

Abstract
Nucleoside reverse transcriptase (RT) inhibitors (NRTIs) are the backbone of current antiretroviral treatments. However, the emergence of viral resistance against NRTIs is a major threat to their therapeutic effectiveness. In HIV-1, NRTI resistance-associated mutations either reduce RT-mediated incorporation of NRTI triphosphates (discrimination mechanism) or confer an ATP-mediated nucleotide excision activity that removes the inhibitor from the 3′ terminus of DNA primers, enabling further primer elongation (excision mechanism). In HIV-2, resistance to zidovudine (3′-azido-3′-deoxythymidine (AZT)) and other NRTIs is conferred by mutations affecting nucleotide discrimination. Mutations of the excision pathway such as M41L, D67N, K70R, or S215Y (known as thymidine-analogue resistance mutations (TAMs)) are rare in the virus from HIV-2–infected individuals. Here, we demonstrate that mutant M41L/D67N/K70R/S215Y HIV-2 RT lacks ATP-dependent excision activity, and recombinant virus containing this RT remains susceptible to AZT inhibition. Mutant HIV-2 RTs were tested for their ability to unblock and extend DNA primers terminated with AZT and other NRTIs, when complexed with RNA or DNA templates. Our results show that Met73 and, to a lesser extent, Ile75 suppress excision activity when TAMs are present in the HIV-2 RT. Interestingly, recombinant HIV-2 carrying a mutant D67N/K70R/M73K RT showed 10-fold decreased AZT susceptibility and increased rescue efficiency on AZT- or tenofovir-terminated primers, as compared with the double-mutant D67N/K70R. Molecular dynamics simulations reveal that Met73influences β3–β4 hairpin loop conformation, whereas its substitution affects hydrogen bond interactions at position 70, required for NRTI excision. Our work highlights critical HIV-2 RT residues impeding the development of excision-mediated NRTI resistance.
Keywords: reverse transcription, drug resistance, human immunodeficiency virus (HIV), nucleoside/nucleotide analogue, DNA replication, antiretroviral drugs, ATP-dependent excision
Introduction
Although the human immunodeficiency virus type 1 (HIV-1) is responsible for most of the global AIDS pandemic, it has been estimated that one to two million people are infected with HIV type 2 (HIV-2) worldwide (1). HIV-2 is endemic in West Africa, but it is also found in regions of Western Europe, India, Brazil, and South Africa. HIV-2 has a longer asymptomatic phase and shows slower progression to AIDS in comparison with HIV-1 (2). However, fewer treatment options are available for those infected with HIV-2. Non-nucleoside reverse transcriptase (RT)3 inhibitors, the fusion inhibitor enfuvirtide and several protease inhibitors approved for treatment of HIV-1 infection, are ineffective against HIV-2 (3, 4). In contrast, HIV-1 and HIV-2 show similar susceptibilities to nucleoside RT inhibitors (NRTIs) (5). Those drugs constitute the backbone of current therapies against infections caused by both types of HIV (6).
HIV RTs are heterodimeric enzymes that convert the single-stranded RNA genome into a double-stranded DNA that can be integrated in the host cell genome (7, 8). RTs have two catalytic activities as follows: (i) a DNA polymerase activity that uses RNA or DNA as templates, and (ii) a ribonuclease H (RNase H) activity that degrades RNA from RNA/DNA hybrids. NRTIs are converted to triphosphate derivatives inside the cell and are incorporated into the DNA by the polymerase activity of the RT. Further DNA synthesis is then blocked due to the absence of a 3′-OH in the ribose moiety of the NRTI (9). Although HIV-1 and HIV-2 share some NRTI resistance mutations, clinical studies have shown that NRTI resistance emergence is faster in HIV-2.
In HIV-1, NRTI resistance can be achieved through a discrimination mechanism (i.e. changes in RT's ability to incorporate nucleoside analogues versus natural dNTP substrates) or by an excision mechanism by which NRTI-resistant RTs are able to remove the inhibitor from the 3′-end of the primer, thereby allowing the extension of the unblocked DNA chain (6, 9–11). The excision reaction requires a pyrophosphate donor that in most cells is ATP. It is most efficient for primers terminated with thymidine analogues zidovudine (3′-azido-3′-deoxythymidine (AZT)), stavudine (2′,3′-didehydro-2′,3′-dideoxythymidine (d4T)), or tenofovir (12–14). Amino acid substitutions conferring resistance to NRTIs through the excision pathway are M41L, D67N, K70R, L210W, T215F or T215Y, and K219E or K219Q (15), which are widely known as thymidine analogue resistance mutations (TAMs). In patients receiving long-term therapy, TAMs appear in two clusters: TAM1 (M41L, L210W, and T215Y) and TAM2 (D67N, K70R, K219E/K219Q, and sometimes T215F) (16). M41L/T215Y in TAM1 or D67N/K70R in TAM2 are needed to achieve significant ATP-dependent excision activity (11, 17). Amino acid changes in the HIV-1 RT that antagonize the effects of TAMs in excision while increasing NRTI susceptibility have been found at the dNTP-binding site (e.g. K65R, L74V, V75I, and M184V), the non-nucleoside RT inhibitor-binding site (e.g. L100I or Y181C), or have been related to foscarnet resistance (e.g. W88G, E89K, L92I, and S156A) (6).
K65R, Q151M (sometimes accompanied by V111I), and M184V are the major mutations conferring resistance to NRTIs in HIV-2 (18, 19). The combination K65R/Q151M/M184V confers class-wide nucleoside analogue resistance (20). Amino acid substitutions in this complex affect residues of the dNTP-binding site in the DNA polymerase domain of the RT, and biochemical studies carried out with the HIV-1 enzyme showed that those changes interfered with the incorporation of nucleotide analogues (9). In contrast, TAMs are difficult to select in cell culture after HIV-2 passage in the presence of AZT (21) and are found with a very low prevalence in HIV-2 obtained from patients treated with AZT and other nucleoside analogues (22–24). HIV-1 and HIV-2 RTs have similar levels of DNA polymerase activity, but the HIV-2 enzyme incorporates AZT-triphosphate (AZTTP) less efficiently than HIV-1 RT and shows a much lower ability to excise the inhibitor in the presence of ATP (25). Moreover, substituting Tyr for Ser215 in the HIV-2 RT did not increase its ATP-dependent phosphorolytic activity but had a detrimental effect on polymerase activity (26). In those studies, the authors speculated on the possibility that selection of TAMs would be detrimental for HIV-2 RT function, and this would justify why the excision mechanism has a negligible influence in HIV-2 resistance to NRTIs. Furthermore, the presence of Ile75 in HIV-2 RT instead of Val as in HIV-1 RT could be responsible in part for the lack of the excision-based resistance mechanism, as suggested by the antagonistic effect of V75I when combined with TAMs in the context of an excision-proficient HIV-1 RT (27). The presence of Ile75 is expected to facilitate the selection of Q151M, because this substitution together with A62V, V75I, F77L, and F116Y forms the “Q151M complex” that in HIV-1 confers resistance to all approved NRTIs except tenofovir (28, 29).
In this work, we show that HIV-2ROD RT with the four classical TAMs (M41L/D67N/K70R/S215Y) has negligible ATP-dependent excision activity. Despite the high level of amino acid sequence similarity between HIV-1 and HIV-2 RTs, there are important differences in their primary structures that affect residues in the β3–β4 hairpin loop (i.e. at positions 63–73), and around Tyr215, in addition to residues elsewhere that in HIV-1 have an antagonistic effect on TAMs. The systematic replacement of HIV-2 RT residues by those found in the HIV-1 RT and the subsequent analysis of the ATP-dependent excision activity of the resulting RTs, using different NRTIs and template–primers, shows that Met73 in HIV-2 RT (instead of Lys as found in HIV-1 RT) acts as a molecular barrier that curbs the possibility of developing HIV-2 resistance through the accumulation of TAMs.
Results
HIV-1BH10 and HIV-2ROD RTs share 61% amino acid sequence identity. The crystal structures of AZT-resistant HIV-1 RT containing TAMs M41L, D67N, K70R, T215Y, and K219Q in its unliganded form, in complex with AZT-terminated DNA/DNA template–primers, and with double-stranded DNA and AZT adenosine dinucleoside tetraphosphate (AZTppppA) have been reported (30). In those studies, Tu et al. (30) showed that TAMs at positions 67, 70, and 219 contribute to the proper orientation of the pyrophosphate moiety during ribonucleotide-dependent nucleotide excision, whereas Tyr215 interacts with the adenine moiety in AZTppppA and defines the ATP-binding site. Based on the available crystal structures and previous work with mutant RTs having insertions and deletions in the β3–β4 hairpin loop (31, 32), we hypothesized that the major determinants of the excision activity in AZT-resistant RTs locate within residues 60–76 (β3–β4 hairpin loop and adjacent residues of β-strands 3 and 4), and around position 215. The amino acid sequences of HIV-1 and HIV-2 RTs show differences at those regions (Fig. 1a). In addition, HIV-1 and HIV-2 RTs differ at specific positions that were previously shown to modulate the excision reaction in the presence of TAMs. Thus, previous studies demonstrated that by themselves V75I, W88G, and Y181C decrease the ATP-dependent excision activity of HIV-1 RTs carrying TAMs (6). The wildtype (WT) HIV-2ROD RT contains Ile75, Thr88, and Ile181 instead of Val, Trp, and Tyr, as found in the HIV-1 RT. These differences could be also responsible for the suppression of the excision activity in HIV-2 RTs. Based on those observations, we obtained HIV-2ROD RT variants whose amino acid sequences are given in Fig. 1b. They included enzymes where the sequence of the β3–β4 hairpin loop of the HIV-1BH10 RT was introduced in the HIV-2ROD RT together with TAMs M41L and T215Y (e.g. 5M_STK), others that contained different substitutions in the 210–215 sequence (e.g. 5M_LRWL), or combinations of mutations, including potential antagonists of TAMs such as I75V, T88W, or I181Y. The enzymes were purified and tested for their ability to remove thymidine analogues and tenofovir, in rescue assays carried out with RNA/DNA and DNA/DNA template–primers.
Figure 1.

Amino acid sequence differences within residues 1–250 of HIV-1BH10 and HIV-2ROD RTs and mutant HIV-2ROD RTs analyzed in this study. a, sequence alignments showing differences between HIV-1 and HIV-2 RTs at positions associated with the excision pathway of resistance to thymidine analogues. Differences between both RTs are highlighted in red. Amino acids shown in blue were introduced in one or more HIV-2ROD mutant RTs generated in this study. b, HIV-2ROD RTs analyzed in this study and their abbreviated designations.
Identification of suppressors of the ATP-dependent unblocking activity of DNA primers terminated with AZT or tenofovir in the HIV-2 RT
The ability of HIV-2ROD RTs to rescue AZTMP-terminated primers in the presence of ATP (3.2 mm) was first assessed with the RNA/DNA template–primer 31Trna/21P (Fig. 2). Reactions were carried out in two steps. The RT and the RNA/DNA hybrid were incubated in the presence of inhibitor (AZTTP) to generate a blocked 22-nucleotide primer. Then, the addition of dNTPs and ATP (the pyrophosphate donor) facilitated unblocking and further extension of the primer to obtain a product of 31 nucleotides. In these assays, the HIV-1BH10 RT that contained TAMs M41L and T215Y (LY_HIV-1) showed relatively high levels of excision activity. In contrast, the WT HIV-2ROD RT and the quadruple mutant M41L/D67N/K70R/S215Y (4M RT) showed almost negligible activity in the presence of 3.2 mm ATP.
Figure 2.

ATP-mediated excision of AZTMP from RNA/DNA template–primers by WT and mutant RTs. Assays were carried out with 31/21-mer RNA–DNA complexes (sequences shown below the upper left panel). The inhibitor was first incorporated at position +1 (indicated with an asterisk) of the 21-nucleotide primer (lane P) to generate a 22-nucleotide product (lane B). Excision of the inhibitor and further primer extension in the presence of 3.2 mm ATP and a mixture of dNTPs led to the formation of a fully extended 31-nucleotide product. Aliquots were removed 2, 4, 6, 8, 10, 12, 15, and 20 min after the addition of ATP. Time courses of the excision reactions are shown in three panels, each one containing data from different sets of related RTs. Time courses obtained with WT HIV-2 RT and mutant 5M are included in all panels for comparison. All dNTPs in the assays were supplied at 200 μm, except for dATP whose concentration was 2 μm. Template–primer and active RT concentrations in these assays were 30 and 24 nm, respectively. Represented values (means ± S.D., error bars) were obtained from at least three independent experiments.
Although Asp67 and Lys70 are conserved in HIV-1 and HIV-2 RTs, there are differences at four residues of the β3–β4 hairpin loop region of both polymerases. By introducing the amino acids found at positions 68, 69, 73, and 75 in HIV-1BH10 RT in the sequence context of the 4M HIV-2 RT, we obtained enzymes with high ATP-dependent excision activity (Fig. 2). The substitution of Lys for Met73 in the HIV-2ROD RT had the largest impact on rescue activity among all tested polymerases. Thus, mutants 5M_STK and 5M_SK showed efficiencies that were almost 2-fold higher than those obtained with the reference HIV-1BH10 RT mutant M41L/T215Y, and well above the efficiencies of the WT HIV-2ROD RT. Other RTs showed lower ATP-dependent excision activities. However, substituting Val75 for Ile in the 4M RT produced a modest but significant increase in rescue efficiencies. The subsequent addition of K68S and/or N69T, several combinations of mutations in the vicinity of Tyr215, or substitutions with a potential antagonistic effect on TAMs (i.e. T88W and I181Y) had a minor effect on DNA primer rescue catalyzed by the 5M RT mutant enzyme.
Similar assays carried out with a DNA–DNA complex containing an AZTMP-terminated primer also revealed that the substitution of Lys for Met73 in the HIV-2ROD RT had a large impact in the rescue activity of the polymerase (Fig. S1). Mutant derivatives containing M73K (i.e. 5M_SK and 5M_STK) rescued between 30 and 35% of the AZTMP-terminated primers after incubating the template–primer and the RT for 20 min in the presence of ATP and dNTPs. In contrast, 5M, 5M_K68S, and 5M_ST RTs had lower excision activity (<14%) under the same conditions. Nevertheless, these three enzymes showed a modest increase in rescue activity in comparison with the 4M RT. This could be attributed to the substitution of Val for Ile75 that could facilitate excision by neutralizing the small but significant antagonistic effect of Ile75. As in the case of experiments carried out with the RNA/DNA hybrid, T88W and I181Y had a minor effect on DNA primer rescue when combined with I75V and other TAMs, although we observed a slight increase with the substitution I181Y. In contrast, none of the mutations located in the vicinity of Tyr215 seemed to affect the rescue activity of the 5M mutant, because all tested HIV-2 RT variants showed rescue activities below 14%, after a 20-min incubation in the presence of ATP and dNTPs.
In general, removal of AZTMP and further extension of the unblocked primer were more efficient with the RNA template than with the DNA template for all tested RTs. Analysis of the efficiencies of the rescue reactions with different RTs and the 31Trna–21P complex confirms the suppressor effect of Ile75, because mutant 5M shows higher rescue activity than 4M, although the addition of K68S and N69T produced a slight but non-significant increase in rescue efficiency. In any case, the effects of those substitutions were relatively small in comparison with the addition of M73K.
The ATP-dependent rescue activity was also tested with all mutants using tenofovir-terminated primers and the DNA/DNA hybrid D38T/25PGA (Fig. S2). The observed ranking of rescue efficiencies was similar to those obtained with AZT-terminated primers, supporting the notion that the inhibitor had a minor impact on the relative excision activity of the RTs. The amounts of rescued primer obtained with the WT HIV-2 RT and its mutant derivative 4M were very small. Again, the presence of the additional substitution I75V produced a significant increase in tenofovir excision and further extension of the unblocked primer. This effect was reduced if other β3–β4 hairpin substitutions such as K68S or N69T were added to the mutational complex. However, in those cases, addition of M73K rendered excision-proficient RTs with rescue efficiencies higher than those observed for the well-characterized HIV-1BH10 RT mutant M41L/T215Y (LY_HIV-1).
ATP-dependent excision activities of WT and mutant HIV-2 RTs
Assays described above have consistently shown that M73K and to a lesser extent I75V facilitate the rescue of NRTI-terminated primers by TAM-containing HIV-2 RTs. However, those assays combine excision with DNA primer extension. In Fig. 3, we show that I75V confers some ATP-dependent excision activity on primers terminated with AZT when added to the 4M complex, whereas the subsequent addition of M73K (in combination with K68S) results in a further enhancement of the excision activity. Interestingly, the effects of I75V were not observed in ATP-dependent excision reactions carried out with primers terminated with 2′,3′-didehydro-2′,3′-dideoxythymidine monophosphate (d4TMP). Mutants 5M, 5M_K68S, and 4M and the WT HIV-2 enzyme showed negligible activity (kobs < 1.6 × 10−3 min−1). However, addition of the M73K substitution to the 5M_K68S complex rendered an RT with significant excision activity and showed a kobs of 5.8 × 10−3 min−1. Taken together, the enzymatic assays reveal the strong effect of Met73 in the context of HIV-2ROD RTs bearing TAMs by suppressing NRTI excision.
Figure 3.

Kinetics of the ATP-dependent excision of AZTMP and d4TMP from DNA/DNA template–primer (D38/25PGA). Time courses of the excision reactions of AZTMP- and d4TMP-terminated primers (26-mer) annealed to their corresponding 38-nucleotide DNA templates (30 nm) were determined in the presence of 3.2 mm ATP (a and b, respectively). The excision reactions were catalyzed by WT and mutant RTs (40 nm active-site concentration). Aliquots were removed 2, 5, 10, 15, 20, 30, 45, 60, and 80 min after the addition of ATP. Represented values were obtained from at least three independent experiments.
M73K restores ATP-dependent rescue activity of HIV-2 RT bearing TAMs D67N and K70R
Unlike HIV-1 (11, 32), the double mutant D67N/K70R HIV-2 RT has very low or negligible rescue activity on primers terminated with AZT either in RNA/DNA or in DNA/DNA template–primer complexes (Fig. 4, a and b). However, adding the M73K substitution produces a large increase in rescue efficiency that was observed both with AZT-terminated (Fig. 4, a and b) and tenofovir-terminated primers (Fig. 4c). As expected, M73K alone had no effect on the unblocking and extension reactions and showed a phenotype similar to that obtained with the WT HIV-2 RT. The three RTs showed significant DNA polymerization activity in single-nucleotide incorporation assays, carried out with template–primer D38/25PGA, under steady-state conditions. In those reactions, the turnover rates (kcat) for WT HIV-2 RT and mutants D67N/K70R and D67N/K70R/M73K were 7.19 ± 0.82, 3.87 ± 0.43, and 3.42 ± 0.19 min−1, respectively.
Figure 4.

ATP-mediated excision of nucleoside analogues from RNA/DNA and DNA/DNA template–primers by WT HIV-2 RT and mutants M73K, D67N/K70R, and D67N/K70R/M73K. Assays were carried out with 31/21-mer RNA–DNA complexes (a) or 38/25-mer DNA–DNA complexes (b and c) (sequences shown above excision reaction time courses). The inhibitors (AZT in a and b, and tenofovir in c) were first incorporated at position +1 (indicated by asterisks) to generate 22- or 26-nucleotide products. Excision of the inhibitor and further primer extension in the presence of 3.2 mm ATP and a mixture of dNTPs led to the formation of fully extended primers of 31 or 38 nucleotides. Aliquots were removed at the appropriate times after ATP addition. Time courses of the excision reactions are shown below. The dNTPs in the assays carried out with RNA–DNA complexes were supplied at 200 μm, except for dATP whose concentration was 2 μm. In assays using DNA–DNA complexes, the dNTP concentration was kept at 100 μm, except for dATP (AZT-terminated primers) or dTTP (tenofovir-terminated primers) which was supplied at 1 μm. Template–primer and active RT concentrations in these assays were 30 and 24 nm, respectively. Represented values (means ± S.D. error bars) were obtained from at least three independent experiments.
The only available crystal structure of HIV-2 RT (Protein Data Bank (PDB) code 1MU2) has an overall folding similar to that of the HIV-1 RT (33) but has no ligands and is incomplete, lacking several key loops. Therefore, for modeling purposes, we used the crystal structure of a ternary complex of HIV-1 RT, double-stranded DNA and dTTP (PDB code 1RTD). Structural models of catalytically competent WT and mutant HIV-1 RTs were obtained after refinement by molecular dynamics. In HIV-1 RT, residue 73 is Lys instead of Met as occurs in the HIV-2 enzyme (Fig. 1). The structural models of mutant HIV-1 RTs D67N/K70R and D67N/K70R/K73M were compared with the one previously obtained for the catalytically competent WT HIV-1 RT (34).
As shown in Fig. S3A, root-mean-square deviations (r.m.s.d.) corresponding to the backbone Cα atoms of the modeled ternary complexes remained below 2.4 Å. Superimpositions of the simulated structures were consistent with the results of the r.m.s.d. analysis, although significant differences were observed between the conformation of the β3–β4 hairpin loop of D67N/K70R/K73M and the equivalent structures of the WT HIV-1 RT and mutant D67N/K70R (Fig. S4A). Mutations did not affect critical interactions required to maintain the catalytic attack distance between the 3′-OH of the primer and the α-phosphorus (Fig. S3B). At the end of the simulation, interactions in the catalytic site of the ternary complex containing two Mg2+ ions were similar for the three studied RTs (Fig. S4).
Molecular dynamics simulations showed that the position of Arg70 was largely affected by substitutions at the β3–β4 hairpin loop. Interestingly, in the double mutant D67N/K70R, distances between the ζ-carbon of Arg70 and the γ-phosphorus of the dNTP remained stable and below 4.5 Å, and they were compatible with the establishment of hydrogen-bonding interactions between the amido groups of Arg70 and the hydroxyl substituents of the dNTP γ-phosphate (32). These interactions were not predicted for RTs devoid of excision activity, such as the WT enzyme or a mutant RT having a single amino acid deletion at position 69 (32). Interestingly, in the D67N/K70R/K73M HIV-1 RT model, the side chain of Arg70 moved away from the dNTP-binding site (Fig. 5) leading to the loss of the hydrogen bonding network involved in the excision reaction. The side chain of Lys73 points toward the incoming nucleotide in the structure of the double mutant. However, its substitution by Met73 produces an internal rearrangement in the fingers subdomain of the RT that results from the establishment of hydrophobic interactions between the side chains of Val60 and Met73. These interactions affect the positioning of β3 and β4 strands and influence folding at the tip of the hairpin loop.
Figure 5.
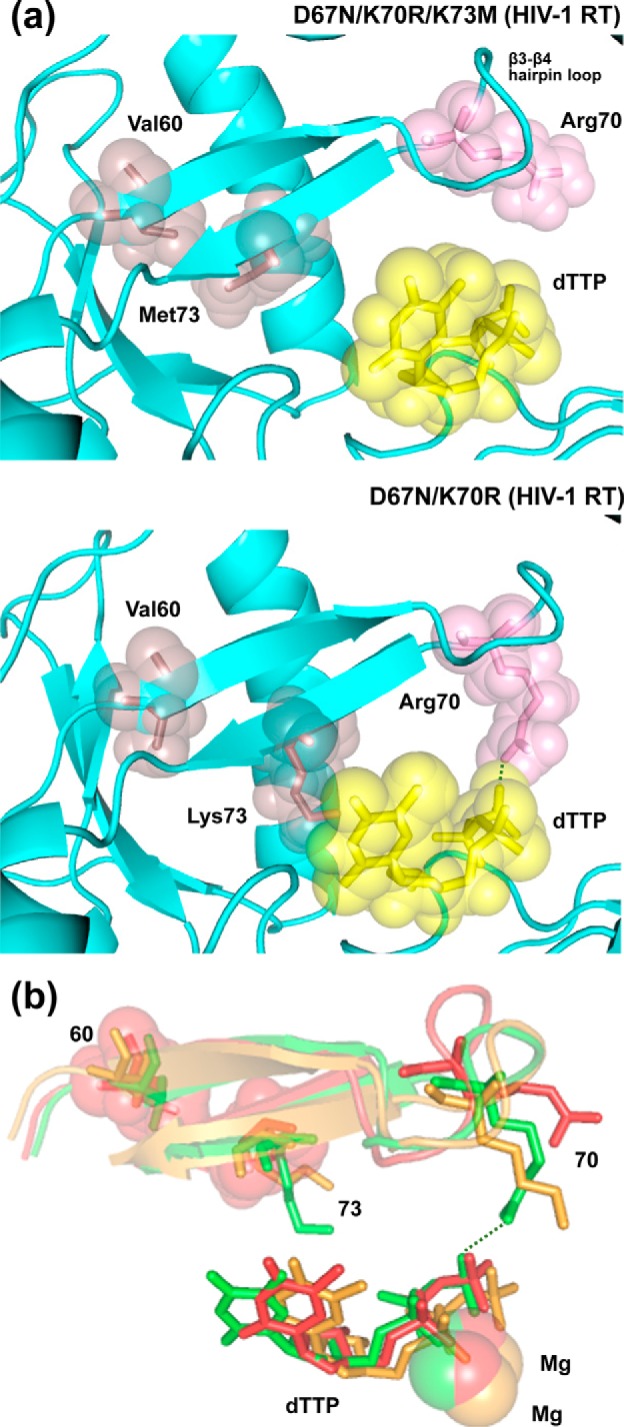
Comparison of the structural models of WT and mutant HIV-1 RTs D67N/K70R/K73M and D67N/K70R complexed with double-stranded DNA and an incoming dNTP. a, ribbon representation of the polypeptide backbone around the nucleotide-binding site of the RT. The side chains of Val60, Arg70, and Met73/Lys73 and the incoming dNTP are shown with stick and sphere representations. In the double-mutant, molecular dynamics simulations predict the formation of a hydrogen bond between amido and hydroxyl groups of the RT (Arg70) and the incoming dNTP, respectively (dotted line). The distance between the relevant atoms involved is 2.4 Å in this structure, but in the D67N/K70R/K73M RT model, this value increases up to 6.2 Å. b, view of the β3–β4 hairpin loop, the incoming dNTP, and the two Mg2+ ions, based on the structural alignment of the three modeled RTs. The catalytically competent WT HIV-1 RT is shown in orange, the D67N/K70R RT in green, and the D67N/K70R/K73M RT in red. Side chains at positions 60 (Val in all cases), 70 (Lys/Arg), and 73 (Met/Lys) are represented with sticks. Spheres are used to indicate the location of Val60 and Met73 in the structure of the triple mutant. The hydrogen bond between Arg70 and the incoming dNTP, predicted for the D67N/K70R RT, is shown with a dotted line.
Further support of the role of Lys73 in nucleotide excision was obtained by comparing the rescue efficiencies of HIV-1 RT mutants D67N/K70R and D67N/K70R/K73M. As shown in Fig. 6, the triple mutant had almost negligible activity, whereas D67N/K70R rescued about 16% of the AZTMP-terminated primers after incubating the blocked D38/25PGA hybrid for 20 min in the presence of 3.2 mm ATP. These rescue efficiencies were observed for the mutant RT D67N/K70R at ATP concentrations as low as 1 mm.
Figure 6.
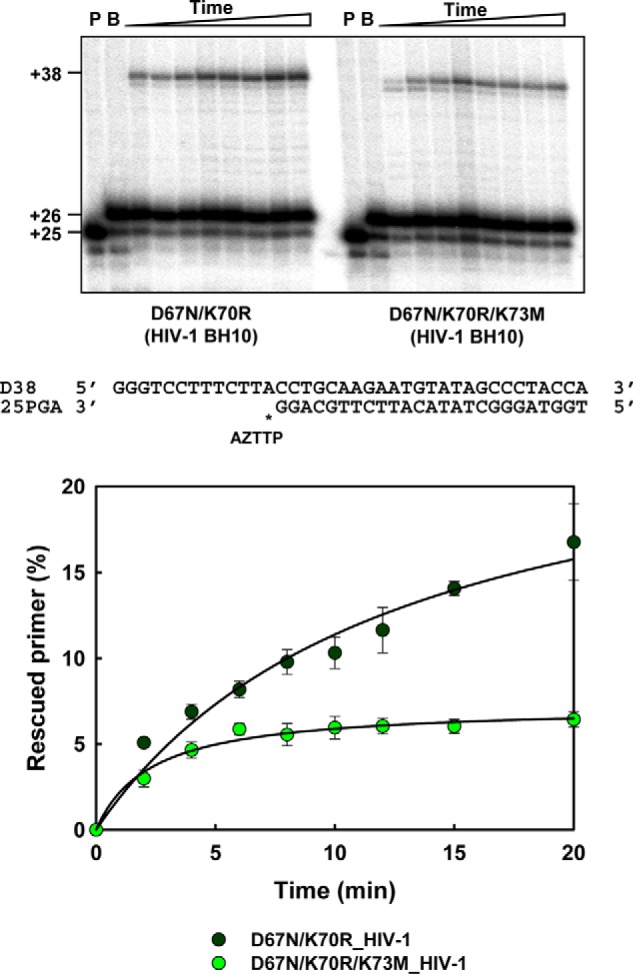
ATP-mediated excision of AZTMP from DNA/DNA template–primers by HIV-1 RT mutants D67N/K70R and D67N/K70R/K73M. Assays were carried out with 38/25-mer DNA–DNA complexes (sequences shown below the upper panel). The inhibitor was first incorporated at position +1 (indicated with an asterisk) of the 25-nucleotide primer (lane P) to generate a 26-nucleotide product (lane B). Excision of the inhibitor and further primer extension in the presence of 3.2 mm ATP and a mixture of dNTPs led to the formation of a fully extended 38-nucleotide product. Aliquots were removed 2, 4, 6, 8, 10, 12, 15, and 20 min after the addition of ATP. Time courses of the excision reactions are shown below. All dNTPs in the assays were supplied at 100 μm, except for dATP whose concentration was 1 μm. Template–primer and active RT concentrations in these assays were 30 and 24 nm, respectively. Represented values (means ± S.D. error bars) were obtained from at least three independent experiments.
Antiretroviral drug susceptibility of recombinant HIV-2
Susceptibility to AZT was determined in MT-4 cells for selected HIV-2 mutants to test whether the results obtained in rescue and excision assays correlated with phenotypic drug susceptibility of recombinant HIV-2. As shown in Table 1, the presence of the four classical TAMs (M41L/D67N/K70R/S215Y) in HIV-2 did not produce an AZT-resistant virus (i.e. compare 4M versus WT HIV-2ROD). However, substituting Val75 for Ile (5M mutant) decreased AZT susceptibility almost four times. Other substitutions in the β3–β4 hairpin loop had no significant effect on thymidine analogue resistance, although we were unable to show the effects of M73K when multiple amino acid substitutions were introduced in the HIV-2 RT. The recombinant HIV-2 with RTs 5M_SK and 5M_STK was not viable and was unable to replicate in MT-4 cells. Nevertheless, a 10-fold increase in the IC50 for AZT was observed for the triple mutant D67N/K70R/M73K HIV-2 RT when compared with the double mutant D67N/K70R. These data are consistent with the biochemical assays showing that amino acid substitutions M73K and to a lesser extent I75V confer resistance through increased primer-unblocking activity on AZT-terminated primers.
Table 1.
Susceptibility of HIV constructs to AZT and darunavir
| Virus | RTa | IC50 (nm)b |
|
|---|---|---|---|
| AZT | Darunavir | ||
| HIV-2ROD | WT | 13.0 ± 3.1 | 3.5 ± 0.5 |
| 4M (M41L/D67N/K70R/S215Y) | 19.3 ± 9.6 (1.5) | 4.3 ± 1.7 (1.2) | |
| 5M (M41L/D67N/K70R/I75V/S215Y) | 74.6 ± 24.6 (5.8)* | 4.3 ± 1.4 (1.2) | |
| 5M_K68S (M41L/D67N/K68S/K70R/I75V/S215Y) | 24.1 ± 8.1 (1.9) | 5.5 ± 0.7 (1.6) | |
| 5M_ST (M41L/D67N/K68S/N69T/K70R/I75V/S215Y) | 52.0 ± 5.2 (4.0)* | 5.4 ± 0.4 (1.6) | |
| 5M_SK (M41L/D67N/K68S/K70R/M73K/I75V/S215Y) | NDc | ND | |
| 5M_STK (M41L/D67N/K68S/N69T/K70R/M73K/I75V/S215Y) | ND | ND | |
| D67N/K70R | 14.4 ± 3.7 (1.1) | 3.6 ± 1.9 (1.0) | |
| D67N/K70R/M73K | 135.7 ± 63.9 (10.5)* | 17.1 ± 10.9 (4.9) | |
| HIV-1BH10 | WT | 7.9 ± 0.9 (0.6) | 1.2 ± 0.3 (0.3) |
| LY_HIV-1 (M41L/T215Y) | 26.0 ± 4.0 (2.0)* | 1.3 ± 0.2 (0.4) | |
a Amino acid substitutions in the RT relative to the corresponding WT strain are indicated in parentheses.
b The IC50 values shown are averages ± S.D. of at least three tests, with each one performed six times. The fold increase in IC50 relative to wild-type HIV-2ROD control is shown in parentheses. The significance of the differences of HIV-2ROD and HIV-1BH10 mutants with their corresponding WT strains were determined by using a Student t test (*, p < 0.05).
c ND, not determined (recombinant viruses were not viable).
Discussion
In HIV-1, high-level AZT resistance develops through the accumulation of TAMs in the viral RT. These amino acid substitutions confer an excision activity that enables the polymerase to unblock DNA primers terminated with AZT and other NRTIs, and resume DNA synthesis. The excision reaction takes place in the presence of a pyrophosphate donor (usually ATP). However, in HIV-2 this pathway is suppressed, and TAMs are rarely observed in treated individuals (23, 35–39). In HIV-2 RT, AZT resistance is usually conferred by Q151M despite the fact that S215F/S215Y, a key substitution in the excision mechanism, requires only one nucleotide change, in contrast to HIV-1, where two mutations are needed to generate the equivalent substitution (i.e. T215F/T215Y). Q151M occurs at the nucleotide-binding site and affects the enzyme's ability to discriminate between NRTI triphosphates and dNTPs (40).
Our results demonstrate that the major determinants that suppress resistance through the excision pathway in HIV-2 are located adjacent to the β3–β4 hairpin loop, at positions 73 and 75 of the RT. HIV-2 RT bearing the four major TAMs (M41L/D67N/K70R/S215Y) lacks ATP-dependent excision activity and fails to unblock and extend primers terminated with AZT or tenofovir. However, when the HIV-1 RT sequence NSTRWRKLV (residues 67–75) was introduced at the equivalent positions of the HIV-2 RT in combination with TAMs M41L and S215Y, the enzyme was turned into an excision-proficient RT. Interestingly, our mutagenesis analysis shows that major contributors to this phenotype are Lys73 and to a lesser extent Val75. The presence of Ile75 in HIV-2 may favor the acquisition of resistance through the discrimination pathway because V75I is part of the multi-NRTI resistance Q151M complex of HIV-1 RT (28) and V75I antagonizes the effect of TAMs in HIV-1 RT (27). As in HIV-1, development of Q151M in HIV-2 requires at least two nucleotide changes with potential viable intermediates bearing amino acid substitutions Q151I or Q151L (38, 41, 42). Residues in the vicinity of Tyr215 or amino acid substitutions with a potential antagonistic effect on TAMs do not seem to have a major impact on HIV-2 NRTI resistance through the excision mechanism. These results are in agreement with previous findings showing that HIV-2 RT mutants S215Y and F214L/S215Y had negligible excision activity on NRTI-terminated DNA primers (26).
Met73 in HIV-2 RT plays a critical role in the suppression of the excision pathway. All HIV-2 RT mutants having the substitution M73K were excision-proficient in biochemical assays. However, HIV-2 variants carrying this substitution in the RT showed impaired viral replication, rendering non-viable virus in those cases where HIV-2 contained mutant RTs 5M_SK and 5M_STK. Although HIV-2 RT variants with the substitution M73K were active in biochemical assays, we found that those mutated enzymes were prone to degradation, and therefore additional purification steps had to be introduced to obtain enzymes of highest purity. In HIV-1 RT, random mutagenesis studies have shown that Lys73 can be replaced by many different amino acids (even non-conservative substitutions) while maintaining catalytic activity, although Met was not identified among the recovered active mutants (43). However, Lys73 is absolutely conserved in all groups and subtypes of HIV-1, whereas Met73 is conserved in all HIV-2 groups as well as in major simian immunodeficiency virus (SIV) species (e.g. mac, mne, and smm) (44).
Our results showing that M73K could confer HIV-2 RT-significant ATP-dependent rescue activity in enzymatic assays in the presence of D67N/K70R and decreased AZT susceptibility to the corresponding recombinant virus raise the question of how Lys73 could affect the conformation of the β3–β4 hairpin loop and the interactions needed for NRTI removal. Previous molecular dynamics studies with insertion- and deletion-containing HIV-1 RTs had revealed that changes at positions 67 and 70 affected critical hydrogen bond interactions between amido groups of Arg70 and the hydroxyl substituents of the dNTP γ-phosphate (32). These interactions that could be established in the mutant D67N/K70R RT were consistent with the formation of equivalent hydrogen bonds in the crystal structure of the excision-proficient HIV-1 RT M41L/D67N/K70R/T215Y/K219Q, complexed with AZT-terminated DNA/DNA template–primers, and AZTppppA (30).
In contrast, when we replaced Met for Lys73 in the HIV-1 RT D67N/K70R model and performed molecular dynamics simulations, there were remarkable conformational changes that eventually led to the loss of the potential hydrogen bond between Arg70 and the incoming dNTP. This proposal is consistent with the results of rescue assays carried out with AZTMP-terminated primers that showed that unlike the double-mutant D67N/K70R, the HIV-1BH10 RT D67N/K70R/K73M had negligible ATP-dependent phosphorolytic activity (Fig. 6).
According to the molecular models, the hydrophobic side chain of Met73 moves away from the dNTP-binding site into the hydrophobic core of the RT to interact with Val60. Interestingly, Val or Ile are found at this position in all HIV-1 strains, whereas Thr60 is highly conserved in all HIV-2 and SIV variants. The interaction between Met73 and Thr60 is likely to be stabilized through hydrogen bonding and/or hydrophobic interaction of their side chains. Therefore, the presence of Thr60 in the HIV-2 RT would be an additional restriction to the development of NRTI resistance through the excision pathway, if wildtype Met73 is replaced by Lys.
Apart from their influence on nucleoside analogue resistance, differences in the conformation of the β3–β4 hairpin loop may also affect other properties of the HIV-2 RT DNA polymerase activity. Recently, we showed that the tenofovir resistance-associated mutation K65R that confers increased fidelity of DNA synthesis in HIV-1 group M and group O RTs had no effect on the accuracy of the enzyme when introduced in the HIV-2 RT (45). This result was consistent with a previous report showing relative minor differences in the mutation rates determined for WT and K65R RT-containing SIVmac239 infecting pigtailed macaques (46). Interestingly, the amino acid sequence of the β3–β4 hairpin loop region is identical in HIV-2 and SIVmac239 RTs.
Despite having similar NRTI susceptibilities, our work underlines the important differences between HIV-1 and HIV-2 RTs, including the development of resistance through different pathways. However, these differences can be mapped to a couple of residues and suggest that genotypic monitoring for the emergence of mutations affecting the β3–β4 hairpin loop will be needed in the long run to prevent the eventual emergence of HIV-2 NRTI resistance through the excision pathway. In any case, continuing efforts in the development of anti-HIV-2 drugs are needed to overcome limitations imposed by the reduced armamentarium of antiretroviral drugs available to combat HIV-2 infection.
Experimental procedures
Expression and purification of RTs
WT heterodimeric (p66/p51) HIV-1BH10 RT and mutants M41L/T215Y, D67N/K70R and D67N/K70R/K73M were purified as described previously (47–49). Histidine-tagged WT and mutant HIV-2ROD RTs were obtained as p68/p54 heterodimers after co-expression of the p68-coding region of the HIV-2ROD RT together with the HIV-2 protease, using constructs derived from plasmid pT5m (50). HIV-2 RT heterodimers were purified by immobilized metal affinity chromatography on Ni2+-nitrilotriacetic acid-agarose, followed by ion-exchange chromatography (45). Before dialysis against 50 mm Tris-HCl, pH 7.0, containing 25 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol (DTT), and 10% glycerol, some RTs were further purified by heparin-Sepharose (GE Healthcare) affinity chromatography. For this purpose, columns (1 ml) were equilibrated with 50 mm sodium phosphate buffer, pH 6.8, containing 0.1 m NaCl, and the RTs were eluted with a 0.1–0.8 m NaCl gradient. After dialysis and concentration, the purity of the enzyme was tested by SDS-PAGE. Before biochemical studies, RT concentrations were determined by active-site titration (51, 52). Steady-state kinetics of nucleotide incorporation were determined with 32P-labeled D38/25PGA template-primers, under conditions described previously (53).
Mutagenesis
Site-directed mutagenesis was carried out by using the standard QuikChangeTM (Stratagene) protocol. The pT5m plasmid encoding the large subunit of the HIV-2ROD RT was used as template (50) to obtain the HIV-2 RT mutants. Complementary mutagenic primers (Table S1) were used to amplify the entire pT5m(RODRT) plasmid in a thermocycling reaction carried out with high-fidelity Pfu DNA polymerase. Mutant RTs were constructed in an orderly manner. At first, we obtained the double-mutant D67N/K70R that was used as template to generate the M41L/D67N/K70R/S215Y mutant. The I75V substitution was introduced in this construct to obtain M41L/D67N/K70R/I75V/S215Y, a quintuple-mutant that was then use to generate derivatives containing one additional mutation, such as K68S, T88W, I181Y, N210L, or N210W. The complex M41L/D67N/K68S/K70R/I75V/S215Y was used as template to introduce additional mutations such as N69T, M73K, or N69T/M73K. The combination G211R/L212W was introduced in the context of M41L/D67N/K70R/I75V/N210L/S215Y, and the obtained plasmid was used as template to generate the mutant M41L/D67N/K70R/I75V/N210L/G211R/L212W/F214L/S215Y that contained the F214L substitution. HIV-1BH10 mutants D67N/K70R and D67N/K70R/K73M were obtained by following the same protocol, but using plasmid p66RTB as template (48, 49) and the corresponding mutagenic primers listed the Table S1. All constructs were verified by DNA sequencing.
Nucleotides and template–primers
Stock solutions (100 mm) of dNTPs and rNTPs were obtained from GE Healthcare. AZTTP was purchased from Trilink Biotechnologies (San Diego). Tenofovir diphosphate (tenofovir-DP) and stavudine triphosphate (d4TTP) were obtained from Toronto Research Chemicals (Toronto, Canada) and Sierra Bioresearch (Tucson, AZ), respectively. Before use, nucleoside triphosphates were treated with inorganic pyrophosphatase (Roche Applied Science) to remove traces of pyrophosphate, as described (13). DNA oligonucleotides 21P (5′-ATACTTTAACCATATGTATCC-3′), 25PGA (5′-TGGTAGGGCTATACATTCTTGCAGG-3′), D38 (5′-GGGTCCTTTCTTACCTGCAAGAATGTATAGCCCTACCA-3′), and D38T (5′-GGGTCCTTTCAATCCTGCAAGAATGTATAGCCCTACCA-3′), and the RNA oligonucleotide 31Trna (5′-UUUUUUUUUAGGAUACAUAUGGUUAAAGUAU-3′) were obtained from Life Technologies, Inc., and Sigma. Primer oligonucleotides were labeled at their 5′ termini with 1–2 μCi of [γ-32P]ATP (10 mCi/ml; 3000 Ci/mmol, PerkinElmer Life Sciences) and 5 units of T4 polynucleotide kinase (New England Biolabs) in 70 mm Tris-HCl buffer, pH 7.6, containing 10 mm MgCl2 and 5 mm DTT. Template–primer solutions were prepared at 300 nm in 500 mm Hepes buffer, pH 7.0, containing 150 mm magnesium acetate and 150 mm NaCl.
Chain terminator excision assays
RT-catalyzed DNA rescue reactions were carried out as described previously with template–primers D38/25PGA, D38T/25PGA, and 31Trna/21P (54, 55). Reactions were carried out in 70 mm Hepes buffer, pH 7.0, containing 20 mm magnesium acetate, 20 mm NaCl, 130 mm potassium acetate, 1 mm DTT, and 5% (w/v) polyethylene glycol 6000. The labeled template–primers (75 nm) were preincubated with the corresponding RT (60 nm active enzyme concentration) at 37 °C for 10 min. Primers were then blocked by adding an equal amount of buffer containing AZTTP or tenofovir-DP, at a final concentration of 25 μm, and incubated at 37 °C for 30 min. Rescue reactions were initiated by adding a mixture of all dNTPs in the presence of 3.2 mm ATP. In rescue assays carried out with DNA/DNA template–primers, dNTPs were supplied at 100 μm (final concentration), except dATP (with D38/25PGA) or dTTP (with D38T/25PGA) whose concentration was kept at 1 μm to minimize the inhibitory effect of the next complementary dNTP, caused by the formation of “dead-end” complexes (9, 56). Inhibitor and dNTP concentrations were 2-fold higher in assays carried with the RNA/DNA template–primer 31Trna/21P. After addition of dNTPs and the pyrophosphate donor, reactions were incubated for up to 20 min at 37 °C to facilitate nucleotide excision and primer elongation. Aliquots were removed at appropriate times, and reactions were stopped by adding an equal amount of sample loading buffer (10 mm EDTA in 90% formamide containing 3 mg/ml xylene cyanol FF and 3 mg/ml bromphenol blue). Products were resolved on denaturing 20% (w/v) polyacrylamide, 8 m urea gels, and primer rescue was quantified by phosphorimaging with a BAS 1500 scanner (Fuji), using the program Tina version 2.09 (Raytest Isotopenmessgerate Gmbh, Staubenhardt, Germany).
Kinetics of the ATP-dependent excision reaction
The kinetics of the excision reaction were monitored with template–primers D38/25PGAd4TMP and D38/25PGAAZTMP, obtained after blocking the primer with nucleotide analogues with terminal deoxynucleotidyltransferase (Roche Applied Science) or WT HIV-1BH10 RT, respectively (13, 53). AZTMP and d4TMP excision was determined by using template–primers D38/25PGAAZTMP and D38/25PGAd4TMP, respectively. To ensure single turnover conditions, we used an excess of enzyme over template–primer. Thus, RTs (80 nm) and blocked template–primers (60 nm) were preincubated at 37 °C for 10 min in 0.11 m Hepes buffer, pH 7.0, containing 30 mm NaCl, 30 mm magnesium acetate, 130 mm potassium acetate, 1 mm DTT, and 5% (w/v) polyethylene glycol 6000. Reactions were initiated by adding 1 volume of a solution containing 6.4 mm ATP in 130 mm potassium acetate, 1 mm DTT, and 5% (w/v) polyethylene glycol 6000. Excision reactions were carried out for 0 to 80 min at 37 °C, and aliquots were removed at appropriate times, stopped with sample loading buffer, and analyzed by denaturing PAGE as described above. The amount of hydrolyzed product generated over time was fitted to a single exponential decay equation: [P] = A × e −kobs × t, where kobs is apparent kinetic constant of the excision reaction.
HIV drug susceptibility assays
AZT and darunavir susceptibility assays were carried out with infectious HIV-2 clones generated in this study and with previously described recombinant HIV-1 carrying either the WT BH10 RT or a double-mutant M41L/T215Y RT (49). To generate infectious HIV-2 carrying the different RT variants, we prepared three DNA fragments by PCR covering the full-length HIV-2 proviral DNA. These three PCR DNA fragments overlap 31–35 bp, and after transfection of susceptible MT-4 cells, they were able to generate infectious HIV-2 particles (57). The template for fragments I and III (HIV-2ROD strain nucleotides 1–2421 and 4019–9671, respectively) was proviral DNA obtained from MT-4 cells infected with HIV-2ROD (Centre for AIDS Reagents, NIBSC, ID 0121) (58). Fragment II, corresponding to HIV-2 RT (HIV-2ROD nucleotides 2381–4049), was obtained by PCR from the RT expression plasmids described above. PCR oligonucleotides for fragments I, II, and III were as follows: 1HIV-2RODF (5′-GGTCGCTCTGCGGAGAGGCT-3′, HIV-2ROD nts 1–20); 2HIV-RODR (5′-GGCTTTAGCATTATTTTTATTGGGTCTACTTTGGC-3′, HIV-2RODnts 2384–2421) and 2HIV-2RODF (5′-GCCAAAGTAGACCCAATAAAAATAATGCTAAAGCC-3′, HIV-2ROD nts 2384–2421); 3HIV-2RODR (5′-GTCTGATACCCTGAGTCACTAAATGATCTAC-3′, HIV-2ROD nts 4019–4049) and 3HIV-2RODF (5′-GTAGATCATTTAGTGACTCAGGGTATCAGAC-3′, HIV-2ROD nts 4019–4049); and 5HIV-2RODR (5′-TGCTTCTAACTGGCAGCTTTATTAAGG-3′, HIV-2ROD nts 9645–9671), respectively. Proviral DNA was purified using the QuickExtract DNA extraction protocol (Epicenter Biotechnologies). Cycling parameters were 1 cycle of denaturation at 94 °C for 2 min, followed by 40 cycles of 45 s at 95 °C, 45 s at 55 °C, and 2–6 min at 68 °C, with a final extension step of 68 °C for 7 min. High fidelity Platinum TaqDNA polymerase (Thermo Fisher Scientific) was used for PCR amplifications. After PCR purification (QIAquick PCR purification kit, Qiagen), 100 ng of each of the three PCR fragments were co-transfected into MT-4 cells as we described previously (32). Cell culture supernatants were harvested on days 3, 5, and 7 after transfection when the concentration of HIV-1 p24 antigen (Innotest HIV Antigen mAB, Fujirebio) surpassed 500 ng/ml. If p24 antigen was not detected after 7 days of culture, three blind passages, feeding the cultures with fresh medium and new MT-4 cells, were performed to recover viable virus. The nucleotide sequence of the RT-coding region of the progeny virus was checked for possible reversions or undesired mutations. After three blind passages without p24 antigen detection in the cultured supernatant, the construct was considered non-viable in MT-4 cell culture. Susceptibility to AZT and darunavir was determined in MT-4 cells, as described previously (49), using a multiplicity of infection of 0.003. Viable cells at different drug concentrations were quantified with a tetrazolium-based colorimetric method (59). AZT and darunavir were obtained from the AIDS Research and Reference Reagent Program (National Institutes of Health). HIV-2 shows natural resistance to non-nucleoside RT inhibitors and several protease inhibitors. We used darunavir as a control due to its potent in vitro activity against both types of HIV (4).
Homology modeling and molecular dynamics
A structural model of HIV-1 RT bearing the amino acid substitutions D67N/K70R/K73M bound to double-stranded DNA and dTTP was constructed by standard homology modeling techniques (60), using as reference the one previously obtained for the double-mutant D67N/K70R (32). Molecular dynamics simulations, based on the models, were performed as described previously for the WT HIV-1 RT (34). This model for the catalytically-competent WT RT was based on the X-ray structure determined by Huang et al. (61) (PDB file 1RTD) corresponding to a ternary complex of HIV-1 RT, a dideoxyguanosine-terminated primer–template and dTTP, and containing two Mg2+ ions at the DNA polymerase active site. Atomic charges for dTTP were obtained with the RESP program (62) to fit potentials calculated at 6–31G* level using the Gaussian-03 package (63). The van der Waals parameters for Mg2+ were taken from Allnér et al. (64). The total simulation length was around 10 ns, and the analysis of trajectories was performed as reported previously (34). The SHAKE algorithm was applied allowing for an integration time step of 2 fs (34).
Author contributions
M. A. M. and L. M.-A. designed research; M. Á. and M. N. carried out experimental work; J. M. carried out software and computer analysis; M. Á., M. N., J. M., M. A. M., and L. M.-A. analyzed data; M. Á., M. A. M. and L. M.-A. wrote the paper.
Supplementary Material
Acknowledgment
An institutional grant from the Fundación Ramón Areces to the Centro de Biologia Molecular Severo Ochoa is also acknowledged.
This work was supported in part by Spanish Ministry of Economy, Industry and Competitiveness Grants BIO2013-48788-C2-1-R and BIO2016–76716-R (AEI/FEDER, UE) (to L. M.-A.) and SAF2016-75277-R (to M. A. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4 and Table S1.
- RT
- reverse transcriptase
- NRTI
- nucleoside reverse transcriptase inhibitor
- AZT
- 3′-azido-3′-deoxythymidine
- TAM
- thymidine analogue resistance mutations
- d4T
- 2′,3′-didehydro-2′,3′-dideoxythymidine
- AZTTP
- AZT-triphosphate
- AZTppppA
- AZT adenosine dinucleoside tetraphosphate
- r.m.s.d.
- root-mean-square deviations
- d4TTP
- d4T triphosphate
- tenofovir-DP
- tenofovir diphosphate
- nt
- nucleotide
- PDB
- Protein Data Bank
- d4TMP
- 2′,3′-didehydro-2′,3′-dideoxythymidine monophosphate
- SIV
- simian immunodeficiency virus.
References
- 1. Gottlieb G. S., Eholié S. P., Nkengasong J. N., Jallow S., Rowland-Jones S., Whittle H. C., and Sow P. S. (2008) A call for randomized controlled trials of antiretroviral therapy for HIV-2 infection in West Africa. AIDS 22, 2069–2072 10.1097/QAD.0b013e32830edd44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campbell-Yesufu O. T., and Gandhi R. T. (2011) Update on human immunodeficiency virus (HIV)-2 infection. Clin. Infect. Dis. 52, 780–787 10.1093/cid/ciq248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Menéndez-Arias L., and Tözsér J. (2008) HIV-1 protease inhibitors: effects on HIV-2 replication and resistance. Trends Pharmacol. Sci. 29, 42–49 10.1016/j.tips.2007.10.013 [DOI] [PubMed] [Google Scholar]
- 4. Menéndez-Arias L., and Alvarez M. (2014) Antiretroviral therapy and drug resistance in human immunodeficiency virus type 2 infection. Antiviral Res. 102, 70–86 10.1016/j.antiviral.2013.12.001 [DOI] [PubMed] [Google Scholar]
- 5. Smith R. A., Gottlieb G. S., Anderson D. J., Pyrak C. L., and Preston B. D. (2008) Human immunodeficiency virus types 1 and 2 exhibit comparable sensitivities to zidovudine and other nucleoside analog inhibitors in vitro. Antimicrob. Agents Chemother. 52, 329–332 10.1128/AAC.01004-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Menéndez-Arias L. (2013) Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments. Antiviral Res. 98, 93–120 10.1016/j.antiviral.2013.01.007 [DOI] [PubMed] [Google Scholar]
- 7. Sarafianos S. G., Marchand B., Das K., Himmel D. M., Parniak M. A., Hughes S. H., and Arnold E. (2009) Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 385, 693–713 10.1016/j.jmb.2008.10.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu W. S., and Hughes S. H. (2012) HIV-1 reverse transcription. Cold Spring Harb. Perspect. Med. 2, a006882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Menéndez-Arias L. (2008) Mechanisms of resistance to nucleoside analogue inhibitors of HIV-1 reverse transcriptase. Virus Res. 134, 124–146 10.1016/j.virusres.2007.12.015 [DOI] [PubMed] [Google Scholar]
- 10. Arion D., Kaushik N., McCormick S., Borkow G., and Parniak M. A. (1998) Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 37, 15908–15917 10.1021/bi981200e [DOI] [PubMed] [Google Scholar]
- 11. Meyer P. R., Matsuura S. E., Mian A. M., So A. G., and Scott W. A. (1999) A mechanism of AZT resistance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol. Cell 4, 35–43 10.1016/S1097-2765(00)80185-9 [DOI] [PubMed] [Google Scholar]
- 12. Meyer P. R., Matsuura S. E., Schinazi R. F., So A. G., and Scott W. A. (2000) Differential removal of thymidine nucleotide analogues from blocked DNA chains by human immunodeficiency virus reverse transcriptase in the presence of physiological concentrations of 2′-deoxynucleoside triphosphates. Antimicrob. Agents Chemother. 44, 3465–3472 10.1128/AAC.44.12.3465-3472.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mas A., Vázquez-Alvarez B. M., Domingo E., and Menéndez-Arias L. (2002) Multidrug-resistant HIV-1 reverse transcriptase: involvement of ribonucleotide-dependent phosphorolysis in cross-resistance to nucleoside analogue inhibitors. J. Mol. Biol. 323, 181–197 10.1016/S0022-2836(02)00911-7 [DOI] [PubMed] [Google Scholar]
- 14. White K. L., Chen J. M., Margot N. A., Wrin T., Petropoulos C. J., Naeger L. K., Swaminathan S., and Miller M. D. (2004) Molecular mechanisms of tenofovir resistance conferred by human immunodeficiency virus type 1 reverse transcriptase containing a diserine insertion after residue 69 and multiple thymidine analog-associated mutations. Antimicrob. Agents Chemother. 48, 992–1003 10.1128/AAC.48.3.992-1003.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Larder B. A. (1994) Interactions between drug resistance mutations in human immunodeficiency virus type 1 reverse transcriptase. J. Gen. Virol. 75, 951–957 10.1099/0022-1317-75-5-951 [DOI] [PubMed] [Google Scholar]
- 16. Yahi N., Tamalet C., Tourrès C., Tivoli N., Ariasi F., Volot F., Gastaut J. A., Gallais H., Moreau J., and Fantini J. (1999) Mutation patterns of the reverse transcriptase and protease genes in human immunodeficiency virus type 1-infected patients undergoing combination therapy: survey of 787 sequences. J. Clin. Microbiol. 37, 4099–4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meyer P. R., Matsuura S. E., Tolun A. A., Pfeifer I., So A. G., Mellors J. W., and Scott W. A. (2002) Effects of specific zidovudine resistance mutations and substrate structure on nucleotide-dependent primer unblocking by human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 46, 1540–1545 10.1128/AAC.46.5.1540-1545.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Damond F., Collin G., Matheron S., Peytavin G., Campa P., Delarue S., Taieb A., Bénard A., Chêne G., Brun-Vézinet F., Descamps D., and French ANRS HIV-2 Cohort (ANRS CO 5 VIH-2). (2005) In vitro phenotypic susceptibility to nucleoside reverse transcriptase inhibitors of HIV-2 isolates with the Q151M mutation in the reverse transcriptase gene. Antivir. Ther. 10, 861–865 [PubMed] [Google Scholar]
- 19. Deuzing I. P., Charpentier C., Wright D. W., Matheron S., Paton J., Frentz D., van der Vijver D. A., Coveney P. V., Descamps D., ANRS COS HIV-2 Cohort, Boucher C. A., and Beerens N. (2015) Mutation V111I in HIV-2 reverse transcriptase increases the fitness of the nucleoside analogue-resistant K65R and Q151M viruses. J. Virol. 89, 833–843 10.1128/JVI.02259-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Smith R. A., Anderson D. J., Pyrak C. L., Preston B. D., and Gottlieb G. S. (2009) Antiretroviral drug resistance in HIV-2: three amino acid changes are sufficient for classwide nucleoside analogue resistance. J. Infect. Dis. 199, 1323–1326 10.1086/597802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reid P., MacInnes H., Cong M. E., Heneine W., and García-Lerma J. G. (2005) Natural resistance of human immunodeficiency virus type 2 to zidovudine. Virology 336, 251–264 10.1016/j.virol.2005.03.030 [DOI] [PubMed] [Google Scholar]
- 22. Brandin E., Lindborg L., Gyllensten K., Broström C., Hagberg L., Gisslen M., Tuvesson B., Blaxhult A., and Albert J. (2003) Pol gene sequence variation in Swedish HIV-2 patients failing antiretroviral therapy. AIDS Res. Hum. Retroviruses 19, 543–550 10.1089/088922203322230905 [DOI] [PubMed] [Google Scholar]
- 23. Gottlieb G. S., Badiane N. M., Hawes S. E., Fortes L., Toure M., Ndour C. T., Starling A. K., Traore F., Sall F., Wong K. G., Cherne S. L., Anderson D. J., Dye S. A., Smith R. A., Mullins J. I., Kiviat N. B., et al. (2009) Emergence of multiclass drug-resistance in HIV-2 in antiretroviral-treated individuals in Senegal: implications for HIV-2 treatment in resource-limited West Africa. Clin. Infect. Dis. 48, 476–483; Correction (2009) Clin. Infect. Dis. 48, 848 10.1086/597577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ntemgwa M. L., d'Aquin Toni T., Brenner B. G., Camacho R. J., and Wainberg M. A. (2009) Antiretroviral drug resistance in human immunodeficiency virus type 2. Antimicrob. Agents Chemother. 53, 3611–3619 10.1128/AAC.00154-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boyer P. L., Sarafianos S. G., Clark P. K., Arnold E., and Hughes S. H. (2006) Why do HIV-1 and HIV-2 use different pathways to develop AZT resistance? PLoS Pathog. 2, e10 10.1371/journal.ppat.0020010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boyer P. L., Clark P. K., and Hughes S. H. (2012) HIV-1 and HIV-2 reverse transcriptases: different mechanisms of resistance to nucleoside reverse transcriptase inhibitors. J. Virol. 86, 5885–5894 10.1128/JVI.06597-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matamoros T., Nevot M., Martínez M. A., and Menéndez-Arias L. (2009) Thymidine analogue resistance suppression by V75I of HIV-1 reverse transcriptase: effects of substituting Valine 75 on stavudine excision and discrimination. J. Biol. Chem. 284, 32792–32802 10.1074/jbc.M109.038885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shirasaka T., Kavlick M. F., Ueno T., Gao W.-Y., Kojima E., Alcaide M. L., Chokekijchai S., Roy B. M., Arnold E., Yarchoan R., and Mitsuya H. (1995) Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. U.S.A. 92, 2398–2402 10.1073/pnas.92.6.2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hachiya A., Kodama E. N., Schuckmann M. M., Kirby K. A., Michailidis E., Sakagami Y., Oka S., Singh K., and Sarafianos S. G. (2011) K70Q adds high-level tenofovir resistance to “Q151M complex” HIV reverse transcriptase through the enhanced discrimination mechanism. PLoS ONE 6, e16242 10.1371/journal.pone.0016242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tu X., Das K., Han Q., Bauman J. D., Clark A. D. Jr., Hou X., Frenkel Y. V., Gaffney B. L., Jones R. A., Boyer P. L., Hughes S. H., Sarafianos S. G., and Arnold E. (2010) Structural basis of HIV-1 resistance to AZT by excision. Nat. Struct. Mol. Biol. 17, 1202–1209 10.1038/nsmb.1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cases-González C. E., Franco S., Martínez M. A., and Menéndez-Arias L. (2007) Mutational patterns associated with the 69 insertion complex in multi-drug-resistant HIV-1 reverse transcriptase that confer increased excision activity and high-level resistance to zidovudine. J. Mol. Biol. 365, 298–309 10.1016/j.jmb.2006.09.073 [DOI] [PubMed] [Google Scholar]
- 32. Kisic M., Matamoros T., Nevot M., Mendieta J., Martinez-Picado J., Martínez M. A., and Menéndez-Arias L. (2011) Thymidine analogue excision and discrimination modulated by mutational complexes including single amino acid deletions of Asp-67 or Thr-69 in HIV-1 reverse transcriptase. J. Biol. Chem. 286, 20615–20624 10.1074/jbc.M111.226100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ren J., Bird L. E., Chamberlain P. P., Stewart-Jones G. B., Stuart D. I., and Stammers D. K. (2002) Structure of HIV-2 reverse transcriptase at 2.35-Å resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc. Natl. Acad. Sci. U.S.A. 99, 14410–14415 10.1073/pnas.222366699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mendieta J., Cases-González C. E., Matamoros T., Ramírez G., and Menéndez-Arias L. (2008) A Mg2+-induced conformational switch rendering a competent DNA polymerase catalytic complex. Proteins 71, 565–574 10.1002/prot.21711 [DOI] [PubMed] [Google Scholar]
- 35. Rodés B., Holguín A., Soriano V., Dourana M., Mansinho K., Antunes F., and González-Lahoz J. (2000) Emergence of drug resistance mutations in human immunodeficiency virus type 2-infected subjects undergoing antiretroviral therapy. J. Clin. Microbiol. 38, 1370–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Damond F., Matheron S., Peytavin G., Campa P., Taieb A., Collin G., Delaunay C., Chêne G., Brun-Vézinet F., and Descamps D. (2004) Selection of K65R mutation in HIV-2-infected patients receiving tenofovir-containing regimen. Antivir. Ther. 9, 635–636 [PubMed] [Google Scholar]
- 37. Descamps D., Damond F., Matheron S., Collin G., Campa P., Delarue S., Pueyo S., Chêne G., Brun-Vézinet F., and French ANRS HIV-2 Cohort Study Group. (2004) High frequency of selection of K65R and Q151M mutations in HIV-2 infected patients receiving nucleoside reverse transcriptase inhibitors containing regimen. J. Med. Virol. 74, 197–201 10.1002/jmv.20174 [DOI] [PubMed] [Google Scholar]
- 38. Colson P., Henry M., Tivoli N., Gallais H., Gastaut J.-A., Moreau J., and Tamalet C. (2005) Polymorphism and drug-selected mutations in the reverse transcriptase gene of HIV-2 from patients living in Southeastern France. J. Med. Virol. 75, 381–390 10.1002/jmv.20296 [DOI] [PubMed] [Google Scholar]
- 39. Treviño A., de Mendoza C., Caballero E., Rodríguez C., Parra P., Benito R., Cabezas T., Roc L., Aguilera A., Soriano V., and HIV-2 Spanish Study Group. (2011) Drug resistance mutations in patients infected with HIV-2 living in Spain. J. Antimicrob. Chemother. 66, 1484–1488 10.1093/jac/dkr164 [DOI] [PubMed] [Google Scholar]
- 40. Deval J., Selmi B., Boretto J., Egloff M. P., Guerreiro C., Sarfati S., and Canard B. (2002) The molecular mechanism of multidrug resistance by the Q151M human immunodeficiency virus type 1 reverse transcriptase and its suppression using α-boranophosphate nucleotide analogues. J. Biol. Chem. 277, 42097–42104 10.1074/jbc.M206725200 [DOI] [PubMed] [Google Scholar]
- 41. van der Ende M. E., Guillon C., Boers P. H., Ly T. D., Gruters R. A., Osterhaus A. D., and Schutten M. (2000) Antiviral resistance of biologic HIV-2 clones obtained from individuals on nucleoside reverse transcriptase inhibitor therapy. J. Acquir. Immune Defic. Syndr. 25, 11–18 10.1097/00126334-200009010-00002 [DOI] [PubMed] [Google Scholar]
- 42. Matsumi S., Kosalaraksa P., Tsang H., Kavlick M. F., Harada S., and Mitsuya H. (2003) Pathways for the emergence of multi-dideoxynucleoside-resistant HIV-1 variants. AIDS 17, 1127–1137 10.1097/00002030-200305230-00003 [DOI] [PubMed] [Google Scholar]
- 43. Kim B., Hathaway T. R., and Loeb L. A. (1996) Human immunodeficiency virus reverse transcriptase–functional mutants obtained by random mutagenesis coupled with genetic selection in Escherichia coli. J. Biol. Chem. 271, 4872–4878 10.1074/jbc.271.9.4872 [DOI] [PubMed] [Google Scholar]
- 44. Foley B., Leitner T., Apetrei C., Hahn B., Mizrachi I., Mullins J., Rambaut A., Wolinsky S., and Korber B. (eds) (2014) HIV Sequence Compendium 2014. Los Alamos National Laboratory, Los Alamos, NM [Google Scholar]
- 45. Álvarez M, Sebastián-Martín A., García-Marquina G., and Menéndez-Arias L. (2017) Fidelity of classwide-resistant HIV-2 reverse transcriptase and differential contribution of K65R to the accuracy of HIV-1 and HIV-2 reverse transcriptases. Sci. Rep. 7, 44834 10.1038/srep44384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lloyd S. B., Lichtfuss M., Amarasena T. H., Alcantara S., De Rose R., Tachedjian G., Alinejad-Rokny H., Venturi V., Davenport M. P., Winnall W. R., and Kent S. J. (2016) High fidelity simian immunodeficiency virus reverse transcriptase mutants have impaired replication in vitro and in vivo. Virology 492, 1–10 10.1016/j.virol.2016.02.008 [DOI] [PubMed] [Google Scholar]
- 47. Boretto J., Longhi S., Navarro J.-M., Selmi B., Sire J., and Canard B. (2001) An integrated system to study multiply substituted human immunodeficiency virus type 1 reverse transcriptase. Anal. Biochem. 292, 139–147 10.1006/abio.2001.5045 [DOI] [PubMed] [Google Scholar]
- 48. Matamoros T., Deval J., Guerreiro C., Mulard L., Canard B., and Menéndez-Arias L. (2005) Suppression of multidrug-resistant HIV-1 reverse transcriptase primer unblocking activity by α-phosphate-modified thymidine analogues. J. Mol. Biol. 349, 451–463 10.1016/j.jmb.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 49. Betancor G., Puertas M. C., Nevot M., Garriga C., Martínez M. A., Martinez-Picado J., and Menéndez-Arias L. (2010) Mechanisms involved in the selection of HIV-1 reverse transcriptase thumb subdomain polymorphisms associated with nucleoside analogue therapy failure. Antimicrob. Agents Chemother. 54, 4799–4811 10.1128/AAC.00716-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sevilya Z., Loya S., Hughes S. H., and Hizi A. (2001) The ribonuclease H activity of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2 is affected by the thumb subdomain of the small protein subunits. J. Mol. Biol. 311, 957–971 10.1006/jmbi.2001.4904 [DOI] [PubMed] [Google Scholar]
- 51. Kati W. M., Johnson K. A., Jerva L. F., and Anderson K. S. (1992) Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267, 25988–25997 [PubMed] [Google Scholar]
- 52. Menéndez-Arias L. (1998) Studies on the effects of truncating α-helix E′ of p66 human immunodeficiency virus type 1 reverse transcriptase on template-primer binding and fidelity of DNA synthesis. Biochemistry 37, 16636–16644 10.1021/bi981830g [DOI] [PubMed] [Google Scholar]
- 53. Betancor G., Garriga C., Puertas M. C., Nevot M., Anta L., Blanco J. L., Pérez-Elías M. J., de Mendoza C., Martínez M. A., Martinez-Picado J., Menéndez-Arias L., Resistance Platform of the Spanish AIDS Research Network (ResRIS), et al. (2012) Clinical, virological and biochemical evidence supporting the association of HIV-1 reverse transcriptase polymorphism R284K and thymidine analogue resistance mutations M41L, L210W and T215Y in patients failing tenofovir/emtricitabine therapy. Retrovirology 9, 68 10.1186/1742-4690-9-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mas A., Parera M., Briones C., Soriano V., Martínez M. A., Domingo E., and Menéndez-Arias L. (2000) Role of a dipeptide insertion between codons 69 and 70 of HIV-1 reverse transcriptase in the mechanism of AZT resistance. EMBO J. 19, 5752–5761 10.1093/emboj/19.21.5752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Matamoros T., Franco S., Vázquez-Alvarez B. M., Mas A., Martínez M. A., and Menéndez-Arias L. (2004) Molecular determinants of multi-nucleoside analogue resistance in HIV-1 reverse transcriptases containing a dipeptide insertion in the fingers subdomain. Effect of mutations D67N and T215Y on removal of thymidine nucleotide analogues from blocked DNA primers. J. Biol. Chem. 279, 24569–24577 10.1074/jbc.M312658200 [DOI] [PubMed] [Google Scholar]
- 56. Tong W., Lu C.-D., Sharma S. K., Matsuura S., So A. G., and Scott W. A. (1997) Nucleotide-induced stable complex formation by HIV-1 reverse transcriptase. Biochemistry 36, 5749–5757 10.1021/bi962410z [DOI] [PubMed] [Google Scholar]
- 57. Fujita Y., Otsuki H., Watanabe Y., Yasui M., Kobayashi T., Miura T., and Igarashi T. (2013) Generation of a replication-competent chimeric simian-human immunodeficiency virus carrying env from subtype C clinical isolate through intracellular homologous recombination. Virology 436, 100–111 10.1016/j.virol.2012.10.036 [DOI] [PubMed] [Google Scholar]
- 58. Clavel F., Guétard D., Brun-Vézinet F., Chamaret S., Rey M. A., Santos-Ferreira M. O., Laurent A. G., Dauguet C., Katlama C., Rouzioux C., Klatzmann D., Champalimaud J.-L., and Montagnier L. (1986) Isolation of a new human retrovirus from West African patients with AIDS. Science 233, 343–346 10.1126/science.2425430 [DOI] [PubMed] [Google Scholar]
- 59. Pauwels R., Balzarini J., Baba M., Snoeck R., Schols D., Herdewijn P., Desmyter J., and De Clercq E. (1988) Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 20, 309–321 10.1016/0166-0934(88)90134-6 [DOI] [PubMed] [Google Scholar]
- 60. Kisic M., Mendieta J., Puertas M. C., Parera M., Martínez M. A., Martinez-Picado J., and Menéndez-Arias L. (2008) Mechanistic basis of zidovudine hypersusceptibility and lamivudine resistance conferred by the deletion of codon 69 in the HIV-1 reverse transcriptase-coding region. J. Mol. Biol. 382, 327–341 10.1016/j.jmb.2008.07.030 [DOI] [PubMed] [Google Scholar]
- 61. Huang H., Chopra R., Verdine G. L., and Harrison S. C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282, 1669–1675 10.1126/science.282.5394.1669 [DOI] [PubMed] [Google Scholar]
- 62. Cieplak P., Cornell W. D., Bayly C., and Kollman P. A. (1995) Application of the multimolecule and multiconformational RESP methodology to biopolymers: charge derivation for DNA, RNA and proteins. J. Comput. Chem. 16, 1357–1377 10.1002/jcc.540161106 [DOI] [Google Scholar]
- 63. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Montgomery J. A. Jr., Vreven T., Kudin K. N., Burant J. C., Millam J. M., Iyengar S. S., Tomasi J., Barone V., Mennucci B., et al. (2004) Gaussian 03, Gaussian, Inc., Wallingford, CT [Google Scholar]
- 64. Allnér O., Nilsson L., and Villa A. (2012) Magnesium ion-water coordination and exchange in biomolecular simulations. J. Chem. Theory Comput. 8, 1493–1502 10.1021/ct3000734 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.