

Abstract
The PI 3-kinase Vps34 (Pik3c3) synthesizes phosphatidylinositol 3-phosphate (PI3P), a lipid critical for both endosomal membrane traffic and macroautophagy. Human genetics have implicated PI3P dysregulation, and endosomal trafficking in general, as a recurring cause of demyelinating Charcot-Marie-Tooth (CMT) peripheral neuropathy. Here, we investigated the role of Vps34, and PI3P, in mouse Schwann cells by selectively deleting Vps34 in this cell type. Vps34-Schwann cell knockout (Vps34SCKO) mice show severe hypomyelination in peripheral nerves. Vps34−/− Schwann cells interact abnormally with axons, and there is a delay in radial sorting, a process by which large axons are selected for myelination. Upon reaching the promyelinating stage, Vps34−/− Schwann cells are significantly impaired in the elaboration of myelin. Nerves from Vps34SCKO mice contain elevated levels of the LC3 and p62 proteins, indicating impaired autophagy. However, in the light of recent demonstrations that autophagy is dispensable for myelination, it is unlikely that hypomyelination in Vps34SCKO mice is caused by impaired autophagy. Endosomal trafficking is also disturbed in Vps34−/− Schwann cells. We investigated the activation of the ErbB2/3 receptor tyrosine kinases in Vps34SCKO nerves, as these proteins, which play essential roles in Schwann cell myelination, are known to traffic though endosomes. In Vps34SCKO nerves, ErbB3 was hyperphosphorylated on a tyrosine known to be phosphorylated in response to Nrg1 exposure. ErbB2 protein levels were also decreased during myelination. Our findings suggest that the loss of Vps34 alters the trafficking of ErbB2/3 through endosomes. Abnormal ErbB2/3 signaling to downstream targets may contribute to the hypomyelination observed in Vps34SCKO mice.
Keywords: Charcot-Marie-Tooth neuropathy, endosomal trafficking, axo-glial interactions, lysosome, phosphoinositides
INTRODUCTION
In eukaryotic cells, phosphoinositides (PIs) are regulated by PI kinases, phosphatases, and lipases, a network of enzymes that control localization and turnover, thereby orchestrating compartmentalized membrane signaling. Phosphorylation and dephosphorylation of PIs both play important roles in the regulation of acute cellular signaling, membrane trafficking and cytoskeletal dynamics (Balla, 2013; Schink et al., 2016). PIs function as inducible binding sites on membranes, as specific phosphorylation states on the inositol head group trigger the recruitment of specific effector proteins (Balla, 2013; Schink et al., 2016).
PI signaling plays an important role in myelination in both the central and peripheral nervous systems (CNS and PNS). Myelin is a multilayer wrapping of specialized plasma membrane that surrounds axons and makes possible efficient saltatory impulse propagation (Nave and Werner, 2014). Oligodendrocytes and Schwann cells are the specialized glial cells which produce myelin in the CNS and PNS, respectively. Phosphatidylinositol 3,4,5-trisphosphate (PI[3,4,5]P3), 3,5-bisphosphate (PI[3,5]P2), and 3-phosphate (PI3P) have each been demonstrated to play important roles in myelination (Goebbels et al., 2010; Sherman et al., 2012; Taveggia et al., 2005; Vaccari et al., 2011). Moreover, mutations in four PI phosphatases, myotubularin-related (MTMR) 2, MTMR5, MTMR13 and FIG4, have been identified as the causes of specific forms of demyelinating Charcot-Marie-Tooth (CMT) peripheral neuropathy (Bolino et al., 2000; Chow et al., 2007; Nakhro et al., 2013; Senderek et al., 2003) (Fig. 1A).
Figure 4.
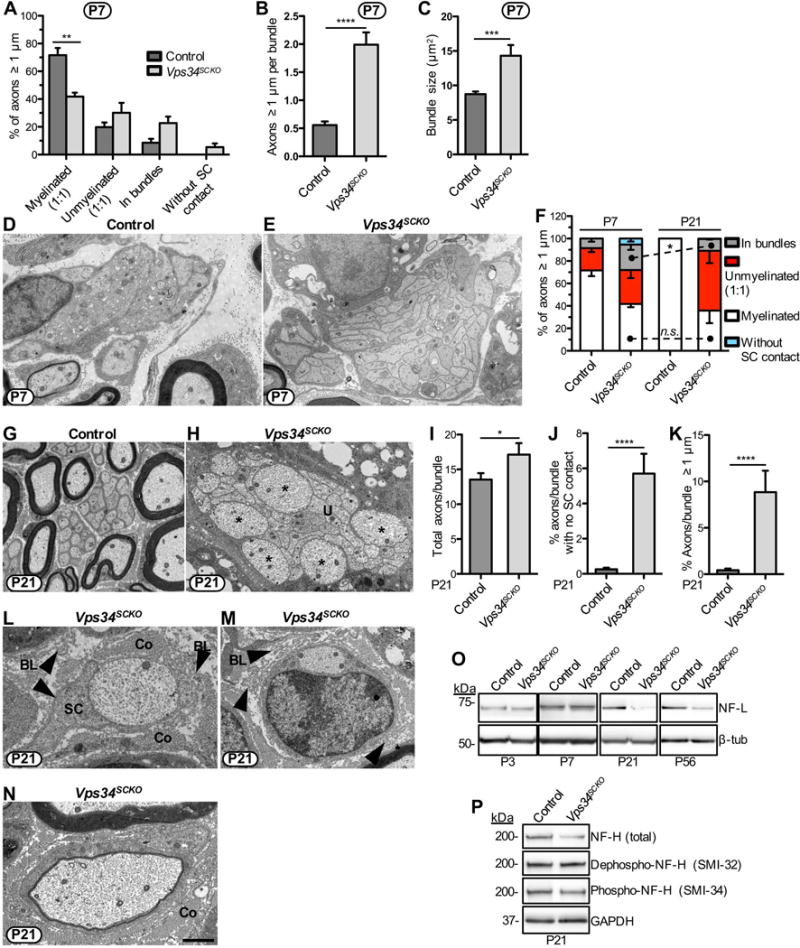
Delayed axonal sorting, persistent hypomyelination and axonal abnormalities when Schwann cells lack Vps34. (A) The status of all axons ≥ 1 μm in diameter was determined in P7 nerves using EM. The percentage of axons myelinated is significantly decreased in Vps34SCKO sciatic nerves (n = 3 mice). (B) Increased numbers of large caliber axons per bundle in Vps34SCKO nerves at P7 (n = 165 bundles pooled from 5 control and 104 bundles pooled from 4 Vps34SCKO mice). (C) Increased axon bundle size in Vps34SCKO nerves at P7 (n = 393 bundles pooled from 5 control and 274 bundles pooled from 4 Vps34SCKO mice, respectively). (D & E) Representative axon bundles from control and Vps34SCKO nerves at P7. (F) Large axons are successfully sorted after P7 in Vps34SCKO nerves, but hypomyelination persists. There is no signification change in the percent of axons myelinated when comparing Vps34SCKO nerves at P7 and P21 (p = 0.6787; n = 3 for P7; n = 4 for P21). (G & H) Axon bundles of Vps34SCKO nerves are poorly invested with Schwann cell processes and contain unsorted (U) axons of both small and large caliber (asterisks). (I) The number of axons per bundle is increased in Vps34SCKO nerves at P21. (J) Increased percentage of bundle axons that lack Schwann cell contact in Vps34SCKO nerves. (K) Large axons are aberrantly retained in bundles in Vps34SCKO nerves. For I-K, n = 152 bundles pooled from 3 control and 97 or 99 bundles pooled from 2 Vps34SCKO mice, respectively. (L & M) Vps34SCKO nerves contain a few axons ≥ 1 μm with limited or no Schwann cell (SC) contact at P21. Redundant basal lamina (BL, arrowheads) are observed in Vps34SCKO nerves, indicating Schwann cell withdrawal from axons. For clarity, collagen (Co) is indicated. (N) A particularly thin myelin sheath indicates either delayed myelination or de/remyelination in a P21 Vps34SCKO nerve. Scale bar: 1.3 μm (D & E), 2 μm (G), 1.57 μm (H), 1.05 μm (L), 1.5 μm (M & N) (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; mean ± SEM). (O) Immunoblot analysis of axonal proteins in sciatic nerves. The level of NF-L is not significantly changed in Vps34SCKO nerves analyzed at P3 or P7. However, by P21, NF-L protein abundance is significantly decreased in Vps34SCKO nerves. (P) Immunoblot analysis of phosphorylated epitopes on NF-H in sciatic nerves of control and Vps34SCKO mice at P21. The SMI-32 antibody detects the dephosphorylation of normally phosphorylated epitopes. The SMI-34 antibody is specific for a distinct set of phosphorylated epitopes on NF-H. Total NF-H levels are decreased in Vps34SCKO nerves, as are the levels of phospho-NF-H (SMI-34) (n = 3 lysates).
Figure 1.
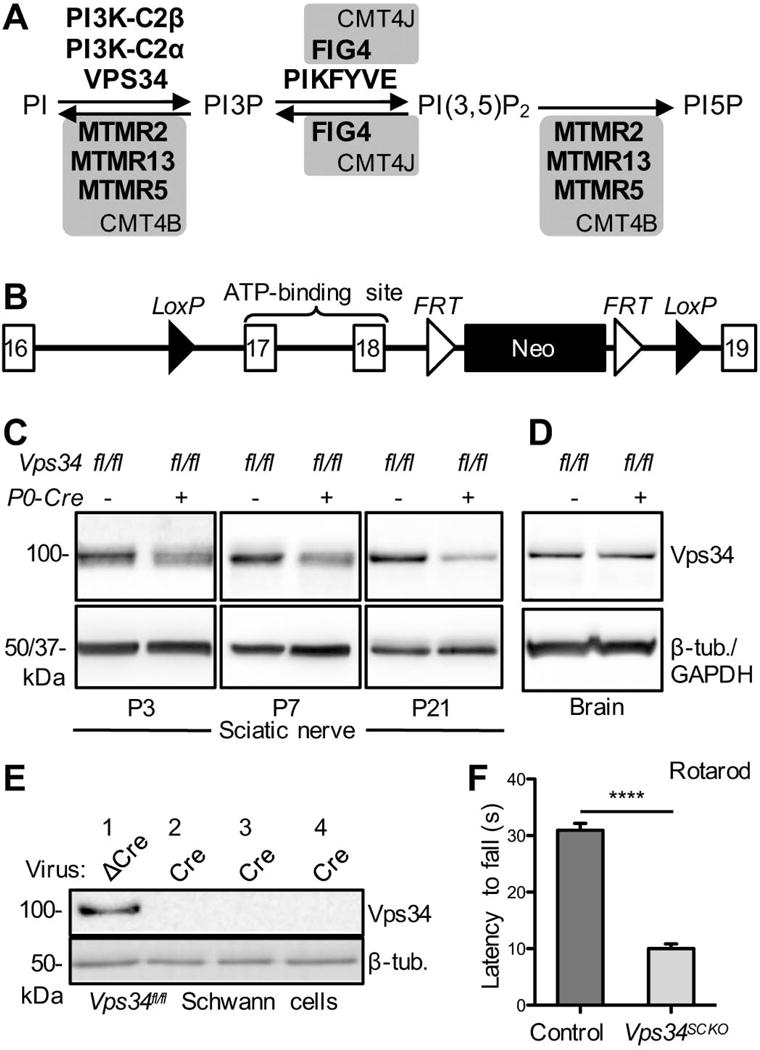
Schwann cell-specific deletion of Pik3c3/Vps34. (A) Biochemical pathway for the regulation of PI3P, PI(3,5)P2, and PI5P abundance by PI kinases and PI phosphatases. Loss-of-function mutations in the phosphatases in grey boxes cause the indicated forms of demyelinating CMT. Although the PI 5-phosphatase FIG4 is capable of dephosphorylating PI(3,5)P2, the predominant role of this enzyme in mammalian cells is the promotion of PIKFYVE kinase activity. (B) The mouse Pik3c3/Vps34 conditional deletion allele used in this study (Zhou et al., 2010) (Supplemental Material). Schwann cell-specific expression of Cre Recombinase from the P0-Cre transgene triggers deletion of Pik3c3/Vps34 exons 17 and 18, as well as the neomycin expression cassette. (C) Immunoblot analysis of Vps34 protein abundance in sciatic nerve extracts from Vps34fl/fl (control) and Vps34fl/fl;P0-Cre+ (Vps34SCKO) mice of the indicated ages, using an antibody specific for the amino-terminus. Arbitrary Vps34 protein levels at P3: 1.00 ± 0.111 vs. 0.561 ± 0.109 for Vps34fl/fl and Vps34fl/fl;P0-Cre+, respectively; p = 0.0472; n = 3 independent extracts for both genotypes. At P7: 1.00 ± 0.035 vs. 0.462 ± 0.052 for Vps34fl/fl and Vps34fl/fl;P0-Cre+, respectively; p < 0.0001; n = 4 and 5 independent extracts for Vps34fl/fl and Vps34fl/fl;P0-Cre+, respectively. At P21: 1.00 ± 0.078 vs. 0.379 ± 0.077 for Vps34fl/fl and Vps34fl/fl;P0-Cre+, respectively; p = 0.0003; n = 5 and 6 independent extracts for Vps34fl/fl and Vps34fl/fl;P0-Cre+, respectively. The protein loading control was either beta-tubulin (P3 and P7) or GAPDH (P21). Each sciatic nerve extract (the biological replicate) was prepared by homogenizing the (pooled) nerves from three mice of the same genotype at P3 or P7, and one or two mice at P21. (D) Immunoblot analysis of brain extracts from control and Vps34SCKO mice at 14 weeks indicated no significant change in Vps34 protein levels (n = 3 mice; beta-tubulin was the protein loading control). (E) Loss of Vps34 protein following lentiviral expression of Cre recombinase in primary Vps34fl/fl Schwann cells. ΔCre is an inactive version of the recombinase. Cultures 1 & 4 and 2 & 3 were infected with 1 ml and 1.5 ml of lentiviral supernatant, respectively. (F) Rotarod testing of control and Vps34SCKO mice (6–7 weeks; accelerating Rotarod). The average time to fall (in seconds [s]) was determined for each mouse (n = 5 and 13 mice for control and Vps34SCKO, respectively; ****p < 0.0001; mean ± SEM).
PI3P plays an important role in both endosomal trafficking and autophagy (Roberts and Ktistakis, 2013; Schink et al., 2016). Highly enriched on early endosomes, PI3P recruits specific effector proteins, many of which contain FYVE or PX domains (Raiborg et al., 2013). Depletion of PI3P delays the traffic of proteins through endosomes (Schink et al., 2016). PI3P also plays a key role in autophagosome formation (Roberts and Ktistakis, 2013). In mammalian cells, the majority of PI3P is thought to be generated via the phosphorylation of PI by the class II and III PI 3-kinases (PI3Ks) (Devereaux et al., 2013; Schink et al., 2016) (Fig. 1A).
CMT-causing mutations and studies with gene knockout mice have indicated that PI3P dysregulation and endosomal-lysosomal dysfunction may be a common trigger for dysmyelination in the PNS (Jerath and Shy, 2015). However, it is unclear which isoforms of PI3K are involved in generating PI3P in Schwann cells (Fig. 1A). Moreover, a number of distinct pools of PI3P may be present in these cells. To begin to elucidate the roles of PI3P, endosomal trafficking, and autophagy in myelinating Schwann cells, we have examined the role of the PI3K Vps34 (Pik3c3) in mice. Vps34 is the sole member of the class III PI3K sub-family; a homolog has been identified in most eukaryotes (Backer, 2008; Raiborg et al., 2013). Vps34 is involved in endosomal-lysosomal trafficking, autophagy, and cellular responses to nutrient availability (Backer, 2008). The role of Vps34 in specialized mammalian cells and tissues, however, remains unclear. Recently, investigators have begun using conditional alleles of Vps34 to examine protein function in specific mouse cell types (Bechtel et al., 2013; Devereaux et al., 2013; Jaber et al., 2012; Willinger and Flavell, 2012; Zhou et al., 2010).
Given the potential roles of PI3P, endosomal trafficking, and autophagy in myelinating Schwann cells, we sought to determine the function of Vps34 in this unique cell type. We generated a mouse line in which Vps34 is specifically deleted in Schwann cells and show that this mutation leads to delayed radial sorting of axons and arrested myelination. Despite reaching the promyelinating stage, Schwann cells that lack Vps34 are inefficient at generating myelin sheaths. We also found that both the autophagic and endosomal pathways are disturbed in Schwann cells that lack Vps34. Lastly, we assessed the impact of Vps34 loss on the activation of the ErbB2/3 receptor tyrosine kinases, which play essential roles in Schwann cell myelination, and are known to traffic though endosomes. ErbB2/3 showed abnormal posttranslational modifications in Vps34-deficient nerves, suggesting that deranged endosomal trafficking of these kinases has altered their signaling output, a change that may contribute to the observed hypomyelination.
MATERIALS AND METHODS
Schwann cell-specific inactivation of Vps34
The mouse Pik3c3 (Vps34) gene consists of 25 exons strung over about 75 kilobases (kb) on the forward strand of chromosome 18. Pik3c3/Vps34flox/flox mice were a generous gift from Fan Wang. Vps34flox/flox mice, initially a 50:50 mix of 129 and C57BL/6 backgrounds, had been backcrossed thrice onto C57BL/6 (generation N4). This line of mice contains an engineered allele of Pik3c3 in which exons 17 and 18, which encode the ATP binding sequences of the kinase, are flanked by loxP sites (Zhou et al., 2010). In the presence of Cre recombinase, exons 17 and 18 are removed and the Vps34 protein is not expressed (Devereaux et al., 2013; Zhou et al., 2010). Genotyping of Vps34flox/flox mice was carried out using PCR (40 cycles) using primers ALO-18 (Vps34 F; GCCACCCATTGCTGCCT) and ALO-21 (Vps34 R; CCCTCACTGGCTGTGGC). To generate mice with a Schwann cell-specific deletion of Vps34, Vps34fl/fl mice were crossed with mP0TOTA(Cre) (P0-Cre) transgenic mice (Feltri et al., 1999), which were congenic on the C57BL/6 background. The P0-Cre transgene was detected by PCR with oligonucleotides ALO-5 (P0-F; CACCACCTCTCCATTGCAC) and ALO-6 (P0-R; GCTGGCCCAAATGTTGCTGG). Vps34fl/fl mice were previously found to be indistinguishable from wild type mice (Zhou et al., 2010). Control mice were either Vps34fl/fl (P0-Cre−) or Vps34+/fl (P0-Cre−) littermates of Vps34fl/fl P0-Cre+ (Vps34SCKO) mutant mice. Phenotyped mice were the products of crosses between Vps34fl/fl (N4 on C57BL/6) and Vps34+/fl P0-Cre+ (N5 on C57BL/6) mice. Phenotypic analyses were performed on littermates, whenever possible. Mice of either sex were used in all experiments. All work with animals was approved by and conformed to the standards of the Oregon Health & Science University Institutional Animal Care and Use Committee.
Morphology
Preparation of mouse sciatic nerves for electron microscopy (EM), or for light microscopy of toluidine blue-stained nerve sections, was as previously described (Ng et al., 2013). EM and light microscopy imaging were also as previously described (Ng et al., 2013). Toluidine blue sections were imaged at 63× on a Zeiss ApoTome microscope. Two cross sections of the sciatic nerve (one from each nerve) were analyzed per mouse; 4–7 mice of each genotype were analyzed per time point. The entire sciatic nerve cross-section was imaged using a tiling function and the acquired images were stitched together into a single file.
Toluidine blue stained cross-sections were used to determine g ratio by measuring the axonal circumference and dividing that number by the circumference of the outer edge of the myelin sheath. The diameter of each corresponding axon was found by dividing axon circumference by π. For g ratio analysis, axons from throughout the entire nerve cross-section were sampled; 100 axons per mouse). In P3 nerves, g ratio on all possible round, myelinated axons was measured (60–100 axons per mouse).
For total axon counts, toluidine blue stained cross sections were analyzed. At P56, four cross sectional areas (2 each from the left and right nerve) of 100 μm2 per animal were analyzed (4–5 animals of each genotype). Axons ≥ 1 μm in diameter and in a 1:1 relationship with a Schwann cell were counted, whether myelinated or not. For P56, about 20–28% of the entire transverse fascicular area (TFA) was counted. For P21 axon counts, total axons ≥ 1 μm were counted in ~30% of the complete TFA of each mouse (n = 4) on two cross sections (left and right sciatic nerve). For axon counts in P3 and P7 mice, the entire TFA was counted in 4–7 mice per genotype. Every axon that was in a 1:1 relationship with a Schwann cell was counted in two cross-sections (right and left sciatic nerve), and the average of the two sections was calculated. An additional analysis of total large caliber axon number at P7 was performed using EM images (890x magnification; 8–16 images per animal) (Fig. S3F). The total number of axons in 1:1 relationships with Schwann cells (myelinated or not) was determined. On average, 42% of the TFA was analyzed for each mouse (n = 5 for control; n = 4 for Vps34SCKO). Once the axon count for a significant fraction of the nerve was established, this number was scaled based on total nerve area to generate a predicted total axon count for the nerve.
For analysis of axon bundles at P7, EM images of 890x magnification were analyzed. A bundle was defined as any assortment of axons in contact with, but not in a 1:1 relationship with a Schwann cell. The area of the axonal portion of the bundle was measured; the area of the associated Schwann cell nucleus, cytoplasm and any significant processes was excluded. An average of 74 bundles (from about seven 890x images) were measured per mouse (n = 5 for control; n = 4 for Vps34SCKO). The number of large caliber axons per bundle was also determined in P7 sciatic nerves. An average of 30 bundles per mouse were analyzed from five control and four Vps34SCKO mice. A large caliber axon was defined as such if the average of long and short axes was above 1 μm and the smallest of these two measures was above 0.35 μm. EM images were analyzed using Fiji (Schindelin et al., 2012).
To assess axon sorting at P3, randomly selected 1400x EM images were analyzed from three control and three Vps34SCKO mice. The axons contained in an average of 24 bundles from each mouse were counted and then pooled by genotype to determine the average number of axons per bundle. To assess axon bundle morphology at P3, EM images of 4800 or 6800x from three control and four Vps34SCKO mice were examined. An average of 19 randomly selected bundles from each mouse were qualitatively analyzed for the following three abnormal characteristics: (1) Schwann cell projections away from the axons, (2) Failure to encircle at least 75% of the axon bundle circumference with Schwann cell processes, and (3) Presence of axons ≥ 1 μm in the center of a bundle.
To assess the status of axons at P7, all axons in four randomly selected 1400x EM images or eleven such 2900x EM images (per mouse) from three control and three Vps34SCKO mice were counted and scored for their relationships with Schwann cells. Thus, 5–6% of the entire TFA of the sciatic nerve was analyzed in each mouse. Axon density (axons/μm2) was derived from this sampling as well. Bundle morphology in P7 nerves were qualitatively assessed by examining an average of 17 randomly selected EM images of 4800 or 6800x magnification (per mouse) from three control and three Vps34SCKO mice. To assess the status of axons ≥ 1 μm at P21, the axons in an average of twelve 1200x EM images (per mouse) from three control and four Vps34SCKO mice were counted and scored for their relationships with Schwann cells. Thus, an average of 14% of the entire TFA of the sciatic nerve was analyzed in each mouse. The numbers of total axons ≥ 1 μm and total myelinated axons per sciatic nerve were predicted from these axon counts by extrapolation based on the area sampled, with a correction for the increased cross-sectional area of P21 Vps34SCKO nerves.
To assess axon content in bundles at P21, 1200x EM images were used. 152 randomly selected bundles from three control and 95 randomly selected bundles from two Vps34SCKO mice were analyzed. Remyelination and Schwann cell-axon interactions in P21 nerves were qualitatively assessed by examining an average of 21 randomly selected EM images of 1200–2800x (per mouse) from three control and four Vps34SCKO mice.
Rotarod test
Six to seven week-old male and female mice were placed on an elevated, accelerating, rotating rod (Economex model 0207-003M, Columbus Instruments; rod diameter, 7 cm), which began rotating at 5 rpm at the start of the test. The rotation speed of the rod accelerated at a rate of 0.5 rpm/sec and the latency to fall (in seconds) was recorded. Each mouse underwent three trials per day, with no delay between trials, on three consecutive days. On the 3rd day, the three trials were averaged and plotted on a bar graph (n = 5 control; n = 13 for Vps34SCKO).
Immunofluorescence
P21 mice were perfused with 4% paraformaldehyde (PFA) and nerves were subsequently processed as described previously (Ng et al., 2013), with the exception that 10 μm thick longitudinal sections were analyzed. Primary antibodies used were guinea pig anti-Sox10, a gift from Dr. Michael Wegner, and rabbit anti-Krox20 (Covance). Images were acquired on a Zeiss Apotome microscope using the tiling function to image the entire nerve. Krox20-positive nuclei, Sox10-positive nuclei, and total nuclei (4′,6-diamidino-2-phenylindole [DAPI]-positive) were counted in 7–8 entire nerve sections from each mouse (both left and right nerve) from 2 mice per genotype. The area of the entire nerve was determined using MetaMorph to allow for calculation of the number of nuclei/μm2. A similar approach was taken for immunofluorescence of P7 and P14 sciatic nerves using rabbit anti-Ki67 (Cell Signaling Technology).
Immunoblotting
Immunoblotting of sciatic nerve and brain extracts was performed as previously described (Ng et al., 2013). Rabbit monoclonal antibodies (mAb) for Vps34, EEA1, Rab5, ErbB3, pErbB3 (Tyr1289), ErbB2, S6, pS6 (Ser235/236), pAkt (Ser473), pAkt (Thr308), and pJNK (Thr183/Tyr185) were from Cell Signaling Technology. Rabbit polyclonal antibodies (pAb) for pErbB2 (Tyr1248), Akt, Erk1/2, and JNK were from Cell Signaling Technology. Additional antibodies used were rabbit pAbs to MAG and p62 (Santa Cruz Biotechnology), mouse mAb to FAK (BD Bioscience), rabbit pAb to ppErk1/2 (pTpY185/187) (Invitrogen), mouse mAb to SMI-34 (Biolegend), chicken mAb to NF-H (Abcam), rat mAb to Lamp1 (1D4B), mouse mAb to β-tubulin (E7) (Developmental Studies Hybridoma Bank), rabbit pAb to LC3 (Novus Biologicals), mouse mAbs to GAPDH and SMI-32, rat mAb to MBP, rabbit mAb to NF-L, rabbit pAb to pFAK (Tyr397) (Millipore), and rabbit mAb to Oct6 (Abcam). For each P0, P3, and P7 nerve extract, three mice of the same genotype (6 nerves total) were pooled. For P21, the nerves from one or two mice were used to generate each independent extract. At P56, a single mouse (2 nerves) was used for each extract. Each time point/antibody combination was run in triplicate (3 lysates per genotype per time point). 17–25 μg of protein per lane was resolved in 4–12% NuPAGE Bis-Tris gels (Invitrogen), and chemiluminescent quantitation of immunoblots was as previously described (Ng et al., 2013).
Statistics
GraphPad Prism 5 was used for all statistical analyses. Unpaired t-tests (two-tailed) were used to evaluate significance, except in Figure 8, where unpaired t-tests with Welch’s correction were used.
Figure 8.
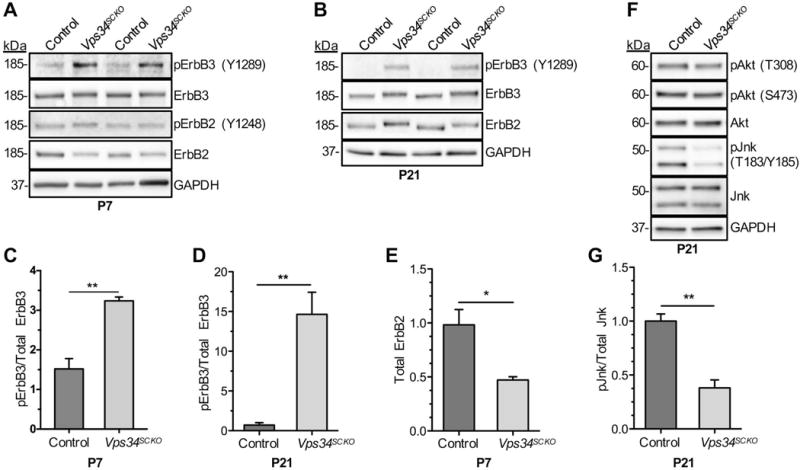
Altered ErbB2/3 receptor signaling in Vps34SCKO nerves. (A & B) The activation of ErbB2/3 was assessed by immunoblotting P7 and P21 sciatic nerve extracts with the indicated antibodies and quantifying the relative amounts of activated (phosphorylated) forms compared to total protein levels. Two (replicate) lanes for each genotype are shown. At P7 and P21, the ErbB2 and ErbB3 proteins from Vps34SCKO nerve extracts migrated more slowly in SDS-PAGE gels than the same proteins from control extracts, suggesting altered post-translational modifications. (C & D) The ratio of activated ErbB3 (pY1289) to total ErbB3 was significantly increased in Vps34SCKO nerve extracts at both P7 and P21. Activated ErbB2 (Y1248) was undetectable in P21 nerve extracts. (E) Vps34SCKO nerves contained significantly decreased levels of total ErbB2 protein at P7. (F) The activation of downstream targets of ErbB2/3, Akt and Jnk, was assessed by immunoblotting P21 sciatic nerve extracts and quantifying the relative level of activated (phosphorylated) forms compared to total protein levels. Akt phosphorylation at Thr308 or Ser473 was not significantly changed in Vps34SCKO nerves. In contrast, the level of doubly-phosphorylated Jnk (Thr183-Tyr185) was significantly reduced in Vps34SCKO nerves. (G) Quantification of suppressed Jnk phosphorylation. Each sciatic nerve extract (the biological replicate) was prepared by homogenizing the (pooled) nerves from three mice of the same genotype at P7, and one or two mice at P21. For P7, n = 4 and 3 independent nerve extracts from control and Vps34SCKO mice, respectively. For P21, n = 3 and 4 independent nerve extracts from control and Vps34SCKO mice, respectively (n.s., not significant; *p < 0.05; **p < 0.01; mean ± SEM).
RESULTS
Schwann cell-specific deletion of Vps34/Pik3c3
To selectively inactivate Vps34 in Schwann cells, Vps34flox/flox mice (Zhou et al., 2010) were crossed with myelin protein zero (P0)-Cre transgenic mice, which express Cre recombinase in Schwann cells beginning at embryonic (E) day 14.5 (Feltri et al., 1999) (Fig. 1B–C) (Materials and Methods, Supp. Fig. S1). Vps34flox/flox P0-Cre+ mice are hereafter referred to as Vps34-Schwann cell knockout (Vps34SCKO). Loss of Vps34 protein in sciatic nerve extracts was confirmed by immunoblotting (Fig. 1C). Vps34 protein was reduced in mutant nerves at postnatal day 0 (P0) and thereafter (Fig. 1C & Supp. Fig. S1). At P21, Vps34 protein was reduced to about 37% of control levels in Vps34SCKO sciatic nerves (Fig. 1C & Supp. Fig. S1). The remaining Vps34 protein in Vps34SCKO nerves can be accounted for by cell types other than Schwann cells, including axons, fibroblasts, and endothelial cells. A similar degree of targeted protein persistence in sciatic nerves has been reported when using Schwann cell-specific Cre drivers and whole nerve extracts (Beirowski et al., 2014; Gomez-Sanchez et al., 2015; Jang et al., 2015; Nodari et al., 2007; Sherman et al., 2012). Vps34 protein levels in mutant brain tissue were similar to controls (Fig. 1D), consistent with the documented Schwann cell specificity of the P0-Cre transgene (Feltri et al., 1999). We also examined the efficiency of Cre recombination of the floxed Vps34 allele in cultured cells; lentiviral expression of Cre in primary Vps34fl/fl Schwann cells resulted in a complete loss of Vps34 protein (Fig. 1E).
Although Vps34SCKO mice appear normal at birth, minor motor deficits, including a wide-based gait, become apparent by 21 days. By 56 days, mutant mice display an unsteady gait, hind limb weakness, and tremor (Supp. Movie). RotaRod testing confirmed significant motor impairment in Vps34SCKO mice (Fig. 1F).
Loss of Schwann cell Vps34 leads to arrested myelination
To evaluate the role of Vps34 in myelination, we examined sciatic nerves from control and Vps34SCKO mice at postnatal (P) days 3, 7, 21 and 56 (Fig. 2A). In control nerves examined at P3, significant radial sorting has occurred, a majority of the large caliber axons are in 1:1 relationships with Schwann cells, and many large axons have thin myelin sheaths (Grove et al., 2007; Nodari et al., 2007) (Fig. 2A). At P3, Vps34SCKO nerves were similar to those from control animals (Fig. 2A). Neither myelin thickness (g ratio), nor the number of myelinated axons in the nerve were significantly altered (Fig. 3A, B, Supp. Fig. S3F, and data not shown). Nerve pathology becomes apparent in Vps34SCKO mice at P7, when vacuoles are observed in the cytoplasm of many Schwann cells (Fig. 2A, B). Accumulation of large, late endosomal/lysosomal vacuoles is a well-established hallmark of Vps34 loss and PI3P depletion (Futter et al., 2001; Johnson et al., 2006; Schu et al., 1993).
Figure 2.
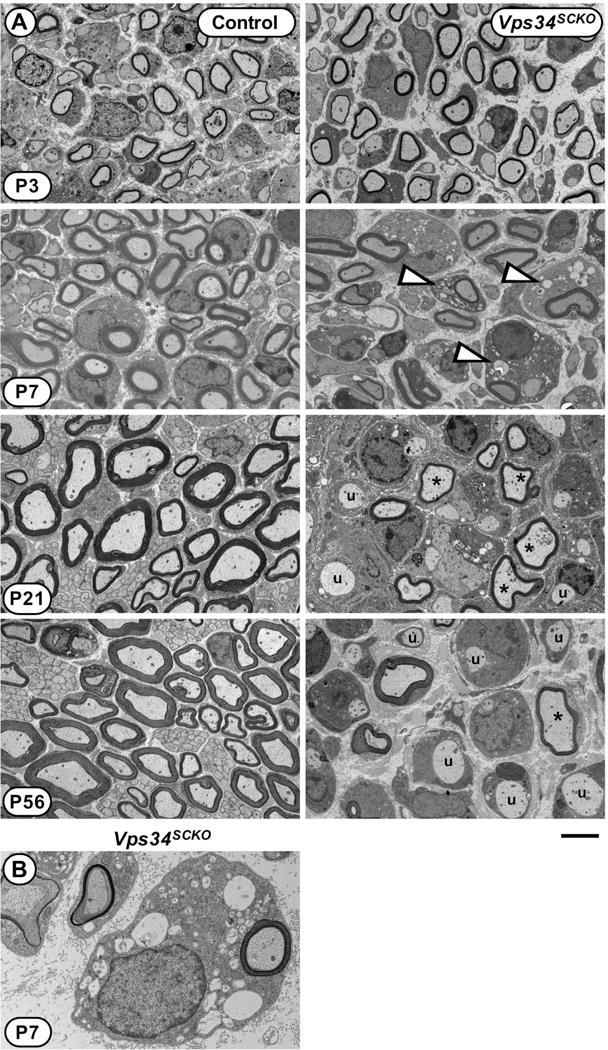
Defective myelination in Vps34SCKO nerves. (A) EM analysis of mid-sciatic nerve cross sections of control and Vps34SCKO mice at P3, P7, P21, and P56. At P3, Vps34SCKO nerves appear normal when compared to controls (n = 7 mice). By P7, nerve pathology is evident, notably vacuoles (white arrowheads) in the cytoplasm of some Schwann cells (n = 4 or 5 mice). At P21 and P56, hypomyelinated (asterisks) and unmyelinated (u) axons are evident in mutant nerves (n = 4 or 5 mice). (B) An example of a Vps34−/− Schwann cell with numerous endosomal-lysosomal vacuoles within its cytoplasm. Scale bar: 4 μm (A), 1.3 μm (B).
Figure 3.
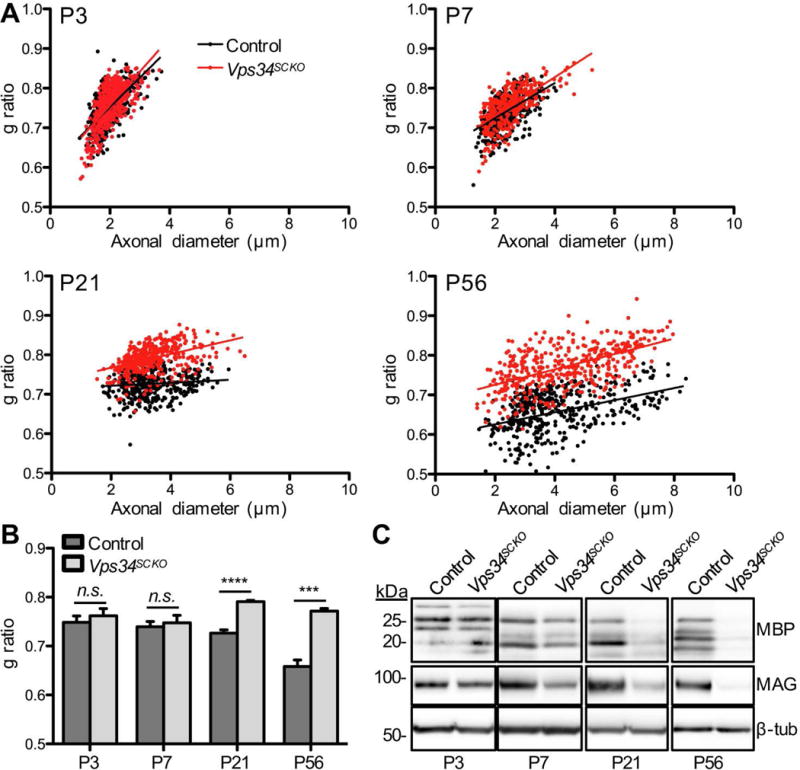
Peripheral nerve hypomyelination in the absence of Schwann cell Vps34. (A) G ratio analysis of myelin thickness in control and Vps34SCKO sciatic nerves at P3, P7, P21, and P56. The best-fit line is shown. (B) Average g ratios. At P3 and P7 there is no significant difference in average g ratio between control and Vps34SCKO mice (n = 7 mice at P3; n = 4–5 mice at P7). At both P21 and P56, there is a significant increase in the average g ratio of Vps34SCKO mice, indicating hypomyelination (n = 4 mice at P21; n = 4–5 mice at P56). (C) Immunoblot analysis of myelin proteins in sciatic nerves. At P3, the levels of MBP and MAG are similar in control and Vps34SCKO nerves. By P7, mutant nerves contain slightly reduced MBP and significantly decreased MAG. Both MBP and MAG are significantly reduced at P21 and P56 (n = 3 lysates). The data are mean ± SEM (n.s., not significant; ***p < 0.001; ****p < 0.0001)
We next examined Vps34SCKO nerves at 21 days, when myelination is normally complete, with nearly all axons 1 μm or larger myelinated. At P21, most Vps34SCKO Schwann cells contain cytoplasmic vacuoles, and myelin sheaths are thinner than in controls (Fig. 2A), as indicated by a significant increase in g ratio (Fig. 3A, B). Nerve pathology progressively worsens in adult Vps34SCKO mice; at 56 days, myelin sheaths are thinner, and about 56% of the large axons are completely unmyelinated (Fig. 2A, Fig. 3A, B, and data not shown). In accord with our morphological findings, at P7 and thereafter the levels of myelin basic protein (MBP) and myelin-associated glycoprotein (MAG) were both decreased in Vps34SCKO nerves (Fig. 3C).
Abnormal interactions between Vps34-deficient Schwann cells and axons
To elucidate how the loss of Schwann cell Vps34 leads to hypomyelination, we examined postnatal Schwann cell-axon interactions in the mutant mice. Radial axonal sorting, a developmental process by which Schwann cells select larger axons for myelination, is ongoing between embryonic day 12.5 and P10 (Feltri et al., 2016; Grove et al., 2007; Jessen et al., 2015; Monk et al., 2015). In wild type P3 nerves, bundles comprised mostly of small axons are encircled by Schwann cells, which are generating basal laminae (Feltri et al., 2016; Grove et al., 2007; Jessen et al., 2015; Monk et al., 2015). Axons 1 μm or larger, if associated with bundles at P3-P5, are generally at the periphery of the bundle, and are often in direct contact with invading Schwann cell processes. Schwann cells will subsequently extract such axons from bundles, an event required for myelination (Feltri et al., 2016; Grove et al., 2007; Jessen et al., 2015; Monk et al., 2015; Nodari et al., 2007). In P3 Vps34SCKO sciatic nerves, we found radial sorting and axo-glial interactions to be largely normal (Supp. Fig. S2), consistent with our finding that endo-lysosomal vacuole formation was not observed at this stage (Fig. 2A). The only abnormality that we discerned at P3 was a limited number of giant axon bundles in most, but not all, Vps34SCKO nerves (Supp. Fig. S2). These abnormal bundles, which were not observed in control littermates, likely indicate a subtle or incipient defect in Schwann cell-axon interactions.
By day 7, there is a significant reduction in the fraction of large axons that have acquired myelin in Vps34SCKO nerves (Fig. 4A). This decrease in myelination is associated with increases in the proportions of unmyelinated axons in 1:1 relationships with Schwann cells, axons ≥ 1 μm retained in bundles, and similar axons which are not in contact with Schwann cells (Fig. 4A). In mutant nerves, signs of axonal stress (increased density of neurofilaments, irregular axolemmal profiles) are observed in the few large axons that lacked Schwann cell contact (Supp. Fig. S2I). However, the overall number of axons in Vps34SCKO nerves is not decreased at P7 (Supp. Fig. S2J). Direct analysis of bundles confirmed abnormal retention of large axons and increased cross-sectional areas of bundles in Vps34SCKO nerves (Fig. 4B–E). Thus, P7 nerves from Vps34SCKO mice show impaired Schwann cell-axon interactions.
Delayed radial sorting and persistent hypomyelination in the absence of Schwann cell Vps34
We considered whether the abnormal axo-glial associations and delayed myelination observed at P7 would be resolved as Vps34SCKO mice matured. Strikingly, only about 35% of large axons have acquired myelin by P21 in mutant nerves (Fig. 4F). Most of the unmyelinated large axons in Vps34SCKO nerves are in 1:1 relationships with Schwann cells by P21; the remainder are retained in bundles (Fig. 4F, H, K). The overall number of large axons in Vps34SCKO nerves is not decreased at P21 (Supp. Fig. S3C). Between P7 and P21 in Vps34SCKO nerves, the percentage of axons myelinated does not change significantly (Fig. 4F). However, the percentage of large axons that are aberrantly retained in bundles decreases, indicating that significant axonal sorting occurred after P7 in Vps34SCKO mice (Fig. 4F). Additional examinations of Vps34SCKO nerves at P21 revealed that axon bundles remain poorly invested with Schwann cell processes (Fig. 4H, J).
Hypomyelination in Vps34SCKO nerves could result from delayed or arrested initial myelination, or alternatively, from demyelination/remyelination. Redundant basal laminae, indicative of the pathological retraction of Schwann cell processes from axons, were observed in mutant nerves at P21 (Fig. 4L, M), and were more prevalent at P56 (not shown). Basal lamina proliferation is a feature of congenital hypomyelinating neuropathies, which typically also show fewer “onion bulb” Schwann cell processes than neuropathies characterized by demyelination/remyelination (Wrabetz et al., 2004). We did not observe onion bulbs in the nerves of Vps34SCKO mice at P21, P56, or 9 months (not shown). Moreover, myelin debris was very rare in Vps34SCKO nerves (Supp. Fig. S4), suggesting that the observed hypomyelination is not a consequence of demyelination. A modest number of particularly thin myelin sheaths, suggesting either delayed initial myelination or remyelination, were observed in mutant nerves at P21, (Fig. 4N). In aggregate, the data lead us to conclude that the unmyelinated and hypomyelinated axons in mutant nerves at P21 and P56 are more likely the result of failed or weak initial myelination, rather than demyelination followed by remyelination.
Changes to axons, including axonal degeneration, are a feature of some forms demyelinating CMT caused by mutations in myelin proteins (Martini, 2001; Watson et al., 1994). Likewise, genetic mutations in mice that cause hypomyelination or other Schwann cell abnormalities trigger decreased neurofilament phosphorylation, altered stoichiometry of axonal neurofilament proteins, impaired axonal transport, and axonal degeneration (de Waegh et al., 1992; Martini, 2001; Yin et al., 1998). We therefore considered whether the absence of myelin (or hypomyelination) might lead to changes in the axons of Vps34SCKO mice. Although normal at P3/P7, NF-L levels were significantly lower in Vps34SCKO nerves at P21 and P56 (Fig. 4O). The levels of total NF-H, as well as those of phosphorylated NF-H, detected using the SMI-34 antibody, were reduced to a similar extent in mutant nerves (Fig. 4P). We also employed the SMI-32 antibody, which reports the abnormal dephosphorylation of NF-H at a distinct epitope. The level of dephosphorylated NF-H detected in mutant nerves was not altered (Fig. 4P). However, when one considers the reduction in total NF-H protein in mutant nerves, our data suggest that NF-H is hypophosphorylated on SMI-32 epitopes. Thus, the absence of Vps34 in Schwann cells triggers changes in axons, presumably due to the absence of myelin or other Schwann cell abnormalities. Despite the observed changes in neurofilaments, our morphological studies indicate no loss of large axons at these times points (Supp. Fig. S3C). A comprehensive analysis of axon diameter in P21 nerves showed a trend toward decreased axon caliber in mutants, suggesting that hypomyelination in Vps34SCKO nerves has limited the radial growth of axons (Supp. Fig. S5). The decreased levels of neurofilament proteins in Vps34SCKO nerves at P21 and P56 likely result from reduced axon caliber, a known consequence of demyelination (Friede and Samorajski, 1970; Hoffman et al., 1984; Windebank et al., 1985).
Hyperproliferation of Vps34-deficient Schwann cells
Appropriately regulated Schwann cell proliferation and differentiation are critical for myelination (Jessen et al., 2015). Given the stalled myelination in Vps34SCKO nerves, we examined the proliferation of Schwann cells using Ki67 immunofluorescence (Fig. 5). The percentage of proliferating Schwann cells in mutant nerves was not significantly different from that of controls at P7, but was significantly elevated by P14 (Fig. 5A–C). Consistent with excessive Schwann cell proliferation and growth after P7 in Vps34SCKO nerves, ribosomal protein S6, and its phosphorylated form (pS6), were not significantly altered at P7, but were dramatically upregulated by P21 (Fig. 5D).
Figure 5.
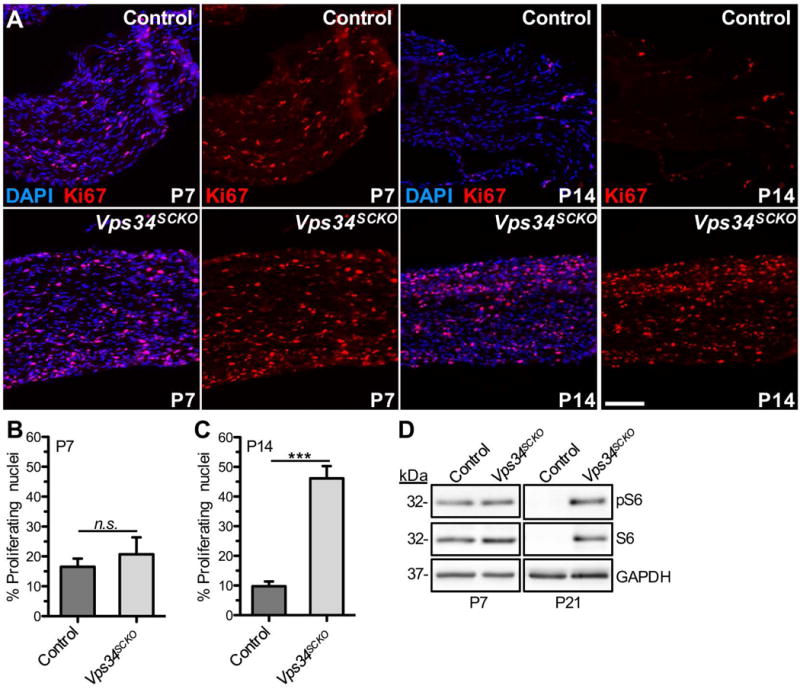
Vps34-deficient Schwann cells are hyperproliferative. (A) Longitudinal sections of sciatic nerves from control (upper) and Vps34SCKO (lower) mice were examined by immunofluorescence with an antibody for Ki67 (red) and DAPI (blue) staining at the indicated ages. (B) At P7, the percentage of proliferating (Ki67+) nuclei was not significantly different in control and Vps34SCKO nerves (p = 0.5372; n = 3 nerves for each genotype). (C) However, by P14, significant hyperproliferation of Schwann cells is evident in Vps34SCKO nerves (n = 3 and 4 for control and Vps34SCKO mice, respectively; n.s., not significant; ***p < 0.001; mean ± SEM). Scale bar: 100 μm. (D) Upregulation of ribosomal protein S6 in the nerves of Vps34SCKO mice. Phospho-S6 (Ser235/236) and total S6 protein levels in sciatic nerves were compared by immunoblotting. At P7, the levels of phospho-S6 and total S6 were not significantly different in the nerves of control and Vps34SCKO mice. Total S6 protein (arbitrary units); p = 0.1806; phospho-S6 protein (arbitrary units); p = 0.4390; n = 4 and 3 lysates for control and Vps34SCKO, respectively. However, by P21, both total S6 and phospho-S6 protein were significantly increased in Vps34SCKO nerves compared to controls (n = 2 and 3 lysates for control and Vps34SCKO, respectively).
Transcriptional regulation of myelination in Schwann cells lacking Vps34
To assess the capacity of Vps34−/− Schwann cells for differentiation to the myelinating phenotype, we examined the expression of Sox10, Oct6, and Krox20/Egr2, three transcription factors required for myelination. Sox10 is a key regulator of the differentiation of pluripotent neural crest cells into peripheral glia; mice lacking Sox10 fail to form Schwann cells (Kuhlbrodt et al., 1998). Oct6 is specifically expressed by promyelinating Schwann cells, and is down-regulated as myelination proceeds (Arroyo et al., 1998). Together with Oct6, Sox10 regulates progression of immature Schwann cells through the promyelinating stage, and into the myelinating stage, by inducing the expression of Krox20/Egr2 (Svaren and Meijer, 2008). Sox10 expression is maintained in myelinating Schwann cells; immature Schwann cells express Sox10, but not Krox20 (Svaren and Meijer, 2008). Schwann cells lacking Krox20 do not express late myelin genes, such as P0 and MBP, and fail to form myelin altogether (Le et al., 2005; Topilko et al., 1994). At P21, the number of Krox20+ nuclei was not significantly different in Vps34SCKO and control nerves (Fig. 6A, B), suggesting that mutant Schwann cells transition to the myelinating phenotype efficiently. However, Vps34SCKO nerves showed a striking, 10-fold increase in Sox10+ cells lacking Krox20 expression (Fig. 6A, C). Moreover, the level of Oct6, which is normally significantly down-regulated by P16 (Jaegle et al., 2003), was dramatically elevated in Vps34SCKO nerves at P21 (Fig. 6D). Thus, while Vps34−/− Schwann cells are capable of differentiation to the promyelinating phenotype and the initiation of myelination, these cells are likely unable to fully elaborate or maintain a myelin sheath. The persistent expression of Oct6 in Vps34SCKO nerves indicates that many Schwann cells lacking Vps34 are arrested at (or have reverted to) the promyelinating stage.
Figure 6.
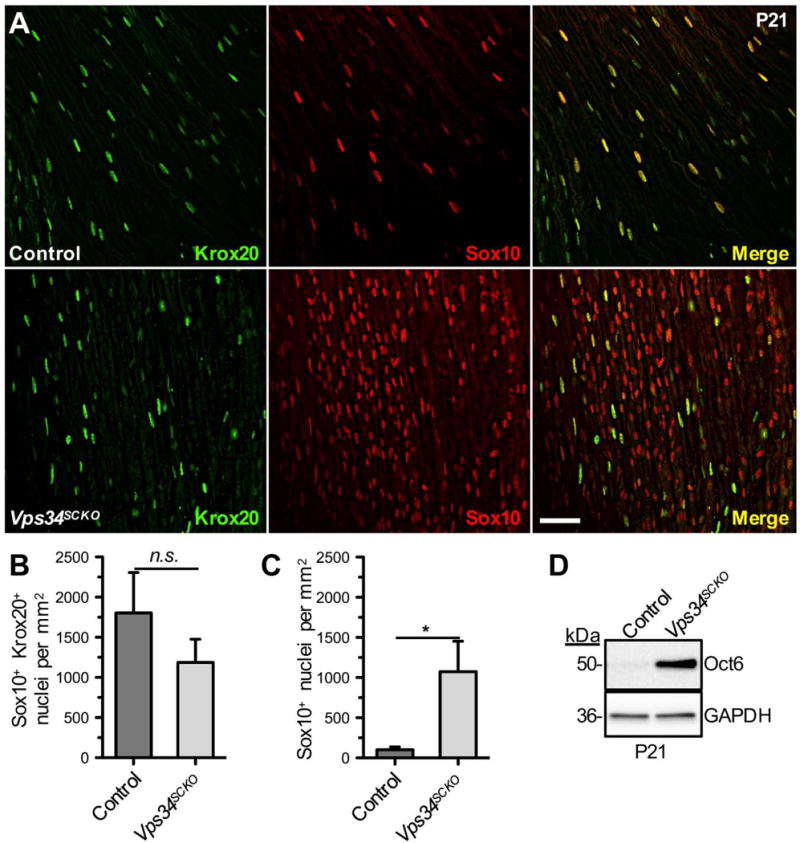
Loss of Vps34 does not preclude myelinating Schwann cell differentiation, but leads to accumulation of promyelinating Schwann cells. (A) Longitudinal sections of sciatic nerves of P21 control (upper) and Vps34SCKO (lower) mice were examined by immunofluorescence for Krox20 (green) and Sox10 (red). Scale bar: 40 μm. (B) The number of (Sox10+ Krox20+) nuclei/mm2 is not significantly different between control and Vps34SCKO nerves (p = 0.29). (C) Significantly increased abundance of immature or promyelinating (Sox10+) Schwann cells in Vsp34SCKO nerves at P21 (*p = 0.0335; n = 7–8 whole nerve sections per genotype, 2 animals per genotype; mean ± SEM). (D) The promyelinating transcription factor Oct6 is dramatically upregulated in Vps34SCKO sciatic nerves nerves at P21 (representative immunoblot data from one of three biological replicates are shown).
In summary, Vps34−/− Schwann cells have an impaired capacity for myelination by P7. After failing to myelinate a large axon, Vps34−/− Schwann cells likely withdraw from the axon, possibly de-differentiating and reducing Krox20 expression. Thereafter, mutant cells proliferate, likely as immature or promyelinating (Oct6+ Krox20−) cells, such that by P21 there is a dramatic excess of Krox20− Schwann cells in nerves (Fig. 6). Our findings are in accord with other work on Schwann cell proliferation. First, unmyelinated axons are believed to provide a mitogenic signal to Schwann cells (Atanasoski et al., 2002). Moreover, Schwann cell hyperproliferation is a prominent feature in the nerves of CMT patients, as well as in mouse models of demyelinating CMT1A (Atanasoski et al., 2002; Robertson et al., 2002; Suter et al., 1992).
Although we found no evidence of axonal degeneration in Vps34SCKO nerves, EM analyses revealed the presence of a modest number of monocytes/macrophages within the endoneurium at P21 (Supp. Fig. S4). Most macrophages appeared to be relatively quiescent, without significant upregulation of phagocytic structures or lysosomal membranes. Large, digestively active macrophages engaged in the degradation of myelin debris/ovoids (or axons) were not observed in Vps34SCKO nerves. Given that neither axonal degeneration, nor myelin debris were observed in Vps34SCKO nerves, we propose that other, less well-defined signals are beginning to recruit macrophages by P21. Hyperproliferative Vps34−/− Schwann cells (deprived of axonal contact) may themselves attract macrophages, a notion consistent with other published work (Klein and Martini, 2016; Tofaris et al., 2002). Additionally, the apparent axonal stress observed in Vps34SCKO nerves (reduced axon caliber, abnormal neurofilament levels and phosphorylation) might also contribute to the recruitment of macrophages.
Autophagic abnormalities in Schwann cells lacking Vps34
Having concluded that Vps34 plays an important role in myelination and Schwann cell-axon interactions, we considered how potential disturbances in endosomal trafficking or autophagy - two processes requiring Vps34-generated PI3P (Backer, 2008) - might lead to these phenotypes. At an early step in macroautophagy, PI3P is generated by Vps34 on pre-autophagosomal membrane structures (Roberts and Ktistakis, 2013). On these membranes, PI3P brings about the recruitment of the Atg5-Atg12-Atg16 complex, which catalyzes the lipidation of LC3-I and its subsequent localization to the expanding autophagosomal membrane (Roberts and Ktistakis, 2013). Previous work has demonstrated that loss of Vps34 leads to a significant reduction in the formation of autophagosomes (Devereaux et al., 2013; Roberts and Ktistakis, 2013), but this area has not been investigated in Schwann cells. To evaluate autophagy in Vps34SCKO sciatic nerves, we examined the levels of the LC3 and p62/SQSTM1 proteins. When autophagy is activated, LC3-I is coupled to phosphatidylethanolamine, yielding the membrane-associated LC3-II isoform, which is incorporated into growing autophagosomes, and is proportional to the number of autophagosomes (Klionsky et al., 2012). The levels of LC3-I/II were significantly increased in Vps34SCKO nerves beginning at P7 (Fig. 7B–D). P62 is a cargo receptor which promotes the degradation of ubiquitinated proteins by the autophagosome (Klionsky et al., 2012). We observed elevated levels of p62 in the nerves of Vps34SCKO mice beginning at P7 (Fig. 7B–D).
Figure 7.
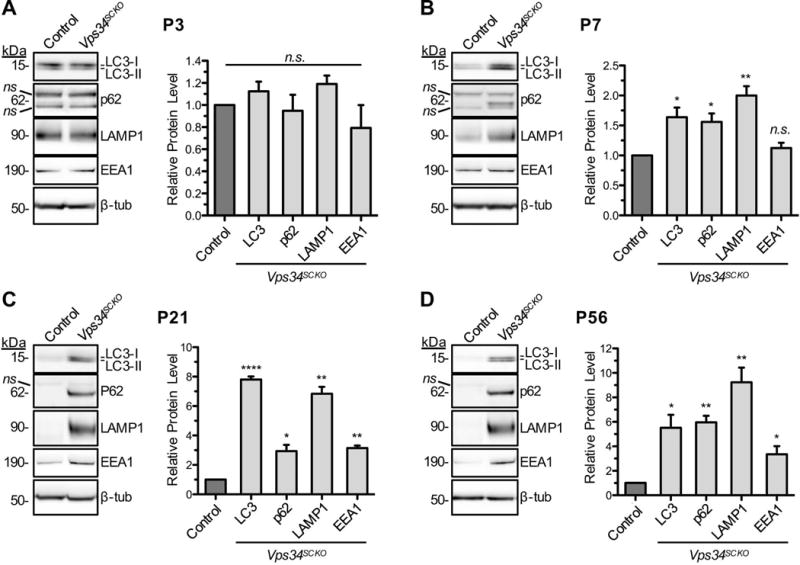
Upregulation of endosomal-lysosomal membranes and impaired autophagy in Vps34SCKO nerves. The levels of LC3-I and -II, p62, Lamp1, and EEA1 in sciatic nerve extracts were examined by immunoblotting at P3, P7, P21 and P56 (A–D). The abundance of each protein relative to that of the control is presented graphically. (A) At P3, the levels of the examined proteins were not significantly altered in mutant nerves. (B) By P7, there is a significant increase in LC3-I/II, p62, and Lamp1 levels in Vps34SCKO nerve extracts (EEA1: p = 0.27). (C & D) At P21 and P56, significant increases in the levels of LC3-I/II, p62, Lamp1, and EEA1 protein were observed in Vps34SCKO nerves. At each age, the average control value for a given protein was normalized to 1 (n.s., not significant; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; mean ± SEM). For all ages, n = 3 independent extracts per genotype. Each sciatic nerve extract (the biological replicate) was prepared by homogenizing the (pooled) nerves from three mice of the same genotype at P3 or P7, and one mouse at P21 and P56.
Our results indicate that autophagy is significantly impaired in Vps34−/− Schwann cells. In the absence of PI3P, autophagic isolation membranes do not mature into functional autophagosomes, despite LC3-II formation (Roberts and Ktistakis, 2013). Without functional autophagosomes, LC3 and p62 (substrates consumed by autophagy) accumulate in the nerves of Vps34SCKO mice. However, we believe that this defect is unlikely to be the primary cause of the abnormal axo-glial interactions and significant hypomyelination that we observe. Several recent studies have examined the role of autophagy in Schwann cells by selectively deleting Atg7, an essential autophagy protein which acts immediately downstream of Vps34 (Gomez-Sanchez et al., 2015; Jang et al., 2015). In contrast to what we observe in Vps34SCKO mice, radial sorting and myelination were found to be normal in Atg7SCKO mice (Gomez-Sanchez et al., 2015; Jang et al., 2015). Given that genetic inactivation of autophagy has no impact on Schwann cell myelination, the phenotypes we observe in Vps34SCKO nerves are unlikely to result from impaired autophagy.
Schwann cell Vps34 is essential for endosomal-lysosomal membrane homeostasis
To assess the importance of Vps34 and PI3P for trafficking in Schwann cells, we compared the levels of early and late endosomal/lysosomal proteins in the nerves of control and mutant mice. The Rab5 effector, early endosome antigen 1 (EEA1), binds to PI3P and facilitates homotypic fusion of early endosomes, a process essential for trafficking (Raiborg et al., 2013). Lysosomal-associated membrane protein 1 (LAMP1) is integral to late endosomes (LE) and lysosomes. At P3, EEA1 and LAMP1 levels were similar in control and Vps34SCKO sciatic nerves (Fig. 7A). In contrast, mutant nerves analyzed at P7 and thereafter contained significantly elevated levels of LAMP1 (Fig. 7B–D). EEA1 levels were also significantly elevated in Vps34SCKO nerves by P21 (Fig. 7C,D). Thus, the loss of Schwann cell Vps34 disturbs membrane traffic through the endosomal-lysosomal pathway. The accumulation of such membranes is likely triggered by the absence of PI3P, which leads to the slowing of specific membrane fusion and budding events along the pathway from initial endocytosis to the lysosome (Raiborg et al., 2013) (Fig. 9; discussed further below).
Figure 9.
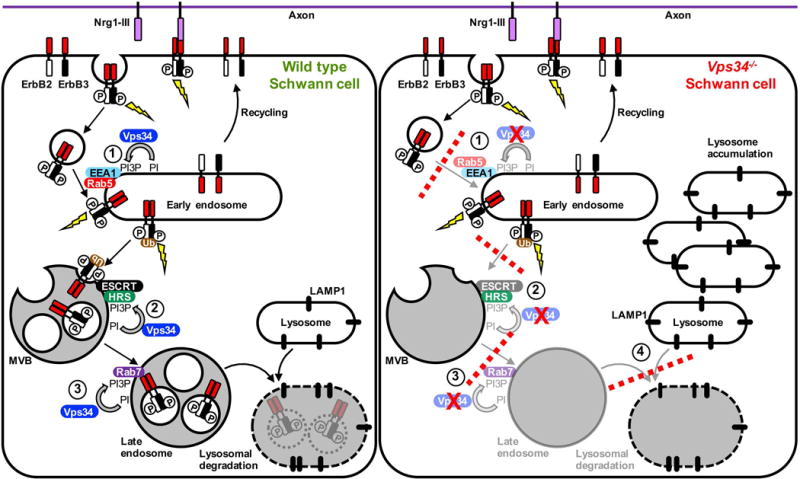
Proposed model for the role of Vps34 in endo-lysosomal trafficking of ErbB2/3 in Schwann cells. Binding of axonal Nrg1-III to ErbB3 induces receptor heterodimerization and stimulates ErbB2 kinase activity. ErbB2 phosphorylates both itself and ErbB3, creating phosphotyrosine-based docking sites for downstream signaling proteins (lightning bolt). Activation of ErbB2/3 triggers internalization by clathrin-mediated endocytosis. Fusion of ErbB2/3 containing vesicles with early endosomes requires PI3P, which is produced by Vps34, and requires EEA1 and Rab5 (Step 1) (Simonsen et al., 1998). Internalized ErbB2/3 heterodimers likely continue to signal from endosomal compartments, and can be recycled from endosomes to the cell membrane. On endosomes, ubiquitination (Ub) of ErbB2/3 targets the heterodimer for sorting into multivesicular bodies (MVBs). In a second Vps34-dependent step, PI3P recruits Hrs, which binds concomitantly to ubiquitin on ErbB2/3. Hrs recruits the ESCRT complex, which drives the budding event that generates the receptor-containing internal vesicles of MVBs (Step 2) (Fernandez-Borja et al., 1999). Once in MVBs, ErbB2/3 signaling is terminated (Lee et al., 2017). PI3P also mediates early-to-late endosome maturation through the recruitment of Rab7 (Poteryaev et al., 2010) (Step 3). ErbB2/3 receptors are degraded when late endosomes fuse with LAMP1-positive lysosomes (Lee et al., 2017). We propose that, in Vps34−/− Schwann cells the depletion of PI3P results in a slowing or blockade of trafficking Steps 1, 2, and 3 (red dashed lines). Such trafficking defects are predicted to reduce the fusion of ErbB2/3-bearing endocytic vesicles with early endosomes (Step 1), and prevent the sorting of phosphorylated ErbB2/3 into MVBs (Step 2). This proposed abnormal trafficking of ErbB2/3 is consistent with our observations of both enhanced phosphorylation of ErbB3 on Y1289 and altered posttranslational modification of ErbB2 in Vps34SCKO peripheral nerves. Vps34−/− Schwann cells are also likely deficient in Rab7-dependent late endosome maturation (Step 3). Finally, abnormal MVBs/late endosomes may fuse inefficiently with lysosomes (Step 4), resulting in lysosome accumulation.
Altered ErbB2/3 signaling in the absence of Schwann cell Vps34
As endosomal-lysosomal abnormalities are first observed in Vps34−/− Schwann cells at P7, we reasoned that the initial deficits in the trafficking of membrane proteins might be discernable at this time point. Further, given the stalled myelination and altered axo-glial interactions observed at P7, we considered whether the loss of Vps34 might alter the trafficking and signaling of Schwann cell membrane proteins critical to myelination, such as ErbB2/3 and integrins, which are known to traffic through endosomes (Feltri et al., 2016; Newbern and Birchmeier, 2010).
Given the critical role of the ErbB2-ErbB3 receptor tyrosine kinases (RTK) in myelination, we investigated whether the activation or trafficking of these kinases might be altered in Vps34SCKO mice. Phosphorylation of ErbB3 on Tyr1289, which correlates with neuregulin 1 (Nrg1) exposure and ErbB2 activation, was significantly increased in Vps34SCKO nerves (Fig. 8A–D). Consistently, the relative phosphorylation of ErbB2 on Tyr1248 was increased, albeit not to a statistically significant extent (Fig. 8A, Fig. S6D). A more notable change in ErbB2 was a decrease of about 50% in the overall abundance of the protein at P7, consistent with altered kinase turnover (Fig. 8A, E). Strikingly, both ErbB2 and ErbB3 possessed higher apparent molecular weights in mutant nerves, a feature which was subtle at P7, but readily apparent at P21 (Fig. 8A, B).
Collectively, our findings suggest that the loss of Vps34 has altered the trafficking of ErbB2/3 through endosomes. In Vps34−/− Schwann cells, PI3P-depleted endosomes likely allow the persistence of hyper-phosphorylated forms of ErbB2/3, which would normally be sorted into multivesicular bodies (MVBs) by the PI3P-dependent Hrs/Vps27-ESCRT pathway for degradation in the lysosome (Futter et al., 2001; Raiborg et al., 2013) (Fig. 9). Other posttranslational modifications, such as ubiquitination, might also be partially responsible for the increased apparent molecular weights of ErbB2/3 in Vps34SCKO nerves. The endosomal sorting of ErbB receptors into the MVB/LE pathway to the lysosome involves the tagging of receptors with ubiquitin (Sorkin and Goh, 2009). Hrs/Vps27, which binds simultaneously to PI3P and ubiquitin, recruits the ESCRT complex to drive sequestration of activated receptors in the lumen of the MVB, thus terminating kinase signaling and routing the receptor to the lysosome (Sorkin and Goh, 2009) (Fig. 9). Lastly, impaired endosomal recycling of ErbB receptors could also contribute to the abnormal posttranslational modifications we observe, perhaps by preventing the normal dephosphorylation of ErbB2/3 (Fig. 9) (Sorkin and Goh, 2009).
As a downstream target of ErbB2/3, the PI3K-Akt kinase pathway plays a key role in Schwann cell myelination (reviewed in (Norrmen and Suter, 2013; Salzer, 2015; Wood et al., 2013)). However, we found that Akt activation, as reported by phosphorylation of Thr308 and Ser473, was not significantly altered in Vps34SCKO nerves (Fig. 8F, Supp. Fig. S6). The activation of another downstream signaling pathway, Erk1/2, was also unaltered in Vps34SCKO nerves (Supp. Fig. S6). The c-Jun N-terminal kinase/stress-activated protein kinase (Jnk/Sapk) pathway is also activated downstream of ErbB2/3 in Schwann cells; Jnk activation has been shown to promote Schwann cell proliferation and migration (Parkinson et al., 2004; Yamauchi et al., 2008). We found that Jnk activation was significantly decreased in nerve extracts from Vps34SCKO mice (Fig. 8F, G and Supp. Fig. S6), suggesting that signaling from ErbB2/3 to Jnk is deficient in the absence of Vps34.
Given the crucial role of the integrin pathway in radial sorting (reviewed in (Feltri et al., 2016; Monk et al., 2015)), we considered whether deranged endosomal trafficking in Vps34−/− Schwann cells might lead to abnormal integrin trafficking and reduced focal adhesion kinase (FAK) activation. However, we found that FAK phosphorylation on Tyr397, which correlates with kinase activation, was similar in Vps34SCKO and control nerves (Supp. Fig. S6), suggesting that the FAK pathway is not affected by the loss of Vps34 and associated trafficking defects. In summary, our results suggest that the loss of Vps34 alters the trafficking of a subset of Schwann cell membrane proteins through endosomes.
DISCUSSION
Relatively little is known about how the endosomal trafficking of proteins and lipids regulates Schwann cell myelination. However, mutations in eight different endosomal proteins cause demyelinating CMT (Jerath and Shy, 2015). Although the specific functions of FRABIN/FGD4, SH3TC2, MTMR2, MTMR5, MTMR13, FIG4, SIMPLE/LITAF, and Dynamin 2 are still being determined, their association with human disease suggests that Schwann cell myelination is susceptible to subtle disturbances in endosomal-lysosomal trafficking (Jerath and Shy, 2015; Lee et al., 2017; Scherer and Wrabetz, 2008). To explore this enigmatic aspect of myelination, we examined the role of a key endosomal phosphoinositide, PI3P, by selectively deleting the PI 3-kinase Vps34 in mouse Schwann cells. Vps34 is believed to be responsible for generating about 65% of cellular PI3P (Devereaux et al., 2013). To our knowledge, our study is the first investigation of the role of Vps34 in glia. Vps34SCKO mice show abnormal Schwann cell-axon interactions and persistent hypomyelination in peripheral nerves. Immature or promyelinating Schwann cells proliferate dramatically in mutant nerves, likely because of their poor capacity for myelination. Endosomal-lysosomal membranes accumulate in Vps34−/− Schwann cells during initial myelination, indicating abnormal trafficking. In addition, Vps34SCKO nerves show altered post-translational modification of ErbB2/3, a tyrosine kinase heterodimer known to traffic through endosomes (Sorkin and Goh, 2009). Here, we discuss cellular mechanisms by which Vps34 deletion may cause the observed peripheral nerve pathology.
Endosomal trafficking of transmembrane signaling receptors
One mechanism by which endocytic trafficking may regulate myelination is by controlling which signaling and adhesion proteins Schwann cells express on their plasma membranes before and during myelination. This “tuning” of the extracellular face of the cell membrane will likely influence both the nature and amplitude of proliferation and differentiation signals that Schwann cells receive from axons, and from the basal lamina. Such signals will also likely influence the cells capacity for elaborating myelin membranes after committing to this phenotype (Salzer, 2015).
There is in vitro evidence for the essentiality of Schwann cell endocytosis for myelination (Sidiropoulos et al., 2012). Mutations in Dynamin 2 (Dnm2) cause dominant intermediate CMT (DI-CMT). Dynamins are membrane-associated GTPases which facilitate the scission of endocytic vesicles from the plasma membrane (Schink et al., 2016). The expression of DI-CMT-causing variants of Dnm2 in Schwann cell-dorsal root ganglion co-cultures was shown to impair endocytosis, and to suppress myelination (Sidiropoulos et al., 2012). Moreover, the endocytosis of several key membrane proteins (β1-integrin, ErbB2, and transferrin receptor) was reduced following the expression of disease-causing variants of Dnm2 in Schwann cell lines.
We explored how the in vivo signaling of surface receptors might be affected when Schwann cell endosomal trafficking is disturbed by Vps34 deletion. In mutant nerves, we observed hyperphosphorylation of ErbB3 on a tyrosine residue known to be phosphorylated by its binding partner, ErbB2, following exposure to axonal Nrg1 (Sorkin and Goh, 2009). ErbB2 levels and post-translational modifications were also altered in Vps34SCKO nerves. We propose that these alterations to ErbB2/3 result from abnormal trafficking of the heterodimer through endosomes.
The described alterations to ErbB2/3 were observed both during initial myelination (P7) and when myelination is normally complete (P21). Thus, in Vps34−/− Schwann cells, the ErbB2/3 heterodimer may have signaled abnormally during a period when its activity is critical for myelination (P7 and before) (Newbern and Birchmeier, 2010). Although the persistent hyperphosphorylation of ErbB3 formally implies that the heterodimer may be hyperactive, several other findings are consistent with decreased ErbB2/3 signaling. First, ErbB2 protein levels are decreased by about 50% at P7. Second, Jnk activation is suppressed in Vps34SCKO nerves. Moreover, while the activation of several other downstream targets of ErbB2/3, the Akt and Erk1/2 kinases, was not significantly changed, these pathways were clearly not hyperactive. Altered trafficking of ErbB2/3 might have prevented access to key downstream targets, thereby causing an overall decrease in ErbB2/3 biological activity. We speculate that suppressed ErbB2/3 signaling to downstream targets, such as Jnk, contributes to the hypomyelination observed in Vps34SCKO mice.
Given the importance of ErbB2/3 signaling to Schwann cell myelination, abnormal trafficking of these receptors has been proposed as a common pathogenic mechanism linking multiple subtypes of demyelinating CMT (Lee et al., 2017). An example of such dysregulation is found in SH3TC2, a likely endosomal scaffold protein which is mutated in CMT4C. SH3TC2 has been shown to bind to ErbB2 and alter ErbB2/3 signaling (Gouttenoire et al., 2013). We anticipate that Vps34SCKO mice will be useful for learning more about ErbB2/3 trafficking and activation in Schwann cells, as altered trafficking of the receptor complex in these mice appears to have trapped ErbB2/3 in specific states of post-translational modification.
We also noted altered Schwann cell-axon interactions and moderately delayed radial sorting in Vps34SCKO nerves, suggesting that endocytosis of receptors critical for interactions with the basal lamina might be altered. Accordingly, we postulated that FAK activation, which is triggered by integrin signaling, might be altered in Vps34−/− Schwann cells, particularly since endocytosis of the α6β1 integrin dimer potentates FAK activation in cancer cell lines (Alanko et al., 2015). However, FAK activation was not altered in Vps34SCKO nerves. Thus, Vps34 may not regulate the trafficking of integrins through Schwann cell endosomes. Alternatively, integrin trafficking might be disturbed, but in a manner that effects downstream targets other than FAK; we have yet to examine the impact of loss of Vps34 on ILK, Rac1 and p38 MAP kinase (Monk et al., 2015). Finally, additional adhesion molecules known to be involved in radial sorting, such as dystroglycan or Gpr126, could potentially be affected by the altered endosomal trafficking of Vps34−/− Schwann cells (Berti et al., 2011; Mogha et al., 2013). It is important to note that the delayed radial sorting we observe in Vps34SCKO mice is less significant than the radial sorting arrest caused by Schwann cell-specific deletion of ILK, FAK, β1-integrin, or Gpr126 (Feltri et al., 2002; Grove et al., 2007; Mogha et al., 2013; Pereira et al., 2009), suggesting that the signaling pathways controlling radial sorting may be subtly affected in our mutant mice. Further studies will be required to determine how disturbed endosomal trafficking in Vps34−/− Schwann cells leads to delayed radial sorting.
Loss of function mutations in more than 30 mouse genes have been shown to trigger radial sorting defects (Feltri et al., 2016). Mutations affecting different stages of radial sorting lead to distinct nerve pathologies. The hallmark of an early radial sorting delay/arrest is the presence of large bundles of axons of mixed caliber (Feltri et al., 2016). In contrast, small bundles with large axons retained at the periphery, indicates a late radial sorting arrest/delay. Polyaxonal myelination, in which Schwann cells aberrantly myelinate bundles of small axons, results from premature differentiation of Schwann cells (Feltri et al., 2016). Finally, radial sorting deficits can be specific to Remak bundles. In this case, defective differentiation of non-myelinating Schwann cells is thought to preclude membrane process insertion into bundles of small axons (Feltri et al., 2016). The sorting defect we observe in Vps34SCKO mice is best described as a moderate, late radial sorting delay.
In summary, our findings suggest that the loss of Vps34, and the associated depletion of PI3P, affects the trafficking of a subset of Schwann cell membrane proteins through endosomes. Abnormal post-translational modification of ErbB2/3, consistent with altered endosomal trafficking, was observed, and the activation of a downstream kinase, Jnk, was suppressed. In contrast, FAK phosphorylation, which is influenced by the trafficking of integrins through endosomes, was not discernibly altered in Vps34SCKO nerves. Our study with Vps34SCKO mice provides an in vivo complement to in vitro work on Dynamin 2, which demonstrated the importance of endocytosis for myelination (Sidiropoulos et al., 2012). Loss of Vps34 disrupts or delays several steps in the flow of cargo proteins through endosomes, all of which are downstream of Dynamin’s function in liberating endocytic pits from the plasma membrane (Fig. 9) (Schink et al., 2016). Continued study of Vps34SCKO mice will likely provide additional insights into the role of endosomal trafficking in myelination.
Bulk endosomal-lysosomal transport of myelin membranes
A second mechanism by which Vps34 deletion may inhibit myelination is by disturbing the overall balance of endocytosis and exocytosis of myelin membranes, which are enriched in specialized lipids and proteins. This aspect of myelin biogenesis has been largely studied in oligodendrocytes, and it remains to be determined if similar mechanisms operate in Schwann cells (Simons and Trotter, 2007). Proteolipid protein (PLP) is the most abundant protein in CNS myelin. In oligodendrocytes, some myelin membrane components, such as PLP, cholesterol, and galactosylceramide, are preassembled into small membrane domains as they traffic though the Golgi apparatus (Simons and Trotter, 2007). Such membranes are thought to be delivered to the growing myelin sheet by secretory vesicular trafficking. This vectorial delivery of partially assembled myelin membranes from the trans-Golgi to the plasma membrane is believed to occur in Schwann cells as well (Simons and Trotter, 2007). After delivery to the plasma membrane, small myelin domains are thought to be clustered together through the action of MBP (Simons and Trotter, 2007).
It has been proposed that regulating the balance of endocytosis and exocytosis provides an additional mode of myelin production (Simons and Trotter, 2007). In immature oligodendrocytes, plasma membrane-associated PLP is endocytosed and accumulates in late endosomes (Trajkovic et al., 2006). Exposure of these cells to axons suppresses endocytosis and triggers exocytosis of PLP-laden membranes from late endosomes into growing myelin membranes (Trajkovic et al., 2006).
It is unclear whether an analogous mechanism for endosomal regulation of myelination operates in Schwann cells. However, myelin protein zero (P0), the most abundant myelin protein in the PNS, has been reported to be enriched in late endosomes/lysosomes at the onset of myelination (P1) (Chen et al., 2012). The same study found that elevating intracellular Ca2+ triggers exocytosis of lysosomes in cultured Schwann cells. Lysosome exocytosis in Schwann cells was found to be regulated by the Rab27a GTPase; loss of Rab27a in mice inhibited remyelination in injured adult nerves (Chen et al., 2012). Thus, secretory lysosomes containing P0 may contribute to myelin delivery to the plasma membrane; this mechanism may be analogous to the regulated delivery of PLP from late endosomes in oligodendrocytes (Chen et al., 2012; Trajkovic et al., 2006).
The altered endosomal-lysosomal membranes that we observe in Vps34−/− Schwann cells suggest that the endocytosis of myelin components, such as lipids and P0, might be impaired. Moreover, the transport of endocytosed myelin components from early endosomes to late endosomes/lysosomes would very likely be suppressed in Vps34−/− Schwann cells, given the established role of PI3P in MVB formation and endosome maturation (Schink et al., 2016) (Fig. 9). Consistently, we observe accumulation of EEA1, an early endosomal protein that appears to accumulate when MVB formation is impaired (Fig. 9). If the transport of myelin membranes and proteins to late endosomal/lysosomal depots was inadequate in Vps34−/− Schwann cells, such components would be unavailable for exocytosis into growing myelin membranes. We speculate that such a mechanism could contribute to the hypomyelination we observe in Vps34SCKO nerves. However, further studies will be required to clarify the roles of Vps34 and PI3P in the trafficking of myelin membrane components.
In summary, we demonstrate that Vps34, a kinase that generates PI3P, is essential for proper myelination by Schwann cells. Depletion of PI3P leads to enlarged late endosomal/lysosomal vacuoles and suppressed trafficking in Schwann cells. Suppressed endosomal trafficking likely causes changes in the abundance and posttranslational modification of ErbB2/3, a signaling defect that may contribute to arrested myelination in Vps34SCKO nerves. The observed defects in endosomal-lysosomal membranes, when considered in the light of established roles of Vps34 in trafficking, suggest that Vps34 could also be key to controlling bulk myelin membrane production by maintaining a balance between endo- and exocytosis. Our study has yielded a novel disease-relevant model that will likely provide further insight into those forms of demyelinating CMT that involve endosome dysfunction.
Supplementary Material
Main points.
To myelinate axons, Schwann cells require the PI 3-kinase Vps34, a regulator of endosomal trafficking. The ErbB2-ErbB3 tyrosine kinase receptor, which traffics through endosomes and is key to myelination, is altered when Schwann cells lack Vps34.
Acknowledgments
Deconvolution microscopy and analysis were carried out in the OHSU Advanced Light Microscopy Core at The Jungers Center for Neurosciences Research. The authors wish to thank Aurelie Snyder and Stefanie Kaech Petrie of the Advanced Light Microscopy Core for expert advice. The DeltaVison CoreDV microscope was purchased with a Shared Instrumentation Grant from the National Institutes of Health-National Center for Research Resources (S10-RR023432 to Thomas Keller). The authors wish to thank Robert Kayton, Lisa Dirling Vecchiarelli, Mellissa Williams and Sue Aicher for assistance with electron microscopy, nerve preparation and expert advice. The electron microscope was purchased through a grant from the Murdock Charitable Trust (to Sue Aicher). The authors wish to thank Dr. Michael Wegner for the Sox10 antibody, and Dr. Thomas Sudhof for the lentiviral plasmids Lenti-EGFP-CreNLS and Lenti-EGFP-∆CreNLS. The 1D4B (LAMP-1) and E7 (β-tubulin) monoclonal antibodies, developed by Drs. J. Thomas August and Michael Klymkowsky, respectively, were obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institutes of Health-National Institute of Child Health & Human Development, and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242.
This work was supported by National Institutes of Health – National Institute of Neurological Disorders and Stroke grants NS057903 and NS086812 (to F.L.R.) and the OHSU Neuroscience Imaging Center P30 grant (NS061800; To Sue Aicher), as well as an Oregon Brain Institute Neurobiology of Disease Graduate Fellowship (to A.M.L.), and through the philanthropy of Frank and Julie Jungers.
Footnotes
Conflict of Interest Statement. None declared
References
- Alanko J, Mai A, Jacquemet G, Schauer K, Kaukonen R, Saari M, Goud B, Ivaska J. Integrin endosomal signalling suppresses anoikis. Nat Cell Biol. 2015;17:1412–1421. doi: 10.1038/ncb3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo EJ, Bermingham JR, Jr, Rosenfeld MG, Scherer SS. Promyelinating Schwann cells express Tst-1/SCIP/Oct-6. J Neurosci. 1998;18:7891–7902. doi: 10.1523/JNEUROSCI.18-19-07891.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasoski S, Scherer SS, Nave KA, Suter U. Proliferation of Schwann cells and regulation of cyclin D1 expression in an animal model of Charcot-Marie-Tooth disease type 1A. J Neurosci Res. 2002;67:443–449. doi: 10.1002/jnr.10133. [DOI] [PubMed] [Google Scholar]
- Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. The Biochemical journal. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. 2013;93:1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel W, Helmstadter M, Balica J, Hartleben B, Kiefer B, Hrnjic F, Schell C, Kretz O, Liu S, Geist F, et al. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. J Am Soc Nephrol. 2013;24:727–743. doi: 10.1681/ASN.2012070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Golden JP, Chen YJ, Yang K, Gross RW, Patti GJ, Milbrandt J. Metabolic regulator LKB1 is crucial for Schwann cell-mediated axon maintenance. Nat Neurosci. 2014;17:1351–1361. doi: 10.1038/nn.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti C, Bartesaghi L, Ghidinelli M, Zambroni D, Figlia G, Chen ZL, Quattrini A, Wrabetz L, Feltri ML. Non-redundant function of dystroglycan and beta1 integrins in radial sorting of axons. Development. 2011;138:4025–4037. doi: 10.1242/dev.065490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolino A, Muglia M, Conforti FL, LeGuern E, Salih MAM, Georgiou DM, Christodoulou K, Hausmanowa-Petrusewicz I, Mandich P, Schenone A, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nature Genetics. 2000;25:17–19. doi: 10.1038/75542. [DOI] [PubMed] [Google Scholar]
- Chen G, Zhang Z, Wei Z, Cheng Q, Li X, Li W, Duan S, Gu X. Lysosomal exocytosis in Schwann cells contributes to axon remyelination. Glia. 2012;60:295–305. doi: 10.1002/glia.21263. [DOI] [PubMed] [Google Scholar]
- Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, Szigeti K, Shy ME, Li J, Zhang X, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-d. [DOI] [PubMed] [Google Scholar]
- Devereaux K, Dall’Armi C, Alcazar-Roman A, Ogasawara Y, Zhou X, Wang F, Yamamoto A, De Camilli P, Di Paolo G. Regulation of mammalian autophagy by class II and III PI 3-kinases through PI3P synthesis. PloS one. 2013;8:e76405. doi: 10.1371/journal.pone.0076405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, D’Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Annals of the New York Academy of Sciences. 1999;883:116–123. [PubMed] [Google Scholar]
- Feltri ML, Graus Porta D, Previtali SC, Nodari A, Migliavacca B, Cassetti A, Littlewood-Evans A, Reichardt LF, Messing A, Quattrini A, et al. Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. The Journal of cell biology. 2002;156:199–209. doi: 10.1083/jcb.200109021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, Poitelon Y, Previtali SC. How Schwann Cells Sort Axons: New Concepts. Neuroscientist. 2016;22:252–265. doi: 10.1177/1073858415572361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Borja M, Wubbolts R, Calafat J, Janssen H, Divecha N, Dusseljee S, Neefjes J. Multivesicular body morphogenesis requires phosphatidyl-inositol 3-kinase activity. Curr Biol. 1999;9:55–58. doi: 10.1016/s0960-9822(99)80048-7. [DOI] [PubMed] [Google Scholar]
- Friede RL, Samorajski T. Axon caliber related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec. 1970;167:379–387. doi: 10.1002/ar.1091670402. [DOI] [PubMed] [Google Scholar]
- Futter CE, Collinson LM, Backer JM, Hopkins CR. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J Cell Biol. 2001;155:1251–1264. doi: 10.1083/jcb.200108152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Oltrogge JH, Kemper R, Heilmann I, Bormuth I, Wolfer S, Wichert SP, Mobius W, Liu X, Lappe-Siefke C, et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. Journal of Neuroscience. 2010;30:8953–8964. doi: 10.1523/JNEUROSCI.0219-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sanchez JA, Carty L, Iruarrizaga-Lejarreta M, Palomo-Irigoyen M, Varela-Rey M, Griffith M, Hantke J, Macias-Camara N, Azkargorta M, Aurrekoetxea I, et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol. 2015;210:153–168. doi: 10.1083/jcb.201503019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouttenoire EA, Lupo V, Calpena E, Bartesaghi L, Schupfer F, Medard JJ, Maurer F, Beckmann JS, Senderek J, Palau F, et al. Sh3tc2 deficiency affects neuregulin-1/ErbB signaling. Glia. 2013;61:1041–1051. doi: 10.1002/glia.22493. [DOI] [PubMed] [Google Scholar]
- Grove M, Komiyama NH, Nave KA, Grant SG, Sherman DL, Brophy PJ. FAK is required for axonal sorting by Schwann cells. J Cell Biol. 2007;176:277–282. doi: 10.1083/jcb.200609021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman PN, Griffin JW, Price DL. Control of axonal caliber by neurofilament transport. J Cell Biol. 1984;99:705–714. doi: 10.1083/jcb.99.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109:2003–2008. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaegle M, Ghazvini M, Mandemakers W, Piirsoo M, Driegen S, Levavasseur F, Raghoenath S, Grosveld F, Meijer D. The POU proteins Brn-2 and Oct-6 share important functions in Schwann cell development. Genes Dev. 2003;17:1380–1391. doi: 10.1101/gad.258203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang SY, Shin YK, Park SY, Park JY, Rha SH, Kim JK, Lee HJ, Park HT. Autophagy is involved in the reduction of myelinating Schwann cell cytoplasm during myelin maturation of the peripheral nerve. PLoS One. 2015;10:e0116624. doi: 10.1371/journal.pone.0116624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerath NU, Shy ME. Hereditary motor and sensory neuropathies: Understanding molecular pathogenesis could lead to future treatment strategies. Biochimica et Biophysica Acta. 2015;1852:667–678. doi: 10.1016/j.bbadis.2014.07.031. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R, Lloyd AC. Schwann Cells: Development and Role in Nerve Repair. Cold Spring Harb Perspect Biol. 2015;7:a020487. doi: 10.1101/cshperspect.a020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EE, Overmeyer JH, Gunning WT, Maltese WA. Gene silencing reveals a specific function of hVps34 phosphatidylinositol 3-kinase in late versus early endosomes. J Cell Sci. 2006;119:1219–1232. doi: 10.1242/jcs.02833. [DOI] [PubMed] [Google Scholar]
- Klein D, Martini R. Myelin and macrophages in the PNS: An intimate relationship in trauma and disease. Brain Res. 2016;1641:130–138. doi: 10.1016/j.brainres.2015.11.033. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlbrodt K, Herbarth B, Sock E, Hermans-Borgmeyer I, Wegner M. Sox10, a novel transcriptional modulator in glial cells. J Neurosci. 1998;18:237–250. doi: 10.1523/JNEUROSCI.18-01-00237.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Araki T, Schmidt RE, Milbrandt J. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci U S A. 2005;102:2596–2601. doi: 10.1073/pnas.0407836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Chin LS, Li L. Dysregulation of ErbB Receptor Trafficking and Signaling in Demyelinating Charcot-Marie-Tooth Disease. Mol Neurobiol. 2017;54:87–100. doi: 10.1007/s12035-015-9668-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R. The effect of myelinating Schwann cells on axons. Muscle Nerve. 2001;24:456–466. doi: 10.1002/mus.1027. [DOI] [PubMed] [Google Scholar]
- Mogha A, Benesh AE, Patra C, Engel FB, Schoneberg T, Liebscher I, Monk KR. Gpr126 functions in Schwann cells to control differentiation and myelination via G-protein activation. J Neurosci. 2013;33:17976–17985. doi: 10.1523/JNEUROSCI.1809-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk KR, Feltri ML, Taveggia C. New insights on Schwann cell development. Glia. 2015;63:1376–1393. doi: 10.1002/glia.22852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhro K, Park JM, Hong YB, Park JH, Nam SH, Yoon BR, Yoo JH, Koo H, Jung SC, Kim HL, et al. SET binding factor 1 (SBF1) mutation causes Charcot-Marie-Tooth disease type 4B3. Neurology. 2013;81:165–173. doi: 10.1212/WNL.0b013e31829a3421. [DOI] [PubMed] [Google Scholar]
- Nave KA, Werner HB. Myelination of the nervous system: mechanisms and functions. Annual review of cell and developmental biology. 2014;30:503–533. doi: 10.1146/annurev-cellbio-100913-013101. [DOI] [PubMed] [Google Scholar]
- Newbern J, Birchmeier C. Nrg1/ErbB signaling networks in Schwann cell development and myelination. Semin Cell Dev Biol. 2010;21:922–928. doi: 10.1016/j.semcdb.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng AA, Logan AM, Schmidt EJ, Robinson FL. The CMT4B disease-causing phosphatases Mtmr2 and Mtmr13 localize to the Schwann cell cytoplasm and endomembrane compartments, where they depend upon each other to achieve wild-type levels of protein expression. Human Molecular Genetics. 2013;22:1493–1506. doi: 10.1093/hmg/dds562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nodari A, Zambroni D, Quattrini A, Court FA, D’Urso A, Recchia A, Tybulewicz VL, Wrabetz L, Feltri ML. Beta1 integrin activates Rac1 in Schwann cells to generate radial lamellae during axonal sorting and myelination. The Journal of cell biology. 2007;177:1063–1075. doi: 10.1083/jcb.200610014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrmen C, Suter U. Akt/mTOR signalling in myelination. Biochem Soc Trans. 2013;41:944–950. doi: 10.1042/BST20130046. [DOI] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Droggiti A, Dickinson S, D’Antonio M, Mirsky R, Jessen KR. Krox-20 inhibits Jun-NH2-terminal kinase/c-Jun to control Schwann cell proliferation and death. J Cell Biol. 2004;164:385–394. doi: 10.1083/jcb.200307132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JA, Benninger Y, Baumann R, Goncalves AF, Ozcelik M, Thurnherr T, Tricaud N, Meijer D, Fassler R, Suter U, et al. Integrin-linked kinase is required for radial sorting of axons and Schwann cell remyelination in the peripheral nervous system. J Cell Biol. 2009;185:147–161. doi: 10.1083/jcb.200809008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poteryaev D, Datta S, Ackema K, Zerial M, Spang A. Identification of the switch in early-to-late endosome transition. Cell. 2010;141:497–508. doi: 10.1016/j.cell.2010.03.011. [DOI] [PubMed] [Google Scholar]
- Raiborg C, Schink KO, Stenmark H. Class III phosphatidylinositol 3-kinase and its catalytic product PtdIns3P in regulation of endocytic membrane traffic. The FEBS journal. 2013;280:2730–2742. doi: 10.1111/febs.12116. [DOI] [PubMed] [Google Scholar]
- Roberts R, Ktistakis NT. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays in biochemistry. 2013;55:17–27. doi: 10.1042/bse0550017. [DOI] [PubMed] [Google Scholar]
- Robertson AM, Perea J, McGuigan A, King RH, Muddle JR, Gabreels-Festen AA, Thomas PK, Huxley C. Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A. J Anat. 2002;200:377–390. doi: 10.1046/j.1469-7580.2002.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer JL. Schwann cell myelination. Cold Spring Harb Perspect Biol. 2015;7:a020529. doi: 10.1101/cshperspect.a020529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56:1578–1589. doi: 10.1002/glia.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nature methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schink KO, Tan KW, Stenmark H. Phosphoinositides in Control of Membrane Dynamics. Annu Rev Cell Dev Biol. 2016;32:143–171. doi: 10.1146/annurev-cellbio-111315-125349. [DOI] [PubMed] [Google Scholar]
- Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- Senderek J, Bergmann C, Weber S, Ketelsen UP, Schorle H, Rudnik-Schoneborn S, Buttner R, Buchheim E, Zerres K. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Human Molecular Genetics. 2003;12:349–356. doi: 10.1093/hmg/ddg030. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Krols M, Wu LM, Grove M, Nave KA, Gangloff YG, Brophy PJ. Arrest of myelination and reduced axon growth when Schwann cells lack mTOR. Journal of Neuroscience. 2012;32:1817–1825. doi: 10.1523/JNEUROSCI.4814-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidiropoulos PN, Miehe M, Bock T, Tinelli E, Oertli CI, Kuner R, Meijer D, Wollscheid B, Niemann A, Suter U. Dynamin 2 mutations in Charcot-Marie-Tooth neuropathy highlight the importance of clathrin-mediated endocytosis in myelination. Brain. 2012;135:1395–1411. doi: 10.1093/brain/aws061. [DOI] [PubMed] [Google Scholar]
- Simons M, Trotter J. Wrapping it up: the cell biology of myelination. Curr Opin Neurobiol. 2007;17:533–540. doi: 10.1016/j.conb.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Simonsen A, Lippe R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh BH, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–498. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009;315:683–696. doi: 10.1016/j.yexcr.2008.07.029. [DOI] [PubMed] [Google Scholar]
- Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, Billings-Gagliardi S, Sidman RL, Shooter EM. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992;356:241–244. doi: 10.1038/356241a0. [DOI] [PubMed] [Google Scholar]
- Svaren J, Meijer D. The molecular machinery of myelin gene transcription in Schwann cells. Glia. 2008;56:1541–1551. doi: 10.1002/glia.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, et al. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47:681–694. doi: 10.1016/j.neuron.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofaris GK, Patterson PH, Jessen KR, Mirsky R. Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J Neurosci. 2002;22:6696–6703. doi: 10.1523/JNEUROSCI.22-15-06696.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, Babinet C, Charnay P. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–799. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- Trajkovic K, Dhaunchak AS, Goncalves JT, Wenzel D, Schneider A, Bunt G, Nave KA, Simons M. Neuron to glia signaling triggers myelin membrane exocytosis from endosomal storage sites. J Cell Biol. 2006;172:937–948. doi: 10.1083/jcb.200509022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccari I, Dina G, Tronchere H, Kaufman E, Chicanne G, Cerri F, Wrabetz L, Payrastre B, Quattrini A, Weisman LS, et al. Genetic interaction between MTMR2 and FIG4 phospholipid phosphatases involved in Charcot-Marie-Tooth neuropathies. PLoS genetics. 2011;7:e1002319. doi: 10.1371/journal.pgen.1002319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson DF, Nachtman FN, Kuncl RW, Griffin JW. Altered neurofilament phosphorylation and beta tubulin isotypes in Charcot-Marie-Tooth disease type 1. Neurology. 1994;44:2383–2387. doi: 10.1212/wnl.44.12.2383. [DOI] [PubMed] [Google Scholar]
- Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci U S A. 2012;109:8670–8675. doi: 10.1073/pnas.1205305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windebank AJ, Wood P, Bunge RP, Dyck PJ. Myelination determines the caliber of dorsal root ganglion neurons in culture. J Neurosci. 1985;5:1563–1569. doi: 10.1523/JNEUROSCI.05-06-01563.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood TL, Bercury KK, Cifelli SE, Mursch LE, Min J, Dai J, Macklin WB. mTOR: a link from the extracellular milieu to transcriptional regulation of oligodendrocyte development. ASN Neuro. 2013;5:e00108. doi: 10.1042/AN20120092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrabetz L, Feltri ML, Kleopa KA, Scherer SS. Inherited Neuropathies: Clinical, Genetic, and Biological Features. In: Lazzarini RA, editor. Myelin Biology and Disorders. San Diego, CA: Elsevier Academic Press; 2004. pp. 905–951. [Google Scholar]
- Yamauchi J, Miyamoto Y, Chan JR, Tanoue A. ErbB2 directly activates the exchange factor Dock7 to promote Schwann cell migration. J Cell Biol. 2008;181:351–365. doi: 10.1083/jcb.200709033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P, Lee VM, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang L, Hasegawa H, Amin P, Han BX, Kaneko S, He Y, Wang F. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9424–9429. doi: 10.1073/pnas.0914725107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.