

Abstract
The heme-containing enzyme myeloperoxidase (MPO) is critical for optimal antimicrobial activity of human neutrophils. We recently discovered that the bacterium Staphylococcus aureus expresses a novel immune evasion protein, called SPIN, that binds tightly to MPO, inhibits MPO activity, and contributes to bacterial survival following phagocytosis. A co-crystal structure of SPIN bound to MPO suggested that SPIN blocks substrate access to the catalytic heme by inserting an N-terminal β-hairpin into the MPO active-site channel. Here, we describe a series of experiments that more completely define the structure/function relationships of SPIN. Whereas the SPIN N terminus adopts a β-hairpin confirmation upon binding to MPO, the solution NMR studies presented here are consistent with this region of SPIN being dynamically structured in the unbound state. Curiously, whereas the N-terminal β-hairpin of SPIN accounts for ∼55% of the buried surface area in the SPIN–MPO complex, its deletion did not significantly change the affinity of SPIN for MPO but did eliminate the ability of SPIN to inhibit MPO. The flexible nature of the SPIN N terminus rendered it susceptible to proteolytic degradation by a series of chymotrypsin-like proteases found within neutrophil granules, thereby abrogating SPIN activity. Degradation of SPIN was prevented by the S. aureus immune evasion protein Eap, which acts as a selective inhibitor of neutrophil serine proteases. Together, these studies provide insight into MPO inhibition by SPIN and suggest possible functional synergy between two distinct classes of S. aureus immune evasion proteins.
Keywords: neutrophil, myeloperoxidase, protein–protein interaction, innate immunity, Staphylococcus aureus (S. aureus), immune evasion, SPIN
Introduction
Neutrophils are the most abundant white blood cells in human circulation and key players in the first line of defense against invading bacteria (1). Upon activation, neutrophils phagocytose pathogens whereby their various intracellular granules fuse with the maturing phagosome (2, 3). Neutrophils' azurophilic granules (sometimes referred to as primary granules) contain high concentrations of antibacterial peptides and proteins/enzymes. The most abundant component of azurophilic granules is the enzyme myeloperoxidase (MPO).3 In the presence of halides, MPO converts hydrogen peroxide (H2O2) into bactericidal hypohalous acids, such as HOCl and HOBr (4). Azurophilic granules also contain high concentrations of chymotrypsin-like proteases (NSPs), which can directly attack certain bacterial cells and/or the secreted and surface-retained proteins these cells produce (5–7). Although the activities of MPO and NSPs are typically viewed as independent entities, some studies have indicated that these two systems have synergistic effects with one another inside the phagosome (8). Thus, the concerted action of MPO and NSPs forms a foundation of neutrophil-mediated defense against potentially infectious bacteria.
Invading pathogens are subjected to a nearly instantaneous assault by their host's innate immune system. As a consequence, there is heavy selective pressure for these organisms to evolve the molecular wherewithal that provides for escape from the innate immune response. Although extensive study of various pathogens has cataloged an array of these so-called immune evasion strategies, the Gram-positive bacterium Staphylococcus aureus appears to be particularly adept at attempting to block the events that lead to its opsonization with complement components and subsequent phagocytosis by neutrophils (9–12). Recent work has shown that a portion of those S. aureus cells that undergo phagocytosis survive, leading to speculation that these leukocytes might serve as “Trojan Horses” for bacterial dissemination in vivo (13). For this to be the case, we believe that S. aureus must be engaged in an elaborate immune evasion program that acts intracellularly, and likely within the maturing phagosome. Indeed, our discovery of S. aureus extracellular adherence proteins (EAPs) as highly selective NSP inhibitors that promote virulence is consistent with this hypothesis (14).
We recently deployed a phage display strategy to identify secreted S. aureus proteins that interact with putative evasion targets found within neutrophils (15). Because MPO is the most abundant component of azurophilic granules (4, 16), it represented a promising initial candidate for this approach. We succeeded in identifying a novel protein of 8.4 kDa that binds and inhibits human MPO, and we named it SPIN, for staphylococcal peroxidase inhibitor (17). Gene expression studies revealed that spn transcription is up-regulated following phagocytosis of S. aureus cells by neutrophils, suggesting that the SPIN protein operates primarily within the phagosomal compartment (17). Through subsequent studies, we found that SPIN protects S. aureus from MPO-mediated killing and thereby established SPIN as a novel immune evasion protein of S. aureus (17).
SPIN shows no sequence similarity to other characterized proteins, which initially prevented understanding of its effects on MPO. To circumvent this limitation, we solved a 2.4 Å resolution crystal structure of SPIN bound to a recombinant form of human MPO (i.e. rMPO) (17). This structure suggested that SPIN acts by occluding the exchange of substrate, product, and bulk solvent with the reactive heme that lies within the MPO active site. Although a majority of the SPIN protein adopts a three α-helical bundle fold (Fig. S1), inspection of the crystal structure suggested that this helical bundle might not be responsible per se for inhibiting MPO. Instead, an ∼10-residue extension at the SPIN N terminus assumes a β-hairpin structure that almost completely occupies the MPO active-site channel (17). Further examination of the SPIN structure suggested that this N-terminal β-hairpin might not be intrinsically stable in the absence of its MPO ligand, however, as it lacks features such as disulfide bonds, etc., that would constrain these two short β-strands into their MPO-bound conformation.
In this report, we present the outcome of a series of experiments designed to assess the structure/function relationships of SPIN. We initially carried out a solution NMR study to characterize the SPIN N terminus in the absence of MPO. We then prepared and characterized a panel of deletion and site-directed mutants of SPIN to define the contributions of its two discrete regions in MPO binding and inhibition. Together, our results suggest that the SPIN N terminus is dynamically structured in the absence of MPO, but is absolutely required to inhibit MPO activity even though it contributes only modestly to binding. Intriguingly, we found that the SPIN N terminus is susceptible to site-specific proteolysis by NSPs but is protected in the presence of an S. aureus EAP protein. This suggests that functional synergy may exist between these two distinct classes of intracellularly-acting innate immune evasion proteins. Collectively, our data provide insights into SPIN structure/function relationships and deepen our appreciation of the biomolecular events that underlie S. aureus innate immune evasion.
Results
N-terminal region of SPIN is dynamically structured in the absence of MPO
Although we previously solved a 2.4-Å resolution crystal structure of SPIN bound to rMPO, our attempts to crystallize SPIN on its own were unsuccessful. We therefore explored solution NMR spectroscopy as an alternative approach to obtain insight into the structure of SPIN in its unbound state. Because the 1H-15N HSQC spectrum of isotopically-enriched SPIN exhibited excellent dispersion (Fig. S2), we collected a standard suite of two- and three-dimensional NMR spectra to permit assignment of the SPIN backbone resonances (18). Initially, 97% of the resonances identified in the 1H-15N HSQC spectrum collected at 700 MHz were assigned, leaving only those arising from Phe48 and Leu49 unaccounted for. These chemical shift assignments have been deposited in the BioMagResBank (accession number 27069) and are described elsewhere (18).
We used the TALOS-N platform to calculate the secondary structure of SPIN on the basis of backbone chemical shift values (19), and we additionally determined both the longitudinal (R1) and transverse (R2) relaxation rates for the backbone resonances assigned in the 1H-15N HSQC spectrum (Fig. 1, A–C). Because the relaxation data were recorded on an 500-MHz spectrometer, we collected an additional 1H-15N HSQC spectrum at this lower field strength (Fig. S2); due to the decreased resolution at 500 MHz, the resonances of Gln37 and Glu50 were no longer detected. From these data, we inferred that the four unassigned residues from the SPIN backbone (i.e. Gln37, Phe48, Leu49, and Glu50) are all located within an apparently flexible region that composes the N terminus up to the start of the first α-helix (i.e. His51–Asp61). For internally rigid proteins, the values of R1 and R2 are expected to have homogeneous values. However, we found that the N-terminal residues of SPIN displayed higher and lower values for R1 and R2, respectively, when compared with the entire protein on average. For these parameters, deviation from the average is often associated with sites that present backbone flexibility and undergo fast internal dynamics (20). In the case of SPIN, the overall average value for R1 was 2.49 ± 0.58 with a value of 2.90 ± 0.78 for the N-terminal region, whereas R2 rates overall averaged 8.97 ± 1.29 with a value of 7.64 ± 1.33 in the N-terminal region. Similarly, the overall average value of R2/R1 was 3.72 ± 0.73, whereas residues in the N-terminal region averaged 2.76 ± 0.59.
Figure 1.

N-terminal region of SPIN is dynamically structured in the absence of ligand. NMR spectroscopy was used to characterize the solution structure of SPIN in the absence of MPO. A series of solution dynamics studies was performed following assignment of the resonances identified in a 1H-15N HSQC spectrum of isotopically-enriched SPIN. A summary of 15N longitudinal (R1) relaxation time constants (A), transverse (R2) relaxation time constants (B), and R2/R1 values (C) are presented as a measure of site-specific rotational diffusion motion. The highlighted regions (light blue) illustrate the TALOS-N prediction of SPIN secondary structure in the free form based upon NMR chemical shift data, as represented above the top of panel in rectangular boxes (α-helical regions). The mean value for each parameter (R1, R2, or R2/R1) for residues within either the N-terminal region or the protein overall is represented a dashed blue line. Chemical shift index variance plots are shown for Cα (D) and Cβ (E) resonances, respectively. The values were calculated as the difference between the experimentally determined chemical shift and BMRB statistical chemical shift values for residues found in unstructured regions. F, three-dimensional superposition of MPO-bound SPIN and a chemical shift-derived model for SPIN in the solution state. The model in blue represents the bound form of SPIN as determined by the SPIN–rMPO crystal structure, whereas the model in orange represents the structure of SPIN calculated by the Chemical Shift Rosetta server derived from solution NMR measurements.
To gain further insight into the solution structure of SPIN, we also collected three-dimensional HCCONH, CCCONH, HCCH-TOCSY, 15N HSQC-TOCSY, and 15N-edited NOESY spectra to facilitate side-chain assignments. These efforts resulted in assignment of 95% of the 13Cα resonances and 92% of the 13Cβ resonances for SPIN (18). We compared the chemical shift index (CSI) values for these SPIN resonances to the BMRB statistical database values for CSI (Fig. 1, D and E). For these parameters, residues that approach a neutral CSI value are associated with a random coil-like structure (21); residues within α-helical structure have positive values for Cα and negative values for Cβ, whereas residues within β-strands are characterized by negative values for Cα and positive values for Cβ (21). Significantly, the CSI variance for residues in the SPIN N-terminal region was markedly lower than that of the protein overall and was consistent with the absence of regular secondary structure.
Finally, we used the CS-Rosetta server to calculate an ensemble of 40,000 three-dimensional structural models compatible with the SPIN chemical shift data. Comparison of the entire ensemble versus the lowest-energy structure gave an r.m.s.d. of 1.13 ± 0.32 Å. When we carried out similar calculations using an option where flexible regions were truncated automatically (i.e. residues 33–53), the r.m.s.d. of the ensemble relative to the lowest-energy structure was 0.80 ± 0.16 Å. Superposition of this lowest-energy model onto the structure of MPO-bound SPIN (17) yielded an r.m.s.d. of 1.47 Å for all backbone atoms, and it demonstrated otherwise good agreement between structural data obtained for the α-helical bundle region SPIN in solution versus the crystalline form (Fig. 1F). In summary, although SPIN in the bound state displays a β-hairpin structure at its N terminus, our studies presented here strongly suggest that this region is dynamic and has a random coil character prior to MPO binding.
N-terminal region of SPIN is dispensable for MPO binding
The crystal structure of SPIN bound to rMPO buries ∼1600 Å2 of SPIN surface area (17), as judged by the EBI-PISA server (22). Although the SPIN–rMPO interface is contiguous on the SPIN surface, it is useful for purposes of analysis to consider it composed of two distinct binding sites (17). The first site accounts for ∼45% of the buried SPIN surface area and is derived from residues within the α-helical bundle; the second binding site accounts for the remaining ∼55% of buried SPIN surface area and arises from residues within the N-terminal β-hairpin (17). The number of potential hydrogen bonds and salt bridges at the SPIN–rMPO interface is distributed roughly equally across these two binding sites, although the α-helical bundle appears to contribute more hydrogen bonds and the β-hairpin seems to predominate in salt bridges. Given the inconclusive nature of this interface analysis, we determined that structural information alone could not be used to establish which binding site was more important for forming and/or maintaining the SPIN–MPO interaction. However, because the N-terminal β-hairpin of SPIN does not appear to form until after MPO binding has occurred (Fig. 1), we hypothesized that the α-helical bundle site might be of primary importance in initially binding to MPO.
To test this hypothesis, we prepared and characterized both a deletion and site-directed mutant that altered the SPIN N terminus (Figs. S1 and S2). The deletion mutant removed the entire N-terminal region and consisted of residues Ala46–Lys105 inclusive (i.e. SPIN(46–105)). The site-directed mutant was prepared to assess the contributions of His43, Asp44, and Asp45 and exchanged each of these residues for Ala in the full-length SPIN background (i.e. SPIN43–45→AAA). These residues are essentially invariant across SPIN sequences from non-aureus Staphylococcus spp. and also form polar interactions with MPO side chains in the SPIN–rMPO co-crystal structure (17).
We examined the ability of these SPIN mutants to bind both native MPO and recombinant human MPO (rMPO) using a surface plasmon resonance approach (Fig. 2, A–C). We found that SPIN(46–105) is ∼3.2-fold weakened in its affinity for native human MPO relative to full-length SPIN (Table 1). Similarly, SPIN43–45→AAA is 3.4-fold diminished in its affinity for native human MPO (Table 1). For both mutants, the decreased affinity for MPO was associated with an enhanced dissociation rate of the complex relative to wildtype SPIN. Nevertheless, these mutants still bind MPO with low nanomolar KD values near 30 nm and retain the potent MPO-binding capacity intrinsic to wildtype SPIN (17). This latter feature is reflected in a 2.3 Å resolution crystal structure of SPIN(46–105) bound to rMPO, which we solved and refined to Rwork/Rfree values of 17.9 and 22.9%, respectively (Fig. 2D and Table S1). In this regard, the structure of SPIN(46–105)–rMPO is largely indistinguishable from SPIN–rMPO, as the Cα positions from the two models superimpose upon one another with an r.m.s.d. of 0.16 Å (Fig. 2E).
Figure 2.

N-terminal region of SPIN is dispensable for binding to MPO. The ability of SPIN proteins and SPIN-derived peptides to bind MPO was examined using a combination of biochemical and structural methods. A 2-fold dilution series of SPIN proteins ranging from 1.35 to 2000 nm was injected over a biosensor surface prepared from randomly immobilized MPO. Representative sensorgram series are shown for native MPO binding to full-length SPIN (A), SPIN(46–105) (B), and SPIN43–45→AAA (C). Additional curve fitting and analysis parameters are presented in Table 1. D, representation of a 2.3 Å resolution co-crystal structure of SPIN(46–105) bound to a recombinant form of human MPO. SPIN(46–105) is depicted as a ribbon diagram and is colored with its N terminus in blue and its C terminus in red, and MPO is rendered as a gray surface. The location of the MPO active-site heme is indicated by a green ball-and-stick for the purposes of reference. E, superposition of the MPO-bound forms of SPIN(46–105) and full-length SPIN, as judged by X-ray crystallography. Proteins are depicted as wire diagrams, where the N-terminal β-hairpin of full-length SPIN is shown at the top right. F, MPO-binding properties of two different synthetic peptides corresponding to the SPIN N-terminal region were assessed by SPR. Peptide binding was investigated using both a direct binding approach (single injection at 50 μm) and through a competition format where peptides were co-injected along with SPIN (50 nm). Neither approach showed any evidence for binding between these peptides and MPO. A representative sensorgram series is shown.
Table 1.
Surface plasmon resonance assessment of SPIN proteins binding to various forms of MPO
| Analyte | Surface | KD | kon | Error kon | koff | Error koff | χ2 |
|---|---|---|---|---|---|---|---|
| nm | m−1s−1 | m−1s−1 | s−1 | s−1 | |||
| SPIN | Native MPO | 9.3 | 5.37 × 105 | 3.5 × 103 | 4.99 × 10−3 | 3.0 × 10−5 | 0.148 |
| SPIN | Human rMPO | 11.8 | 4.46 × 105 | 1.5 × 103 | 5.24e × 10−3 | 1.5 × 10−5 | 0.128 |
| SPIN(46–105) | Native MPO | 29.8 | 6.19 × 105 | 2.3 × 103 | 1.85 × 10−2 | 3.0 × 10−5 | 0.102 |
| SPIN(46–105) | Human rMPO | 35.1 | 4.30 × 105 | 1.2 × 103 | 1.51 × 10−2 | 2.0 × 10−5 | 0.067 |
| SPIN43–45→AAA | Native MPO | 31.4 | 5.27 × 105 | 3.3 × 103 | 1.65 × 10−2 | 1.0 × 10−4 | 0.170 |
| SPIN43–45→AAA | Human rMPO | 32.6 | 4.88 × 105 | 1.2 × 103 | 1.59 × 10−2 | 1.8 × 10−5 | 0.043 |
Our studies with these SPIN mutants indicated that the N-terminal region of SPIN is dispensable for MPO binding. However, to examine this issue through an alternative approach, we synthesized two short peptides designed to mimic the SPIN N terminus (Fig. S1). The first such peptide corresponded to the 13 N-terminal-most residues of SPIN (i.e. SPIN-p1), flanked by two Ser residues. The second contained the same residues as the first, plus a pair of Cys at the respective termini (i.e. SPIN-p2) that were oxidized to constrain the peptide into the β-hairpin conformation adopted by this region in the SPIN–rMPO crystal structure (17). Because of the small size of these peptides, we examined their MPO-binding properties through both conventional SPR methods as well as a competition-based assay wherein wildtype SPIN would be impeded from binding to the immobilized MPO surface. We found that neither SPIN-p1 nor SPIN-p2 gave evidence for direct or competitive binding to MPO at concentrations up to 50 μm (Fig. 2F). Thus, the experiments with these synthetic peptides were consistent with our mutagenesis and crystallographic studies in defining the α-helical region as the primary MPO-binding site of SPIN.
N-terminal region of SPIN is necessary but not sufficient for inhibition of MPO
Whereas the SPIN N terminus could be removed from the protein with only an ∼3-fold loss of affinity for MPO, the SPIN–rMPO crystal structure implied that this region of SPIN likely makes important contributions to inhibition of MPO (17). Moreover, although the α-helical region of SPIN is responsible for MPO binding (Fig. 2 and Table 1), it remained unknown whether this portion of SPIN retained any MPO inhibitory capacity on its own. To investigate these questions, we characterized our SPIN mutants using an MPO activity assay wherein H2O2 reduction was linked to oxidation of o-dianisidine (17). Because previous analysis of our SPIN mutants showed that they have affinities within the 9–35 nm range for MPO (Table 1), we used a single concentration for each potential inhibitor (i.e. 74 nm) sufficiently higher than the KD value to ensure high occupancy of the respective complexes. Whereas full-length SPIN significantly inhibited MPO activity under these conditions, we found that SPIN(46–105) failed to inhibit MPO when compared with the negative control, BSA (Fig. 3A). Intriguingly, SPIN43–45→AAA also failed to significantly block MPO activity, even though it binds MPO with a KD ∼30 nm and has a full-length N-terminal region (Fig. S1).
Figure 3.

N-terminal region of SPIN is necessary but not sufficient for inhibition of MPO. The functional consequences of deletion or mutation within the SPIN N-terminal region were assessed using three independent assay formats. A, effect of SPIN proteins on MPO activity was investigated using a spectrophotometric assay. Various SPIN proteins (74 nm final concentration) were incubated with MPO for 1 h prior to addition of a substrate mixture. Peroxidase activity was then measured at A450 nm relative to a negative control. Although full-length SPIN inhibits MPO activity, loss of either the entire N terminus (SPIN(46–105)) or positions 43–45 (SPIN43–45→AAA) rendered SPIN inactive. Significance relative to the negative control was determined by one-way ANOVA with Dunnett's post-test correction for multiple comparisons. Bars express S.D. with n = 3. B, influence of SPIN proteins on a coupled glucose oxidase-MPO system that kills S. aureus strain Newman was also investigated. Whereas addition of full-length SPIN protects S. aureus from MPO-dependent killing, neither SPIN(46–105) nor SPIN43–45→AAA retained this property, as judged by recovery of CFU counts compared with GO + MPO only. As a control, the MPO inhibitor 4-ABAH also led to S. aureus survival. Significance relative to GO + MPO only (a negative control for inhibition) was determined by one-way ANOVA with Dunnett's post-test correction for multiple comparisons. Bars express S.D. with n = 3. C, S. aureus cells constitutively expressing a cytosolic form of GFP were opsonized by human serum and incubated with 50 μm SPIN, 50 μm SPIN(46–105), or buffer alone (CTRL). The mixture of opsonized bacteria was added to freshly isolated human neutrophils to allow for phagocytosis to occur. The geometric mean of MFI for each sample was determined by flow cytometry at 2 h following inception of phagocytosis and normalized to the signal for DPI-treated neutrophils at time 0. Whereas full-length SPIN significantly protected the GFP-expressing S. aureus cells from MPO-mediated bleaching, SPIN(46–105) gave only slight but not significant protection against bleaching relative to control. Levels of significance relative to buffer control were determined by one-way ANOVA with Dunnett's post-test correction for multiple comparisons. Bars express S.D. with n = 6. D, analogous experiment to that shown in C, except that the properties of 50 μm SPIN, 50 μm SPIN43–45→AAA, or buffer alone (CTRL) were compared. SPIN43–45→AAA gave only slight but not significant protection against bleaching relative to control. Levels of significance relative to buffer control were determined by one-way ANOVA with Dunnett's post-test correction for multiple comparisons. Bars express S.D. with n = 4. Because SPIN(46–105) and SPIN43–45→AAA do not inhibit MPO in activity (A) or killing assays (B), the signal observed in C and D likely reflects competition between the inactive SPIN protein and GFP for reaction with HOCl generated in the phagosome, thereby blocking bleaching of GFP indirectly. Experiments in C and D are presented separately due to variability in the assay, as they were conducted with neutrophils isolated at different times from different donors. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; ns, non-significant.
As an independent test of SPIN function, we examined the effects of our mutants in an in vitro assay designed to replicate HOCl-dependent killing of bacteria within the phagosomal compartment (17, 23). In this model, the enzyme glucose oxidase (GO) is employed as a surrogate H2O2-generating system for MPO in lieu of the multipartite NADPH oxidase that assembles within the phagosomal membrane. Whereas full-length SPIN added in trans increased bacterial survival, neither SPIN(46–105) nor SPIN43–45→AAA had any significant influence on the killing of S. aureus via bactericidal levels of HOCl generated by MPO (Fig. 3B). By contrast, the MPO inhibitor 4-ABAH restored the survival of the bacteria to normal levels. A similar trend was also seen in a neutrophil-bleaching assay, which measures the loss of fluorescence by GFP-expressing S. aureus cells following phagocytosis (24). Here, full-length SPIN added in trans significantly preserved the bacterially-derived GFP signal, whereas neither SPIN(46–105) (Fig. 3C) nor SPIN43–45→AAA (Fig. 3D) added at an identical concentration was significantly more effective than buffer control. The slight reduction in bleaching observed for both SPIN(46–105) and SPIN43–45→AAA likely resulted from these proteins acting competitively as a target for HOCl, rather than as inhibitors of MPO activity per se (Fig. 3, A, C and D). Together, these results established that the N-terminal region of SPIN is necessary but not sufficient for blocking MPO activity even though it makes only relatively minor contributions to MPO binding.
N-terminal region of SPIN is a target for degradation by NSPs
The phagosomal compartment matures in a stepwise fashion following uptake of opsonized bacteria. Early on in this process, azurophilic granules fuse with the phagosome whereby their contents are released into its lumen. This results in the accumulation of high levels of antimicrobial enzymes in the phagosome, with concentrations of MPO estimated to approach 1 mm (25). Concentrations in this range have also been proposed for NSPs (7, 26–28), which render the maturing phagosomal compartment not only highly oxidizing but exceptionally digestive in character. In light of this, the dynamically-structured nature of the SPIN N-terminal region suggested that it might be susceptible to proteolysis. As an initial test of this hypothesis, we treated full-length SPIN with the relatively non-specific protease, subtilisin. Indeed, characterization of the reaction products by SDS-PAGE and MALDI-TOF mass spectrometry revealed that the 10 N-terminal residues of SPIN were removed following a 30-min exposure to catalytic levels of enzyme (Fig. S3).
To examine the protease sensitivity of SPIN under more biologically relevant conditions, we tested whether the three canonical NSPs neutrophil elastase (NE), cathepsin G (CG), and proteinase-3 (PR3) might also degrade the SPIN N terminus. Moreover, because this region of SPIN is required for SPIN to inhibit MPO (Fig. 3), we investigated the functional consequences of such proteolysis using an MPO activity assay. We found that an excess of NE had to be incubated with SPIN to restore MPO activity to appreciable levels (Fig. 4A); this was due to incomplete cleavage by NE and residual full-length SPIN present in the reaction (Fig. S4). By contrast, we observed that incubation of SPIN with either CG (Fig. 4B) or PR3 (Fig. 4C) each resulted in loss of MPO-inhibitory activity in a dose-dependent manner. Significantly, analysis of the SPIN cleavage products after CG and PR3 treatment not only showed clear evidence of proteolysis in the SPIN N-terminal region (Figs. S4 and S5), it was also consistent with the established cleavage preference for these NSPs (5).
Figure 4.
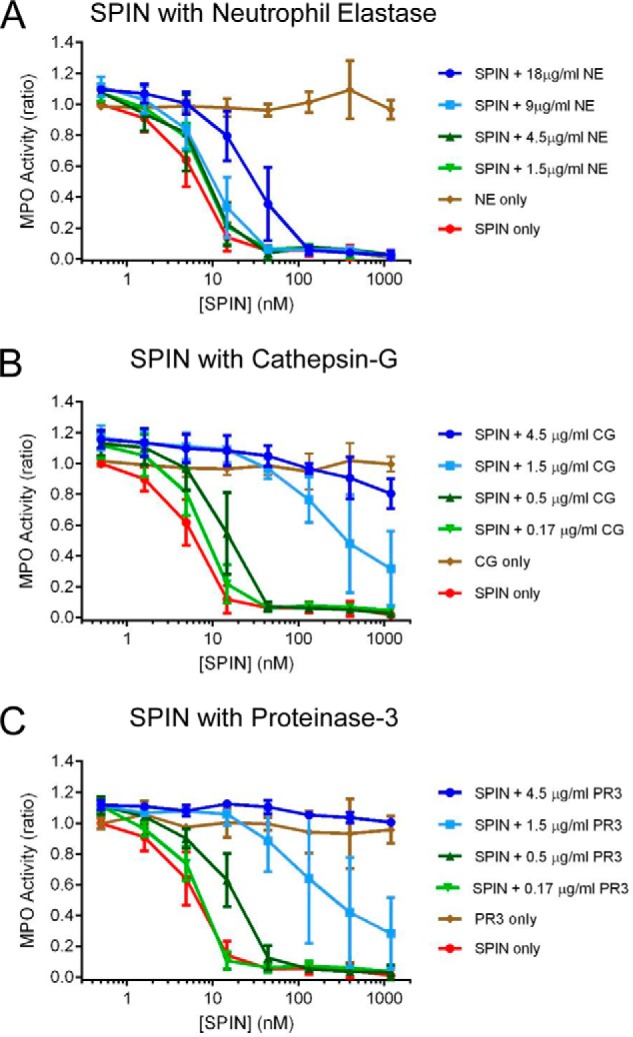
N-terminal region of SPIN is subject to proteolysis by NSPs. The influence of three proteases found in neutrophil azurophilic granules on SPIN activity was investigated using a spectrophotometric assay. Four different concentrations of NE (A), CG (B), and PR3 (C) were incubated with purified SPIN for 1 h after which proteolysis was stopped by adding PMSF. The reaction contents were serially diluted and incubated with MPO for 1 h further, prior to addition of a substrate mixture. Peroxidase activity was then measured at A450 nm relative to a negative control for each set of samples. Significantly, preincubation with all three NSPs examined resulted in a dose-dependent loss of SPIN activity, although this effect was far more pronounced for CG and PR3 when compared with NE. Subsequent characterization of the NSP digestion products of SPIN revealed that loss of activity was due to specific cleavage of the functionally essential SPIN N-terminal region (Figs. S4 and S5). Bars express S.D. with n = 3, and legends are at right.
We previously showed that production of SPIN is up-regulated following phagocytosis of S. aureus cells (17). A similar increase in expression has also been reported for the genes encoding Eap and EapH1 following exposure of S. aureus to neutrophil granule components (29). Because both Eap and EapH1 act as specific inhibitors of NSPs (14), we wondered whether these staphylococcal innate immune evasion proteins might rescue SPIN from deleterious proteolysis by NSPs. To test this possibility, we repeated the assays described above both in the absence and presence of saturating levels of recombinant Eap (14). Consistent with the previously described NSP-inhibitory function of Eap (14), as well as prior results shown here (Fig. 4), we found that Eap protected SPIN from degradation by all three canonical NSPs (Fig. 5). Together, these results show that the N-terminal region of SPIN is subject to proteolysis by NSPs but can be protected from such degradation through the action of the S. aureus NSP inhibitor Eap.
Figure 5.
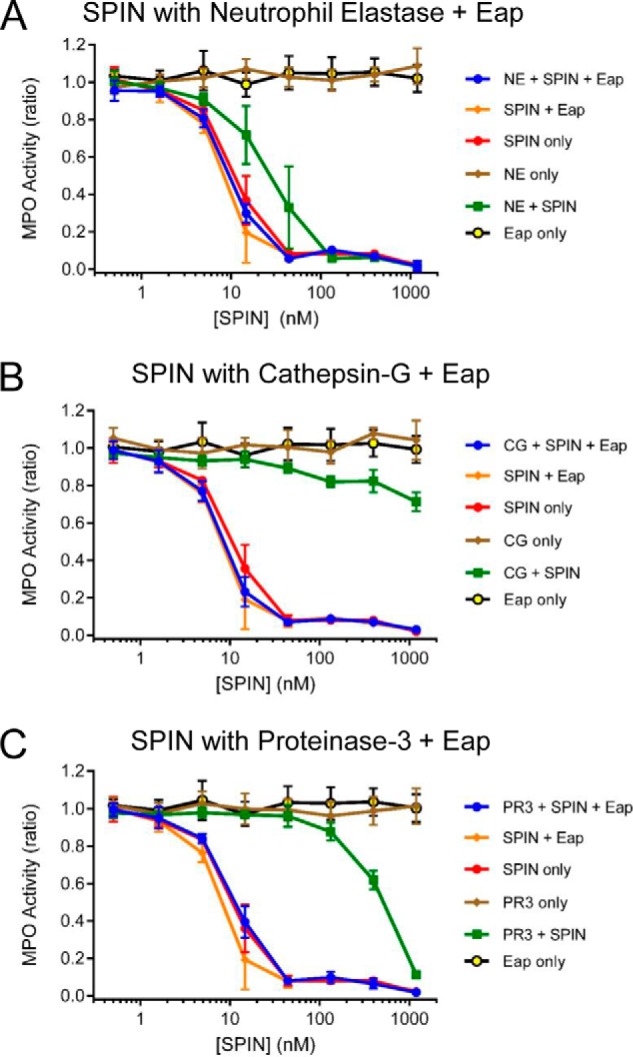
S. aureus Eap protects the N terminus of SPIN from degradation by NSPs. The consequences of including the S. aureus-derived NSP inhibitor, Eap (14), in the SPIN–NSP proteolysis reactions were assessed using an assay format identical to that presented in Fig. 4. The presence of Eap protected the N terminus of SPIN from proteolysis by 18 μg/ml NE (A), 4.5 μg/ml CG (B), and 4.5 μg/ml PR3 (C). Importantly, although SPIN on its own inhibited MPO activity, the addition of Eap was required to fully restore SPIN function in the presence the NSPs. Bars express S.D. with n = 3, and legends are at right.
Discussion
The last decade has witnessed tremendous growth in our understanding of staphylococcal innate immune evasion. In many respects, a majority of the functional, mechanistic, and structural details have been elucidated for those S. aureus secreted proteins that disrupt complement initiation and amplification, neutrophil recruitment, and the initial steps of phagocytosis (9–12). Although the topic of intracellularly-acting immune evasion proteins has received considerably less attention, it is no less relevant; in fact, the ability of S. aureus cells to resist killing within the neutrophil phagosome suggests that additional evasion molecules remain to be discovered (12). Recent work from our groups has led to identification of distinct S. aureus proteins that block function of NSPs (14) and MPO (17), which are key antimicrobial enzyme systems of neutrophils that act primarily within the phagosome (3, 12). Herein, we have further defined the structure/function relationships of the novel MPO inhibitor, SPIN (Figs. 1–3) (17). We have also provided evidence that suggests a potential functional synergy between SPIN and the EAP family of NSP inhibitors (Figs. 4 and 5) (14). Together, this work significantly furthers our understanding of an only recently discovered aspect of S. aureus immune escape.
Our previous crystal structure of SPIN–rMPO identified two MPO-binding sites within the SPIN protein (17). Although the relative contributions of each potential site to MPO binding could not be parsed directly from the structure, the biochemical data we present here have largely resolved this ambiguity. We found that loss of the SPIN N-terminal region has a minimal overall effect on its MPO-binding properties, as the KD of 29.8 nm for SPIN(46–105)–MPO compares favorably with that of 9.3 nm for SPIN–MPO (Fig. 2 and Table 1). Still, it is noteworthy that the observed 3.2-fold decrease in MPO affinity of SPIN(46–105) relative to its full-length counterpart is attributable almost entirely to a faster dissociation rate constant for its complex (3.7-fold enhancement (Table 1)). This result might be expected if the apparent interaction site involving the SPIN N-terminal region only forms after an initial binding event has occurred via the SPIN α-helical bundle. Consistent with this premise, synthetic peptides corresponding to both linear and constrained forms of the SPIN N-terminal region failed to bind MPO, even at concentrations some 3 orders of magnitude above the KD for SPIN–MPO (Fig. 2 and Table 1). These data, along with successful determination of a co-crystal structure of SPIN(46–105) bound to rMPO, argue that the primary MPO-binding site of SPIN lies within its α-helical bundle domain.
Although the α-helical bundle is responsible for driving SPIN binding to MPO, we found that the SPIN N-terminal region is absolutely required for inhibiting MPO activity (Fig. 3). SPIN(46–105) binds MPO with relatively high affinity (Fig. 2 and Table 1), yet it has no inhibitory capacity on its own (Fig. 3). This result appears to eliminate the possibility that SPIN binding inhibits MPO via perturbation of its heme redox chemistry, as has been described for the endogenous MPO inhibitor ceruloplasmin (30). In light of the data currently available, we favor a two-step steric/competitive model for SPIN action, whereby the initial MPO-binding event is driven by the α-helical bundle region and then the inhibitory β-hairpin folds and inserts into MPO-active site. In this manner, SPIN acts as a molecular plug, as suggested by our initial SPIN–rMPO crystal structure (17).
Although deletion of the N terminus yielded a SPIN protein that could not inhibit MPO, simply having these residues in the context of full-length SPIN was not sufficient to manifest inhibition on its own (Fig. 3). Our studies investigating residues His43–Asp45 were particularly helpful in addressing this issue, as loss of these conserved side chains caused only a 3.4-fold loss of affinity for MPO, yet it rendered the SPIN43–45→AAA mutant incapable of inhibiting MPO (Fig. 3). Examination of the SPIN–rMPO (17) structure reveals that both His43 and Asp44 form salt bridges with Asp379 and Arg271 of rMPO, respectively. Because these SPIN residues do not seem to contribute significantly to MPO binding (Fig. 2 and Table 1), we suggest that His43–Asp45 might help guide insertion of the inhibitory N-terminal β-hairpin into the MPO active-site channel.
It seems that SPIN faces a rather precarious existence inside the phagosomal compartment. Although SPIN is absolutely dependent on its N terminus for function (Fig. 3), this region of the protein is dynamic in the absence of its binding partner (Fig. 1) and sensitive to proteolysis by NSPs in a manner that yields biologically inactive SPIN fragments (Fig. 4 and Figs. S4 and S5). Although the high concentration of MPO within the maturing phagosome would appear to promote stability of SPIN by effectively chaperoning its N terminus, the abundance of NSPs in the same environment suggests that a significant portion of SPIN molecules could be cleaved soon after secretion. In light of this, our observation that S. aureus Eap preserves SPIN function by inhibiting CG, NE, and PR3 is a potentially significant finding (Fig. 5). All three S. aureus EAP domain-containing proteins act as tightly binding, non-covalent inhibitors of NSPs (14), and they have been shown to protect against NSP-mediated cleavage of key staphylococcal virulence factors in vitro and in vivo (31). Although that previous study examined virulence proteins that act in the extracellular environment (31), our results here suggest that EAP domains can serve a similarly protective role for SPIN and perhaps other molecules whose primary sites of action lie within the phagosomal compartment. Although these results highlight the potential for functional synergy among various S. aureus immune evasion proteins, much work remains to be done if we are to truly understand the complex relationships of these proteins with one another. In this regard, long-standing evidence describing functional synergy between MPO and NSPs (8) serves as an important reminder that the numerous biochemical events inside the phagosome are best considered as part of an overall process, rather than reactions in isolation.
Experimental procedures
Human neutrophil protein preparations
Forms of human MPO were purchased from various commercial sources. For structural studies, a recombinant form of MPO bearing a C-terminal His10 tag (catalogue no. 3174-MP-250) was obtained from R&D Systems (Minneapolis, MN). For biochemical and functional analyses, native MPO isolated from purulent human sputum (catalogue no. MY862) was obtained from Elastin Products Corp. (Owensville, MO). The neutrophil serine proteinases neutrophil elastase (catalogue no. SE563), cathepsin G (catalogue no. SG623), and proteinase-3 (catalogue no. ML734) were likewise isolated from purulent human sputum and obtained from Elastin Products Corp. (Owensville, MO). All materials were reconstituted and handled according to the manufacturer's suggestions unless otherwise noted.
Recombinant protein expression and purification
A codon-optimized form of the S. aureus SPIN open reading frame from strain Newman that lacks the N-terminal signal sequence was synthesized using gBlocks Gene Fragments (Integrated DNA Technologies). This coding fragment was subcloned into the SalI and EcoRI sites of a modified form of the prokaryotic expression vector pT7HMT (32). All other expression vectors for site-directed mutants of SPIN were derived from this parental plasmid and were constructed using standard mutagenic PCR approaches. Each plasmid was verified by DNA sequencing prior to use.
All recombinant SPIN proteins were expressed as N-terminally His6-tagged precursors and prepared according to standard techniques. Briefly, recombinant strains of Escherichia coli BL21(DE3) bearing a plasmid of interest were grown in 1 liter of selective Terrific Broth at 37 °C, prior to inducing protein expression with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 18 °C overnight. Cells from the induced culture were harvested by centrifugation, resuspended, lysed by microfluidization, and processed for nickel-nitrilotriacetic acid–affinity chromatography as described previously (33). Following initial purification, the recombinant SPIN proteins were digested with tobacco etch virus protease to remove the affinity tag as described elsewhere (32). Final purification was achieved by gel-filtration chromatography in a buffer of PBS (pH 7.4) using a Superdex 75 26/60 column connected to an AKTA FPLC system (GE Healthcare).
Uniformly 15N and 15N/13C double-labeled SPIN proteins were prepared for NMR spectroscopy studies. Both forms of wildtype SPIN were expressed in E. coli BL21(DE3) cells grown in minimal medium (M9) enriched with 15NH4Cl and [13C]glucose as described before (34).
Samples of recombinant Eap (S. aureus strain Mu50) were expressed and purified from E. coli strain BL21(DE3), as described previously (35).
Synthetic peptide mimics of the SPIN N terminus
Peptides SKVYSQNGLVLHDDS (i.e. SPIN-p1) and CKVYSQNGLVLHDDC (i.e. SPIN-p2) were synthesized at >90% purity by GenScript (Piscataway, NJ). Peptide SPIN-p2 was chemically oxidized to form a disulfide bond between its two cysteine residues. The identity of each product was confirmed by MALDI-TOF prior to use. Peptide stock solutions were prepared at 5 mm in double distilled H2O.
Solution NMR spectroscopy
All NMR measurements were performed at 25 °C on either a Varian 500 NMR (499.84 MHz for 1H frequency) or a Bruker Avance (700.11 MHz for 1H frequency) spectrometer, both of which were equipped with cryogenic probes. The 1H chemical shifts were referenced to the external standard 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS) at 25 °C, and the 13C and 15N chemical shifts were referenced indirectly from DSS. The purified, isotopically enriched SPIN proteins were dissolved at 0.75–1.0 mm final concentration in 50 mm sodium phosphate (pH 6.5) supplemented with 5% (v/v) D2O as a lock solvent prior to spectral collection.
Backbone and side-chain resonances for SPIN were assigned through standard double and triple resonance spectra. Triple-resonance NMR spectra corresponding to HNCA, HN(CO)CA, HNCACB, CBCA(CO)NH, and HNCO were recorded on uniformly 15N/13C-labeled SPIN samples to facilitate backbone assignment. Two-dimensional 13C HSQC and three-dimensional HCCONH, CCCONH, HCCH-TOCSY, 15N HSQC-TOCSY (Tm = 80 ms) and 15N-edited NOESY (Tm = 100 ms) spectra were recorded for side-chain assignment. NMR data were processed using NMRPipe (35), and spectra were analyzed and visualized using CARA (http://www.nmr.ch/)4 (36). The chemical shift assignments have been deposited in the BioMagResBank under the accession number 27069, and described in more detail elsewhere (18).
Longitudinal (R1) and transverse (R2) relaxation time constants for 15N amide atoms were determined using the steady-state, inversion-recovery, and Carr-Purcell-Meiboom-Gill methods, respectively (37, 38). For R1 determination, 15N HSQC spectra with the following relaxation delays were collected: 0.03, 0.05, 0.07, 0.12, 0.15, 0.23, 0.35, 0.80, 1.00, 1.20, 15.0, and 1.80 s. For R2 determination, 15N HSQC spectra with the following delays were collected: 0.03, 0.05, 0.07, 0.13, 0.15, 0.17, 0.19, 0.21, and 0.23 s. The R1 and R2 time constants were calculated by measuring the intensity of the peak corresponding to each assigned 1H-15N pair from the 15N HSQC spectra and fitting the resulting decay as a function of time delay (t) to a two-parameter exponential decay described by I(t) = Ioe−tR, where I(t) is the intensity of the peak as a function of relaxation delay time t. Io is the normalized peak intensity at t = 0. All such calculations were performed using the rate analysis feature of NMRView 9.2 (One Moon Scientific, Westfield, NJ) (39).
The experimentally-derived chemical shift values for SPIN resonances were used to determine solution structural features of SPIN in the absence of MPO. The secondary structure of SPIN was predicted using the TALOS-N platform (18, 19). Likewise, the CS-Rosetta Server (Rosetta version 3.8, CS-Rosetta Toolbox version 3.3) was used to generate 40,000 independent models for three-dimensional structure of SPIN (40, 41) consistent with the chemical shift data. This approach was used because the NMR spectra collected on SPIN were not sufficient to permit experimental determination of its structure.
Surface plasmon resonance
Direct binding of SPIN proteins to various forms of MPO was assessed by SPR using a Biacore T-200 instrument (GE Healthcare) at 25 °C (17). All experiments used a running buffer of HBS-T (20 mm HEPES (pH 7.4), 140 mm NaCl, and 0.005% (v/v) Tween 20) and a flow rate of 20 μl min−1. Experimental biosensors were created on CMD 200M sensor chips (XanTec Bioanalytics GmbH; Düsseldorf, Germany) by coupling native human MPO (1183 resonance units) and rMPO (1718 resonance units) to separate flow cells using random amine chemistry, whereas a reference surface was prepared by activation followed by immediate inactivation with ethanolamine. A concentration series of each SPIN protein was injected over the surfaces for 3 min and allowed to dissociate for 4 min. Regeneration to baseline was achieved by three consecutive 30-s injections of 100 mm glycine (pH 10.0). Kinetic analysis of each reference-subtracted injection series was performed using Biacore T-200 Evaluation Software version 3.0 (GE Healthcare) using a 1:1 binding model and fitting Rmax locally.
The ability of SPIN peptides to compete with SPIN–MPO complex formation was assessed using an SPR-based competition assay. All experiments were performed on a Biacore T-200 at 25 °C using a flow rate of 30 μl min−1 in HBS-T running buffer. Injections were performed for 1 min, and dissociation was monitored for 3 min at which point baseline regeneration was achieved as described above. SPIN (50 nm) or SPIN peptides (50 μm) were injected either alone or as a mixture (i.e. SPIN + peptide) over the MPO surface, and the response was monitored. Residual response of SPIN during co-injections was calculated by subtracting the response of the peptide-only injections. Sensorgram overlays were prepared using Biacore T-200 Evaluation Software and GraphPad Prism.
Crystallization, structure determination, refinement, and analysis
A sample of SPIN(46–105) bound to recombinant human MPO (R&D Systems) was prepared by mixing stoichiometric amounts of each protein and concentrating to 5 mg/ml total protein in a buffer of 5 mm Tris (pH 7.4), 50 mm NaCl. Crystals were obtained as initially described for the SPIN–rMPO complex (17). Single crystals were harvested and cryopreserved in precipitant solution supplemented with an additional 10% (v/v) PEG-400.
X-ray diffraction data were collected at 1.0 Å wavelength using beamline 22-BM of the Advanced Photon Source (Argonne National Laboratory). The reflections were processed using the HKL-2000 package (42), and the structure was solved by molecular replacement using the refined coordinates of full-length SPIN–rMPO (PDB code 5UZU) as a search model (17) and PHASER (43) as implemented within the PHENIX software suite (44, 45). The final model was constructed by both automated and manual rebuilding, followed by refinement using PHENIX.REFINE (44, 45). 94.26% of the modeled polypeptide residues lie in favored regions of the Ramachandran plot, with only 1.26% of residues found in areas classified as outliers. A quantitative description of the cell constants, diffraction data quality, and properties of the final model for the SPIN(46–105)–rMPO complex can be found in Table S1.
The rMPO used for these structural studies binds SPIN similarly to native human MPO (Table 1), but it has a lower occupancy of the heme prosthetic group when compared with MPO produced by human neutrophils. This resulted in weak electron density for the heme in the SPIN(46–105)–rMPO co-crystal structure, and it precluded inclusion of heme in the final crystallographic model. When necessary, the location of the heme prosthetic group was inferred from the coordinates of halide-bound, native human MPO (PDB code 1CXP) (46). All structural analyses, including calculation of buried surface areas and identification of potential hydrogen bonds, were performed using EBI-PISA. Representations of protein structures were generated by PyMOL.
MPO activity assays
MPO activity in the presence of H2O2 as a substrate and o-dianisidine as a redox indicator were monitored at 450 nm (17). Briefly, either 0.2 unit/ml MPO isolated from human sputum (Elastin Products Co.) was incubated with 74 nm of various SPIN proteins for 1 h at 37 °C in 96-well plates to allow protein–protein interactions to form. A substrate mixture composed of 45 mm phosphate buffer (pH 6.0), 0.5 mm H2O2, and 15 μg of o-dianisidine dihydrochloride (Sigma catalogue no. D9154) was then added. The A450 nm was measured continuously every 45 s for 1 h at 37 °C using a FLUOstar Omega microplate reader. The slope of each trial before saturation was calculated via GraphPad Prism 6 and defined as MPO activity. BSA was used as a negative control for inhibition.
The influence of NSPs on MPO inhibition by SPIN was also examined through a similar approach. Specifically, full-length SPIN (1200 nm) was preincubated for 1 h at 37 °C with either 18 μg/ml NE, 4.5 μg/ml CG, or 4.5 μg/ml PR3 after which 1 mm PMSF was added to stop protease activity. The SPIN digestion reaction was then serially diluted and assayed for MPO activity as described above. Where indicated, NSPs were also preincubated for 5 min at room temperature with 37.5 μg/ml EAP prior to adding this mixture to full-length SPIN.
MPO bactericidal assay
S. aureus cells were grown to logarithmic phase (A660 ∼0.5) in Todd Hewitt Broth (THB), washed twice, and resuspended to an A660 ∼0.5 in sterile Hanks' balanced salt solution (HBSS). 100 μl of bacterial suspension were diluted into 10 ml of substrate solution containing 300 mm glucose in HBSS. 50 μl of this mixture were diluted with an equal part of enzyme solution containing 4 ng/ml glucose oxidase from Aspergillus (Sigma) and 2.3 nm MPO with or without the addition of 40 nm SPIN proteins. 4-ABAH was included in a separate sample at a final concentration of 500 μm and served as a control for MPO-inhibited conditions. The enzyme/bacteria mixture was incubated for 1 h at 37 °C, and peroxidase activity was stopped by addition of 10 mg/ml catalase from bovine liver (Sigma). Samples were serially diluted in PBS and plated onto Todd Hewitt Agar (THA) plates. The final number of colony-forming units (CFU) was counted the next day following an overnight incubation at 37 °C.
Photobleaching of GFP-expressing S. aureus by neutrophils
The capacity of SPIN proteins to inhibit MPO activity within neutrophil phagosomes was examined. Human neutrophils were isolated from venous blood, as described previously (47). Written consent was obtained from each volunteer in accordance with both the Declaration of Helsinki and a protocol approved by the Institutional Review Board for Human Subjects at the University of Iowa. Isolated neutrophils were then fed opsonized cells of S. aureus strain USA300 expressing superfolded GFP (multiplicity of infection 1:1) as described previously (24), either in the absence or presence of 50 μm SPIN proteins. Samples identical to these except for the presence of 10 μm diphenyleneiodonium (DPI) were run in parallel and were included to inhibit NADPH oxidase-dependent bleaching of GFP (48). After 10 min of phagocytosis, undigested bacteria were removed, and neutrophils laden with S. aureus were incubated at 37 °C for 0 or 120 min. Samples were analyzed by flow cytometry for loss of GFP fluorescence, as reported previously (24). The mean fluorescence intensity (MFI) for each sample was calculated as the geometric mean of the GFP-positive population multiplied by the percent of cells gated. The percent MFI was normalized to values obtained for DPI-treated neutrophils.
Author contributions
N. W. d. J., N. T. P., K. X. R., B. L. G., A. I. H., O. P., W. N., K. v. K., J. v. S., and B.V.G. conceptualization; N. W. d. J., N. T. P., K. X. R., B. L. G., A. I. H., O. P., B. B. K., K. G. L., W. N., K. v. K., J. v. S., and B. V. G. formal analysis; N. W. d. J., N. T. P., K. X. R., B. L. G., A. I. H., B. B. K., K. G. L., and K. v. K. investigation; N. W. d. J., N. T. P., B. L. G., K. v. K., J. v. S., and B. V. G. visualization; N. W. d. J., N. T. P., K. X. R., B. L. G., A. I. H., O. P., B. B. K., K. G. L., K. v. K., and B. V. G. methodology; N. W. d. J., N. T. P., J. v. S., and B. V. G. writing-original draft; N. W. d. J., N. T. P., J. v. S., and B. V. G. writing-review and editing; N. T. P., K. X. R., B. L. G., A. I. H., O. P., K. G. L., W. N., K. v. K., J.v.S., and B. V. G. data curation; N. T. P., K. X. R., B. L. G., A. I. H., O. P., and B. V. G. validation; K. X. R., B. L. G., O. P., W. N., K. v. K., J. v. S., and B. V. G. supervision; O. P., W. N., J. v. S., and B. V. G. resources; W. N., J. v. S., and B. V. G. funding acquisition; B. V. G. project administration.
Supplementary Material
Acknowledgments
X-ray diffraction data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. Use of the facilities at the Iowa City, IA, Department of Veterans Affairs Medical Center, was supported by a Merit Review award.
This work was supported by ZonMw Grant 205200004 from the Netherlands Organization for Health Research and Development (to J. A. G. v. S.), National Institutes of Health Grants AI111203 and GM121511 (to B. V. G.) and AI116546 (to W. M. N.), and a Merit Review Award (to the Nauseef laboratory). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Table S1.
The atomic coordinates and structure factors (code 6AZP) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Chemical shift assignments have been deposited in the BioMagResBank under accession number 27069.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MPO
- myeloperoxidase
- NSP
- neutrophil serine protease
- rMPO
- recombinant MPO
- 4-ABAH
- 4-aminobenzoic acid hydrazide
- CG
- cathepsin G
- NE
- neutrophil elastase
- HBSS
- Hanks' balanced salt solution
- ANOVA
- analysis of variance
- EAP
- extracellular adherence protein
- CSI
- chemical shift index
- r.m.s.d.
- root mean square deviation
- DSS
- 2,2-dimethyl-2-silapentane-5-sulfonic acid
- HSQC
- heteronuclear single quantum coherence
- BMRB
- BioMagResBank
- MFI
- mean fluorescence intensity
- DPI
- diphenyleneiodonium
- CFU
- colony-forming unit
- PMSF
- phenylmethylsulfonyl fluoride
- GO
- glucose oxidase
- SPR
- surface plasmon resonance.
References
- 1. Nauseef W. M. (2007) How human neutrophils kills and degrade microbes: an integrated view. Immunol. Rev. 219, 88–102 10.1111/j.1600-065X.2007.00550.x [DOI] [PubMed] [Google Scholar]
- 2. Faurschou M., and Borregaard N. (2003) Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 5, 1317–1327 10.1016/j.micinf.2003.09.008 [DOI] [PubMed] [Google Scholar]
- 3. Amulic B., Cazalet C., Hayes G. L., Metzler K. D., and Zychlinsky A. (2012) Neutrophil function: from mechanisms to disease. Annu. Rev. Immunol. 30, 459–489 10.1146/annurev-immunol-020711-074942 [DOI] [PubMed] [Google Scholar]
- 4. Nauseef W. M. (2014) Myeloperoxidase in human neutrophil host defense. Cell. Microbiol. 16, 1146–1155 10.1111/cmi.12312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Korkmaz B., Horwitz M. S., Jenne D. E., and Gauthier F. (2010) Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human disease. Pharmacol. Rev. 62, 726–759 10.1124/pr.110.002733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pham C. T. (2006) Neutrophil serine proteases: specific regulators of inflammation. Nat. Rev. Immunol. 6, 541–550 10.1038/nri1841 [DOI] [PubMed] [Google Scholar]
- 7. Stapels D. A., Geisbrecht B. V., and Rooijakkers S. H. (2015) Neutrophil serine proteases in antibacterial defense. Curr. Opin. Microbiol. 23, 42–48 10.1016/j.mib.2014.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Odeberg H., and Olsson I. (1976) Microbicidal mechanisms of human granulocytes: synergistic effects of granulocyte elastase and myeloperoxidase or chymotrypsin-like cationic protein. Infect. Immun. 14, 1276–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garcia B. L., Zwarthoff S. A., Rooijakkers S. H., and Geisbrecht B. V. (2016) Novel evasion mechanisms of the classical complement pathway. J. Immunol. 197, 2051–2060 10.4049/jimmunol.1600863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim H. K., Thammavongsa V., Schneewind O., and Missiakas D. (2012) Recurrent infections and immune evasion strategies of Staphylococcus aureus. Curr. Opin. Microbiol. 15, 92–99 10.1016/j.mib.2011.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lambris J. D., Ricklin D., and Geisbrecht B. V. (2008) Complement evasion by human pathogens. Nat. Rev. Microbiol. 6, 132–142 10.1038/nrmicro1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spaan A. N., Surewaard B. G., Nijland R., and van Strijp J. A. (2013) Neutrophils versus Staphylococcus aureus: a biological tug of war. Annu. Rev. Microbiol. 67, 629–650 10.1146/annurev-micro-092412-155746 [DOI] [PubMed] [Google Scholar]
- 13. Thwaites G. E., and Gant V. (2011) Are bloodstream leukocytes trojan horses for the metastasis of Staphylococcus aureus? Nat. Rev. Microbiol. 9, 215–222 10.1038/nrmicro2508 [DOI] [PubMed] [Google Scholar]
- 14. Stapels D. A., Ramyar K. X., Bischoff M., von Köckritz-Blickwede M., Milder F. J., Ruyken M., Eisenbeis J., McWhorter W. J., Herrmann M., van Kessel K. P., Geisbrecht B. V., and Rooijakkers S. H. (2014) Staphylococcus aureus secretes a novel class of neutrophil-serine-protease inhibitors that promote bacterial infection. Proc. Natl. Acad. Sci. U.S.A. 111, 13187–13192 10.1073/pnas.1407616111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fevre C., Bestebroer J., Mebius M. M., de Haas C. J., van Strijp J. A., Fitzgerald J. R., and Haas P.-J. (2014) Staphylococcus aureus proteins SSL6 and SEIX interact with neutrophil receptors as identified using secretome phage display. Cell. Microbiol. 16, 1646–1665 10.1111/cmi.12313 [DOI] [PubMed] [Google Scholar]
- 16. Schultz J., and Kaminker K. (1962) Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch. Biochem. Biophys. 96, 465–467 10.1016/0003-9861(62)90321-1 [DOI] [PubMed] [Google Scholar]
- 17. de Jong N. W. M., Ramyar K. X., Guerra F. E., Nijland R., Fevre C., Voyich J. M., McCarthy A. J., Garcia B. L., van Kessel K. P. M., van Strijp J. A. G., Geisbrecht B. V., and Haas P. A. (2017) Immune evasion by a Staphylococcal inhibitor of myeloperoxidase. Proc. Natl. Acad. Sci. U.S.A. 114, 9439–9444 10.1073/pnas.1707032114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ploscariu N. T., Herrera A. I., Jayanthi S., Suresh Kumar T. K., Geisbrecht B. V., and Prakash O. (2017) Backbone and side-chain 1H, 15N, and 13C resonance assignments of a novel Staphylococcal inhibitor of myeloperoxidase. Biomol. NMR Assign. 11, 285–288 10.1007/s12104-017-9764-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen Y., and Bax A. (2013) Protein backbone and side-chain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 10.1007/s10858-013-9741-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kleckner I. R., and Foster M. P. (2011) An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 1814, 942–968 10.1016/j.bbapap.2010.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wishart D. S., Sykes B. D., and Richards F. M. (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222, 311–333 10.1016/0022-2836(91)90214-Q [DOI] [PubMed] [Google Scholar]
- 22. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 23. Denys G. A., Grover P., O'Hanley P., and Stephens J. T. (2011) In vitro antibacterial activity of E-101 solution, a novel myeloperoxidase-mediated antimicrobial, against Gram-positive and Gram-negative pathogens. J. Antimicrob. Chemother. 66, 335–342 10.1093/jac/dkq429 [DOI] [PubMed] [Google Scholar]
- 24. Schwartz J., Leidal K. G., Femling J. K., Weiss J. P., and Nauseef W. M. (2009) Neutrophil bleaching of GFP-expressing staphylococci: probing the intraphagosomal fate of individual bacteria. J. Immunol. 183, 2632–2641 10.4049/jimmunol.0804110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winterbourn C. C., Hampton M. B., Livesey J. H., and Kettle A. J. (2006) Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome. J. Biol. Chem. 281, 39860–39869 10.1074/jbc.M605898200 [DOI] [PubMed] [Google Scholar]
- 26. Reeves E. P., Lu H., Jacobs H. L., Messina C. G., Bolsover S., Gabella G., Potma E. O., Warley A., Roes J., and Segal A. W. (2002) Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416, 291–297 10.1038/416291a [DOI] [PubMed] [Google Scholar]
- 27. Campbell E. J., Campbell M. A., and Owen C. A. (2000) Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibition. J. Immunol. 165, 3366–3374 10.4049/jimmunol.165.6.3366 [DOI] [PubMed] [Google Scholar]
- 28. Campbell E. J., Silverman E. K., and Campbell M. A. (1989) Elastase and cathepsin G of human monocytes. Quantification of cellular content, release in response to stimuli, and heterogeneity in elastase-mediated proteolytic activity. J. Immunol. 143, 2961–2968 [PubMed] [Google Scholar]
- 29. Palazzolo-Ballance A. M., Reniere M. L., Braughton K. R., Sturdevant D. E., Otto M., Kreiswirth B. N., Skaar E. P., and DeLeo F. R. (2008) Neutrophil microbicides induce a pathogen survival response in community-associated methicillin-resistant Staphylococcus aureus. J. Immunol. 180, 500–509 10.4049/jimmunol.180.1.500 [DOI] [PubMed] [Google Scholar]
- 30. Chapman A. L., Mocatta T. J., Shiva S., Seidel A., Chen B., Khalilova I., Paumann-Page M. E., Jameson G. N., Winterbourn C. C., and Kettle A. J. (2013) Ceruloplasmin is an endogenous inhibitor of myeloperoxidase. J. Biol. Chem. 288, 6465–6477 10.1074/jbc.M112.418970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stapels D. A., Kuipers A., von Köckritz-Blickwede M., Ruyken M., Tromp A. T., Horsburgh M. J., de Haas C. J., van Strijp J. A., van Kessel K. P., and Rooijakkers S. H. (2016) Staphylococcus aureus protects its immune-evasion proteins against degradation by neutrophil serine proteases. Cell. Microbiol. 18, 536–545 10.1111/cmi.12528 [DOI] [PubMed] [Google Scholar]
- 32. Geisbrecht B. V., Bouyain S., and Pop M. (2006) An optimized system for the expression and purification of secreted bacterial proteins. Protein Expr. Purif. 46, 23–32 10.1016/j.pep.2005.09.003 [DOI] [PubMed] [Google Scholar]
- 33. Barta M. L., Skaff D. A., McWhorter W. J., Herdendorf T. J., Miziorko H. M., and Geisbrecht B. V. (2011) Crystal structures of Staphylococcus epidermidis mevalonate diphosphate decarboxylase bound to inhibitory analogs reveal new insight into substrate binding and catalysis. J. Biol. Chem. 286, 23900–23910 10.1074/jbc.M111.242016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Woehl J. L., Takahashi D., Herrera A. I., Geisbrecht B. V., and Prakash O. (2016) Backbone and side-chain 1H, 15N, and 13C resonance assignments of Staphylococcus aureus extracellular adherence protein domain 4. Biomol. NMR Assign. 10, 301–305 10.1007/s12104-016-9688-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 36. Keller R. (2004) The Computer Aided Resonance Assignment Tutorial. CANTINA Verlag, Goldau, Switzerland [Google Scholar]
- 37. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., and Kay L. E. (1994) Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 10.1021/bi00185a040 [DOI] [PubMed] [Google Scholar]
- 38. Kay L. E., Nicholson L. K., Delaglio F., Bax A., and Torchia D. A. (1992) Pulse sequences for the removal of the effects of cross correlation between dipolar and chemical-shift anisotropy relaxation mechanisms on the measurement of heteronuclear T1 and T2 values in proteins. J. Magn. Reson. 97, 359–375 10.1016/0022-2364(92)90320-7 [DOI] [Google Scholar]
- 39. Johnson B. A., and Blevins R. A. (1994) NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 10.1007/BF00404272 [DOI] [PubMed] [Google Scholar]
- 40. Lange O. F., Rossi P., Sgourakis N. G., Song Y., Lee H.-W., Aramini J. M., Ertekin A., Xiao R., Acton T. B., Montelione G. T., and Baker D. (2012) Determination of solution structures of protein up to 40 kDa using CS-rosetta with sparse NMR data from deuterated samples. Proc. Natl. Acad. Sci. U.S.A. 109, 10873–10878 10.1073/pnas.1203013109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen Y., Bryan P. N., He Y., Orban J., Baker D., and Bax A. (2010) De novo structure generation using chemical shifts for proteins with high-sequence identity but different folds. Protein Sci. 19, 349–356 10.1002/pro.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 43. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adams P. D., Grosse-Kunstleve R. W., Hung L.-W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 10.1107/S0907444902016657 [DOI] [PubMed] [Google Scholar]
- 45. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fiedler T. J., Davey C. A., and Fenna R. E. (2000) X-ray crystal structure and characterization of halide-binding sites of human myeloperoxidase at 1.8 A resolution. J. Biol. Chem. 275, 11964–11971 10.1074/jbc.275.16.11964 [DOI] [PubMed] [Google Scholar]
- 47. Nauseef W. M. (2014) Isolation of human neutrophils from venous blood. Methods Mol. Biol. 1124, 13–18 10.1007/978-1-62703-845-4_2 [DOI] [PubMed] [Google Scholar]
- 48. Greenlee-Wacker M. C., Rigby K. M., Kobayashi S. D., Porter A. R., DeLeo F. R., and Nauseef W. M. (2014) Phagocytosis of Staphylococcus aureus by human neutrophils prevents efferocytosis and induces programmed necrosis. J. Immunol. 192, 4709–4717 10.4049/jimmunol.1302692 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.