

Abstract
Aggregation of huntingtin protein arising from expanded polyglutamine (polyQ) sequences in the exon-1 region of mutant huntingtin plays a central role in the pathogenesis of Huntington's disease. The huntingtin aggregation pathways are of therapeutic and diagnostic interest, but obtaining critical information from the physiologically relevant htt exon-1 (Httex1) protein has been challenging. Using biophysical techniques and an expression and purification protocol that generates clean, monomeric Httex1, we identified and mapped three distinct aggregation pathways: 1) unseeded in solution; 2) seeded in solution; and 3) membrane-mediated. In solution, aggregation proceeded in a highly stepwise manner, in which the individual domains (N terminus containing 17 amino acids (N17), polyQ, and proline-rich domain (PRD)) become ordered at very different rates. The aggregation was initiated by an early oligomer requiring a pathogenic, expanded Gln length and N17 α-helix formation. In the second phase, β-sheet forms in the polyQ. The slowest step is the final structural maturation of the PRD. This stepwise mechanism could be bypassed by seeding, which potently accelerated aggregation and was a prerequisite for prion-like spreading in vivo. Remarkably, membranes could catalyze aggregation even more potently than seeds, in a process that caused significant membrane damage. The N17 governed membrane-mediated aggregation by anchoring Httex1 to the membrane, enhancing local concentration and promoting collision via two-dimensional diffusion. Considering its central roles in solution and in membrane-mediated aggregation, the N17 represents an attractive target for inhibiting multiple pathways. Our approach should help evaluate such inhibitors and identify diagnostic markers for the misfolded forms identified here.
Keywords: amyloid, electron paramagnetic resonance (EPR), Huntington disease, polyglutamine, protein aggregation, huntingtin exon-1
Introduction
Huntington's disease (HD)2 is an autosomal dominant disorder caused by polyglutamine (polyQ) expansions in the exon 1 region of mutant huntingtin protein (Httex1) (1). Mutant Httex1 is expressed in disease, readily misfolds into amyloid fibrils, and is sufficient to cause toxicity in cell and animal models of HD, suggesting that the exon1 fragment plays a central role in pathogenesis (2–7). The length of the polyQ expansions is important for pathogenesis, as increasing polyQ lengths enhance aggregation and cause earlier disease onset (8–12).
Because of its role in disease and its potential importance in the development of therapeutics, the mechanism of Httex1 aggregation has been of significant interest (13–18). Httex1 has three domains, an N terminus containing 17 amino acids (N17), a polyQ region of variable length (polyQ), and a C-terminal proline-rich domain (PRD). EPR and solid-state NMR experiments have revealed some of the structural features of Httex1 fibrils (19–23). Although the N17 forms the α-helical structure, the polyQ takes up a cross-β-structure (12, 14). According to the “bottle-brush” model, the PRD forms bristled polyproline II helix-rich structures that project away from the fibril core (20).
Studies using small Httex1-mimicking peptides reveal that the N17 can accelerate the overall aggregation propensity (15, 17, 21). A potential mechanism is N17 helix bundle formation that could bring polyQ regions from different proteins into close proximity (20, 21, 24–26). Other studies, however, have challenged such a mechanism as substituting the N17 with a coiled-coil helical bundle reduced aggregation (27, 28). In contrast to the N17, the PRD generally seems to retard aggregation (15, 29, 30), but the molecular underpinnings of this inhibitory function remain poorly understood. Compared with the body of work on Httex1-mimetic peptides, little is known about the aggregation of intact Httex1 protein. Such studies require seed-free, monomeric Httex1 as starting material, but typical aggregation protocols start with a fusion protein and trigger aggregation by enzymatic cleavage of the solubilizing fusion partner. Unfortunately, this in situ generation of monomeric Httex1 produces monomers in an unsynchronized manner over an extended period of time, and it is also prone to contaminations from oligomers and other misfolded species. An additional problem has been the highly repetitive sequence of Httex1 that has complicated site-specific spectroscopic analyses.
These issues have also hampered efforts to evaluate the potential effects of aggregation modulators, such as lipid membranes. Studies from other amyloid proteins revealed that lipid membranes (31–34) as well as lipid-like risk factor molecules (33) can strongly promote aggregation. Httex1 can interact with membranes via an amphipathic helix formed by N17 (35–38), and huntingtin has several membrane-mediated functions, including intracellular vesicle trafficking and autophagy (39, 40). Interestingly, Httex1 has been shown to co-aggregate with lipids in transgenic mouse models (39, 41). Considering the prevalence of Htt membrane interaction in health and disease, it is important to understand whether membranes modulate the aggregation of Httex1.
To study the aggregation of Httex1 in solution and on membranes, we 1) developed a Httex1 expression and purification protocol that does not require enzymatic cleavage to trigger aggregation and that yields clean monomeric proteins. 2) Moreover, we adapted a combination of biophysical techniques, including EPR and fluorescence, to obtain site-specific temporal information of the aggregation process. This approach enabled us to map the stepwise aggregation landscapes in solution and on membranes, which, despite being entirely different, are governed by the N17. Thus, the N17 is a pivotal target for inhibiting multiple aggregation pathways.
Results
EPR kinetics reveal domain-specific aggregation behavior for Httex1
Httex1 derivatives were first prepared as an N-terminal thioredoxin fusion protein. The thioredoxin fusion partner was then removed enzymatically, and the resulting Httex1 was purified using reversed phase chromatography (Fig. S1a). The protein gave rise to a single band on a gel (Fig. S1b) with a position similar to that previously reported (42) and with its correct molecular weight confirmed by mass spectrometry. According to fluorescence correlation spectroscopy (FCS), freshly generated Httex1 with 46 Gln residues (Httex1(Q46)) was initially monomeric at a concentration of 15 μm (Fig. S1c). This notion was further supported by nearly superimposable FCS traces obtained at sub-saturating Httex1 concentrations (43). ThT fluorescence indicated, however, that the monomer was only metastable at 15 μm, as Httex1(Q46) slowly misfolded over a period of hours (Fig. S2a). These kinetics were not significantly affected by incorporation of spin labels. In agreement with prior studies (42, 44), we found that the aggregation of Httex1 can be fitted to single exponential decay kinetics (see under “Experimental procedures”). Such fits resulted in rather similar rate constant (kf) ranging from 0.13 to 0.22/h. The mean kf (estimated via averaging the kf for individual mutants) and the kf for unlabeled Httex1(Q46) were nearly identical at ∼0.17 and ∼0.18/h, respectively (Fig. S2b). Fibril formation was further verified by electron microscopy (Fig. S2c).
To examine the structural changes of different regions during aggregation, we monitored the EPR signals of Httex1(Q46) derivatives with spin labels (Fig. 1a) either in the N17 (3R1, 9R1, and 15R1), the polyQ (35R1 and 48R1), or the PRD (81R1 and 101R1). The readout was based on the reduction in EPR signal amplitude that occurs as different regions of the dynamic monomer oligomerize and become less dynamic (19). Representative EPR spectra for different labeling positions at t = 0 min (blue) and t = 20 h (orange) are shown in Fig. S3a. The time-dependent line broadening and amplitude loss in these spectra are consistent with a loss in mobility as the dynamic monomer (t = 0 min) aggregates over time. Overall, this effect was most pronounced in the N17 and the polyQ, consistent with our prior studies indicating that these regions are predominantly becoming ordered upon oligomerization and fibril formation (19–21).
Figure 1.
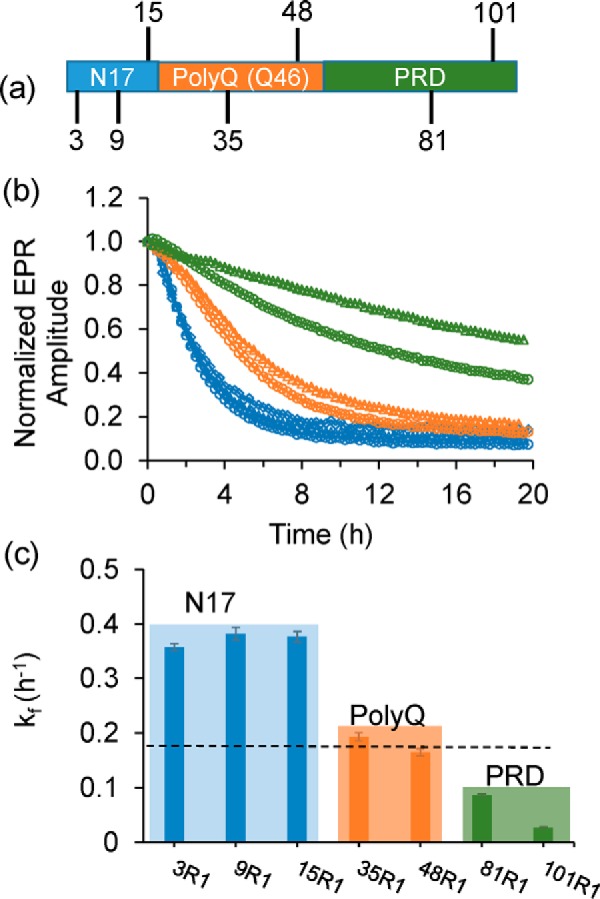
Aggregation time course of Httex1(Q46) monitored via EPR spectroscopy. a, schematic representation of the domain organization of Httex1(Q46) highlighting the positions at which the spin-labeled side chain R1 was introduced. b, time-dependent normalized EPR amplitudes for R1-containing spin-labeled Httex1(Q46) derivatives are given as fraction of initial amplitude. The traces for the N-terminal labeling sites, 3R1 (squares), 9R1 (triangles), and 15R1 (circles), are shown in blue and have a faster signal decay than the polyQ sites, 35R1 (circles) and 48R1 (triangles), which are given in orange. The slowest signal change is observed for labeling sites in the PRD (green), 81R1 (circles) and 101R1 (triangles). c, corresponding rate constants were obtained by fitting the kinetic traces in b to a single exponential decay. The dashed line represents the rate constant of native, unlabeled Httex1 obtained from ThT measurements (Fig. S2a). Error bar represents standard deviation.
Next we sought to time-resolve this process. As shown in Fig. 1b, the amplitudes decreased at different rates for the different labeling positions. The fastest kinetics were observed for sites in the N17 region, where detectable structural changes were nearly complete after 4 h with rate constants (kf) from 0.36 to 0.38/h. In contrast, the slowest kinetics were seen for sites in the PRD, where the spectral changes were still not complete during the time scale of the measurements (20 h) giving rise to kf values from 0.02 to 0.08/h. The kinetics for sites in the polyQ (kf values from ∼0.16 to 0.19/h) were in between those of the N17 and the PRD. Interestingly, the rates of the structural changes in the polyQ region are closest to those obtained from ThT measurements which, according to the fits of the data in Fig. S2a, were in the range of ∼0.13 to 0.22/h for all derivatives (Fig. S2b) and ∼0.18/h for native, unlabeled Httex1(Q46) (dashed line in Fig. 1c). These data suggest that maturation of the polyQ region is responsible for the structural changes that result in an enhanced ThT fluorescence, in agreement with the notion that the polyQ region harbors the β-sheet containing core of the fibrils. Collectively, these data indicate that Httex1(Q46) misfolds in a stepwise process, where the structuring of the N17 precedes that of the polyQ, whereas the PRD continues to mature even after the β-sheet is formed.
To evaluate whether Httex1 with non-pathogenic Gln length undergoes similar stepwise structural transitions, we repeated the EPR experiments with spin-labeled Httex1(Q25) derivatives. Interestingly, no significant EPR spectral changes could be observed in the N17 or the polyQ within a 20-h time frame (Fig. S3b), indicating that the aforementioned steps in the aggregation pathway are Gln length–dependent. Thus, even the early transitions, in which the N17 became ordered, were stabilized by an expanded polyQ that was not yet completely structured.
α-Helical oligomers form prior to β-sheet formation
To determine the secondary structure changes during aggregation, we performed circular dichroism measurements. At t = 0 min, Httex1(Q46) yielded a CD spectrum with a minimum at ∼205 nm (Fig. 2a), consistent with previously reported CD data, which revealed a mixture of random coil, polyproline II, and α-helical structure for monomeric Httex1(Q46) (45, 46). Over a period of 20 h, the signal intensity gradually decreased, and the minimum shifted to 214 nm. This spectrum was nearly identical to that previously reported for Httex1(Q46) fibrils (19). Those spectra had been fitted using the DichroWeb suite (47) of fitting programs giving rise to estimates of 35–45% β-sheet (assigned to polyQ) and 5–10% α-helical structure (assigned to N17). The remainder was attributed to random coil and polyproline II (assigned to PRD) (19).
Figure 2.
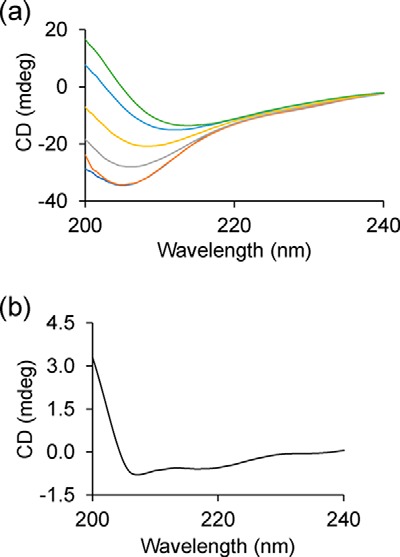
Secondary structural changes during Httex1(Q46) aggregation monitored by CD. a, CD spectra of native, unlabeled Httex1 obtained at different time points (0 min, dark blue; 30 min, orange; 2.5 h, gray; 5 h, yellow; 10 h, light blue; 20 h, green). b, difference spectrum for the t = 0-min and t = 30-min time points shows α-helical character as judged by the peaks at 208 and 222 nm. The concentration was 15 μm. The data are given in millidegrees and were not converted to MRE, as the starting concentrations do not accurately describe the behavior during self-association and aggregation.
To characterize the early structural changes during N17 ordering, the difference spectrum for the two earliest time points (t = 0 min versus t = 30 min, Fig. 2b) was created. The generation of this difference spectrum was facilitated by the fact that the aggregation-dependent loss in CD signal intensity, which is commonly observed during fibril formation (48), had not yet occurred in the first 30 min. Inasmuch as the ordering in the N17 was only ∼10% complete after 30 min according to the EPR data, it was not surprising that the change in the CD spectra was relatively small at this early time point. Nonetheless, this difference spectrum exhibited clearly detectable minima at 208 and 222 nm. These changes are characteristic of the α-helical structure, indicating that the predominant structural change in the first 30 min is α-helix formation. Based on a change in the MRE at 222 nm of −261.11 degrees cm2 dmol−1 and a 10% completion of structuring in the N17 after 30 min, we estimate that on the order of 10 amino acids become helical (also see “Experimental procedures”). Considering that the N17 already contains residual helical structure in the monomer, these data are consistent with a significant fraction (if not all) of the N17 becoming α-helical. Together with the EPR data, this result suggests that the initial structuring of the N17 corresponds to α-helix formation and that the β-sheet formation in the polyQ region occurs at a later time.
Next, we used FCS to further characterize the oligomerization state at early time points. As shown in Fig. S4a, FCS revealed the emergence of increasing amounts of oligomers during the first 2 h. Comparison of the diffusion times of these oligomers (220–260 μs) to those of the monomer (115 μs) indicated that oligomers contain 7–11 subunits. This size estimate from FCS is in reasonable agreement with negative stain EM images that reveal oligomeric species predominantly with an average diameter of 6.7 ± 1.1 nm (Fig. S4b). Assuming an average protein density and spherical oligomer structure, the average size corresponds to ∼10 subunits. The generation of an oligomeric species during the early stages of N17 α-helix formation is also consistent with the EPR spectra. Although the EPR spectra at t = 0 h, like those previously described for the Httex1 monomers (19), were devoid of strongly immobilized components, the later time points were very different. To investigate the structural features of intermediates, we generated difference spectra for early time points (30 min and 2 h, Fig. S5a) by subtraction of the t = 0 time points. The corresponding spectra for 3R1, 9R1, and 15R1 had significantly immobilized components with spectral widths over 60 G. The strongly reduced rotational movement indicated by these spectral widths would not be expected for a fast tumbling, relatively small Httex1 monomer (∼14 kDa). Rather, the spectra are consistent with oligomer formation. In fact, the difference spectra at early time points are highly similar to those from the later time points (Fig. S5b) suggesting that the overall structural features within the N17 remain largely unchanged during the subsequent aggregation steps.
External seeds promote aggregation of Httex1
Seeding has become an important aspect of many amyloid diseases as it could promote aggregation and cell-to-cell spreading (49–51). Thus, seeding could be of significant pathological significance. To test for the effect of seeding in our case and to investigate whether and how it will impact the stepwise mechanism described above, we repeated the experiments in the presence of seeds. As shown in Fig. 3, a and b, the addition of 10% (1.5 μm) seeds significantly sped up the ThT and EPR kinetics for the 35R1 derivative, giving rise to nearly identical rates (0.63 and 0.64/h, respectively). Thus, external seeds strongly accelerated aggregation. Interestingly, the EPR kinetics of a labeling site in the N17 (9R1) were nearly superimposable with those obtained for 35R1 (Fig. 3c). Thus, the stepwise mechanism, where the N17 becomes ordered prior to β-sheet formation, is circumvented in the presence of seeds.
Figure 3.

Effect of seeding on Httex1(Q46) aggregation. a, ThT fluorescence kinetics for the 15 μm 35R1 Httex1(Q46) derivative in the presence (red) and absence (black) of 10% (1.5 μm) seeds. b, acceleration of Httex1(Q46) aggregation in the presence of seeds (red) monitored via EPR kinetics for 35R1 derivative. The control without seed is shown in black. c, EPR-based kinetics for spin-labeled Httex1(Q46) derivatives (9R1, blue curve; 35R1, red curve) show very similar kinetics, indicative of similar rates of aggregation in the respective regions.
Membranes catalyze Httex1(Q46) aggregation by altering the pathway
Next, we tested whether membranes affect the aggregation of Httex1(Q46). For these measurements, we focused on the 35R1 derivative, which reports on β-sheet formation in the polyQ core region. As shown in Fig. 4a, the ThT fluorescence in the presence of membranes (25% POPS/75% POPC) increased rapidly within just 1 h. This time course is mirrored by the change in the EPR amplitude over time (Fig. 4b). Remarkably, the kinetics observed by ThT and EPR were not only ∼6-fold faster than those in solution, but they were even faster than those in the presence of exogenous seeds.
Figure 4.
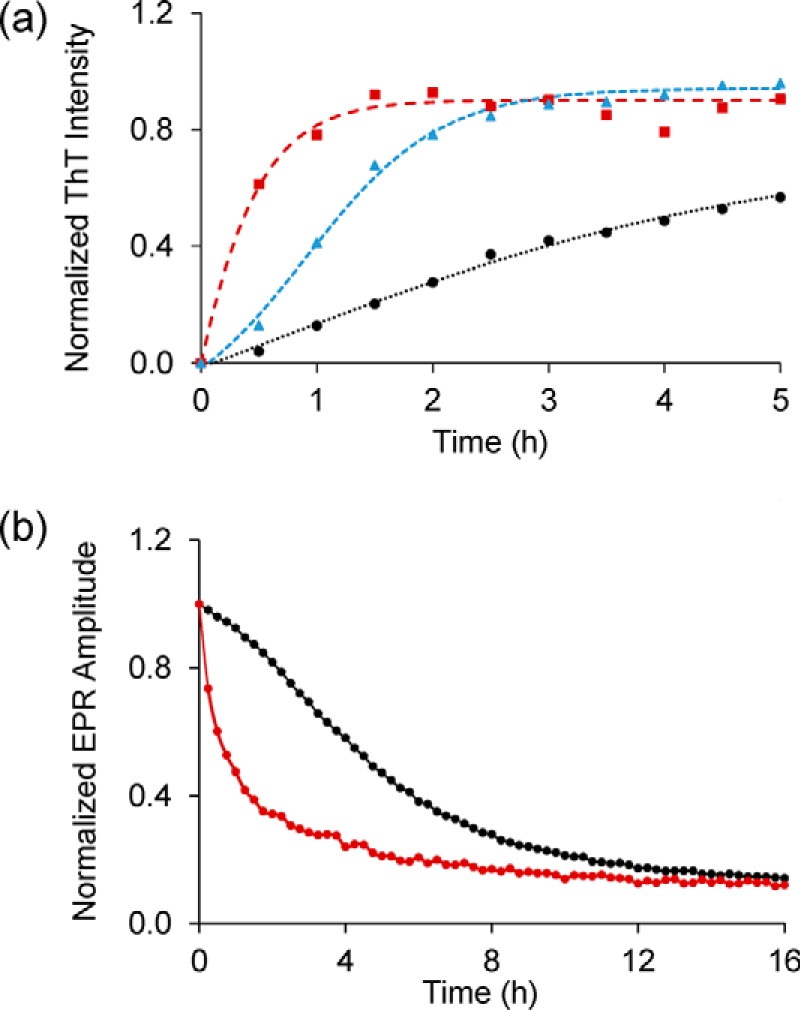
Membranes strongly enhance aggregation of Httex1(Q46). a, ThT fluorescence kinetics for 35R1 Httex1(Q46) derivative in the presence (red) or absence (black) of vesicles (25% POPS/75% POPC, 375 μm total lipid concentration). Also shown for comparison are the seeded kinetics from Fig. 3 (blue). b, membrane-mediated enhancement in aggregation can also be detected by EPR using the 35R1 derivative of Httex1(Q46). The control without membranes is shown in black, whereas the EPR data obtained in the presence of 25% POPS/75% POPC vesicles (375 μm) are in red. The protein concentration was 15 μm in all cases.
Prior studies using isolated N17 peptides indicated that this region can bind to membranes by forming an amphipathic α-helix (36, 52–54). To test whether the N17 can anchor Httex1 to the membrane and initiate aggregation, we compared the EPR spectra for freshly prepared N17-labeled derivatives (3R1, 9R1, and 15R1) in the presence and absence of membranes. Detectable differences could be seen for all sites (Fig. S6a) with membrane-binding inducing spectral components similar to those previously observed in amphipathic, membrane-bound helices of α-synuclein and IAPP (55–57). In contrast, no early spectral changes could be observed for 35R1 or 81R1, indicating that these regions did not partake in the initial ordering on membranes (Fig. S6b). Similar results were obtained for Httex1(Q25), which does not undergo subsequent aggregation under the present conditions (Fig. S7, a and b). To further verify α-helix formation, we performed CD measurements. Although the rapid increase in light scattering complicated such experiments for Httex1(Q46), CD of Httex1(Q25) showed that the incubation of the protein with membranes enhanced helicity (Fig. S7, c and d). Collectively, these data support the notion that early membrane interaction of Httex1 is mediated by an α-helical, membrane-bound N17.
To further verify that rapid aggregation occurred in the presence of membranes, we investigated sample morphology after 3 h by electron microscopy (Fig. S8). Fibrils with a clustered appearance were commonly observed at this early time point. Vesicles were remarkably absent, however, indicating that the addition of Httex1(Q46) leads to the disruption of vesicle integrity (37). Instead of vesicles, amorphous structures that could be remnants of lipid membranes (Fig. S8, black arrows) were frequently observed in the proximity of fibrils.
To examine whether the membrane-mediated enhancement is affected by lipid composition, we reduced the amounts of the negatively charged POPS to 10%. The change strongly reduced the effect of membrane-mediated aggregation (Fig. S6c) indicating that lipid composition, and presumably negative charge density, plays an important role.
Discussion
We present a comprehensive analysis of three different Httex1 aggregation pathways. In solution, aggregation is relatively slow, but it can be strongly accelerated by pre-formed seeds or lipid membranes. All of these reactions proceed via very different pathways, where the N17 plays entirely different roles.
In the absence of seeds and membranes, the N17 is the first region to become ordered. This ordering coincides with oligomerization (according to FCS and EPR) and an increase in α-helical structure (according to CD), suggesting that the early oligomer is stabilized by N17 α-helices. The process of early oligomer formation is Gln length–dependent as no time-dependent structural changes could be observed for Httex1(Q25). The N17 remains ordered throughout the aggregation process with only minor EPR spectral changes upon transition from the helical intermediate to fibrils. This strongly suggests that the early helix-rich intermediate is on-pathway to fibril formation (Fig. 5). This notion is consistent with the reported α-helical structure of the N17 in fibrils (17, 21). Although it is not known exactly how the helical bundles are arranged in oligomers or fibrils, it is clear that the N17 bundles promote aggregation more efficiently than the previously tested coiled coils (28). This could be caused by different arrangements of the polyQ regions with respect to each other. It is also possible that the larger size (7–11 subunits estimated by FCS) could facilitate aggregation by bringing more polyQ regions into close proximity. Regardless of the precise details by which the transition from α-helix-rich oligomers to early α-helix and β-sheet oligomers occurs, our seeding data indicate that the activation barriers associated with the first two steps can be circumvented by aggregation seeds (Fig. 5).
Figure 5.

Representation of three different Httex1 aggregation pathways. a, Httex1(Q46) aggregation in solution is initiated by an α-helix-rich early oligomer, in which the N17 (blue) increases in α-helicity. This early oligomer contains 7–11 subunits as estimated by FCS. Subsequently, this early oligomer gradually transforms into α-helix/β-sheet-rich intermediates where the polyQ region takes on β-sheet structure. The size distribution of this oligomer was not established in this study and is arbitrarily depicted as a 10-mer. The formation of fibrils and bundled fibrils enhances the crowding for the PRD bristles (green), leading to a slow reduction in mobility. b, presence of seeds circumvents the need for intermediate steps shown in a and leads to enhanced aggregation. c, membrane-mediated aggregation begins with the membrane insertion of the N17, leading to an increase in local protein concentration and enabling encounters via two-dimensional diffusion.
In contrast to the early ordering of the N17, the maturation of the PRD represents the slowest phase, which continues long after the ordering, and β-sheet formation in the polyQ has been completed. Solid-state NMR and EPR data suggest that the PRD structure in monomers and fibrils is remarkably similar, containing large amounts of polyproline II helical structure in both cases (19–21). Thus, the reduction in EPR amplitude for sites in the PRD is likely dominated by changes in dynamics due to crowding rather than significant secondary structure changes. Increased crowding of the PRD region could occur at multiple steps, including the transition from smaller β-sheet-containing oligomers to fibrils as well as the formation of more bundled fibrils (21, 22). These crowding steps are likely to be important for the aggregation-inhibiting properties of the PRD.
The N17 also plays an important role in the presence of membranes, where it forms an amphipathic helix (35–38) that initially anchors Httex1 to the membrane. The enhanced local concentration and the two-dimensional diffusion on membranes then enhances encounters between aggregation-prone polyQ regions from different molecules. This mechanism, which is related to that of IAPP and α-synuclein aggregation in the presence of membranes (31, 58, 59), affords an astonishing enhancement of the aggregation kinetics. In addition to generating misfolded and potentially toxic species, membrane-mediated aggregation also caused membrane disruption as judged by EM. Such disruption of membrane integrity could further contribute to toxicity and has been invoked as a mechanism of pathogenesis in Huntington's disease as well as other amyloid diseases (37, 60–62). Considering that Httex1 interacts with a number of cellular membranes and that Htt-lipid co-aggregates are found in transgenic mouse models (39, 41), membrane-mediated aggregation and the concurrent membrane damage is likely to occur in vivo.
Our results illustrate the complexity of Httex1 aggregation (Fig. 5) and the challenges this could represent for the development of therapeutics aimed at inhibiting aggregation. In the case of Tafamidis, a misfolding and aggregation inhibitor for transthyretin-related amyloidosis, it proved advantageous to target an early aggregation step (63). For aggregation in solution, this means that there are three potential steps of interest: 1) helix bundle formation; 2) conversion into a seeding competent structure; and 3) elongation from a seed (Fig. 5). However, even if those steps are blocked, membrane-mediated aggregation could still occur. It may therefore be necessary to simultaneously block various aggregation pathways. One feature that both the solution and membrane-mediated aggregation pathways have in common is their reliance on the N17, making this region an ideal target for blocking multiple pathways at once. Phosphorylation at positions 13 and 16 in the N17 has been shown to protect from toxicity (64, 65). Prior studies have already shown that phosphorylation at these sites slows down aggregation in solution (64, 65). It is very likely that the addition of two negatively charged phosphates in the N17 also strongly reduces the N17's affinity for negatively charged membranes, thereby inhibiting membrane-mediated aggregation and membrane damage.
The methodology and the comprehensive aggregation map presented here will enable future studies aimed at determining how post-translational modifications or other modulators affect aggregation. Not only will it be possible to map which pathways potential aggregation modulators affect most, but it will also be possible to determine which specific step in a given pathway they act on. This should help decipher the relative importance of the various pathways as well as facilitate future therapeutic efforts aimed at inhibiting aggregation, especially if it will become possible to determine the detailed three-dimensional structures of the various misfolded intermediates.
Experimental procedures
Expression and purification of Httex1 fusion protein
Thioredoxin fusion proteins of Httex1 with 46- or 25-amino acid-long glutamine stretches (Q46 or Q25, respectively) were expressed and purified using a slightly modified version of a previous expression protocol (19, 20). Httex1 does not naturally contain Cys residues. The native Cys-free protein as well as single Cys mutants for subsequent labeling to give spin-labeled side chain R1 were generated by mutagenesis and expressed using pET32a-HD46Q. The plasmids were transformed into BL21(DE3) cells, and starter cultures were grown at 37 °C for 4 h. These cultures were then used to inoculate a 50-fold excess of LB media and allowed to grow further at 37 °C in an incubator shaker at 225 rpm to A600 nm = 0.7–0.8. Protein expression was induced overnight by the addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside at 18 °C. Cells were harvested by centrifugation at 4500 × g and resuspended in 20 mm Tris-HCl, pH 7.4, 300 mm NaCl, and 10 mm imidazole containing 1% Triton X-100 (Sigma). After incubation for 20 min at room temperature on a rocker in the presence of 1 mm DTT, lysates were separated from cell debris by centrifugation at 19,000 × g for 20 min and incubated with NiHis60 superflow resin (Clontech) for 45 min. The resin was washed with 5–6 column volumes of 20 mm Tris-HCl, pH 7.4, 300 mm NaCl, 20 mm imidazole, 1 mm DTT. The final washing was performed using the same buffer, except without DTT. Finally, the protein was eluted with 20 mm Tris-HCl, pH 7.4, 300 mm NaCl, 300 mm imidazole buffer. Immediately after elution, the recombinant protein was spin-labeled using a 5-fold molar excess of the 1-oxyl-2,2,5,5 tetramethyl-Δ3-pyrroline-3-methylmethanethiosulfonate spin label for 45 min at room temperature to give the spin-labeled side chain R1, as described earlier (19, 20). Excess spin label was removed using HiTrap Q XL ion-exchange chromatography (GE Healthcare).
Preparation and purification of Httex1 monomers and seeds
To cleave the thioredoxin fusion tag, 5 μm solutions of spin-labeled or unlabeled Httex1 fusion proteins were digested with EKMax (Invitrogen) for 50 min. 1 unit of EKMax was used for each milliliter of reaction volume. The reaction was stopped by the addition of 4 m urea. The subsequent isolation of Httex1 from the mixture was achieved on a reversed phase Phenomenex C4 column (15 μm, 300 Å, 250 × 4.60 mm) using an AKTA FPLC system (Amersham Biosciences) with buffer A (99.9% water, 0.1% trifluoroacetic acid) and buffer B (90% acetonitrile, 9.9% water, 0.1% trifluoroacetic acid). The Httex1 fractions were collected (Fig. S1a) and subsequently lyophilized. The Httex1 powder was stored in a vacuum desiccator in low protein-binding tubes (Eppendorf) until further use. The correct mass of the purified Httex1 was confirmed by electrospray ionization-mass spectrometry, and the purity was confirmed via SDS-PAGE. We noticed that over-digestion of Httex1 can occur for protein concentration above 10 μm, but it can be avoided at the lower concentrations used here. Regardless, our purification method allowed us to purify the correct product and separate it from any over-digested species as they had significantly different migration properties on the Phenomenex C4 column. Seeds were prepared from unlabeled Cys-free Httex1(Q46) fibrils harvested at 15,000 × g for 15 min, fragmented using sonication, and stored at 4 °C. The subsequent experiments to monitor the aggregation pathway of Httex1 variants prepared in this section were performed at 25 °C.
Vesicle preparation
Lipid vesicles were made by mixing 25% of POPS and 75% of POPC (Avanti Polar Lipids). Chloroform was removed by a gentle stream of N2 gas and vacuum-desiccated overnight. The lipid film was resuspended into 20 mm Tris, 150 mm NaCl, pH 7.4, to a final concentration of 8 mg/ml and subjected to 10 freeze-thaw cycles. The size of the vesicles was standardized using a mini-extruder containing a polycarbonate filter (Avanti) of 100 nm diameter. The protein to lipid molar ratio for all experiments was 1:25. For circular dichroism experiments using membrane-bound Httex1(Q25), vesicles were sonicated to reduce scattering.
Thioflavin T(ThT) fluorescence
The lyophilized samples in a low protein-binding tube were treated with 0.5% TFA (v/v) in methanol to solubilize and disaggregate the Httex1 powder. The organics were removed under a gentle stream of N2 gas to obtain a thin film of Httex1 at the bottom of the tube. The film was dissolved in 20 mm Tris, 150 mm NaCl, pH 7.4, to a desired concentration of 15 μm. This protocol resulted in clean monomeric protein (also see under “Results”). The aggregation kinetics of purified Httex1 derivatives were measured by ThT fluorescence using an Eppendorf Plate Reader AF2200 equipped with appropriate filters in a Falcon 96-well (clear flat bottom) plate, over a period of 20 h at 25 °C. The excitation and emission wavelengths were kept at 440 and 484 nm, respectively, with a slit width of 20 nm each. The fluorescence of all samples was recorded at least in triplicate. The ThT concentration was kept at 50 μm in all cases. The data were baseline-corrected using a no protein control and fitted to a single exponential using Origin 7.0. The data were fitted using y = A × exp(−x/t) + y0, where y0 = Y offset, A = amplitude, t = time constant (h). The rate constant of fibril formation (Kf) was estimated by the following equation: Kf= (1/t). x and y were treated as variables in the fit. Origin 7.0 fitting wizard automatically adjust the data set for the abovementioned equation.
Continuous wave-EPR measurements
The samples for EPR measurements were prepared from lyophilized powder as described for ThT sample preparation. Samples were loaded into Boro Glass Tubing (0.6 mm inner diameter × 0.84 mm outer diameter, Vitro-Com, Mt. Lakes, NJ), and the EPR spectra were obtained using an X-band Bruker EMX spectrometer equipped with a Bruker ER4119HS resonator at 25 °C. The scan width for all spectra was 100 G with an incident power of 12.7 milliwatts. The central line amplitude was obtained from the EPR spectra using WinAcquisit (Bruker Biospin). EPR was also used to verify the concentrations of all the labeled samples. This was done by double integration of the EPR spectra and comparison with concentration standards. Like the ThT data, the EPR kinetics were fitted to a single exponential function in Origin 7.0.
Far UV-CD
The far UV-CD measurements were performed using a Jasco-810. The spectra were accumulated at 15 μm protein concentration in 20 mm Tris, 150 mm NaCl, pH 7.4, between 200 and 240 nm at a scan rate of 50 nm/min using a quartz cuvette of 0.1-cm path length. Difference spectra were obtained using the Spectra Analysis tool provided with the Jasco-810. The same program was also used for smoothing of the data. To fit the percent α-helicity changes at 30 min, we employed the same method previously employed for IAPP and Httex1 α-helix formation (31, 46). Helicity changes were estimated using fH = (θ222 − θC)/(θH − θC). The resulting change was converted into percent helicity and based on a total number of amino acids of 119, the percent increase in α-helicity was converted into number of α-helical amino acids.
Transmission electron microscopy
10 μl of each sample was adsorbed onto 150 mesh carbon-coated copper grids. The grids containing samples were then negatively stained using 1% uranyl acetate for 1 min. The images were acquired using JEOL JEM-1400 transmission electron microscope at 100 kV.
Fluorescence correlation spectroscopy (FCS)
FCS measurements were recorded on a laser-scanning confocal microscope Zeiss LSM 780 (Zeiss, Jena, Germany). Samples were excited with a 488-nm argon-ion laser line using a water-immersion objective (Zeiss C-Apochromat, 40×/1.2 W UV-Vis-IR, Zeiss, Jena, Germany). The pinhole diameter was set to 1 Airy unit, and pinhole x-y positions were adjusted for maximum fluorescence intensity before measurements. Fluorescence was separated from excitation using an MBS488 beam splitter. Emission was recorded between 505 and 580 nm using a built-in detector. To avoid detector saturation, laser intensity was kept at low levels, and the unlabeled to rhodamine 110-labeled protein ratio was kept low (300:1). However, for Httex1 monomer recordings at sub-saturated nanomolar concentrations, the sample was fully labeled. A dilute solution of rhodamine 110 (τD of ∼47 μs) was used for obtaining the structural parameter of the confocal volume that was used for subsequent fitting of correlation curves of protein samples. For each sample, at least 10 traces, each 30 s, were recorded. The FCS data were fitted to one- or two-component models using the open source program PyCorrFit (66). The size of the oligomers compared with that of monomer was estimated using the dependence of τD on the cube root of molecular weight as described in the literature (67).
Author contributions
R. L. and N. K. P. designed research; N. K. P., J. M. I., A. R., R. V. L., J. L., and P. P. performed research; R. L., A. R., and N. K. P. analyzed data; R. L. and N. K. P. wrote the paper.
Supplementary Material
This work was supported by National Institutes of Health Grants NS084345, NS089932, and GM115736. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S8 and supporting Refs. 1–2.
- HD
- Huntington's disease
- polyQ
- polyglutamine
- N17
- N terminus containing 17 amino acids
- PRD
- proline-rich domain
- FCS
- fluorescence correlation spectroscopy
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- ThT
- thioflavin T
- MRE
- mean residue ellipticity
- IAPP
- islet amyloid polypeptide.
References
- 1. MacDonald M. E., Ambrose C. M., Duyao M. P., Myers R. H., Lin C., Srinidhi L., Barnes G., Taylor S. A., James M., Groot N., MacFarlane H., Jenkins B., Anderson M. A., Wexler N. S., Gusella J. F., et al. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- 2. Sathasivam K., Neueder A., Gipson T. A., Landles C., Benjamin A. C., Bondulich M. K., Smith D. L., Faull R. L., Roos R. A., Howland D., Detloff P. J., Housman D. E., and Bates G. P. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U.S.A. 110, 2366–2370 10.1073/pnas.1221891110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ross C. A. (2002) Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron 35, 819–822 10.1016/S0896-6273(02)00872-3 [DOI] [PubMed] [Google Scholar]
- 4. Saunders H. M., and Bottomley S. P. (2009) Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins. Protein Eng. Des. Sel. 22, 447–451 10.1093/protein/gzp033 [DOI] [PubMed] [Google Scholar]
- 5. Mangiarini L., Sathasivam K., Seller M., Cozens B., Harper A., Hetherington C., Lawton M., Trottier Y., Lehrach H., Davies S. W., and Bates G. P. (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87, 493–506 10.1016/S0092-8674(00)81369-0 [DOI] [PubMed] [Google Scholar]
- 6. DiFiglia M., Sapp E., Chase K. O., Davies S. W., Bates G. P., Vonsattel J. P., and Aronin N. (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 10.1126/science.277.5334.1990 [DOI] [PubMed] [Google Scholar]
- 7. Legleiter J., Lotz G. P., Miller J., Ko J., Ng C., Williams G. L., Finkbeiner S., Patterson P. H., and Muchowski P. J. (2009) Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J. Biol. Chem. 284, 21647–21658 10.1074/jbc.M109.016923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morley J. F., Brignull H. R., Weyers J. J., and Morimoto R. I. (2002) The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U.S.A. 99, 10417–10422 10.1073/pnas.152161099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scherzinger E., Sittler A., Schweiger K., Heiser V., Lurz R., Hasenbank R., Bates G. P., Lehrach H., and Wanker E. E. (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology. Proc. Natl. Acad. Sci. U.S.A. 96, 4604–4609 10.1073/pnas.96.8.4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen S., Berthelier V., Yang W., and Wetzel R. (2001) Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J. Mol. Biol. 311, 173–182 10.1006/jmbi.2001.4850 [DOI] [PubMed] [Google Scholar]
- 11. Lee J. M., Ramos E. M., Lee J. H., Gillis T., Mysore J. S., Hayden M. R., Warby S. C., Morrison P., Nance M., Ross C. A., Margolis R. L., Squitieri F., Orobello S., Di Donato S., Gomez-Tortosa E., et al. (2012) CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology 78, 690–695 10.1212/WNL.0b013e318249f683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kar K., Jayaraman M., Sahoo B., Kodali R., and Wetzel R. (2011) Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat. Struct. Mol. Biol. 18, 328–336 10.1038/nsmb.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramdzan Y. M., Trubetskov M. M., Ormsby A. R., Newcombe E. A., Sui X., Tobin M. J., Bongiovanni M. N., Gras S. L., Dewson G., Miller J. M., Finkbeiner S., Moily N. S., Niclis J., Parish C. L., Purcell A. W., et al. (2017) Huntingtin inclusions trigger cellular quiescence, deactivate apoptosis, and lead to delayed necrosis. Cell Rep. 19, 919–927 10.1016/j.celrep.2017.04.029 [DOI] [PubMed] [Google Scholar]
- 14. Shen K., Calamini B., Fauerbach J. A., Ma B., Shahmoradian S. H., Serrano Lachapel I. L., Chiu W., Lo D. C., and Frydman J. (2016) Control of the structural landscape and neuronal proteotoxicity of mutant huntingtin by domains flanking the polyQ tract. Elife 5, e18065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crick S. L., Ruff K. M., Garai K., Frieden C., and Pappu R. V. (2013) Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U.S.A. 110, 20075–20080 10.1073/pnas.1320626110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jayaraman M., Mishra R., Kodali R., Thakur A. K., Koharudin L. M., Gronenborn A. M., and Wetzel R. (2012) Kinetically competing huntingtin aggregation pathways control amyloid polymorphism and properties. Biochemistry 51, 2706–2716 10.1021/bi3000929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jayaraman M., Kodali R., Sahoo B., Thakur A. K., Mayasundari A., Mishra R., Peterson C. B., and Wetzel R. (2012) Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J. Mol. Biol. 415, 881–899 10.1016/j.jmb.2011.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Büning S., Sharma A., Vachharajani S., Newcombe E., Ormsby A., Gao M., Gnutt D., Vöpel T., Hatters D. M., and Ebbinghaus S. (2017) Conformational dynamics and self-association of intrinsically disordered huntingtin exon 1 in cells. Phys. Chem. Chem. Phys. 19, 10738–10747 10.1039/C6CP08167C [DOI] [PubMed] [Google Scholar]
- 19. Bugg C. W., Isas J. M., Fischer T., Patterson P. H., and Langen R. (2012) Structural features and domain organization of huntingtin fibrils. J. Biol. Chem. 287, 31739–31746 10.1074/jbc.M112.353839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Isas J. M., Langen R., and Siemer A. B. (2015) Solid-state nuclear magnetic resonance on the static and dynamic domains of huntingtin exon-1 fibrils. Biochemistry 54, 3942–3949 10.1021/acs.biochem.5b00281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sivanandam V. N., Jayaraman M., Hoop C. L., Kodali R., Wetzel R., and van der Wel P. C. (2011) The aggregation-enhancing huntingtin N terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 133, 4558–4566 10.1021/ja110715f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoop C. L., Lin H. K., Kar K., Hou Z., Poirier M. A., Wetzel R., and van der Wel P. C. (2014) Polyglutamine amyloid core boundaries and flanking domain dynamics in huntingtin fragment fibrils determined by solid-state nuclear magnetic resonance. Biochemistry 53, 6653–6666 10.1021/bi501010q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Isas J. M., Langen A., Isas M. C., Pandey N. K., and Siemer A. B. (2017) Formation and structure of wild type Huntingtin exon-1 fibrils. Biochemistry 56, 3579–3586 10.1021/acs.biochem.7b00138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thakur A. K., Jayaraman M., Mishra R., Thakur M., Chellgren V. M., Byeon I. J., Anjum D. H., Kodali R., Creamer T. P., Conway J. F., Gronenborn A. M., and Wetzel R. (2009) Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct. Mol. Biol. 16, 380–389 10.1038/nsmb.1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tam S., Spiess C., Auyeung W., Joachimiak L., Chen B., Poirier M. A., and Frydman J. (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat. Struct. Mol. Biol. 16, 1279–1285 10.1038/nsmb.1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Atwal R. S., Desmond C. R., Caron N., Maiuri T., Xia J., Sipione S., and Truant R. (2011) Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 7, 453–460 10.1038/nchembio.582 [DOI] [PubMed] [Google Scholar]
- 27. Kokona B., Johnson K. A., and Fairman R. (2014) Effect of helical flanking sequences on the morphology of polyglutamine-containing fibrils. Biochemistry 53, 6747–6753 10.1021/bi501066q [DOI] [PubMed] [Google Scholar]
- 28. Kokona B., Rosenthal Z. P., and Fairman R. (2014) Role of the coiled-coil structural motif in polyglutamine aggregation. Biochemistry 53, 6738–6746 10.1021/bi500449a [DOI] [PubMed] [Google Scholar]
- 29. Darnell G. D., Derryberry J., Kurutz J. W., and Meredith S. C. (2009) Mechanism of cis-inhibition of polyQ fibrillation by polyP: PPII oligomers and the hydrophobic effect. Biophys. J. 97, 2295–2305 10.1016/j.bpj.2009.07.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duennwald M. L., Jagadish S., Muchowski P. J., and Lindquist S. (2006) Flanking sequences profoundly alter polyglutamine toxicity in yeast. Proc. Natl. Acad. Sci. U.S.A. 103, 11045–11050 10.1073/pnas.0604547103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jayasinghe S. A., and Langen R. (2005) Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 44, 12113–12119 10.1021/bi050840w [DOI] [PubMed] [Google Scholar]
- 32. Relini A., Cavalleri O., Rolandi R., and Gliozzi A. (2009) The two-fold aspect of the interplay of amyloidogenic proteins with lipid membranes. Chem. Phys. Lipids 158, 1–9 10.1016/j.chemphyslip.2008.11.003 [DOI] [PubMed] [Google Scholar]
- 33. Okada A. K., Teranishi K., Isas J. M., Bedrood S., Chow R. H., and Langen R. (2016) Diabetic risk factors promote islet amyloid polypeptide misfolding by a common, membrane-mediated mechanism. Sci. Rep. 6, 31094 10.1038/srep31094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Varkey J., and Langen R. (2017) Membrane remodeling by amyloidogenic and non-amyloidogenic proteins studied by EPR. J. Magn. Reson. 280, 127–139 10.1016/j.jmr.2017.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kegel K. B., Sapp E., Yoder J., Cuiffo B., Sobin L., Kim Y. J., Qin Z. H., Hayden M. R., Aronin N., Scott D. L., Isenberg G., Goldmann W. H., and DiFiglia M. (2005) Huntingtin associates with acidic phospholipids at the plasma membrane. J. Biol. Chem. 280, 36464–36473 10.1074/jbc.M503672200 [DOI] [PubMed] [Google Scholar]
- 36. Michalek M., Salnikov E. S., Werten S., and Bechinger B. (2013) Membrane interactions of the amphipathic amino terminus of huntingtin. Biochemistry 52, 847–858 10.1021/bi301325q [DOI] [PubMed] [Google Scholar]
- 37. Burke K. A., Hensal K. M., Umbaugh C. S., Chaibva M., and Legleiter J. (2013) Huntingtin disrupts lipid bilayers in a polyQ-length dependent manner. Biochim. Biophys. Acta 1828, 1953–1961 10.1016/j.bbamem.2013.04.025 [DOI] [PubMed] [Google Scholar]
- 38. Chaibva M., Burke K. A., and Legleiter J. (2014) Curvature enhances binding and aggregation of huntingtin at lipid membranes. Biochemistry 53, 2355–2365 10.1021/bi401619q [DOI] [PubMed] [Google Scholar]
- 39. Valencia A., Reeves P. B., Sapp E., Li X., Alexander J., Kegel K. B., Chase K., Aronin N., and DiFiglia M. (2010) Mutant huntingtin and glycogen synthase kinase 3-β accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington's disease. J. Neurosci. Res. 88, 179–190 10.1002/jnr.22184 [DOI] [PubMed] [Google Scholar]
- 40. Martin D. D., Ladha S., Ehrnhoefer D. E., and Hayden M. R. (2015) Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 38, 26–35 10.1016/j.tins.2014.09.003 [DOI] [PubMed] [Google Scholar]
- 41. Suopanki J., Götz C., Lutsch G., Schiller J., Harjes P., Herrmann A., and Wanker E. E. (2006) Interaction of huntingtin fragments with brain membranes–clues to early dysfunction in Huntington's disease. J. Neurochem. 96, 870–884 10.1111/j.1471-4159.2005.03620.x [DOI] [PubMed] [Google Scholar]
- 42. Vieweg S., Ansaloni A., Wang Z. M., Warner J. B., and Lashuel H. A. (2016) An intein-based strategy for the production of tag-free huntingtin exon 1 proteins enables new insights into the polyglutamine dependence of Httex1 aggregation and fibril formation. J. Biol. Chem. 291, 12074–12086 10.1074/jbc.M116.713982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Crick S. L., Jayaraman M., Frieden C., Wetzel R., and Pappu R. V. (2006) Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. U.S.A. 103, 16764–16769 10.1073/pnas.0608175103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Perevozchikova T., Stanley C. B., McWilliams-Koeppen H. P., Rowe E. L., and Berthelier V. (2014) Investigating the structural impact of the glutamine repeat in huntingtin assembly. Biophys. J. 107, 411–421 10.1016/j.bpj.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singer D., Zauner T., Genz M., Hoffmann R., and Zuchner T. (2010) Synthesis of pathological and nonpathological human exon 1 huntingtin. J. Pept. Sci. 16, 358–363 [DOI] [PubMed] [Google Scholar]
- 46. Fodale V., Kegulian N. C., Verani M., Cariulo C., Azzollini L., Petricca L., Daldin M., Boggio R., Padova A., Kuhn R., Pacifici R., Macdonald D., Schoenfeld R. C., Park H., Isas J. M., et al. (2014) Polyglutamine- and temperature-dependent conformational rigidity in mutant huntingtin revealed by immunoassays and circular dichroism spectroscopy. PLoS ONE 9, e112262 10.1371/journal.pone.0112262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Whitmore L., and Wallace B. A. (2008) Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers 89, 392–400 10.1002/bip.20853 [DOI] [PubMed] [Google Scholar]
- 48. Jiang D., Rauda I., Han S., Chen S., and Zhou F. (2012) Aggregation pathways of the amyloid β(1–42) peptide depend on its colloidal stability and ordered β-sheet stacking. Langmuir 28, 12711–12721 10.1021/la3021436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang W., Dunlap J. R., Andrews R. B., and Wetzel R. (2002) Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum. Mol. Genet. 11, 2905–2917 10.1093/hmg/11.23.2905 [DOI] [PubMed] [Google Scholar]
- 50. Ren P. H., Lauckner J. E., Kachirskaia I., Heuser J. E., Melki R., and Kopito R. R. (2009) Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 11, 219–225 10.1038/ncb1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tan Z., Dai W., van Erp T. G., Overman J., Demuro A., Digman M. A., Hatami A., Albay R., Sontag E. M., Potkin K. T., Ling S., Macciardi F., Bunney W. E., Long J. D., Paulsen J. S., et al. (2015) Huntington's disease cerebrospinal fluid seeds aggregation of mutant huntingtin. Mol. Psychiatry 20, 1286–1293 10.1038/mp.2015.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Atwal R. S., Xia J., Pinchev D., Taylor J., Epand R. M., and Truant R. (2007) Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Genet. 16, 2600–2615 10.1093/hmg/ddm217 [DOI] [PubMed] [Google Scholar]
- 53. Michalek M., Salnikov E. S., and Bechinger B. (2013) Structure and topology of the huntingtin 1–17 membrane anchor by a combined solution and solid-state NMR approach. Biophys. J. 105, 699–710 10.1016/j.bpj.2013.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang L., Kang H., Vázquez F. X., Toledo-Sherman L., Luan B., and Zhou R. (2017) Molecular mechanism of stabilizing the helical structure of huntingtin N17 in a micellar environment. J. Phys. Chem. B 121, 4713–4721 10.1021/acs.jpcb.7b01476 [DOI] [PubMed] [Google Scholar]
- 55. Jao C. C., Der-Sarkissian A., Chen J., and Langen R. (2004) Structure of membrane-bound α-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. U.S.A. 101, 8331–8336 10.1073/pnas.0400553101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jao C. C., Hegde B. G., Chen J., Haworth I. S., and Langen R. (2008) Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U.S.A. 105, 19666–19671 10.1073/pnas.0807826105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Apostolidou M., Jayasinghe S. A., and Langen R. (2008) Structure of α-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem. 283, 17205–17210 10.1074/jbc.M801383200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jayasinghe S. A., and Langen R. (2007) Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta 1768, 2002–2009 10.1016/j.bbamem.2007.01.022 [DOI] [PubMed] [Google Scholar]
- 59. Dikiy I., and Eliezer D. (2012) Folding and misfolding of α-synuclein on membranes. Biochim. Biophys. Acta 1818, 1013–1018 10.1016/j.bbamem.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Quist A., Doudevski I., Lin H., Azimova R., Ng D., Frangione B., Kagan B., Ghiso J., and Lal R. (2005) Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U.S.A. 102, 10427–10432 10.1073/pnas.0502066102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fändrich M. (2012) Oligomeric intermediates in amyloid formation: structure determination and mechanisms of toxicity. J. Mol. Biol. 421, 427–440 10.1016/j.jmb.2012.01.006 [DOI] [PubMed] [Google Scholar]
- 62. Bemporad F., and Chiti F. (2012) Protein misfolded oligomers: experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem. Biol. 19, 315–327 10.1016/j.chembiol.2012.02.003 [DOI] [PubMed] [Google Scholar]
- 63. Bulawa C. E., Connelly S., Devit M., Wang L., Weigel C., Fleming J. A., Packman J., Powers E. T., Wiseman R. L., Foss T. R., Wilson I. A., Kelly J. W., and Labaudinière R. (2012) Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U.S.A. 109, 9629–9634 10.1073/pnas.1121005109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mishra R., Hoop C. L., Kodali R., Sahoo B., van der Wel P. C., and Wetzel R. (2012) Serine phosphorylation suppresses huntingtin amyloid accumulation by altering protein aggregation properties. J. Mol. Biol. 424, 1–14 10.1016/j.jmb.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gu X., Greiner E. R., Mishra R., Kodali R., Osmand A., Finkbeiner S., Steffan J. S., Thompson L. M., Wetzel R., and Yang X. W. (2009) Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 64, 828–840 10.1016/j.neuron.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Müller P., Schwille P., and Weidemann T. (2014) PyCorrFit-generic data evaluation for fluorescence correlation spectroscopy. Bioinformatics 30, 2532–2533 10.1093/bioinformatics/btu328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sahoo B., Drombosky K. W., and Wetzel R. (2016) Fluorescence correlation spectroscopy: a tool to study protein oligomerization and aggregation in vitro and in vivo. Methods Mol. Biol. 1345, 67–87 10.1007/978-1-4939-2978-8_5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.