

Abstract
Nuclear factor-κB (NF-κB) is a family of transcription factors that play a key role in cell survival and proliferation in many hematological malignancies, including multiple myeloma (MM). Bortezomib, a proteasome inhibitor used in the management of MM, can inhibit both canonical and noncanonical activation of NF-κB in MM cells. However, we previously reported that a significant fraction of freshly isolated MM cells harbor bortezomib-resistant NF-κB activity. Here, we report that hyaluronan and proteoglycan link protein 1 (HAPLN1) is produced in bone marrow stromal cells from MM patients, is detected in patients' bone marrow plasma, and can activate an atypical bortezomib-resistant NF-κB pathway in MM cells. We found that this pathway involves bortezomib-resistant degradation of the inhibitor of NF-κB (IκBα), despite efficient bortezomib-mediated inhibition of proteasome activity. Moreover, HAPLN1 can also confer bortezomib-resistant survival of MM cells. We propose that HAPLN1 is a novel pathogenic factor in MM that induces an atypical NF-κB activation and thereby promotes bortezomib resistance in MM cells.
Keywords: drug resistance, NF-kappa B (NF-KB), multiple myeloma, molecular biology, extracellular matrix protein, bortezomib, hyaluronan and proteoglycan link protein 1 (HAPLN1), matrikine
Introduction
Multiple myeloma (MM)4 is the second most common hematologic malignancy with over 30,000 new cases diagnosed annually in the United States. Currently, MM is generally considered an incurable disease with a median survival of 5–7 years from diagnosis (1). MM is characterized by the uncontrolled proliferation and accumulation of antibody-secreting plasma cells primarily in the bone marrow (1). It is a complex disease with respect to clinical features, cytogenetic abnormalities, and cellular and molecular factors that contribute to disease development and progression (1–4). High throughput whole-genome and whole-exome sequencing efforts have identified frequent MM cell-intrinsic genetic alterations in the components of several signaling pathways, including RAS/MEK/MAPK, JAK2/STAT3, and nuclear factor-κB (NF-κB) (5–8). In addition to cell-intrinsic changes, the tumor microenvironment (TME) further provides cancer cell–extrinsic influences on MM cells, promoting growth, survival, migration, and drug resistance (9). For example, bone marrow stromal cells (BMSCs) can secrete cytokines, growth factors, and additional factors to create a niche for MM cells to thrive by promoting local expansion, immune evasion, and metastasis (4, 10–12).
Proteasome inhibition by bortezomib/Velcade has been a key strategy in the treatment of both newly diagnosed and relapsed MM. Bortezomib is believed to suppress multiple signaling pathways, including stabilization of cell cycle–inhibitory proteins p21 and p27 (13), tumor-suppressor protein p53 (14), and the inhibitor of NF-κB, IκBα (15). In addition, bortezomib is known to induce the unfolded protein response, which monitors endoplasmic reticulum stress to maintain quality control of proteins and homeostasis (16). MM cells are highly susceptible to proteasome inhibition in vivo, possibly arising from the excessive demand of the endoplasmic reticulum system to maintain the high secretion load of immunoglobulins (17–19). Although the widespread use of proteasome inhibitors has improved response and survival, the majority of MM patients eventually develop refractory disease (1–4). Multiple distinct mechanisms of bortezomib resistance have been described, including point mutations of the 26S PSMB5 (proteasome subunit β type 5), decreased expression of unfolded protein response proteins, and changes in critical pathways, such as β-catenin, insulin growth factor receptor, MAPK, AKT, KRAS, and JAK/STAT (5, 6, 8, 20, 21). Activation of NF-κB has also been implicated in bortezomib resistance (22, 23).
NF-κB is a family of transcription factors that has emerged as a critical pathogenic factor in MM development and drug resistance (24–27). Normally, NF-κB (p50-RelA or cRel complexes) is sequestered in the cytoplasm by its inhibitor proteins, including IκBα and IκBβ. “Canonical” activation of NF-κB requires the engagement of the IκB kinase complex composed of IKKα, IKKβ, and IKKγ/NEMO. The IKK complex then causes site-specific phosphorylation of IκB proteins to cause degradation by the ubiquitin-26S proteasome pathway (28, 29). Alternatively, the “noncanonical” NF-κB activation pathway involves the action of NF-κB–inducing kinase and IKKα and limited processing of NFκB2/p100 to p52 by the 26S proteasome to selectively activate p52-RelB complex (30). Accumulated data, including the analysis of gene expression signatures (31) and NF-κB-RelA immunostaining (32), have shown that NF-κB signaling is constitutively active in MM cells of ∼90% of patients. Although MM cancer cell genome and exome sequencing has identified genetic aberrations in canonical and noncanonical NF-κB signaling components in up to ∼23% of MM cases (5, 6, 8, 31, 33), the mechanisms of NF-κB activation in the remaining 70% of MM cases remain unclear. Such an activation could be dependent on extrinsic factors, such as those derived from the TME.
We previously reported that the constitutive NF-κB activity present in freshly isolated patient-derived MM cells is often resistant to inhibition by bortezomib in vitro (34), and such a bortezomib-resistant NF-κB activity could be further induced in MM cells via a factor that is secreted by BMSCs obtained directly from MM patients (35). This bortezomib-resistant NF-κB activity seems to embody an atypical NF-κB pathway, termed proteasome inhibitor-resistant (PIR), which involves IκBα degradation and NF-κB activation that are highly resistant to inhibition by a variety of proteasome inhibitors, including bortezomib (36–38). Paradoxically, bortezomib has also been shown to cause NF-κB activation while blocking the proteasome activity (34, 39), unlike NF-κB activity in MM cells that had often been attributed to bortezomib-sensitive pathways (26, 27).
In this current study, we identified a BMSC-secreted factor, hyaluronan and proteoglycan link protein 1 (HAPLN1), that can induce bortezomib-resistant NF-κB activity in MM cells. Specifically, HAPLN1 proteoglycan tandem repeat (PTR) domain 1 and 2 fragments have strong PIR NF-κB–inducing activities. This was surprising, because HAPLN1 is an extracellular matrix (ECM) protein well-known for its role in structural support in cartilage formation and other tissue ECM (40, 41), but with no previously characterized direct signaling functions through the PTR domains. Importantly, HAPLN1 PTR fragments confer bortezomib-resistant survival in some MM cells and are frequently detectable in MM patient bone marrow aspirates. Our study reveals a novel inducer of drug-resistant NF-κB activity in MM, which could represent a novel therapeutic target for this currently incurable disease.
Results
HAPLN1 PTR domains activate NF-κB in myeloma cells
We previously demonstrated that MM patient-derived BMSCs can cause bortezomib-resistant NF-κB activity in MM cells through a secreted soluble factor (35). Partial purification of this factor from a stromal cell line was described previously (35). Following SDS-PAGE, Coomassie Blue staining, and mass spectrometry analysis of the enriched fraction F3 (Fig. 1A), we identified four potential factors: calumenin, osteonectin, galectin-3-binding protein, and HAPLN1. cDNAs encoding these proteins were isolated from MM patient BMSCs, cloned into a pSecTag2a vector for efficient secretion, sequence-verified, and transiently transfected into HEK293 cells. Conditioned media (CMs) from transfected HEK293 cells were applied to a MM cell line, RPMI8226, to assay for NF-κB activation by an electrophoretic mobility shift assay (EMSA), whereas the presence of corresponding proteins was verified by immunoblot analysis of the CM (Fig. 1, B–E). Only the HAPLN1-containing CM induced NF-κB activation (Fig. 1E). Importantly, this NF-κB activation was highly resistant to bortezomib treatment (Fig. 1E).
Figure 1.

Identification of HAPLN1 as a factor capable of inducing NF-κB activity in myeloma cells. A, SDS-PAGE and Coomassie Blue staining of the enriched fractions with NF-κB–inducing activity previously identified by Markovina et al. (35). Three bands (*) from fraction 3 (F3) that contained the highest NF-κB–inducing activity were excised and analyzed by nano-LC-MS/MS. Shown are identified factors with the number of unique peptides identified in parentheses. B, EMSA analysis of RPMI8226 cells incubated with 10 ng/ml TNFα for 15 min or CM collected from HEK293 transiently transfected with vector containing C-terminally MYC-tagged proteins: calumenin, osteonectin, or galectin-3–binding protein (Gal-3BP) for 2 h. Results are representative of at least three independent experiments. C, representative immunoblot analysis (IB), using anti-MYC antibody, of CM from HEK293 cells transiently transfected with calumenin, osteonectin, or galectin-3–binding protein expression vector showing the presence of corresponding secreted factors. D, immunoblot analysis, using anti-MYC antibody, of HAPLN1 expression in CM collected from transiently transfected HEK293 cells with the HAPLN1 expression vector positive for induction of NF-κB activity. Smaller HAPLN1 (H1) fragments are indicated *, larger immunoreactive bands. E, EMSA analysis of RPMI8226 cells treated with CM from transiently transfected HEK293 cells containing HAPLN1 for 2 h in the absence or presence of 100 nm bortezomib (Bort). Induced NF-κB activity is highly variable in more than five independent experiments, possibly dependent on the variability of smaller H1 species seen in D.
HAPLN1 is a 45–52-kDa ECM protein containing a signal peptide (SP), one immunoglobulin-like (IG), and two PTR domains (PTR1 and PTR2) (Fig. 2A) (42, 43). We found that full-length human HAPLN1 (FL-H1) (Fig. 2B) was unable to induce NF-κB activation in MM cells (Fig. 2C). ECM proteins can sometimes generate smaller active signaling factors, matrikines, via proteolysis (44). We observed smaller species of HAPLN1 in HEK293 CM (Fig. 1D); therefore, we tested whether subdomains of HAPLN1 were sufficient to cause NF-κB activation. We expressed HAPLN1 subdomains as GST-fused recombinant fragments in Escherichia coli and purified them by GSH-Sepharose chromatography (Fig. 2B). We found that the HAPLN1 PTR1 (H1-P1) and PTR2 (H1-P2) domains activated NF-κB ∼6.6-fold and ∼6.8-fold, respectively (Fig. 2C). The IG domain (H1-IG) caused NF-κB activation but to a much lesser degree than that induced by H1-P1 (Fig. 2C) and therefore was not further pursued here. To rule out the possibility that NF-κB activation was due to the presence of contaminating endotoxin, we measured the amounts of endotoxin in H1-P1 preparations. The levels of contaminating endotoxin were found to be <0.2 ng/ml, below the sensitivity of the NF-κB activation assay in RPMI8226 cells (Fig. 2, D and E).
Figure 2.

Generation and characterization of recombinant HAPLN1 proteins. A, diagram of HAPLN1 domains. The numbers indicate the amino acid positions. SP, signal peptide; IG, immunoglobulin-like domain; PTR1, proteoglycan tandem repeat 1 domain; PTR2, proteoglycan tandem repeat 2 domain. B, SDS-PAGE and GelCodeTM staining of GST-fused HAPLN1 domain proteins. Full-length (FL) HAPLN1 (not GST-tagged) is from R&D. *, band of interest. C (top), representative EMSA analysis of NF-κB and Oct-1 activities in RPMI8226 cells following incubation with FL-H1, GST-fused H1-IG, H1-P1, and H1-P2 at 100 nm for 4 h or TNFα (10 ng/ml) for 15 min. Bottom, graph represents results of the mean -fold NF-κB activation ± S.D. (error bars) of three independent EMSA experiments shown above. *, p < 0.05; **, p < 0.01 when compared with appropriate control (untreated or GST only). D, representative EMSA analysis of three independent experiments where RPMI8226 cells were stimulated with increasing doses of lipopolysaccharide (LPS) for 1 h or 10 ng/ml TNFα for 15 min. E, representative graph of three independent endotoxin quantifications using the Pierce LAL chromogenic endotoxin quantification kit (Thermo Fisher Scientific). Depicted is the span of the average amount of endotoxin found in three independent H1-P1 purifications. The eluted fractions were further diluted 50–100-fold when added to culture media for NF-κB activation analyses, making the levels of contaminating lipopolysaccharide at least 3 orders of magnitude below what is detectable for NF-κB activation by EMSA.
We further characterized NF-κB activation in MM cells using H1-P1 domain due to its strong NF-κB activation potential coupled with robust expression and purification relative to other HAPLN1 forms. Additionally, known glycosylation sites lie within the N-terminal region of HAPLN1 far upstream of the PTR domains; therefore, H1-P1–mediated NF-κB activation does not involve glycosylation (45). Dose-response and time course analyses demonstrated that NF-κB activity is induced by as low as 10 nm H1-P1 and saturates at 100 nm (Fig. 3A). Activation occurs within 1 h, peaks at 2–4 h, and persists through 24 h of treatment (Fig. 3B). Supershift analysis using antibodies selective for NF-κB family members demonstrated that p50 and p65 (RelA) are activated within 2 h (Fig. 3C). H1-P1c, which contains no GST tag, was also capable of inducing NF-κB activity (Fig. 3, D and E). However, due to generally poor solubility of the cleaved H1-P1 domain, we employed GST-tagged H1-P1 in the remainder of the study. HAPLN1 is known to be cleaved between residues 31 and 32 by multiple metalloproteinases, including MMP9 (gelatinase B) (46), and removal of the N-terminal 31 amino acids to create HAPLN1(32–354) was also able to cause NF-κB activation (Fig. 3F).
Figure 3.

HAPLN1-PTR1 domain activates NF-κB in myeloma cells. A (top), representative EMSA analysis of RPMI8226 cells incubated with increasing concentrations of H1-P1 for 2 h. Bottom, graph represents results of the mean -fold NF-κB activation ± S.D. (error bars) of three independent EMSA experiments (above). *, p < 0.05; **, p < 0.01 when compared with untreated (0 nm). B (top), representative EMSA analysis of RPMI8226 cells incubated with 100 nm H1-P1 for the times indicated. Bottom, graph represents results of the mean -fold NF-κB activation ± S.D. of three independent EMSA experiments (above). *, p < 0.05; **, p < 0.01 when compared with unstimulated 1-h lane. C, supershift analysis of RPMI8226 cells treated with 100 nm H1-P1 for 2 h using antibodies against the five NF-κB family members. Results are representative of three independent experiments. D, SDS-PAGE and GelCodeTM staining of GST-fused H1-P1 following incubation with GST-fused PreScission protease. H1-P1c, cleaved H1-P1 fragment. E, EMSA analysis of RPMI8226 cells pretreated for 30 min with 100 nm bortezomib as indicated and stimulated with 100 nm H1-P1c or 10 ng/ml TNFα for the times indicated, representative of two independent experiments. F, RPMI8226 cells treated with the indicated concentrations of H1(32–354) for 4 h and analyzed by EMSA, representative of two independent experiments. G, RPMI8226, MM.1S, and H929 cell lines were incubated with 10 ng/ml TNFα for 15 min or 100 nm H1-P1 for 4 h and assayed for induction of NF-κB activity by EMSA, representative of three independent experiments. Where indicated, EMSA was done in the presence of a RelB antibody (RelB Aby) to shift RelB complexes to enable quantification of canonical NF-κB activation. -Fold change of NF-κB DNA binding as measured by phosphor image quantification, corrected for Oct-1 DNA binding control, and normalized to unstimulated but RelB antibody–treated, is indicated below the gel.
H1-P1–induced NF-κB activation was not limited to the RPMI8226 MM cell line, as stimulation of additional myeloma cell lines, such as MM.1S and H929, also showed NF-κB activation (Fig. 3G). EMSA analysis of different cell types stimulated with H1-P1 indicated that H1-P1–mediated NF-κB activity is not limited to MM cell lines but can also be seen in some lymphoma (e.g. mantle cell lymphoma) and leukemia cell lines to varying degrees (summarized in Table 1). Thus, these studies demonstrate that HAPLN1 and its PTR domains possess a previously unprecedented NF-κB signaling potential in MM cell and certain other cancer cell types.
Table 1.
Summary of H1-P1 induction of NF-κB activity in different cell lines of different cell types
Cells were treated with 100 nm H1-P1 for 4 h as analyzed by EMSA. +, a positive induction of NF-κB activity; −, no induction of NF-κB activity; +/−, a very weak induction of NF-κB activity. MM, multiple myeloma; MCL, mantle cell lymphoma; AML, acute myeloid leukemia; ALL, acute lymphoblastic leukemia.
| Cell line | Cell type | H1-P1 |
|---|---|---|
| RPMI8226 | MM | + |
| H929 | MM | + |
| MM.1S | MM | + |
| U266 | MM | + |
| Jeko-1 | MCL | + |
| z138 | MCL | + |
| Loucy | MCL | − |
| 721 | Lymphoblast | − |
| DHL4 | B cell lymphoma | + / − |
| KG-1 | Macrophages (AML) | − |
| RS4;11 | Bone marrow ALL | − |
| J45.01 | T-cell ALL | − |
| REH | ALL lymphocytes (non-B, non-T) | − |
| KIT225 | Lymphocytic leukemia T cells | + |
| U-937 | Histiocytic lymphoma (monocytes) | − |
| HEK293 | Human embryonic kidney | − |
| MEF | Mouse embryonic fibroblasts | − |
HAPLN1-PTR1–induced NF-κB activation does not involve hyaluronic acid binding
HAPLN1 links hyaluronic acid (HA) to specific proteoglycans, such as versican (42, 43). Currently, HA binding is the only known biochemical function of the PTR domains. Thus, it is possible that H1-P1 binds HA to signal through cell surface receptors, such as RHAMM, CD44, and TLR-2/4 to induce NF-κB signaling (4, 47, 48). To test the possible role of HA binding in HAPLN1-mediated signaling, we added low- or high-molecular weight HA forms, alone or in conjunction with H1-P1, but these did not alter NF-κB activation (Fig. 4, A and B). Moreover, treatment with hyaluronidase did not affect H1-P1–induced NF-κB activation (Fig. 4B). Blundell et al. (49) determined critical residues for HA binding in the PTR domain of a H1-P1–related protein, TSG-6. We mutated the 6 conserved residues in H1-P1 to alanines (H1-P1 HABD mt), but these did not affect NF-κB activation in MM cells (Fig. 4C). Taken together, these data suggest that HAPLN1-PTR1 causes NF-κB activation in MM cells in a manner independent of HA binding.
Figure 4.
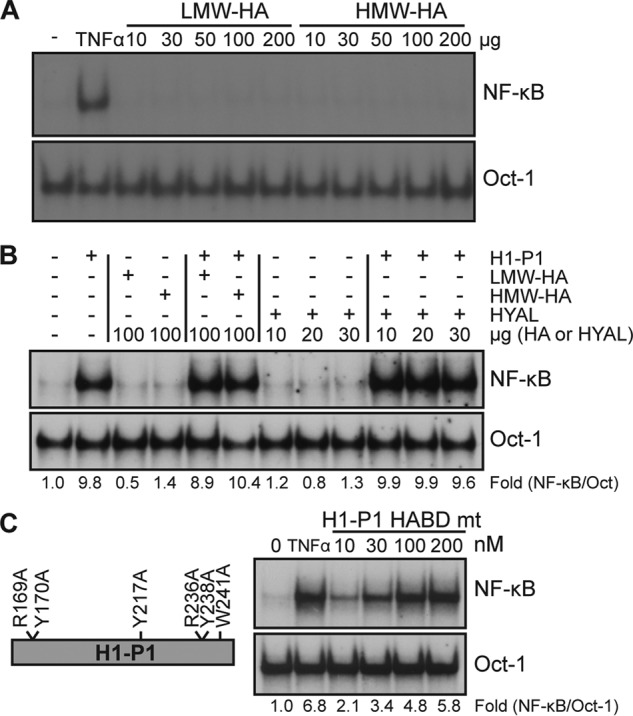
Hyaluronic acid binding is unnecessary for HAPLN1-PTR1–induced NF-κB activation. A, EMSA analysis of NF-κB activity in RPMI8226 cells following incubation with low-molecular weight HA (LMW-HA; 29 kDa) or high molecular weight HA (HMW-HA; 289 kDa) at the microgram amounts indicated for 2 h. Results are representative of two independent experiments. B, EMSA analysis of RPMI8226 cells treated with 100 nm H1-P1, low-molecular weight HA, high-molecular weight HA, or hyaluronidase (HYAL) in the amounts indicated for 4 h. C, H1-P1 HA-binding mutant (H1-P1 HABD mt) was made by mutating the critical residues necessary for HA binding within H1-P1 to alanine where illustrated. RPMI8226 cells were incubated with the indicated concentrations (nm) of recombinant GST-fused H1-P1 HABD mt for 4 h and assayed by EMSA. Results are representative of three independent experiments. -Fold change of NF-κB activity was determined as described previously.
HAPLN1-PTR1 causes bortezomib-resistant IκBα degradation
Whereas canonical and noncanonical activation pathways of NF-κB depend on the proteasome activity, we identified HAPLN1 as a factor capable of inducing bortezomib-resistant NF-κB activity (Fig. 1E). Thus, we next compared the bortezomib dose response for inhibition of NF-κB activity with a proteasome activity assay (Proteasome Glo) (Fig. 5, A–C). Bortezomib showed maximal inhibition of both TNFα-induced NF-κB activity and proteasome activity at 10 nm bortezomib (Fig. 5, B and C). In contrast, H1-P1–induced NF-κB activation was significantly more resistant to bortezomib at doses of 10–100 nm (Fig. 5, A and B). H1-P1 did not affect the ability of bortezomib to block proteasome activity at these doses (Fig. 5C); thus, the lack of NF-κB inhibition was not due to reduced bortezomib efficacy or increased proteasome activity. NF-κB activation by H1-P1 was also resistant to lactacystin, another proteasome inhibitor, unlike TNFα (Fig. 5D). Increasing concentrations of TNFα did not limit the bortezomib-mediated inhibition of NF-κB (Fig. 5E), indicating that bortezomib is capable of fully inhibiting canonical NF-κB activation, no matter what the dose of TNFα used.
Figure 5.

HAPLN1-PTR1 causes bortezomib-resistant NF-κB activation. A, representative EMSA analysis of RPMI8226 cells incubated with 10 ng/ml TNFα or 100 nm H1-P1 in the absence or presence of increasing concentrations (nm) of bortezomib (Bort). B, graph depicts the mean ± S.D. (error bars) of the quantification of three independent replicates of EMSA analysis as in A. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, the proteasome activity in RPMI8226 cells was analyzed following treatment with increasing doses of bortezomib (nm) in the presence and absence of 100 nm H1-P1 using a Proteasome-Glo cell-based assay (Promega) and plotted using the basal proteasome activity in 0 nm bortezomib set as 1. Results represent mean ± S.D. of three independent experiments. D, EMSA analysis of RPMI8226 cells incubated with increasing concentrations of lactacystin (μm) and stimulated with 10 ng/ml TNFα for 15 min or 100 nm H1-P1 for 4 h. Results are representative of two independent experiments. E, EMSA analysis of RPMI8226 cells pretreated with 100 nm bortezomib for 30 min and stimulated with increasing doses of TNFα (ng/ml) for 15 min. -Fold change of NF-κB activity was determined as described previously.
The above observations suggest an atypical mechanism at play in H1-P1–induced NF-κB activation. This activation occurs in the presence of cycloheximide (see below), thus indicating that new protein synthesis is not required for activation. Stimulation with H1-P1 caused degradation of IκBα protein from 1 to 3 h, which was highly resistant to treatment with 100 nm bortezomib (Fig. 6, A and B). Bortezomib alone does not affect IκBα degradation. Canonical NF-κB activation induced by TNFα is accompanied by the accumulation of phosphorylated and ubiquitinated intermediates of IκBα preceding degradation by the proteasome; however, H1-P1–induced IκBα degradation was not readily accompanied by the IκBα intermediates preceding degradation by the proteasome (Fig. 6C). EMSA analysis indicated that bortezomib caused ∼40% inhibition of NF-κB activation induced by H1-P1 (Fig. 5B); however, IκBα continued to be degraded in the presence of bortezomib (Fig. 6, A–B). This suggests that another IκB family member could be involved in this inhibition. Indeed, H1-P1 caused degradation of IκBβ, but IκBβ degradation was more susceptible to inhibition by bortezomib than IκBα degradation (Fig. 6, A and B). Interestingly, IKK16, an IκB kinase (IKK) inhibitor, and MLN4924, a NEDD8-activating enzyme inhibitor that is critical for activation of SCF-type ubiquitin ligases, such as β-TrCP required for IκBα ubiquitination (50, 51), could inhibit H1-P1–mediated NF-κB activation and IκBα degradation (Fig. 6, D–F). H1-P1–induced phospho-IκBα species became readily detectable in the presence of MLN4924 (Fig. 6, E and F). Collectively, these results indicate that HAPLN1-PTR1 causes degradation of both IκBα and IκBβ, but IκBα degradation is resistant to bortezomib, thus explaining significant resistance of H1-P1–induced NF-κB activation to proteasome inhibition in MM cells. However, both IKK and NEDD8-activating enzyme inhibitors blocked IκBα degradation and NF-κB activation induced by H1-P1, suggesting that this activation also involves components of the canonical NF-κB–signaling pathway.
Figure 6.

HAPLN1-PTR1 causes bortezomib-resistant IκBα degradation. A, representative immunoblot analysis of IκBα, IκBβ, and tubulin in RPMI8226 cells pretreated for 30 min with cycloheximide (Cx; 20 μg/ml) or bortezomib (Bort) (100 nm) and stimulated with H1-P1 (100 nm) where indicated. B, three biological replicates of degradation of IκBα and IκBβ as in A were quantified and plotted with mean ± S.D. (error bars). *, p < 0.05. C, immunoblot analysis of RPMI8226 cells pretreated for 30 min with 100 nm bortezomib where indicated and stimulated with 10 ng/ml TNFα for 15 min or 100 nm H1-P1 for 2 h. The top gel image is a longer exposure of the IκBα blot. The positions of IκBα, phospho-IκBα, and IκBα ubiquitin ladders are indicated. Results are representative of at least three independent experiments. D, RPMI8226 cells pretreated for 30 min with 10 μm IKK16 and 20 μg/ml cycloheximide were stimulated with H1-P1 for the indicated times and analyzed by EMSA and immunoblotted (IB) for indicated proteins. Results are representative of three independent experiments. E, EMSA and immunoblot analysis for the indicated proteins of RPMI8226 cells pretreated for 30 min with increasing concentrations MLN4924 (μm) and stimulated with 100 nm H1-P1 for 4 h or 10 ng/ml TNFα for 15 min and analyzed by EMSA and immunoblotting for the indicated proteins. F, results represent mean ± S.D. (*, p < 0.05; **, p < 0.01) of quantification of three independent EMSA and phospho-IκBα immunoblot analyses as in E at 100 μm MLN29424.
HAPLN1 is detectable in myeloma patient BMSCs and BM plasma
We previously showed that BMSC-induced NF-κB activity was highly variable from patient to patient (35). To test whether production of HAPLN1 by myeloma patient–derived bone marrow stromal cells (MM-BMSCs) is also highly variable, we utilized qRT-PCR primers that can detect a common region of six of seven splice variants (ENSEMBL database; the shortest variant contains only the IG domain) to measure the expression of HAPLN1 mRNAs in MM patient–derived BMSCs. HAPLN1 mRNA was observed in different MM-BMSCs at highly variable levels (0.3–180 relative expression) (Fig. 7A).
Figure 7.

HAPLN1 is secreted by BMSCs, and HAPLN1-PTR1 causes bortezomib resistance in myeloma cells. A, mRNA expression of HAPLN1 in myeloma BMSCs analyzed by qRT-PCR in triplicate, normalized to GAPDH mRNA expression; the mean for each sample is presented as a box-and-whisker plot. n = 18 myeloma patient BMSCs. B, schematic illustrating the locations of epitopes for different HAPLN1 antibodies utilized. Dashed lines indicate the uncertainty of the specific epitope regions. C, immunoblot analysis of GST-fused HAPLN1 domains showing the epitope specificity of HAPLN1 antibodies utilized. Four parallel SDS-PAGE analyses and immunoblots were performed to independently determine the reactivity of each antibody. *, species of interest. D, immunoblot analysis of HAPLN1 expression from conditioned media collected from myeloma BMSCs for 48 h. *, HAPLN1 immunoreactivities detected by two independent anti-HAPLN1 antibodies, H-93 and HPA019482 (HPA). Results are representative of three biological replicates. Pt, patient. E, immunoblot analysis of HAPLN1 expression in myeloma patient BM plasma fractions with four different HAPLN1 antibodies detecting PTR1 and PTR2 (K-14 and C-14) and IG (H-93 and HPA019482) domains. One membrane was probed with K-14 and H-93, and a second membrane was probed with C-14 and HPA019482. *, HAPLN1 immunoreactivities detected. NS, non-specific. Results are representative of three independent replicates. F, the indicated myeloma cell lines were cultured at 104 cells (°, except for MM.1S at 5 × 104 cells and 3 nm bortezomib (Bort)) with 100 nm GST or 100 nm H1-P1 alone or in combination with 10 nm bortezomib. After 3 days, cell viability was measured by an MTT assay. Each dot represents the mean of biological replicates performed in triplicate. Bar graphs represent the mean of three biological replicates ± S.D. (error bars). *, p < 0.05; **, p < 0.01. G, CD138+ cells from patients were cultured at 1–1.5 × 104 cells with 100 nm GST or 100 nm H1-P1 alone or in combination with 10 nm bortezomib, as indicated. Following 24 h, cell viability was measured by an MTT assay. Each square represents a technical replicate, and the bar graph represents the mean ± S.D. of triplicates. **, p < 0.01.
Numerous ECM proteins can undergo proteolysis to release biologically active signaling fragments, called matrikines (44, 52–54). We found that FL-H1 was unable to induce NF-κB activation in MM cells (Fig. 2, C and D), but PTR1 and -2 induced NF-κB activation (Fig. 2, C and D). Because HAPLN1 is known to be proteolyzed and multiple subdomains have NF-κB–inducing capabilities, we characterized several HAPLN1 antibodies (Fig. 7, B and C) to determine HAPLN1 expression in BMSC CM. Two antibodies, H-93 and HPA019482 (HPA), that react with the IG domain (Fig. 7, B and C), detected immunoreactivities at ∼40 kDa in four of seven BMSC samples analyzed (Fig. 7D), whereas two additional antibodies that detect PTR1 and PTR2 domains (K-14 and C-14, respectively) did not detect any other species in BMSC CMs (not shown). There was no correlation between HAPLN1 RNA expression and protein detection in CMs, although secretion-positive BMSCs tended to be above the mean RNA expression (Fig. 7A).
We next analyzed HAPLN1 species in BM plasma from aspirates of relapsed/refractory MM patients (Table 2). All four antibodies employed detected immunoreactivities at ∼40–44 kDa in 3 of 15 patient samples analyzed (patients 5, 9, and 15) (Fig. 7E). K-14 and C-14, which detect PTR1 and PTR2 domains, respectively, also detected smaller fragments of 20–25 kDa in 9 of 15 patient BM plasma samples (patients 4, 5, 8–10, and 12–15) (Fig. 7E). This suggests that these smaller species in patient BM samples are HAPLN1 fragments containing the PTR1/2 domains but not the IG domain. Of note, of the 15 patients analyzed for BM plasma, five had progressive disease at the time of biopsy; HAPLN1 was detectable in four of them (patients 10 and 13–15). The detection of HAPLN1 in patients with progressive disease versus others was statistically significantly different (p = 0.04, Wilcox–Mann–Whitney, two-tailed). Overall, our results indicate that HAPLN1 is (i) variably produced by BMSCs, (ii) present in patient BM plasma in both larger forms (∼40–44 kDa) containing all three domains and smaller forms (20–25 kDa) containing PTR1/2 domains but lacking the IG domain, and (iii) potentially associated with progressive disease.
Table 2.
Characteristics of MM patients' samples utilized in bone marrow plasma analysis
Numbers are data gathered for 15 patients.
| Patient age | Male | Female | Lytic bone disease | Average no. of treatments | Autotransplant | Abnormal cytogenetics |
|---|---|---|---|---|---|---|
| years | % | % | % | % | % | |
| 52–86 | 64 | 36 | 94 | 2.4 | 73 | 66 |
HAPLN1-PTR1 causes bortezomib resistance in myeloma cells
We previously showed that BMSC-induced NF-κB activity was associated with bortezomib-resistant survival in RPMI8226 cells (34, 35). Thus, we investigated whether HAPLN1 could cause bortezomib resistance in these cells. Bortezomib is cytotoxic (∼90%) in RPMI8226 cells at 10 nm over 3 days (Fig. 7F). H1-P1 alone did not affect cell viability, but when combined with bortezomib, it induced bortezomib-resistant viability to ∼80% (Fig. 7F). Additional MM cell lines (H929, KMS11, and MM.1S) were also protected from bortezomib-induced toxicity (Fig. 7F). Next, we analyzed primary CD138-positive MM cells from patient bone marrow aspirates. We observed H1-P1–mediated protection from bortezomib-induced toxicity in three of four samples analyzed (Fig. 7G). Of the four patients analyzed, patients a, b, and d had received bortezomib treatment before biopsy, whereas patient c was newly diagnosed with MM and therefore had not received any prior treatment. Overall, our results demonstrate that HAPLN1-matrikine is detectable in MM patient BM microenvironment, and its HAPLN1-PTR1 domain is sufficient to induce resistance to bortezomib-induced cell death in MM cells.
Discussion
In the present study, we identified HAPLN1 as a BMSC-secreted factor that is capable of inducing an atypical, PIR NF-κB signaling pathway and bortezomib-resistant survival in MM cells. To our knowledge, HAPLN1 has not been previously implicated in NF-κB signaling or MM pathogenesis. Our study also suggests the possibility that HAPLN1-mediated NF-κB activity might account for some of the chronic NF-κB activation in MM cells that remains unexplained by genome-sequencing studies (5–7, 31, 33). Full-length HAPLN1 lacked NF-κB signaling activity; only the MMP-processed version, HAPLN1(32–354) (46), and internal PTR domains had strong NF-κB signaling activities (46). ECM components that make up the tissue stroma can be proteolytically processed to produce soluble peptides and products, generally referred to as “matrikines,” that can then signal to different cell types to change their phenotype (4, 44, 53, 54). HAPLN1 is produced by cells in the TME, and that production could be enhanced during disease progression, particularly in patients with progressive disease. We detected HAPLN1 fragments in a considerable fraction of MM patient BM plasma. Thus, it seems plausible that HAPLN1 is produced by BMSCs, and a proteolytic mechanism is associated with the release of NF-κB–activating fragments, matrikines, to confer bortezomib resistance (Fig. 8). Given the lack of detection of smaller PTR1/2 species in BMSC CMs, it seems plausible that MM cells themselves or other cell types could be involved in further processing of HAPLN1 into smaller matrikine fragments in MM patient bone marrow. The development of methodologies to accurately quantify HAPLN1-matrikine levels in individual MM patients could serve as a new biomarker for MM therapy and drug resistance.
Figure 8.

Schematic of proposed role of HAPLN1 in myeloma. MM-BMSCs secrete HAPLN1. Secreted proteases produced by MM-BMSCs, MM cells, or possibly other cell types in the tumor microenvironment process HAPLN1 into PTR1/PTR2-containing fragments to induce an atypical NF-κB signaling pathway in myeloma plasma B-cells leading to bortezomib-resistant survival of MM cells. This pathway involves IKK activation and IκBα phosphorylation, but polyubiquitination of IκBα is hardly detected (denoted by a lighter and shorter ubiquitin chain). In contrast, TNFα activates the canonical NF-κB pathway that is sensitive to bortezomib inhibition, and both IκBα phosphorylation and polyubiquitination are readily detectable.
It is well-established that two major pathways of NF-κB signaling, canonical and noncanonical, share the requirement for the ubiquitin-dependent proteasome pathway to inactivate inhibitor proteins (28, 30). Thus, NF-κB activities present in the tumor cells are generally considered to be sensitive to inhibition by proteasome inhibitors, including bortezomib. However, our laboratory previously reported a case in which a considerable number of freshly isolated MM cells harbor NF-κB activity that is highly resistant to bortezomib (34). We also described an atypical NF-κB activation mechanism, termed the PIR pathway, which is constitutively active in certain B lymphoma cell lines and highly resistant to inhibition by over 10 different types of proteasome inhibitors (37, 38). This PIR NF-κB activation involves selective degradation of IκBα, but not IκBβ, in a manner highly resistant to proteasome inhibitors but is sensitive to inhibition by IKK inhibitors (38). No extracellular ligand capable of inducing the PIR NF-κB signaling pathway was previously known, but our current data demonstrate that HAPLN1 satisfies all of the expected features of such a PIR signaling ligand. HAPLN1 does not modify bortezomib's efficacy to block proteasome activity in MM cells; thus, IκBα degradation occurs despite efficient inhibition of the proteasome activity. Moreover, MLN4924, an NEDD8-activating enzyme inhibitor, could block HAPLN1-induced IκBα degradation and NF-κB activation. Thus, further studies are required to determine how HAPLN1 induces PIR NF-κB signaling (e.g. a cell surface receptor and other signaling components that make this pathway highly resistant to bortezomib inhibition). Our study suggests that HA binding is not required for NF-κB signaling, thus tentatively ruling out the HA receptors as necessary mediators (47, 48).
Beyond the currently discovered role in multiple myeloma disease, HAPLN1 is also expressed in other tissues, including the brain, heart, and digestive tract, and its overexpression has been linked to several human malignancies, including colorectal cancer, breast cancer, and hepatocellular carcinoma (55–57). Our limited analysis of different cell lines demonstrated that NF-κB activation by H1-P1 is not limited to MM cell type but also detected in certain other cancer types. NF-κB is well-known to control inflammatory cytokine production as well as inducing cell survival mediators; therefore, HAPLN1-induced NF-κB signaling could explain some of these HAPLN1-associated pathological processes. The newly uncovered role for HAPLN1 in NF-κB signaling and possible other pathways opens future investigations to greatly deepen our understanding of this ECM protein's role in a multitude of physiological and pathological processes.
Experimental procedures
Antibodies and reagents
Antibodies against cMYC (9E10), HAPLN1 (H-93, K-14, C-14), IκBα (C-21), IκBβ (C-20), IKKα/β (H-470), cRel (C), p65 (C-20), p100/p52 (C-5), p50 (NLS), and RelB (C-19) were obtained from Santa Cruz Biotechnology. Other antibodies were against HAPLN1 (HPA019482, Sigma-Aldrich), phospho-IKKα/β (2697, Cell Signaling Technology), phospho-IκBα (9246, Cell Signaling Technology), and tubulin (CP06, Calbiochem). Reagents purchased include recombinant human TNFα (654205, EMD Millipore), lipopolysaccharide (L2880, Sigma-Aldrich), clasto-lactacystin β-lactone (154226-60-5, Sigma-Aldrich), bortezomib (S1013, Selleckchem), IKK16 (S2882, Selleckchem), MLN4924 (505477, Calbiochem), cycloheximide (C7698, Sigma-Aldrich), recombinant HAPLN1 (2608-HP, R&D Systems), low/high molecular weight hyaluronan (GLR001 and GLR004, R&D Systems), hyaluronidase (75790-208, ProSci), and GelCodeTM Blue Stain (Thermo Fisher Scientific).
Cell line culture
RPMI8226, MM.1S, NCI-H929, and HEK293 cell lines were obtained from American Type Culture Collection (ATCC). KMS11 and L363 cell lines were obtained from Dr. Lixin Rui. All multiple-myeloma cell lines were cultured at 37 °C/5% CO2 in RPMI1640 containing 10% FBS, 2% GlutaMAX (Gibco) + 1% penicillin/streptomycin.
Patient sample collection and processing
In accordance with the University of Wisconsin-Madison Institutional Review Board requirements (HO07403), human bone marrow aspirates (2–10 ml) were obtained with informed consent from multiple myeloma patients at the University of Wisconsin Hospital and Clinics. The samples collected were only accompanied by diagnosis with no further information regarding patient history at the time of analysis. Some aspirates were spun at 400 × g for 5 min at room temperature, and BM plasma fraction was removed and stored at −80 °C for further analysis. Myeloma plasma cells were positively sorted with CD138+ magnetic MACS® beads (Miltenyi Biotec) to >90% purity as described previously (34). CD138-negative fractions were cultured in “BMSC medium” (OptiMEM containing 10% FBS, 2% GlutaMAX + 1× NEAA (Gibco), and 1% penicillin/streptomycin) at 37 °C in a 5% CO2 incubator, and BMSCs were derived by removal of nonadherent cells 24 h after plating. Adherent cells were then expanded until passage 3–4 and cryopreserved as described previously (58).
Conditioned medium mass spectrometry analysis
Partial enrichment of H1-ESC-MSCs-induced factor was described previously (35). Fractions were analyzed by SDS-PAGE and stained with Coomassie Blue. Unique bands were cut, and the protein identification was carried out at ProtTech, Inc. by using nano-LC-MS/MS peptide-sequencing technology. In brief, each protein gel band was destained, cleaned, and digested in-gel with sequencing grade modified trypsin. The resulting peptide mixture was analyzed by a LC-MS/MS system, in which a high-pressure liquid chromatograph with a 75-μm inner diameter reverse phase C18 column was on-line–coupled with an ion trap mass spectrometer. The mass spectrometric data acquired were used to search the most recent non-redundant protein database with ProtTech's proprietary software suite.
HEK293 transient transfection and conditioned media
Multiple myeloma BMSCs were used to isolate total RNAs, and cDNAs encoding each of the mass spectrometry hits in full length were PCR-amplified with the following primers: G3BP, forward (5′-acgaagcttcatgacccctccgaggctcttc-3′) and reverse (5′-agtggatccgtgtccacacctgaggagttgg-3′); calumenin, forward (5′-gtggaattcctatggacctgcgacagtttctt-3′) and reverse (5′-ctcctcgcgtgaactcatcatgccgta-3′); osteonectin, forward (5′-attctgcagaatgagggcctggatcttctttctcc-3′) and reverse (5′-ctctcgagtgatcacaagatccttgtcgatatc-3′); HAPLN1, forward (5′-cgcggatccaatgaagagtctactt-3′) and reverse (5′-gcgactcgaggttgtatgctctgaa-3′). Then they were inserted into a pSecTag2A plasmid and sequence-verified for integrity. pSecTag2A adds a C-terminal Myc tag and His6 tag for detection of secreted factors. HEK293 cells were transiently transfected via a standard calcium phosphate precipitation protocol with each plasmid. CM was collected at 72 h after transfection. Expression and secretion of each of the transfected factors was analyzed by immunoblot using anti-Myc antibody.
CM analysis
CM was prepared by culturing a confluent monolayer of BMSCs, washing monolayer three times with 1× PBS and culturing the cells in serum-free BMSC medium at 37 °C. After 48–72 h, the CM was removed and centrifuged at 3,000 rpm for 5 min to remove cellular debris. Then 0.5 ml of CM was TCA-precipitated and analyzed by SDS-PAGE and immunoblotting.
EMSAs
EMSAs to measure NF-κB activity in myeloma cell lines were performed as described previously (36) using 32P-labeled double-stranded DNA probes with the following sequences: κB, 5′-tcaacagagggactccgagaggcc-3′; Oct-1, 5′-tgtcgaatgcaaatcactagaa-3′. Briefly, cell extracts were made using TOTEX buffer, as described previously (38), and 10 μg of extracts were separated on 4% native polyacrylamide gel, dried, and exposed to a phosphor screen (Amersham Biosciences), followed by quantitation of NF-κB DNA binding through ImageQuant software (GE Healthcare). Each NF-κB lane was normalized to Oct-1 values from the same sample and then to the vehicle-treated control values for each experiment to derive -fold induction.
SDS-PAGE and immunoblot analysis
Myeloma cell lines were pelleted and lysed in TOTEX buffer, as described previously (38). Equal amounts (100 μg) of soluble protein were run on denaturing 10–12.5% SDS-polyacrylamide gel and transferred onto a polyvinylidene fluoride or nitrocellulose membrane (GE Healthcare). The membrane was then incubated with the appropriate antibodies as described previously (36). Immunoblots were analyzed by enhanced chemiluminescence as described by the manufacturer (GE Healthcare). Quantification of immunoblots was performed using ImageJ (National Institutes of Health) to calculate the signal intensity of protein(s) of interest normalized to total protein loading (tubulin or actin) for each lane from the same sample and then to vehicle control values for each experiment.
Purification and expression of GST-tagged proteins
HAPLN1 domains (IG, PTR1, and PTR2) were PCR-amplified with the following primers: IG, forward (5′-cacacaggatccgatcatctttcagac-3′) and reverse (5′-gccgctcgagaagtccagtgctaccac-3′); PTR1, forward (5′-gcgggatccgtggtattcccttac-3′) and reverse (5′-gccgctcgagattgaaattggatgtaa-3′); PTR2, forward (5′-cgcggatccggccgtttttactatc-3′) and reverse (5′-gccgctcgaggttgtatgctctgaag-3′). Double-stranded oligonucleotides encoding HAPLN1(32–354) or H1-P1 HABD mt mutant were purchased from Integrated DNA Technologies and inserted into pGEX6p-1 plasmid and sequence-verified for integrity. Plasmids were transformed into the BL21 Rosetta 2 E. coli strain and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside, followed by lysis and purification by glutathione-agarose beads (G4510, Pierce) and elution by 50 mm reduced glutathione at pH 8.0. Removal of GST from GST-H1-P1 to generate H1-P1c was done by incubation with GST-fused PreScission protease (GE Healthcare) as described previously (59).
Endotoxin quantification
Endotoxin levels were assessed using the Pierce LAL chromogenic endotoxin quantification kit (88282, Thermo Fisher Scientific). Quantification was performed in 96-well tissue culture grade plates and assayed per the manufacturer's instructions.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
MTT (M6494) was purchased from Thermo Fisher Scientific. 104 to 105 cells/well were plated in triplicate in a 96-well clear bottom microassay plate and assayed per the manufacturer's instructions. Cells containing formazan (MTT) were dissolved in 50–75 μl of DMSO, and formazan concentration was measured by absorbance at 540 nm. The assays were run in three biological replicates. The data were normalized to the mean of untreated control.
26S proteasome activity assay
Cells (20,000/well) were plated in a white 96-well microassay plate and treated with varying concentrations of bortezomib in the absence or presence of 100 nm H1-P1 for 4 h. Proteasome activity was assayed using a cell-based Proteasome-Glo assay (G8660, Promega Corp.) according to the manufacturer's instructions, and luminescence was recorded on a luminometer (PerkinElmer Life Sciences). Following subtraction of background, relative light units for the no-drug control indicate 100% proteasome activity, and the ratio of relative light units for each dose of bortezomib over control was used to determine relative chymotryptic activity.
qRT-PCR analysis
Total RNAs from treated cells were purified by a Nucleospin RNA II column (740955, Clontech) according to the manufacturer's instructions. cDNAs were synthesized from the total RNAs using iScript cDNA synthesis kit (1708891, Bio-Rad). qRT-PCR was performed and analyzed using a Bio-Rad CFX Connect real-time system. Relative expression was determined by ΔΔCt calculation and normalized to GAPDH mRNA levels of the same sample. The qRT-PCR primer sequences were as follows: HAPLN1, forward (5′-gatactgttgtggtagcactgg-3′) and reverse (5′-tgctgcgcctcgtgaaaattgag-3′); GAPDH, forward (5′-gaaggtcggagtcaacggattt-3′) and reverse (5′-gaatttgccatgggtggaat-3′).
Bone marrow plasma analysis
Plasma layer from bone marrow aspirates (20 μl) was added to 980 μl of 1× PBS. 200 μl of Affi-Gel Blue gel beads (1537301, Bio-Rad) (1:1 slurry in 1× PBS) were added and tumbled at 4 °C for 2–3 h. Samples were then centrifuged at 3,000 rpm for 5 min, and supernatant was subjected to a second depletion with 100 μl of Affi-Gel Blue beads (3:1 bead slurry in 1× PBS) overnight at 4 °C to remove albumin. 100 μl of protein G-Sepharose 4 Fast Flow (17-0618-01, GE Healthcare) (1:1 slurry in 1× PBS) was added to supernatant and tumbled for 2–3 h at 4 °C. Following centrifugation, supernatant was incubated with 100 μl of protein A Sepharose Cl-4B (17-0963-03, GE Healthcare) (1:1 slurry in 1× PBS) for an additional 2–3 h to remove antibodies. Final supernatant was TCA-precipitated and analyzed by SDS-PAGE and immunoblotting using different HAPLN1 antibodies. To avoid carryover signals, TCA-precipitated supernatant was split into two equal samples, and two SDS-polyacrylamide gels were run in parallel and probed with different antibodies (one membrane probed with K-14 and H-93 and the second membrane probed with C-14 and HPA019482).
Statistical analysis
An unpaired two-sided Student's t test was used to compare two independent groups. Two-way analysis of variance with multiple-comparison test was used to compare bortezomib inhibition and IκB degradation curves. For comparison of HAPLN1 detection and progressive disease, a two-tailed Wilcox–Mann–Whitney test was used. A p value of <0.05 was considered statistically significant. Analysis was performed with GraphPad Prism Software (GraphPad Software Inc.).
Author contributions
M.H., C.P., S. Markovina, N.S.C., K.S.C., P.H., C.H., F.A., L.R., and S. Miyamoto conceptualization; M.H., N.S.C., S.M.W.-D., J.A.K., P.H., C.H., F.A., L.R., and S. Miyamoto resources; M.H., C.P., S. Markovina, N.S.C., K.S.C., S.M.W.-D., D.D.B., J.A.K., P.H., C.H., F.A., and S. Miyamoto data curation; M.H., S. Markovina, N.S.C., K.S.C., S.M.W.-D., D.D.B., P.H., C.H., F.A., L.R., and S. Miyamoto formal analysis; M.H., C.P., S. Markovina, and S. Miyamoto funding acquisition; M.H., D.D.B., J.A.K., and F.A. validation; M.H., C.P., K.S.C., S.M.W.-D., and S. Miyamoto investigation; M.H. and S. Miyamoto visualization; M.H., C.P., N.S.C., D.D.B., J.A.K., P.H., C.H., F.A., L.R., and S. Miyamoto methodology; M.H. and S. Miyamoto writing-original draft; M.H., N.S.C., and S. Miyamoto project administration; M.H., S. Markovina, N.S.C., P.H., F.A., L.R., and S. Miyamoto writing-review and editing; N.S.C., S.M.W.-D., and S. Miyamoto supervision.
Acknowledgments
We thank the members of the Miyamoto, Asimakopoulos, and Rui laboratories for helpful comments on the project and the manuscript.
This study was supported by National Institutes of Health (NIH) Grants R01 CA155192, RO1 CA077474-14S1, and R21 CA194868 and Multiple Myeloma Research Foundation Senior Investigator Awards (to S. Miyamoto); University of Wisconsin Carbone Cancer Center Grant P30 CA014520 and the Trillium Fund (to S. Miyamoto, N. S. C., F. A., and P. H.); NIH Grant T32 CA009135 (to M. H. and C. P); NIH Grant R01 CA077474-14S1 (to M. H.); the SciMed Graduate Research Scholars Fellowship at the University of Wisconsin-Madison (to M. H.); NIH Grant T32 GM008688 (C. P.); and NIH Grant F30 AG029714-02 (to S. Markovina). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MM
- multiple myeloma
- NF-κB
- nuclear factor-κB
- HAPLN1
- hyaluronan and proteoglycan link protein 1
- BM
- bone marrow
- BMSC
- bone marrow stromal cell
- TME
- tumor microenvironment
- ECM
- extracellular matrix
- IKK
- IκB kinase
- IκB
- inhibitor of NF-κB
- TNFα
- tumor necrosis factor α
- PIR
- proteasome inhibitor–resistant
- IG
- immunoglobulin-like
- PTR
- proteoglycan tandem repeat
- H1-P1 and -P2
- HAPLN1-PTR1 and -PTR2 domains, respectively
- EMSA
- electrophoretic mobility shift assay
- CM
- conditioned medium
- GST
- glutathione S-transferase
- HA
- hyaluronic acid
- MM-BMSCs
- myeloma patient–derived bone marrow stromal cells
- qRT-PCR
- quantitative RT-PCR
- PBS
- phosphate-buffered saline
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MAPK
- mitogen-activated protein kinase
- MEK
- mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- JAK
- Janus kinase
- STAT
- signal transducers and activators of transcription
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase.
References
- 1. Bianchi G., and Anderson K. C. (2014) Understanding biology to tackle the disease: multiple myeloma from bench to bedside, and back. CA Cancer J. Clin. 64, 422–444 10.3322/caac.21252 [DOI] [PubMed] [Google Scholar]
- 2. Laubach J., Garderet L., Mahindra A., Gahrton G., Caers J., Sezer O., Voorhees P., Leleu X., Johnsen H. E., Streetly M., Jurczyszyn A., Ludwig H., Mellqvist U.-H., Chng W.-J., Pilarski L., et al. (2016) Management of relapsed multiple myeloma: recommendations of the International Myeloma Working Group. Leukemia. 30, 1005–1017 10.1038/leu.2015.356 [DOI] [PubMed] [Google Scholar]
- 3. Dingli D., Ailawadhi S., Bergsagel P. L., Buadi F. K., Dispenzieri A., Fonseca R., Gertz M. A., Gonsalves W. I., Hayman S. R., Kapoor P., Kourelis T., Kumar S. K., Kyle R. A., Lacy M. Q., Leung N., et al. (2017) Therapy for relapsed multiple myeloma: guidelines from the Mayo stratification for myeloma and risk-adapted therapy. Mayo Clin. Proc. 92, 578–598 10.1016/j.mayocp.2017.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Papadas A., and Asimakopoulos F. (2017) Mechanisms of resistance in multiple myeloma. Handb. Exp. Pharmacol., in press 10.1007/164_2017_10 [DOI] [PubMed] [Google Scholar]
- 5. Lohr J. G., Stojanov P., Carter S. L., Cruz-Gordillo P., Lawrence M. S., Auclair D., Sougnez C., Knoechel B., Gould J., Saksena G., Cibulskis K., McKenna A., Chapman M. A., Straussman R., Levy J., et al. (2014) Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 25, 91–101 10.1016/j.ccr.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lohr J. G., Kim S., Gould J., Knoechel B., Drier Y., Cotton M. J., Gray D., Birrer N., Wong B., Ha G., Zhang C. Z., Guo G., Meyerson M., Yee A. J., Boehm J. S., et al. (2016) Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci. Transl. Med. 8, 363ra147–363ra147 10.1126/scitranslmed.aac7037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mishima Y., Paiva B., Shi J., Park J., Manier S., Takagi S., Massoud M., Perilla-Glen A., Aljawai Y., Huynh D., Roccaro A. M., Sacco A., Capelletti M., Detappe A., Alignani D., et al. (2017) The mutational landscape of circulating tumor cells in multiple myeloma. Cell Rep. 19, 218–224 10.1016/j.celrep.2017.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chapman M. A., Lawrence M. S., Keats J. J., Cibulskis K., Sougnez C., Schinzel A. C., Harview C. L., Brunet J.-P., Ahmann G. J., Adli M., Anderson K. C., Ardlie K. G., Auclair D., Baker A., Bergsagel P. L., et al. (2011) Initial genome sequencing and analysis of multiple myeloma. Nature 471, 467–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kyle R. A., and Rajkumar S. V. (2008) Multiple myeloma. Blood 111, 2962–2972 10.1182/blood-2007-10-078022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Di Marzo L., Desantis V., Solimando A. G., Ruggieri S., Annese T., Nico B., Fumarulo R., Vacca A., and Frassanito M. A. (2016) Microenvironment drug resistance in multiple myeloma: emerging new players. Oncotarget 10.18632/oncotarget.10849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Glavey S. V., Naba A., Manier S., Clauser K., Tahri S., Park J., Reagan M. R., Moschetta M., Mishima Y., Gambella M., Rocci A., Sacco A., O'Dwyer M. E., Asara J. M., Palumbo A., et al. (2017) Proteomic characterization of human multiple myeloma bone marrow extracellular matrix. Leukemia 31, 2426–2434 10.1038/leu.2017.102 [DOI] [PubMed] [Google Scholar]
- 12. Podar K., Chauhan D., and Anderson K. C. (2009) Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 23, 10–24 10.1038/leu.2008.259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu Z., and Hunter T. (2010) Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle 9, 2342–2352 10.4161/cc.9.12.11988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maki C. G., Huibregtse J. M., and Howley P. M. (1996) In vivo ubiquitination and proteasome-mediated degradation of p53(1). Cancer Res. 56, 2649–2654 [PubMed] [Google Scholar]
- 15. DiDonato J., Mercurio F., Rosette C., Wu-Li J., Suyang H., Ghosh S., and Karin M. (1996) Mapping of the inducible IκB phosphorylation sites that signal its ubiquitination and degradation. Mol. Cell. Biol. 16, 1295–1304 10.1128/MCB.16.4.1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 17. Wiita A. P., Ziv E., Wiita P. J., Urisman A., Julien O., Burlingame A. L., Weissman J. S., and Wells J. A. (2013) Global cellular response to chemotherapy-induced apoptosis. Elife 2, e01236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dong H., Chen L., Chen X., Gu H., Gao G., Gao Y., and Dong B. (2009) Dysregulation of unfolded protein response partially underlies proapoptotic activity of bortezomib in multiple myeloma cells. Leuk. Lymphoma 50, 974–984 10.1080/10428190902895780 [DOI] [PubMed] [Google Scholar]
- 19. Nikesitch N., Tao C., Lai K., Killingsworth M., Bae S., Wang M., Harrison S., Roberts T. L., and Ling S. C. W. (2016) Predicting the response of multiple myeloma to the proteasome inhibitor Bortezomib by evaluation of the unfolded protein response. Blood Cancer J. 6, e432 10.1038/bcj.2016.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murray M. Y., Auger M. J., and Bowles K. M. (2014) Overcoming bortezomib resistance in multiple myeloma. Biochem. Soc. Trans. 42, 804–808 10.1042/BST20140126 [DOI] [PubMed] [Google Scholar]
- 21. Oerlemans R., Franke N. E., Assaraf Y. G., Cloos J., van Zantwijk I., Berkers C. R., Scheffer G. L., Debipersad K., Vojtekova K., Lemos C., van der Heijden J. W., Ylstra B., Peters G. J., Kaspers G. L., Dijkmans B. A. C., et al. (2008) Molecular basis of bortezomib resistance: proteasome subunit 5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 112, 2489–2499 10.1182/blood-2007-08-104950 [DOI] [PubMed] [Google Scholar]
- 22. Demchenko Y. N., and Kuehl W. M. (2010) A critical role for the NFκB pathway in multiple myeloma. Oncotarget 1, 59–68 10.18632/oncotarget.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hideshima T., Chauhan D., Richardson P., Mitsiades C., Mitsiades N., Hayashi T., Munshi N., Dang L., Castro A., Palombella V., Adams J., and Anderson K. C. (2002) NF-κB as a therapeutic target in multiple myeloma. J. Biol. Chem. 277, 16639–16647 10.1074/jbc.M200360200 [DOI] [PubMed] [Google Scholar]
- 24. Hayden M. S., and Ghosh S. (2012) NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 10.1101/gad.183434.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ben-Neriah Y., and Karin M. (2011) Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 12, 715–723 10.1038/ni.2060 [DOI] [PubMed] [Google Scholar]
- 26. Staudt L. M. (2010) Oncogenic activation of NF-κB. Cold Spring Harb. Perspect. Biol. 2, a000109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gilmore T. D. (2007) Multiple myeloma: lusting for NF-κB. Cancer Cell 12, 95–97 10.1016/j.ccr.2007.07.010 [DOI] [PubMed] [Google Scholar]
- 28. Chen Z., Hagler J., Palombella V. J., Melandri F., Scherer D., Ballard D., and Maniatis T. (1995) Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin-proteasome pathway. Genes Dev. 9, 1586–1597 10.1101/gad.9.13.1586 [DOI] [PubMed] [Google Scholar]
- 29. Chen Z. J. (2012) Ubiquitination in signaling to and activation of IKK. Immunol. Rev. 246, 95–106 10.1111/j.1600-065X.2012.01108.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun S.-C. (2011) Non-canonical NF-κB signaling pathway. Cell Res. 21, 71–85 10.1038/cr.2010.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Annunziata C. M., Davis R. E., Demchenko Y., Bellamy W., Gabrea A., Zhan F., Lenz G., Hanamura I., Wright G., Xiao W., Dave S., Hurt E. M., Tan B., Zhao H., Stephens O., et al. (2007) Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 12, 115–130 10.1016/j.ccr.2007.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bharti A. C., Shishodia S., Reuben J. M., Weber D., Alexanian R., Raj-Vadhan S., Estrov Z., Talpaz M., and Aggarwal B. B. (2004) Nuclear factor-κB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood 103, 3175–3184 10.1182/blood-2003-06-2151 [DOI] [PubMed] [Google Scholar]
- 33. Keats J. J., Fonseca R., Chesi M., Schop R., Baker A., Chng W.-J., Van Wier S., Tiedemann R., Shi C.-X., Sebag M., Braggio E., Henry T., Zhu Y.-X., Fogle H., Price-Troska T., et al. (2007) Promiscuous Mutations Activate the Noncanonical NF-κB Pathway in Multiple Myeloma. Cancer Cell. 12, 131–144 10.1016/j.ccr.2007.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Markovina S., Callander N. S., O'Connor S. L., Kim J., Werndli J. E., Raschko M., Leith C. P., Kahl B. S., Kim K., and Miyamoto S. (2008) Bortezomib-resistant nuclear factor-κB activity in multiple myeloma cells. Mol. Cancer Res. 6, 1356–1364 10.1158/1541-7786.MCR-08-0108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Markovina S., Callander N. S., O'Connor S. L., Xu G., Shi Y., Leith C. P., Kim K., Trivedi P., Kim J., Hematti P., and Miyamoto S. (2010) Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-κB activity in myeloma cells. Mol. Cancer 9, 176 10.1186/1476-4598-9-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miyamoto S., Seufzer B. J., and Shumway S. D. (1998) Proteolytic pathway in WEHI231 immature B cells. Mol. Cell. Biol. 18, 19–29 10.1128/MCB.18.1.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shumway S. D., and Miyamoto S. (2004) A mechanistic insight into a proteasome-independent constitutive inhibitor κBα (IκBα) degradation and nuclear factor κB (NF-κB) activation pathway in WEHI-231 B-cells. Biochem. J. 380, 173–180 10.1042/bj20031796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O'Connor S., Shumway S. D., Amanna I. J., Hayes C. E., and Miyamoto S. (2004) Regulation of constitutive p50/c-Rel activity via proteasome inhibitor-resistant I B degradation in B cells. Mol. Cell. Biol. 24, 4895–4908 10.1128/MCB.24.11.4895-4908.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hideshima T., Ikeda H., Chauhan D., Okawa Y., Raje N., Podar K., Mitsiades C., Munshi N. C., Richardson P. G., Carrasco R. D., and Anderson K. C. (2009) Bortezomib induces canonical nuclear factor-B activation in multiple myeloma cells. Blood 114, 1046–1052 10.1182/blood-2009-01-199604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hardingham T. E. (1979) The role of link-protein in the structure of cartilage proteoglycan aggregates. Biochem. J. 177, 237–247 10.1042/bj1770237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Binette F., Cravens J., Kahoussi B., Haudenschild D. R., and Goetinck P. F. (1994) Link protein is ubiquitously expressed in non-cartilaginous tissues where it enhances and stabilizes the interaction of proteoglycans with hyaluronic acid. J. Biol. Chem. 269, 19116–19122 [PubMed] [Google Scholar]
- 42. Périn J. P., Bonnet F., Thurieau C., and Jollès P. (1987) Link protein interactions with hyaluronate and proteoglycans: characterization of two distinct domains in bovine cartilage link proteins. J. Biol. Chem. 262, 13269–13272 [PubMed] [Google Scholar]
- 43. Perkins S. J., Nealis A. S., Dudhia J., and Hardingham T. E. (1989) Immunoglobulin fold and tandem repeat structures in proteoglycan N-terminal domains and link protein. J. Mol. Biol. 206, 737–753 10.1016/0022-2836(89)90580-9 [DOI] [PubMed] [Google Scholar]
- 44. Maquart F.-X., Pasco S., Ramont L., Hornebeck W., and Monboisse J.-C. (2004) An introduction to matrikines: extracellular matrix-derived peptides which regulate cell activity. Crit. Rev. Oncol. Hematol. 49, 199–202 10.1016/j.critrevonc.2003.06.007 [DOI] [PubMed] [Google Scholar]
- 45. Seyfried N. T., McVey G. F., Almond A., Mahoney D. J., Dudhia J., and Day A. J. (2005) Expression and purification of functionally active hyaluronan-binding domains from human cartilage link protein, aggrecan and versican: formation of ternary complexes with defined hyaluronan oligosaccharides. J. Biol. Chem. 280, 5435–5448 10.1074/jbc.M411297200 [DOI] [PubMed] [Google Scholar]
- 46. Nguyen Q., Murphy G., Hughes C. E., Mort J. S., and Roughley P. J. (1993) Matrix metalloproteinases cleave at two distinct sites on human cartilage link protein. Biochem. J. 295, 595–598 10.1042/bj2950595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Misra S., Hascall V. C., Markwald R. R., and Ghatak S. (2015) Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 6, 201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scheibner K. A., Lutz M. A., Boodoo S., Fenton M. J., Powell J. D., and Horton M. R. (2006) Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 177, 1272–1281 10.4049/jimmunol.177.2.1272 [DOI] [PubMed] [Google Scholar]
- 49. Blundell C. D., Mahoney D. J., Almond A., DeAngelis P. L., Kahmann J. D., Teriete P., Pickford A. R., Campbell I. D., and Day A. J. (2003) The link module from ovulation- and inflammation-associated protein TSG-6 changes conformation on hyaluronan binding. J. Biol. Chem. 278, 49261–49270 10.1074/jbc.M309623200 [DOI] [PubMed] [Google Scholar]
- 50. Milhollen M. A., Traore T., Adams-Duffy J., Thomas M. P., Berger A. J., Dang L., Dick L. R., Garnsey J. J., Koenig E., Langston S. P., Manfredi M., Narayanan U., Rolfe M., Staudt L. M., Soucy T. A., Yu J., Zhang J., Bolen J. B., and Smith P. G. (2010) MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-κB-dependent lymphoma. Blood 116, 1515–1523 10.1182/blood-2010-03-272567 [DOI] [PubMed] [Google Scholar]
- 51. Godbersen J. C., Humphries L. A., Danilova O. V., Kebbekus P. E., Brown J. R., Eastman A., and Danilov A. V. (2014) The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-κB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin. Cancer Res. 20, 1576–1589 10.1158/1078-0432.CCR-13-0987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maquart F. X., Bellon G., Pasco S., and Monboisse J. C. (2005) Matrikines in the regulation of extracellular matrix degradation. Biochimie 87, 353–360 10.1016/j.biochi.2004.10.006 [DOI] [PubMed] [Google Scholar]
- 53. Jean Baptiste Oudart J. C. M. (2013) Control of tumor progression by extracellular matrix molecule fragments, the matrikines. J. Carcinog. Mutagen. 4, 148 10.4172/2157-518.1000148 [DOI] [Google Scholar]
- 54. Ricard-Blum S., and Salza R. (2014) Matricryptins and matrikines: biologically active fragments of the extracellular matrix. Exp. Dermatol. 23, 457–463 10.1111/exd.12435 [DOI] [PubMed] [Google Scholar]
- 55. Ashktorab H., Schäffer A. A., Daremipouran M., Smoot D. T., Lee E., and Brim H. (2010) Distinct genetic alterations in colorectal cancer. PLoS One 5, e8879 10.1371/journal.pone.0008879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yau C., Esserman L., Moore D. H., Waldman F., Sninsky J., and Benz C. C. (2010) A multigene predictor of metastatic outcome in early stage hormone receptor-negative and triple-negative breast cancer. Breast Cancer Res. 12, R85 10.1186/bcr2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mebarki S., Désert R., Sulpice L., Sicard M., Desille M., Canal F., Dubois-Pot Schneider H., Bergeat D., Turlin B., Bellaud P., Lavergne E., Le Guével R., Corlu A., Perret C., Coulouarn C., et al. (2016) De novo HAPLN1 expression hallmarks Wnt-induced stem cell and fibrogenic networks leading to aggressive human hepatocellular carcinomas. Oncotarget 7, 39026–39043 10.18632/oncotarget.9346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim J., Denu R. A., Dollar B. A., Escalante L. E., Kuether J. P., Callander N. S., Asimakopoulos F., and Hematti P. (2012) Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br. J. Haematol. 158, 336–346 10.1111/j.1365-2141.2012.09154.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hooper C., Jackson S. S., Coughlin E. E., Coon J. J., and Miyamoto S. (2014) Covalent modification of the NF- B essential modulator (NEMO) by a chemical compound can regulate its ubiquitin binding properties in vitro. J. Biol. Chem. 289, 33161–33174 10.1074/jbc.M114.582478 [DOI] [PMC free article] [PubMed] [Google Scholar]