M+HA-hipk3M eye discs, cells proliferated at a high rate, and multiple cells could be seen extruding from the disc into the culture medium (Fig. S3D-F). Furthermore, the extruded cells continued to proliferate after leaving the disc. Within 60 min of imaging this particular disc, 12 cells left the disc into the culture media (Fig. S3F,G) and some continued amplifying to reach a final cell count of 21 over the subsequent 60 min (Fig. S3H-J). We did not observe any such occurrence in control discs (Fig. S3B,C). Together, these data suggest that cells with elevated Hipk can gain the potential to travel away from their original location in the epithelium.
Hipk alters the integrity of the basement membrane and induces EMT
During metastasis, cells extrude from the main epithelium through various mechanisms, including degradation of the basement membrane by matrix metalloproteinases, such as Mmp1 (Beaucher et al., 2007; Page-McCaw et al., 2003; Srivastava et al., 2007). Expression of Hipk using either dpp-Gal4 or flip-out misexpression clones leads to elevated Mmp1 expression in a cell autonomous manner (Fig. 4A,B; Fig. S2B). Hipk was previously shown to induce Mmp1 expression, but only when the smt3 gene encoding Small ubiquitin-related modifier (SUMO; Smt3) was simultaneously knocked down (Huang et al., 2011). Huang et al. (2011) suggested that in the absence of smt3 Hipk translocates to the cytoplasm and induces JNK and its target, Mmp1. In our experimental context, when HA-hipk3M and GFP were expressed by dpp-GAL4 at 29°C, Hipk was largely nuclear (Fig. S4A), suggesting that Mmp1 induction in our assay is probably attributable to another mechanism.
Fig. 4.

Hipk alters epithelial integrity and induces EMT. (A,A′) A control wing disc (A) and a cross-section of the central region of the disc (A′) stained for GFP, Mmp1 and Ndg. (B,B′) dpp>HA-hipk1M+HA-hipk3M+LacZ+GFP stained to detect GFP, Mmp1 and Ndg. (C,C′) dpp>HA-hipk1M+HA-hipk3M+P35+GFP stained to detect GFP, Mmp1 and Ndg. (D,D′) dpp>P35+GFP stained to detect GFP, MMP1 and Ndg. (E) Ndg is expressed in a uniform pattern along the basement membrane. (F-F‴) Gaps in Ndg expression are present in dpp>HA-hipk3M+GFP discs (F′), and higher resolution (F″,F‴) shows that the location of HA-hipk3M+GFP-expressing cells coincides with regions where Ndg is perturbed (arrowheads). (G) Wild-type eye disc stained for Dlg to reveal cell membranes. (H) ey>HA-hipk1M+HA-hipk3M eye disc stained for Dlg showing defects in Dlg stain and apparent cell extensions towards posterior margin of disc (chevrons). (I,J) Cross-sections of dpp>GFP (I) and dpp>HA-hipk3M+GFP-expressing cells (J) in the center of the wing pouch, stained for E-cad. (K) Twi is expressed in the adepithelial myoblasts, located in the notum region of the wing disc. (L) Twi-positive mesenchymal cells are present in the wing pouch region of dpp>HA-hipk3M+GFP discs (arrowheads), and Twi is induced in a swathe of cells along the dpp domain (asterisk). Scale bars: 50 μm. (M) Drosophila Hipk (dHipk) promotes significant proliferation of three cell lines. dHipk-transfected HEK293T cells (mean=2.097, s.e.m.=0.02803) display increased proliferation compared with empty vector transfected (mean=1.161, s.e.m.=0.01619) conditions; t(4)=28.92, ****P<0.0001. dHipk-transfected breast adenocarcinoma MCF7 cells (mean=1.098, s.e.m.=0.01217) display increased proliferation compared with empty vector transfected (mean=0.8671, s.e.m.=0.0035) conditions; t(4)=18.25, ****P<0.0001. dHipk-transfected breast adenocarcinoma MDA-MB-231 cells (mean=1.067, s.e.m.=0.0037) display increased proliferation compared with empty vector transfected (mean=0.6457, s.e.m.=0.0074) conditions; t(4)=50.83, ****P<0.0001. (N) dHipk-transfected MDA-MB-231 cells (mean=1.953, s.e.m.=0.2277) demonstrated significantly increased migration compared with empty vector transfected (mean=0.9999, s.e.m.=0.0375) conditions; t(19)=4.759, ***P=0.0001. (O) qRT-PCR was used to quantify the expression of the human E-cadherin gene (CDH1) after transfection of MDA-MB-231 cells with dHipk [t(2)=34.86, ***P=0.0008]. (P) Western blot analysis of E-cad expression in HEK293T cells after dHipk transfection.
We examined the basement membrane by staining wing imaginal discs for Nidogen (Ndg), an extracellular matrix component (Fig. 4A-F; Wolfstetter et al., 2009). In dpp>HA-hipk3M+GFP discs, disruptions in the Ndg pattern were observed in sections of wing discs (Fig. 4B) and when the basal surface was examined en face (Fig. 4F). Specifically, the location of HA-hipk3M+GFP+ cells and expression of Mmp1 coincided with disruptions in Ndg (Fig. 4B). GFP+ Hipk-expressing cells appeared to be extruding through holes in the basement membrane (Fig. 4F). Cross-sections of discs also revealed that hipk-expressing cells appeared to be intercalated into the basement membrane (Fig. S4K), which is never seen in wild-type discs (Fig. S4I). Consistent with disruptions in Ndg, elevated hipk produced inconsistencies in Collagen IV (Viking, Vkg) in the wing disc (Fig. S4B,C). These data suggest that Hipk promotes Mmp1-mediated degradation of the basement membrane.
Coexpression of P35 with Hipk caused greater abnormalities in disc morphology, with multiple folds and cell layers, owing to the blocking of cell death (Fig. 4C). In this context, GFP+ cells were observed breaking through the disc surface at both the apical and basal surfaces (Fig. 4C′), suggestive of active migration processes rather than cell death being the mechanism driving cell migration and spreading. Cell autonomous alterations in Mmp1 and Ndg (Fig. 4C) were seen in disc sections, and elevated Mmp1 was seen within protrusions of dpp>HA-hipk3M+P35+GFP eye discs (Fig. S4E″). P35 expression alone is also capable of inducing Mmp1, but no defects in Ndg integrity were observed (Fig. 4D′; Rudrapatna et al., 2013).
We also observed abnormal cell behavior after staining eye discs to detect Dlg and Elav to reveal tissue architecture. In a wild-type disc, the posterior margin of the disc displays a tight boundary of photoreceptor cells as detected by Dlg (Fig. 4G) and Elav (Fig. S4F). In ey>HA-hipk1M+HA-hipk3M discs, the cell morphology is altered, and jagged cell extensions can be seen protruding towards the posterior margin using the Dlg antibody stain (Fig. 4H). Overall, altered integrity of the posterior margin is also seen when staining for Elav (Fig. S4G).
EMT occurs naturally in development (Kiger et al., 2007), but it can also be induced inappropriately during tumorigenesis. Characteristics of EMT are increased expression of Mmp1, mesenchymal markers like Twist (Twi) and Snail, and downregulation of E-cadherin (E-cad). In dpp>HA-hipk3M+GFP discs grown at 29°C, levels of E-cad are reduced in a cell autonomous fashion (Fig. 4J). Twist is normally expressed within mesenchymal cells found within the notum region of the wing disc (Herranz et al., 2014; Fig. 4K). When hipk was overexpressed, Twi expression was mildly induced along the dpp domain, and multiple cells within the wing pouch displayed ectopic expression of Twi (Fig. 4L). Others have shown that Hipk promotes epithelial remodeling of the pupal wing through an EMT-like mechanism (Kiger et al., 2007; Link et al., 2007).
Hipk can induce proliferation and cell migration in vertebrate cells
To determine whether the properties of Drosophila Hipk are conserved in a different context, we examined the effects of transient transfection of pCMV-HA-dHipk into a number of human cell lines. We first assayed proliferation induced by Hipk in HEK293T, MCF7 and MDA-MB-231 human cell lines. After transient transfection, we used the MTT assay to measure cell proliferation and found that Hipk significantly stimulated cell proliferation in all three cell lines (Fig. 4M; Table S4). Using a cell migration assay, we found that transfection of Drosophila Hipk caused MDA-MB-231 cells to exhibit a twofold increase in migration relative to control cells (Fig. 4N; Table S3). One crucial aspect of EMT is the downregulation of E-cad expression. In MDA-MB-231 cells transfected with Hipk, E-cad levels were reduced 3.5-fold compared with levels found in control transfected cells (Fig. 4O). Further evidence for decreased E-cad is seen after western blotting of E-cad from HEK293T cells transfected with dHipk (Fig. 4P; Table S2). These observations support our observations from Drosophila tissues that Hipk is a potent factor that can promote proliferation and EMT in different contexts.
Hipk-induced phenotypes cannot be attributed to a single targeted pathway
To investigate genetically the mechanism underlying the ability of Hipk to induce cell spreading, proliferation and migration, we assessed the effects of disruptions of individual pathways on the dpp>HA-hipk3M phenotype in wing discs by assaying the extent of cell proliferation and migration from the dpp expression domain (Fig. 5A). We chose pathways that were previously shown to be promoted by Hipk in various contexts, in addition to conserved tumor pathways. We evaluated the effects on proliferation and migration of the transgenes individually using dpp-Gal4 (Fig. S5). We also validated that each transgene was effective by assaying targets or downstream events specific to each pathway (Fig. S6). To test whether the Hipk phenotype could be reverted, we first used RNAi against hipk and found that it completely rescued the abnormalities seen in dpp>HA-hipk3M discs (Fig. 5B). The proliferative and invasive effects caused by each transgene and their influence on the Hipk-induced phenotype were quantified by measuring the GFP+ cell area relative to total disc area (Fig. S7A; Table S5), and by assigning a ‘relative degree of invasiveness’ score to each disc (Fig. S7B; Table S8).
Fig. 5.

Loss of individual signaling pathway components cannot suppress the Hipk overexpression phenotype. We assessed the ability of knockdown of the activity of individual pathways to suppress phenotypes induced by overexpressed Hipk, by F-actin staining (magenta) to reveal morphology and GFP (green, white) to indicate cells in which genotypes were manipulated. (A,B) As proof of concept, hipkRNAi (B) suppressed effects seen in dpp>HA-hipk3M wing discs (A). The following pathways were targeted with the indicated transgenes: Wg, using (C) UAS-panRNAi [dTCF] and (D) UAS-Axin-GFP; Egfr, using (E) UAS-EgfrDN; JNK, using (F) UAS-bskDN; Hippo, using (G) UAS-ykiRNAi; Notch, using (H) UAS-DlDN; Hedgehog, using (I) UAS-CiRep; JAK/STAT using (J) UAS-hopRNAi and (K) UAS-upd3RNAi. (L) dpp>HA-hipk3M wing disc stained for nuclei (DAPI) and Hipk. (M) Expression of dpp>HA-hipk3M+UAS-ykiRNAi+UAS-bskDN stained to reveal Hipk and F-actin. (N) dpp>UAS-ykiRNAi+UAS-bskDN stained to reveal GFP and F-actin. All crosses were done at 29°C. Scale bars: 50 μm.
The Wg pathway was inhibited through either knockdown of pangolin/TCF (Fig. 5C) or expression of the negative regulator Axin (Fig. 5D). We noticed that hipk-expressing discs with loss of pan (TCF) still displayed the invasive phenotype and even some overgrowth in the notum region. Likewise, expression of Axin failed to suppress the dpp>HA-hipk3M phenotype. Wing discs coexpressing dominant negative Epidermal growth factor receptor (EgfrDN; Fig. 5E) or dominant negative basket (bskDN, Drosophila JNK; Fig. 5F) with hipk were phenotypically indistinguishable from discs expressing hipk alone (dpp>HA-hipk3M). Knockdown of yki could reduce the overgrowth effect to some degree, consistent with the effect of Hipk on Hippo signaling, but the discs still showed ectopic cell migration (Fig. 5G,G′). Expression of dominant negative Delta (DlDN; Fig. 5H) did not appreciably modify the Hipk overexpression phenotype. Interestingly, after inhibition of the Hedgehog pathway through expression of the repressor form of Ci (CiRep; Fig. 5I), the cell spreading phenotype seemed suppressed and the discs displayed only a broad Dpp domain. We also noticed relatively weak GFP expression in the discs, which is most likely to be attributable to the repression of dpp-Gal4 expression, because Hh controls dpp transcription (Basler and Struhl, 1994). Reduction of JAK/STAT signaling through knockdown of hopscotch (hop; Drosophila JAK; Fig. 5J) showed mild reduction of proliferation, whereas knockdown of one of the unpaired ligands, Upd3, appeared to increase proliferation slightly (Fig. 5K). Together, our genetic data show that interfering with individual signaling pathways using expression of RNAi or dominant negative forms of the corresponding key effectors could not effectively suppress Hipk-induced cell proliferation and spreading.
To test whether the effects are attributable to multiple pathways, we simultaneously interfered with the activity of Yki and Bsk by expressing yki RNAi with bskDN in a dpp>HA-hipk3M background (Fig. 5M). Although expression of Hipk induces overproliferation of the dpp-expressing cells, which can be seen by staining for Hipk in dpp>HA-hipk3M, after inhibition of Yki and Bsk, the number of Hipk-expressing cells in the dpp domain is drastically reduced, and no cell spreading is observed. Expression of dpp>ykiRNAi with bskDN alone had a mild effect on the number of GFP+ cells in the dpp stripe (Fig. 5N).
Increasing the activity of individual signaling pathways does not phenocopy Hipk-induced phenotypes
We next examined whether hyperactivation of pathways that are promoted by Hipk, or that are involved in growth and proliferation, can induce similar phenotypes to those caused by overexpression of Hipk (Fig. 6A). This might reveal whether there are certain pathways that play a more dominant role in propagating the Hipk signal. We used UAS-controlled expression of wild type or constitutively active pathway members (Table S1). Wing discs expressing degradation-resistant Arms10 (β-catenin) to promote Wg signaling (Fig. 6B) displayed ectopic wing pouch-like structure in the notum, yet the Dpp stripe appeared relatively normal. Overexpression of Stat92E to elevate JAK/STAT signaling (Fig. 6C) led to oversized discs. Wing discs expressing oncogenic Ras to promote Ras/Erk signaling showed robust overgrowths (Fig. 6D). Stimulation of the JNK pathway using eiger expression primarily caused invasive phenotypes but had little effect on proliferation (Fig. 6E). Inactivation of Hippo signaling by expression of constitutively active Yki (YkiS168A) led to widening of the Dpp domain, and smooth, curved edges along the domain (Fig. 6F). Activated Notch signaling (Nact; Fig. 6G) and ectopic Ci (Fig. 6H), which promotes Hh, both induced very dramatic and unique cellular effects. dpp>Notchact led to phenotypes in the wing disc similar to those previously seen with expression of dpp>Dl (Ferres-Marco et al., 2006). Overall, this assay reveals that activation of different pathways leads to distinct effects on proliferation. These results suggest that Hipk-induced phenotypes are likely to arise as a cumulative effect of stimulating the activity of multiple pathways, because no single pathway can phenocopy the behavior of cells in discs with elevated Hipk in the dpp domain. The proliferative and invasive effects caused by each transgene were quantified by measuring the GFP+ cell area relative to total disc area (Fig. S7C; Table S6), and by assigning a ‘relative degree of invasiveness’ score to each disc (Fig. S7D; Table S9).
Fig. 6.

Overexpression of individual signaling pathway components does not phenocopy the cell-spreading phenotype induced by elevated Hipk. (A) A third instar wing imaginal disc with HA-hipk3M+GFP expressed along the dpp domain serves as the baseline phenotype/control disc. Individual pathway activators were expressed using dpp-Gal4, UAS-GFP, namely: (B) UAS-ArmS10, (C) UAS-Stat92E, (D) UAS-Rasact, (E) UAS-eiger, (F) constitutively active UAS-ykiS168A, (G) UAS-Nact and (H) UAS-Ci. Discs were stained for F-actin (magenta) to reveal tissue morphology and for GFP (green, white) to mark cells in which transgenes were ectopically expressed using dpp-Gal4. Scale bars: 50 μm. All crosses were done at 29°C.
Hipk overexpression synergistically enhances other tumor models
Finally, we assessed whether Hipk expression could synergize with other sensitized tumor models in Drosophila wing discs. We coexpressed Hipk with the same gain of function mutants used in the previous section (Table S1) and assayed proliferation and cell migration. The phenotype of dpp>Arms10 alone was enhanced upon coexpressing HA-hipk3M; notably, the effect was much more pronounced in the notum region of the disc (Fig. 7B). Coexpression of stat92E and hipk resulted in invasive phenotypes (Fig. 7C), whereas discs expressing stat92E alone did not (Fig. 6C). In stark contrast to the phenotype in eiger-expressing discs (Fig. 6E), Hipk cooperated with Eiger to cause a significant increase in migrating cells (Fig. 7E). Despite being smaller than dpp>yki discs, dpp>hipk+yki discs acquired noticeable cell spreading properties (Fig. 7F). Rasact (Fig. 7D) and Notchact (Fig. 7G) both showed a strong synergistic effect with ectopic Hipk, compared with phenotypes seen with either one alone, shown in Fig. 6. The strongest synergy was seen with Ci (Fig. 7H). Of note, the effect with Ci alone was also the most dramatic in these assay conditions. The effects of all transgenes on Hipk-induced phenotypes were quantified by measuring the GFP+ cell area relative to total disc area (Fig. S7E; Table S7) and by quantifying the relative degree of invasiveness (Fig. S7F; Table S10). Thus, Hipk expression can synergize with several well-described Drosophila tumor and metastasis models, supporting its oncogenic properties.
Fig. 7.

Hipk overexpression synergistically enhances other tumor models. Individual pathway activators were expressed by crossing to dpp-Gal4, UAS-GFP; UAS-HA-hipk3M/TM6B, namely: (A) Gal4 titration control crossed to UAS-RFP, (B) UAS-ArmS10, (C) UAS-Stat92E, (D) UAS-Rasact, (E) UAS-eiger, (F) constitutively active UAS-ykiS168A, (G) UAS-Nact and (H) UAS-Ci. Discs were stained for F-actin (magenta) to reveal tissue morphology and for GFP (green, white) to mark cells in which transgenes were ectopically expressed using dpp-Gal4. Scale bars: 50 μm. All crosses were done at 29°C.
DISCUSSION
Accumulating evidence has strongly indicated that mammalian HIPKs are implicated in various diseases and cancers (reviewed by Blaquiere and Verheyen, 2017). However, whether HIPKs act as oncogenes or tumor suppressor genes remains ambiguous, possibly in part because of the genetic heterogeneity of different cancer types. In addition, comprehensive analyses of the four HIPK isoforms are lacking. Given the diverse expression patterns, distinct subcellular localization and potential functional redundancy of HIPK proteins, considerable efforts are needed to identify the roles of individual isoforms in each cell context, not to mention in unstressed or stressed conditions (for example, ultraviolet induction or hypoxia; Schmitz et al., 2014). In light of these complications, we decided to use Drosophila, a simple genetic model organism containing only one well-conserved Hipk, in most of our studies to unravel the roles of Hipk proteins in tumorigenesis.
Our work reveals that elevated expression of a single gene, hipk, in Drosophila tissues is sufficient to produce features of transformed tumors. We provide evidence that Hipk induces hyperplasia in imaginal discs and hemocytes, leading to massive tissue growth and melanotic tumor-like masses, respectively (Figs 1 and 2). Importantly, cells with elevated Hipk display protruding shapes and gain the potential to spread away from their primary site of origin (Figs 3 and 4). Furthermore, we provide evidence that Hipk induces basal invasion through mechanisms such as Mmp1-mediated degradation of the basement membrane and induction of EMT (Fig. 4). We also demonstrate that expression of Drosophila Hipk in the human aggressive breast cancer line MDA-MB-231 can potentiate proliferation, migratory behaviors and, by extension, EMT (Fig. 4).
We speculated that Hipk might trigger EMT in Drosophila tissues and vertebrate cells through conserved molecular mechanisms. Our studies uncover previously unrecognized functions of Drosophila Hipk in mediating metastasis. Our conclusions are in agreement with some studies reporting that human HIPK2 promotes EMT in renal fibrosis (Jin et al., 2012; Huang et al. 2015). HIPK2 expression has been shown to be remarkably upregulated in kidneys of patients with HIV-associated nephropathy, diabetic nephropathy and severe IgA nephropathy (Huang et al., 2015). Moreover, certain human cancers display elevated levels of HIPK2 within tumorous tissue (Al-Beiti and Lu, 2008; Deshmukh et al., 2008; Jacob et al., 2009). We infer that Drosophila Hipk mimics human HIPK2 in these fibrosis and tumor models. By contrast, another study found that in bladder cancer metastasis, downregulation of HIPK2 induced EMT and cell invasion (Tan et al., 2014). The cue for the switch of roles of HIPKs between EMT promotion and EMT suppression requires further investigation.
To elucidate the molecular mechanism by which Hipk can confer both proliferative and migratory properties on cells, we examined genetic interactions between Hipk and tumorigenic pathways that are known, or proposed, to be regulated by Hipk. First, we noticed that interfering with the activity of an individual signaling pathway is not sufficient to suppress both Hipk-mediated cell spreading and invasive phenotypes (Fig. 5). Second, stimulation of single pathways fails to recapitulate all the phenotypes induced by Hipk overexpression (Fig. 6). Of note, we do find that inhibition of individual pathways can suppress a subset of Hipk-induced phenotypes. For example, knockdown of Yki in a Hipk overexpression background inhibited cell proliferation, but did not have a strong effect on cell spreading. Conversely, inhibition of Hedgehog signaling did not affect proliferation, but appeared to reduce cell spreading. We were able to observe that the combined inactivation of Yki and Bsk was able to ameliorate most Hipk phenotypes. We propose that elevation of a single protein kinase, Hipk, even without accumulation of additional mutations, is likely to be potent enough to perturb multiple signaling pathways and, ultimately, have a cumulative effect of oversized, proliferative and protruding phenotypes. This mechanism, in effect, mimics tumor initiation attributable to multiple activating mutations in distinct pathways. Previously described Drosophila tumor models involve concomitant mutations that enhance proliferation, such as activated Ras, along with loss of cell polarity genes, such as scribble, to drive invasive behavior (Pagliarini and Xu, 2003).
Consistent with our proposed mechanism, HIPK2 is thought to mediate EMT by activation of EMT-promoting pathways, including TGFβ, Wnt and Notch (Huang et al. 2015). We believe that, in the future, profiling the transcriptome and the protein/protein and protein/DNA interactions in Hipk-expressing cells will give us an unbiased and thorough analysis of alterations of the signaling network upon Hipk overexpression.
The versatility of Hipk functions raises concerns regarding how we can block Hipk-induced phenotypes efficiently. Although inhibition of multiple downstream effectors of Hipk might be an option, we notice that impeding Hipk expression through hipk-RNAi can strongly reverse the overgrowth and cell spreading phenotypes (Fig. 5B). In line with our suggestion, a previous study proposed that exogenous overexpression of miR-141, which targets the 3′UTR of HIPK2, represented a potential approach to hinder HIPK2-mediated EMT (Jin et al., 2012). Given the large roles of post-translational modifications in Hipk protein turnover and localization (reviewed by Saul and Schmitz, 2013), we consider that mutations in other genes encoding Hipk regulators might also contribute to tumorigenesis even in the absence of Hipk gene mutations or changes in transcription levels of Hipk. Thus, revealing the regulation of Hipk activity is crucial to avoid Hipk-induced deleterious effects and to develop promising therapeutic interventions for Hipk-related disorders.
Lastly, we notice that Hipk is able to cooperate with other sensitized tumor models, probably in both additive and synergistic manners (Fig. 7). This implies that Hipk itself can elicit tumor-like transformations during the early phases of tumorigenesis. During the later phases, when multiple genetic alterations occur, Hipk might play an important role in accelerating tumor progression and metastasis. Future research would need to validate whether the human counterparts, HIPK1-HIPK4, can play roles in cancer initiation and progression in specific cancer types, and whether the functions of HIPK isoforms are redundant or disparate.
MATERIALS AND METHODS
Genetic crosses and fly stocks
Flies were raised on standard media, and w1118 was used as wild type. A commonly used assay for proliferation and cell invasion is the use of ey-Flp to induce clones of tumor suppressors or expression of oncogenes in the eye-antennal disc (Pagliarini and Xu, 2003). We could not use this assay because of the inhibition of early eye specification mediated by Hipk (Blaquiere et al., 2014). All crosses were raised at 29°C to increase the effectiveness of GAL4-driven UAS constructs unless otherwise noted. All genetic interaction studies included controls for GAL4 titration through the use of benign UAS lines such as UAS-GFP, UAS-RFP or UAS-lacZ to match the UAS construct dose in experimental crosses. Fly strains used in this study were as follows: (1) ;;dpp-GAL4/TM6B (Staehling-Hampton et al., 1994); (2) ;vkg-GFP; (Flytrap); (3) ;UAS-HA-hipk1M; (4) ;;UAS-HA-hipk3M [both (3) and (4) are wild-type Hipk transgenes inserted on different chromosomes, which were previously reported as UAS-Hipk (II) and UAS-Hipk (III), respectively (Swarup and Verheyen, 2011)]; (5) ;;dpp-GAL4, UAS-HA-hipk3M/TM6B [recombinant derived from stocks (1) and (4)]; (6) ;UAS-eGFP; (BL#5431); (7) ;;UAS-eGFP (BL#5430); (8) ;UAS-P35; (BL#5072) (Hay et al., 1994); (9) ;hml-GAL4; (BL#30139); (10) ;ey-GAL4; (BL#5535); (11) ;;UAS-Axin-GFP (BL#7225); (12) UAS-EgfrDN (dominant negative) with inserts on both II and III; (13) UAS-bskDN;; (BL# 6409); (14) ;;UAS-ykiRNAi (BL#34067); (15) ;UAS-DlDN; (BL#26697); (16) UAS-CiRep; (17) ;UAS-arms10; generated in our laboratory (Mirkovic et al., 2011); (18) ;UAS-Stat92E;; (19) UAS-Rasact (from H. Richardson); (20) ;;UAS-Eiger; (21) ;UAS-ykiS168A:GFP;; (22) ;;UAS-Nact; and (23) ;UAS-Ci5M;. Strains obtained from the Bloomington Drosophila Stock Center (Bloomington, IN, USA) have BL# stock numbers indicated. The following RNAi lines were primarily obtained from the Vienna Drosophila Resource Center (VDRC): (24) ;UAS-hipkRNAi; (VDRC#108254); (25) ;UAS-hopRNAi; (VDRC#102830); (26) UAS-updRNAi; (27) ;;UAS-panRNAi (TCF; VDRC#3014); (28) y,w,hsflp[122]; sp/CyO; Act>CD2>GAL4,UAS-RFP/TM6B; (29) Dll-lacZ; (30) puc-LacZ; (31) 10xStat92E-GFP (BL#26197) (Bach et al., 2007); and (32) MS1096-GAL4 (BL#8660).
Antibodies and microscopy
Third instar imaginal discs were dissected and stained using standard protocols and, in most cases, we analyzed ≥20 discs per genotype. The following primary antibodies were used: mouse anti-Mmp1 (1:100: 3A6B4, 3B8D12, 5H7B11 DSHB; Rubin, G.M.), rat anti-Ci (1:20; 2A1 DSHB; Holmgren, R.), mouse anti-En (1:10; 4D9 DSHB; Goodman, C.), mouse anti-Dlg (1:100; 4F3 DSHB; Goodman, C.), mouse anti-HA (1:200; ABM), rabbit anti-Cas3 (1:100; 9661S; Cell Signaling), rabbit anti-Ndg [1:500; gift of Anne Hölz; (Wolfstetter et al., 2009)], rabbit anti-Twi [1:3000; gift of Maria Leptin (Roth et al., 1989)], mouse anti-beta-Galactosidase (1:50; 40-1a DSHB; Sanes, J.R.), rabbit anti-CycE (1:100; d-300; Santa Cruz), mouse anti-Wg (1:40; 4D4 DSHB; Cohen, S.M.), mouse anti-Cut (1:50; 2B10 DSHB; Rubin, G.M.), mouse anti-Ptc (1:40; Apa1 DSHB; Guerrero, I.). Rabbit anti-Hipk antibodies were generated in our laboratory and used at 1:200 dilution. The following secondary antibodies were obtained from Jackson ImmunoResearch: DyLight649 anti-rabbit, DyLight649 anti-rat, Cy3 anti-mouse and Cy3 anti-rabbit. Nuclei were detected by staining with DAPI, and F-actin was detected by staining with Rhodamine phalloidin (R-415; Molecular Probes). Immunofluorescent images were acquired using a Nikon Air laser-scanning confocal microscope. For live imaging, dissected eye discs were placed on a depression slide containing insect media and two larval brains. Discs were imaged using differential interference contrast microscopy (DIC) once per minute over 2 h (n=5 for each genotype). Whole larvae were mounted in Hoyer's medium, allowed to sit for 2 min, and imaged with a Canon Rebel T1i. Images were processed with Nikon Elements, Adobe Photoshop, Adobe Illustrator, ImageJ and Helicon Focus. For a subset of fluorescent images, channel colors were converted to accommodate color-blind viewers.
Hemocyte counts
Before hemolymph collection, third instar larvae (hml>GFP+LacZ and hml>HA-hipk3M+GFP) grown at 29°C were washed thoroughly with 1× PBS, dried, and placed in glass dissection wells containing 10 μl of 1× PBS. The cuticle of single larvae was carefully punctured ventrally with forceps, and hemolymph was allowed to drain into the well for 30-60 s. The hemolymph was mixed with a pre-wetted pipet, and 1.5 µl of the hemolymph mixture was transferred to a poly-d-lysine-treated eight-well chamber-slide (BD Falcon CultureSlides; product #354108). Five 1.5 µl droplets were plated per larva, after which they were air dried. The dried samples were washed with 4% formaldehyde for 2 min, washed with PBS, and stained with DAPI. For each sample (n=16), five cell counts were performed from images taken at the center of each droplet at 200× magnification, and means of the five cell counts were plotted; values were subjected to Student's unpaired two-tailed t-test.
Cell culture
MDA-MB-231 (ATCC; CRM-HTB-26), MCF7 (ATCC; HTB-22) and HEK293T (ATCC; CRL-3216) cell lines were grown in DMEM/F-12 (Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12; Gibco; Cat. #: 11330032) with 10% fetal bovine serum (FBS).
Cell transfection
MDA-MB-231 cells were transiently transfected with pCMV-myc empty vector (control) and pCMV-HA-dHipk vector using Lipofectoamine 3000 (Invitrogen) according to the manufacturer's instructions. Two micrograms of plasmid was used for each well in a six-well plate.
Cell proliferation
Cell counts in transfected MDA-MB-231 cells were determined using an MTT proliferation assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Thermo Fisher, M6494). MDA-MB-231 cells were seeded into six-well plates (VWR, Cat# 10062-892) on day 1 to allow cells to attach, and transfections were performed on day 2. On day 3, 5000 cells were seeded into a 96-well plate, and on day 5, 10 μl 12 mM MTT stock solution was added into 100 μl medium in each well and incubated at 37°C for 3 h. Culture medium was then removed and 100 μl DMSO added to each well and incubated for 10 min at 37°C. We then read the optical density at 540 nm. Three replicates were performed for each condition in triplicate. Values were calculated as the mean±s.e.m. Significance between samples was assessed using Student's unpaired two-tailed t-tests. All raw data are provided in Table S4.
Migration assay
MDA-MB-231cells were seeded at 80% confluence into six-well plates for 24 h and then transfected with pCMV-Myc empty vector or pCMV-HA-dHipk for 6 h, after which the medium was changed to starving medium (DMEM/F-12 without FBS) for 24 h. Then transfected cells were trypsinized (0.25% Trypsin-EDTA; Gibco) and counted using Trypan Blue, and 20,000 cells were suspended in 200 μl serum-free DMEM/F-12, seeded into the upper chamber of each insert (24-well insert, pore size of 8 μm; Greiner Bio-one). Eight-hundred microliters of DMED/F-12 containing 50% FBS was added to the bottom wells. After 24 h at 37°C, the culture medium was replaced with 450 μl serum-free medium plus 8 μm calcein-AM, incubated for 45 min at 37°C and then 500 μl trypsin was used to release the cells that had migrated through the membrane, incubating for 10 min. Two-hundred microliters of trypsin solution with detached migrated cells was transferred into a black flat-bottomed 96-well plate, and fluorescence was measured with an excitation wavelength of 485 nm and an emission wavelength of 520 nm. Three replicates were performed for each condition in triplicate. Values were calculated as the mean±s.e.m. Significance between samples was assessed using Student's unpaired two-tailed t-test. All raw data are provided in Table S3.
RNA extraction and qPCR
Total RNA was isolated from cells using RNeasy Mini kits (Qiagen; 74101). First strand cDNA was synthesized from 0.5 μg RNA by PrimeScript First Strand cDNA Synthesis Kit (TaKaRa; 6110A). qPCR was performed using FastStart SYBR Green Master (Roche; 04673484001) on StepOne real-time PCR (ABI). HPRT was used as a housekeeping gene control. Primers used were as follows: hHPRT forward GCTATAAATTCTTTGCTGACCTGCTG; hHPRT reverse AATTACTTTTATGTCCCCTGTTGACTGG; hE-Cad (CDH1) forward GGACTTTGGCGTGGGCCAGG; and hE-Cad (CDH1) reverse CCCTGTCCAGCTCAGCCCGA. Relative fold levels were determined by the 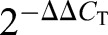 method. Statistical significance was confirmed with Student's unpaired two-tailed t-test, with a theoretical mean set to one. All raw data are provided in Table S2.
method. Statistical significance was confirmed with Student's unpaired two-tailed t-test, with a theoretical mean set to one. All raw data are provided in Table S2.
Western blot
Whole cell lysates were prepared with Cell lysis buffer (Cell Signaling Technology; #9803), supplemented with 1× Protease Inhibitors (Roche; #04693132001) and 1 mM PMSF before use. Protein lysates with 1× SDS sample buffer were subjected to 8% SDS-PAGE, followed by western blotting. The blots were detected by using the Pierce ECL Western Blotting Substrate (#32209). Images were captured with the use of FujiFilm LAS-4000 Chemi-luminescent Scanner. Rabbit anti-E-cadherin (1:1000; #3195; CST) and mouse beta-Actin (1:1000; G043; Abm) were used as primary antibodies. Anti-mouse and anti-rabbit HRP light-chain specific were used as secondary antibodies at 1:5000 (Jackson ImmunoResearch).
Imaginal disc size measurements and invasiveness scoring
Experimental sets from Figs 5-7 were quantified for two parameters. First, the proliferative effects of each transgene were assessed by measuring the area of the GFP+ cells (driven by dpp-GAL4) and dividing it by the total disc area. Area measurements were taken in Image J from the .nd2 file for each disc. Measurements were calculated as ratios of the dpp stripe area to the total disc area (dpp/total; Fig. S7; Tables S5-S7). The difference in ratios was then quantified for Figs 5 and 7 using one-way ANOVA [Fig. S7-5, F(12,136)=41.88, P<0.0001; Fig. S7-7, F(7,81)=49.94, P<0.0001], with the Holm–Sidak post hoc test applied for multiple comparisons to 5A or 7A in Fig. S7-5 or Fig. S7-7, respectively. The scores for the Holm–Sidak tests are depicted in Fig. S7 as ‘ns’=not significant, *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. Second, the invasive effects caused by each transgene were assessed by assigning a ‘relative degree of invasiveness’ score to each disc. We defined the ‘relative degree of invasiveness’ scores as follows: ‘None’=no cells found outside the normal dpp>GFP region; ‘Weak’=a few cells emerging from the dpp>GFP region, not only a widened GFP region attributable to proliferation; ‘Moderate’=extensions of cells that have traveled to edges of the hinge region; or ‘Strong’=all of the above and some solitary GFP masses found distinct from the main dpp>GFP region.
Supplementary Material
Acknowledgements
We are grateful to A. Hölz, M. Leptin, M. Miura, N. Perrimon, H. Richardson, G. Tanentzapf, the Developmental Studies Hybridoma Bank, The Vienna Drosophila Resource Centre, the Bloomington Drosophila Stock Center and Jackson ImmunoResearch Laboratories for providing fly strains and antibodies. We thank J. Parker, A. Kadhim and E. Hall for experimental help. This work was funded by operating grants from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.A.B., K.K.L.W., E.M.V.; Methodology: J.A.B., K.K.L.W.; Validation: J.A.B., K.K.L.W.; Formal analysis: J.A.B., K.K.L.W., S.D.K., J.W.; Investigation: J.A.B., K.K.L.W., S.D.K., J.W.; Writing - original draft: J.A.B., K.K.L.W., E.M.V.; Writing - review & editing: J.A.B., K.K.L.W., S.D.K., E.M.V.; Visualization: J.A.B., K.K.L.W., S.D.K., J.W.; Supervision: E.M.V.; Project administration: E.M.V.; Funding acquisition: E.M.V.
Funding
This work was supported by the Canadian Institutes of Health Research (97835) and the Natural Sciences and Engineering Research Council of Canada (RGPIN/203545).
Supplementary information
Supplementary information available online at http://dmm.biologists.org/lookup/doi/10.1242/dmm.031146.supplemental
References
- Al-Beiti M. A. M. and Lu X. (2008). Expression of HIPK2 in cervical cancer: correlation with clinicopathology and prognosis. Aust. N. Z. J. Obstet. Gynaecol. 48, 329-336. 10.1111/j.1479-828X.2008.00874.x [DOI] [PubMed] [Google Scholar]
- Aldaz S. and Escudero L. M. (2010). Imaginal discs. Curr. Biol. 20, R429-R431. 10.1016/j.cub.2010.03.010 [DOI] [PubMed] [Google Scholar]
- Ayyaz A., Li H. and Jasper H. (2015). Haemocytes control stem cell activity in the Drosophila intestine. Nat. Cell Biol. 17, 736-748. 10.1038/ncb3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach E. A., Ekas L. A., Ayala-Camargo A., Flaherty M. S., Lee H., Perrimon N. and Baeg G.-H. H. (2007). GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr. Patterns 7, 323-331. 10.1016/j.modgep.2006.08.003 [DOI] [PubMed] [Google Scholar]
- Basler K. and Struhl G. (1994). Compartment boundaries and the control of Drosophila limb pattern by hedgehog protein. Nature 368, 208-214. 10.1038/368208a0 [DOI] [PubMed] [Google Scholar]
- Beaucher M., Hersperger E., Page-McCaw A. and Shearn A. (2007). Metastatic ability of Drosophila tumors depends on MMP activity. Dev. Biol. 303, 625-634. 10.1016/j.ydbio.2006.12.001 [DOI] [PubMed] [Google Scholar]
- Berber S., Llamosas E., Thaivalappil P., Boag P. R., Crossley M. and Nicholas H. R. (2013). Homeodomain interacting protein kinase (HPK-1) is required in the soma for robust germline proliferation in C. elegans. Dev. Dyn. 242, 1250-1261. 10.1002/dvdy.24023 [DOI] [PubMed] [Google Scholar]
- Blaquiere J. A. and Verheyen E. M. (2017). Homeodomain-interacting protein kinases: diverse and complex roles in development and disease. Curr. Top. Dev. Biol. 123, 73-103. 10.1016/bs.ctdb.2016.10.002 [DOI] [PubMed] [Google Scholar]
- Blaquiere J. A., Lee W. and Verheyen E. M. (2014). Hipk promotes photoreceptor differentiation through the repression of Twin of eyeless and Eyeless expression. Dev. Biol. 390, 14-25. 10.1016/j.ydbio.2014.02.024 [DOI] [PubMed] [Google Scholar]
- Blaquiere J. A., Wray N. B. and Verheyen E. M. (2016). Hipk is required for JAK/STAT activity and promotes hemocyte-derived tumorigenesis. bioRxiv 10.1101/058156. [DOI] [Google Scholar]
- Brumby A. M. and Richardson H. E. (2005). Using Drosophila melanogaster to map human cancer pathways. Nat. Rev. Cancer 5, 626-639. 10.1038/nrc1671 [DOI] [PubMed] [Google Scholar]
- Chalazonitis A., Tang A. A., Shang Y., Pham T. D., Hsieh I., Setlik W., Gershon M. D. and Huang E. J. (2011). Homeodomain interacting protein kinase 2 regulates postnatal development of enteric dopaminergic neurons and glia via BMP signaling. J. Neurosci. 31, 13746-13757. 10.1523/JNEUROSCI.1078-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. and Verheyen E. M. (2012). Homeodomain-interacting protein kinase regulates yorkie activity to promote tissue growth. Curr. Biol. 22, 1582-1586. 10.1016/j.cub.2012.06.074 [DOI] [PubMed] [Google Scholar]
- Cheng Y., Al-Beiti M. A. M., Wang J., Wei G., Li J., Liang S. and Lu X. (2012). Correlation between homeodomain-interacting protein kinase 2 and apoptosis in cervical cancer. Mol. Med. Rep. 5, 1251-1255. 10.3892/mmr.2012.810 [DOI] [PubMed] [Google Scholar]
- Clark R. I., Woodcock K. J., Geissmann F., Trouillet C. and Dionne M. S. (2011). Multiple TGF-β superfamily signals modulate the adult Drosophila immune response. Curr. Biol. 21, 1672-1677. 10.1016/j.cub.2011.08.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T. K., Sangodkar J., Negre N., Narla G. and Cagan R. L. (2013). Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene 32, 3184-3197. 10.1038/onc.2012.326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh H., Yeh T. H., Yu J., Sharma M. K., Perry A., Leonard J. R., Watson M. A., Gutmann D. H. and Nagarajan R. (2008). High-resolution, dual-platform aCGH analysis reveals frequent HIPK2 amplification and increased expression in pilocytic astrocytomas. Oncogene 27, 4745-4751. 10.1038/onc.2008.110 [DOI] [PubMed] [Google Scholar]
- Doggett K., Turkel N., Willoughby L. F., Ellul J., Murray M. J., Richardson H. E. and Brumby A. M. (2015). BTB-zinc finger oncogenes are required for ras and notch-driven tumorigenesis in drosophila. PLoS ONE 10, e0132987 10.1371/journal.pone.0132987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Orazi G., Sciulli M. G., Di Stefano V., Riccioni S., Frattini M., Falcioni R., Bertario L., Sacchi A. and Patrignani P. (2006). Homeodomain-interacting protein kinase-2 restrains cytosolic phospholipase A2-dependent prostaglandin E2 generation in human colorectal cancer cells. Clin. Cancer Res. 12, 735-741. 10.1158/1078-0432.CCR-05-1557 [DOI] [PubMed] [Google Scholar]
- Ferres-Marco D., Gutierrez-Garcia I., Vallejo D. M., Bolivar J., Gutierrez-Aviño F. J. and Dominguez M. (2006). Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature 439, 430-436. 10.1038/nature04376 [DOI] [PubMed] [Google Scholar]
- Garelli A., Gontijo A. M., Miguela V., Caparros E. and Dominguez M. (2012). Imaginal discs secrete insulin-like peptide 8 to mediate plasticity of growth and maturation. Science 336, 579-582. 10.1126/science.1216735 [DOI] [PubMed] [Google Scholar]
- Gonzalez C. (2013). Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. Cancer 13, 172-183. 10.1038/nrc3461 [DOI] [PubMed] [Google Scholar]
- Hanratty W. P. and Ryerse J. S. (1981). A genetic melanotic neoplasm of Drosophila melanogaster. Dev. Biol. 83, 238-249. 10.1016/0012-1606(81)90470-X [DOI] [PubMed] [Google Scholar]
- Harrison D., Binari R. and Nahreini T. (1995). Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. EMBO J. 14, 2857-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay B., Wolff T. and Rubin G. (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 2129, 2121-2129. [DOI] [PubMed] [Google Scholar]
- Herranz H., Hong X., Hung N. T., Voorhoeve P. M. and Cohen S. M. (2012). Oncogenic cooperation between SOCS family proteins and EGFR identified using a Drosophila epithelial transformation model. Genes Dev. 26, 1602-1611. 10.1101/gad.192021.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz H., Weng R. and Cohen S. M. (2014). Crosstalk between epithelial and mesenchymal tissues in tumorigenesis and imaginal disc development. Curr. Biol. 24, 1476-1484. 10.1016/j.cub.2014.05.043 [DOI] [PubMed] [Google Scholar]
- Herranz H., Eichenlaub T. and Cohen S. M. (2016). Cancer in Drosophila: imaginal discs as a model for epithelial tumor formation. Curr. Top. Dev. Biol. 116, 181-199. 10.1016/bs.ctdb.2015.11.037 [DOI] [PubMed] [Google Scholar]
- Hikasa H. and Sokol S. Y. (2011). Phosphorylation of TCF proteins by homeodomain-interacting protein kinase 2. J. Biol. Chem. 286, 12093-12100. 10.1074/jbc.M110.185280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikasa H., Ezan J., Itoh K., Li X., Klymkowsky M. W. and Sokol S. Y. (2010). Regulation of TCF3 by Wnt-dependent phosphorylation during vertebrate axis specification. Dev. Cell 19, 521-532. 10.1016/j.devcel.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T. G., Stollberg N., Schmitz M. L. and Will H. (2003). HIPK2 regulates transforming growth factor-beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res. 63, 8271-8277. [PubMed] [Google Scholar]
- Hofmann T. G., Jaffray E., Stollberg N., Hay R. T. and Will H. (2005). Regulation of homeodomain-interacting protein kinase 2 (HIPK2) effector function through dynamic small ubiquitin-related modifier-1 (SUMO-1) modification. J. Biol. Chem. 280, 29224-29232. 10.1074/jbc.M503921200 [DOI] [PubMed] [Google Scholar]
- Hofmann T. G., Glas C. and Bitomsky N. (2013). HIPK2: A tumour suppressor that controls DNA damage-induced cell fate and cytokinesis. BioEssays 35, 55-64. 10.1002/bies.201200060 [DOI] [PubMed] [Google Scholar]
- Huang H., Du G., Chen H., Liang X., Li C., Zhu N., Xue L., Ma J. and Jiao R. (2011). Drosophila Smt3 negatively regulates JNK signaling through sequestering Hipk in the nucleus. Development 138, 2477-2485. 10.1242/dev.061770 [DOI] [PubMed] [Google Scholar]
- Huang Y., Tong J., He F., Yu X., Fan L., Hu J., Tan J. and Chen Z. (2015). miR-141 regulates TGF-β1-induced epithelial-mesenchymal transition through repression of Hipk2 expression in renal tubular epithelial cells. Int. J. Mol. Med. 35, 311-318. 10.3892/ijmm.2014.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovelli S., Ciuffini L., Lazzari C., Bracaglia G., Rinaldo C., Prodosmo A., Bartolazzi A., Sacchi A. and Soddu S. (2009). HIPK2 is involved in cell proliferation and its suppression promotes growth arrest independently of DNA damage. Cell Prolif. 42, 373-384. 10.1111/j.1365-2184.2009.00601.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob K., Albrecht S., Sollier C., Faury D., Sader E., Montpetit A., Serre D., Hauser P., Garami M., Bognar L. et al. (2009). Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours. Br. J. Cancer 101, 722-733. 10.1038/sj.bjc.6605179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Ratnam K., Chuang P. Y., Fan Y., Zhong Y., Dai Y., Mazloom A. R., Chen E. Y., D'Agati V., Xiong H. et al. (2012). A systems approach identifies HIPK2 as a key regulator of kidney fibrosis. Nat. Med. 18, 580-588. 10.1038/nm.2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiger J. A., Natzle J. E., Kimbrell D. A., Paddy M. R., Kleinhesselink K. and Green M. M. (2007). Tissue remodeling during maturation of the Drosophila wing. Dev. Biol. 301, 178-191. 10.1016/j.ydbio.2006.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S., Lu Y., Debbas M., Lin A. W., Sarosi I., Itie A., Wakeham A., Tuan J., Saris C., Elliott G. et al. (2003). Characterization of cells and gene-targeted mice deficient for the p53-binding kinase homeodomain-interacting protein kinase 1 (HIPK1). Proc. Natl. Acad. Sci. USA 100, 5431-5436. 10.1073/pnas.0530308100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara A., Sakai H., Xu Y., Itoh Y., Hirabayashi Y. and Gotoh Y. (2014). Tcf3 represses Wnt-β-catenin signaling and maintains neural stem cell population during neocortical development. PLoS ONE 9, e94408 10.1371/journal.pone.0094408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan H.-C., Li H.-J., Lin G., Lai P.-Y. and Chung B. (2007). Cyclic AMP stimulates SF-1-dependent CYP11A1 expression through homeodomain-interacting protein kinase 3-mediated Jun N-terminal kinase and c-Jun phosphorylation. Mol. Cell. Biol. 27, 2027-2036. 10.1128/MCB.02253-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan H.-C., Wu C.-F., Shih H.-M. and Chung B.-C. (2012). Death-associated protein 6 (Daxx) mediates cAMP-dependent stimulation of Cyp11a1 (P450scc) transcription. J. Biol. Chem. 287, 5910-5916. 10.1074/jbc.M111.307603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavra L., Rinaldo C., Ulivieri A., Luciani E., Fidanza P., Giacomelli L., Bellotti C., Ricci A., Trovato M., Soddu S. et al. (2011). The loss of the p53 activator HIPK2 is responsible for galectin-3 overexpression in well differentiated thyroid carcinomas. PLoS ONE 6, e20665 10.1371/journal.pone.0020665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W., Andrews B. C., Faust M., Walldorf U. and Verheyen E. M. (2009a). Hipk is an essential protein that promotes Notch signal transduction in the Drosophila eye by inhbition of the global co-repressor Groucho. Dev. Biol. 325, 263-272. 10.1016/j.ydbio.2008.10.029 [DOI] [PubMed] [Google Scholar]
- Lee W., Swarup S., Chen J., Ishitani T. and Verheyen E. M. (2009b). Homeodomain-interacting protein kinases (Hipks) promote Wnt/Wg signaling through stabilization of beta-catenin/Arm and stimulation of target gene expression. Development 136, 241-251. 10.1242/dev.025460 [DOI] [PubMed] [Google Scholar]
- Link N., Chen P., Lu W.-J., Pogue K., Chuong A., Mata M., Checketts J. and Abrams J. M. (2007). A collective form of cell death requires homeodomain interacting protein kinase. J. Cell Biol. 178, 567-574. 10.1083/jcb.200702125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie S. H., Yang X. Y., Conrad W. H., Muster J., Angers S., Moon R. T. and Cheyette B. N. R. (2009). Modulation of the beta-catenin signaling pathway by the dishevelled-associated protein Hipk1. PLoS ONE 4, e4310 10.1371/journal.pone.0004310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H., Rose P., Barber D., Hanratty W. P., Lee S., Roberts T. M., D'Andrea A. D. and Dearolf C. R. (1997). Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. Mol. Cell. Biol. 17, 1562-1571. 10.1128/MCB.17.3.1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles W. O., Dyson N. J. and Walker J. A. (2011). Modeling tumor invasion and metastasis in Drosophila. Dis. Model. Mech. 4, 753-761. 10.1242/dmm.006908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton C. C., Grusche F. A., Degoutin J. L., Yu E., Dai Q., Lai E. C. and Harvey K. F. (2014). The hippo pathway regulates hematopoiesis in Drosophila melanogaster. Curr. Biol. 24, 2673-2680. 10.1016/j.cub.2014.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkovic I., Gault W. J., Rahnama M., Jenny A., Gaengel K., Bessette D., Gottardi C. J., Verheyen E. M. and Mlodzik M. (2011). Nemo kinase phosphorylates beta-catenin to promote ommatidial rotation and connects core PCP factors to E-cadherin-beta-catenin. Nat. Struct. Mol. Biol. 18, 665-672. 10.1038/nsmb.2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page-McCaw A., Serano J., Santé J. M. and Rubin G. M. (2003). Drosophila matrix metalloproteinases are required for tissue remodeling, but not embryonic development. Dev. Cell 4, 95-106. 10.1016/S1534-5807(02)00400-8 [DOI] [PubMed] [Google Scholar]
- Pagliarini R. A. and Xu T. (2003). A genetic screen in Drosophila for metastatic behavior. Science 302, 1227-1231. 10.1126/science.1088474 [DOI] [PubMed] [Google Scholar]
- Pallavi S. K., Ho D. M., Hicks C., Miele L. and Artavanis-Tsakonas S. (2012). Notch and Mef2 synergize to promote proliferation and metastasis through JNK signal activation in Drosophila. EMBO J. 31, 2895-2907. 10.1038/emboj.2012.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi F., Stefanatos R. K., Strathdee K., Yu Y. and Vidal M. (2014). Transformed epithelia trigger non-tissue-autonomous tumor suppressor response by adipocytes via activation of Toll and Eiger/TNF signaling. Cell Rep. 6, 855-867. 10.1016/j.celrep.2014.01.039 [DOI] [PubMed] [Google Scholar]
- Pierantoni G. M., Bulfone A., Pentimalli F., Fedele M., Iuliano R., Santoro M., Chiariotti L., Ballabio A. and Fusco A. (2002). The homeodomain-interacting protein kinase 2 gene is expressed late in embryogenesis and preferentially in retina, muscle, and neural tissues. Biochem. Biophys. Res. Commun. 290, 942-947. 10.1006/bbrc.2001.6310 [DOI] [PubMed] [Google Scholar]
- Poon C. L. C., Zhang X., Lin J. I., Manning S. A. and Harvey K. F. (2012). Homeodomain-interacting protein kinase regulates Hippo pathway-dependent tissue growth. Curr. Biol. 22, 1587-1594. 10.1016/j.cub.2012.06.075 [DOI] [PubMed] [Google Scholar]
- Potter C. J., Turenchalk G. S. and Xu T. (2000). Drosophila in cancer research. An expanding role. Trends Genet. 16, 33-39. 10.1016/S0168-9525(99)01878-8 [DOI] [PubMed] [Google Scholar]
- Rey C., Soubeyran I., Mahouche I., Pedeboscq S., Bessede A., Ichas F., De Giorgi F. and Lartigue L. (2013). HIPK1 drives p53 activation to limit colorectal cancer cell growth. Cell Cycle 12, 1879-1891. 10.4161/cc.24927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci A., Cherubini E., Ulivieri A., Lavra L., Sciacchitano S., Scozzi D., Mancini R., Ciliberto G., Bartolazzi A., Bruno P. et al. (2013). Homeodomain-interacting protein kinase2 in human idiopathic pulmonary fibrosis. J. Cell. Physiol. 228, 235-241. 10.1002/jcp.24129 [DOI] [PubMed] [Google Scholar]
- Rochat-Steiner V., Becker K., Micheau O., Schneider P., Burns K. and Tschopp J. (2000). FIST/HIPK3: a Fas/FADD-interacting serine/threonine kinase that induces FADD phosphorylation and inhibits fas-mediated Jun NH(2)-terminal kinase activation. J. Exp. Med. 192, 1165-1174. 10.1084/jem.192.8.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S., Stein D. and Nüsslein-Volhard C. (1989). A gradient of nuclear localization of the dorsal protein determines dorsoventral pattern in the Drosophila embryo. Cell 59, 1189-1202. 10.1016/0092-8674(89)90774-5 [DOI] [PubMed] [Google Scholar]
- Rudrapatna V. A., Cagan R. L. and Das T. K. (2012). Drosophila cancer models. Dev. Dyn. 241, 107-118. 10.1002/dvdy.22771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudrapatna V. A., Bangi E. and Cagan R. L. (2013). Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Rep. 14, 172-177. 10.1038/embor.2012.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul V. V. and Schmitz M. L. (2013). Posttranslational modifications regulate HIPK2, a driver of proliferative diseases. J. Mol. Med. (Berl). 91, 1051-1058. 10.1007/s00109-013-1042-0 [DOI] [PubMed] [Google Scholar]
- Schmitz M. L., Rodriguez-Gil A. and Hornung J. (2014). Integration of stress signals by homeodomain interacting protein kinases. Biol. Chem. 395, 375-386. 10.1515/hsz-2013-0264 [DOI] [PubMed] [Google Scholar]
- Shimizu N., Ishitani S., Sato A., Shibuya H. and Ishitani T. (2014). Hipk2 and PP1c cooperate to maintain Dvl protein levels required for Wnt signal transduction. Cell Rep. 8, 1391-1404. 10.1016/j.celrep.2014.07.040 [DOI] [PubMed] [Google Scholar]
- Sinenko S. A. and Mathey-Prevot B. (2004). Increased expression of Drosophila tetraspanin, Tsp68C, suppresses the abnormal proliferation of ytr-deficient and Ras/Raf-activated hemocytes. Oncogene 23, 9120-9128. 10.1038/sj.onc.1208156 [DOI] [PubMed] [Google Scholar]
- Sjölund J., Pelorosso F. G., Quigley D. A., DelRosario R. and Balmain A. (2014). Identification of Hipk2 as an essential regulator of white fat development. Proc. Natl. Acad. Sci. USA 111, 7373-7378. 10.1073/pnas.1322275111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J. J. and Lee Y. J. (2003). Role of the ASK1-SEK1-JNK1-HIPK1 signal in Daxx trafficking and ASK1 oligomerization. J. Biol. Chem. 278, 47245-47252. 10.1074/jbc.M213201200 [DOI] [PubMed] [Google Scholar]
- Sonoshita M. and Cagan R. L. (2017). Modeling human cancers in drosophila. Curr. Top. Dev. Biol. 121, 287-309. 10.1016/bs.ctdb.2016.07.008 [DOI] [PubMed] [Google Scholar]
- Srivastava A., Pastor-Pareja J. C., Igaki T., Pagliarini R. and Xu T. (2007). Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc. Natl. Acad. Sci. USA 104, 2721-2726. 10.1073/pnas.0611666104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staehling-Hampton K., Jackson P. D., Clark M. J. and Brand A. H. and H. F. M. (1994). Specificity of bone morphogenetic protein-related factors: cell fate and gene expression changes in Drosophila embryos induced by decapentaplegic but not 60A. Cell Growth Differ. 5, 585-593. [PubMed] [Google Scholar]
- Swarup S. and Verheyen E. M. (2011). Drosophila homeodomain-interacting protein kinase inhibits the Skp1-Cul1-F-box E3 ligase complex to dually promote Wingless and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 108, 9887-9892. 10.1073/pnas.1017548108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M., Gong H., Zeng Y., Tao L., Wang J., Jiang J., Xu D., Bao E., Qiu J. and Liu Z. (2014). Downregulation of homeodomain-interacting protein kinase-2 contributes to bladder cancer metastasis by regulating Wnt signaling. J. Cell. Biochem. 115, 1762-1767. 10.1002/jcb.24842 [DOI] [PubMed] [Google Scholar]
- Thiery J. P., Acloque H., Huang R. Y. J. and Nieto M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871-890. 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- Trapasso F., Aqeilan R. I., Iuliano R., Visone R., Gaudio E., Ciuffini L., Alder H., Paduano F., Pierantoni G. M., Soddu S. et al. (2009). Targeted disruption of the murine homeodomain-interacting protein kinase-2 causes growth deficiency in vivo and cell cycle arrest in vitro. DNA Cell Biol. 28, 161-167. 10.1089/dna.2008.0778 [DOI] [PubMed] [Google Scholar]
- Wei G., Ku S., Ma G. K., Saito S., Tang A. A., Zhang J., Mao J.-H. H., Appella E., Balmain A. and Huang E. J. (2007). HIPK2 represses beta-catenin-mediated transcription, epidermal stem cell expansion, and skin tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 13040-13045. 10.1073/pnas.0703213104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfstetter G., Shirinian M., Stute C., Grabbe C., Hummel T., Baumgartner S., Palmer R. H. and Holz A. (2009). Fusion of circular and longitudinal muscles in Drosophila is independent of the endoderm but further visceral muscle differentiation requires a close contact between mesoderm and endoderm. Mech. Dev. 126, 721-736. 10.1016/j.mod.2009.05.001 [DOI] [PubMed] [Google Scholar]
- Wu C.-I., Hoffman J. A., Shy B. R., Ford E. M., Fuchs E., Nguyen H. and Merrill B. J. (2012). Function of Wnt/β-catenin in counteracting Tcf3 repression through the Tcf3-β-catenin interaction. Development 139, 2118-2129. 10.1242/dev.076067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Deshmukh H., Gutmann R. J., Emnett R. J., Rodriguez F. J., Watson M. A., Nagarajan R. and Gutmann D. H. (2009). Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology 73, 1526-1531. 10.1212/WNL.0b013e3181c0664a [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.