

Abstract
Radical S-adenosylmethionine (SAM) enzymes exist in organisms from all kingdoms of life, and all of these proteins generate an adenosyl radical via the homolytic cleavage of the S–C(5′) bond of SAM. Of particular interest are radical SAM enzymes, such as heme chaperones, that insert heme into respiratory enzymes. For example, heme chaperones insert heme into target proteins but have been studied only for the formation of cytochrome c-type hemoproteins. Here, we report that a radical SAM protein, the heme chaperone HemW from bacteria, is required for the insertion of heme b into respiratory chain enzymes. As other radical SAM proteins, HemW contains three cysteines and one SAM coordinating an [4Fe–4S] cluster, and we observed one heme per subunit of HemW. We found that an intact iron–sulfur cluster was required for HemW dimerization and HemW-catalyzed heme transfer but not for stable heme binding. A bacterial two-hybrid system screen identified bacterioferritins and the heme-containing subunit NarI of the respiratory nitrate reductase NarGHI as proteins that interact with HemW. We also noted that the bacterioferritins potentially serve as heme donors for HemW. Of note, heme that was covalently bound to HemW was actively transferred to a heme-depleted, catalytically inactive nitrate reductase, restoring its nitrate-reducing enzyme activity. Finally, the human HemW orthologue radical SAM domain–containing 1 (RSAD1) stably bound heme. In conclusion, our findings indicate that the radical SAM protein family HemW/RSAD1 is a heme chaperone catalyzing the insertion of heme into hemoproteins.
Keywords: protein crystallization, protein complex, protein chemistry, protein assembly, bacterial metabolism
Introduction
Radical SAM2 enzymes have been discovered in organisms from all kingdoms of life (1–3). The currently known 114,000 radical SAM proteins catalyze a broad variety of challenging chemical reactions (4–7). For instance, humans possess eight radical SAM proteins: 1) MOCS1 involved in molybdenum cofactor biosynthesis, 2) lipoic-acid synthetase for the formation of lipoic acid, 3) CDK5RAP for 12-methylthio-N5-isopentenyladenosine synthesis, 4) CDKAL1 required for methylthio-N6-threonylcarbamoyladenosine formation, 5) TYW1 for wybutosine biosynthesis, 6) ELP3 for 5-methoxycarbonymethyl uridine, 7) viperin, and 8) RSAD1 (for a review, see Ref. 2). Viperin is involved in the innate antiviral response (8). However, the exact enzymatic functions of human viperin and RSAD1 are currently unknown.
All have in common the generation of an adenosyl radical via the homolytic cleavage of the S–C(5′) bond of SAM. SAM and three cysteine residues generally coordinate a [4Fe–4S] cluster, leading to the typical CX3CX2C protein sequence signature of radical SAM enzymes (1). The first crystal structure of a radical SAM enzyme was solved for an enzyme of bacterial heme biosynthesis called coproporphyrinogen III dehydrogenase (HemN) (9). Three iron atoms of the [4Fe–4S] cluster of HemN are coordinated by the three cysteine residues Cys62, Cys66, and Cys69 of the conserved motif (9, 10). A fourth cysteine (Cys71) is not essential for [4Fe–4S] cluster coordination but for catalysis (10). During the catalytic reaction for the conversion of coproporphyrinogen III into protoporphyrinogen IX, the [4Fe–4S]2+ cluster first gets reduced. This leads to the homolytic cleavage of the SAM S–C(5′) bond and the formation of a 5′-deoxyadenosyl radical. The generated radical then removes stereospecifically one hydrogen atom from a propionate side chain of the substrate to yield 5′-deoxyadenosine and a substrate radical, which in turn leads to the desired decarboxylation reaction (9, 11). However, the presence of HemN proteins (also named coproporphyrinogen III dehydrogenase (CPDH)) carrying the CX3CX2CXC motif is limited to a few classes of bacteria (12). Multiple hemN-like genes encoding proteins of significant amino acid sequence homology were found in most classes of organisms with the exception of fungi and were originally annotated as coproporphyrinogen III oxidase.
Recently, the corresponding Lactococccus lactis protein was observed to bind heme and considered to play a role in maturation of the cytochrome oxidoreductase of the bacterium. It was therefore renamed HemW (13). L. lactis HemW (NP_267295.1) displays high homology to Escherichia coli coproporphyrinogen III dehydrogenase HemN (50% amino acid sequence similarity). Surprisingly, L. lactis HemW did not show CPDH activity in vitro and in vivo (13). In contrast to E. coli HemN, L. lactis HemW is missing 47 N-terminal amino acids and the fourth cysteine residue of the conserved CX3CX2CXC motif (13). E. coli possesses HemN and additionally a HemW-like protein annotated as YggW (NP_417430.1), a protein of hypothetical function. Because of the high degree of amino acid sequence identity of 36% (58% homology) to L. lactis HemW, we renamed YggW to HemW in the present work. Relatedly, the corresponding Pseudomonas aeruginosa protein (WP_003128950) with an amino acid sequence identity of 31% (50% homology) to L. lactis HemW was also renamed to HemW. Similar to L. lactis HemW, the HemWs of E. coli and P. aeruginosa display a truncated N terminus and the conserved cysteine motif lacking the fourth cysteine. A corresponding amino acid sequence alignment is shown in Fig. S1.
The only well characterized systems for the insertion of heme into proteins are the different cytochrome c biogenesis machineries (14). Cytochrome c is involved in multiple electron transport chains. For cytochrome c formation, protoheme IX and the apocytochrome are transported through the membranes of prokaryotes, mitochondria, and chloroplasts. Subsequently, a covalent thioether bond is actively formed between at least one cysteine and a vinyl group of the heme. Currently, five different systems are proposed to perform the processes of heme insertion into a c-type cytochrome; these systems differ in their level of complexity and are found in distinct organisms (14). Sporadically, reports on other heme-binding and potential heme-inserting proteins occur in the literature, as for NikA, an E. coli periplasmic nickel protein (15), or human glyceraldehyde-3-phosphate dehydrogenase (16). Furthermore, a putative role for the protein Surf1 of Paracoccus denitrificans as a heme a chaperone involved in cytochrome c oxidase (COX) biogenesis was described (17). Recently, we described the heme-binding protein HemW from L. lactis, hypothesizing that HemW is involved in heme trafficking (13). Here, we provide biochemical, genetic, and biophysical evidence that the bacterial HemW proteins are heme chaperones for the insertion of heme b into enzymes of respiratory chains.
Results
E. coli HemW has no coproporphyrinogen III dehydrogenase activity in vitro and in vivo
E. coli HemN and HemW amino acid sequences are 33% identical but differ in two major features. E. coli HemN carries an extra 46 N-terminal amino acid residues that have been proposed to be crucial for substrate binding (9). Moreover, the fourth cysteine of the HemN CX3CX2CXC motif is replaced by a phenylalanine in HemW. These differences are found in all HemW-like proteins (13). To investigate whether HemW carries CPDH activity, it was first analyzed in vitro. For this purpose, E. coli HemN and HemW were recombinantly produced and purified to apparent homogeneity. In contrast to HemN (18), E. coli HemW completely failed to catalyze the conversion of coproporphyrinogen III into protoporphyrinogen IX. In a next step, in vivo complementation experiments using the E. coli ΔhemN strain JKW3838 under anaerobic growth conditions were performed. Due to the presence of the oxygen-dependent coproporphyrinogen III oxidase HemF, this mutant grew efficiently under aerobic conditions (data not shown). However, in the absence of oxygen, a severe growth impairment was detected. Minimal remaining growth might result from fermentative energy generation or residual HemF activity. Clearly, E. coli hemN (pET3-hemN) complemented the ΔhemN E. coli strain to wildtype-comparable growth, whereas E. coli pGEXhemW failed to restore anaerobic growth of the mutant (Fig. 1). Both results clearly indicate that E. coli HemW does not harbor coproporphyrinogen III dehydrogenase activity as was also observed for L. lactis HemW (13). These results are consistent with heme auxotrophy reported for a Salmonella enterica serovar Typhimurium hemF/hemN double mutant carrying an intact hemW (19). To test whether the deviating N terminus and the missing fourth cysteine residue were responsible for the observed behavior, we constructed a HemW-F25C protein carrying the fourth cysteine and a HemW-HemN hybrid protein carrying the 46 N-terminal amino acids of HemN fused to HemW-F25C (HemWF25C+46N-term). Nevertheless, HemWF25C+46N-term did not show any coproporphyrinogen III dehydrogenase activity in vitro. In agreement, no complementation of the E. coli ΔhemN strain under anaerobic conditions was observed with any other of the HemW variants. Obviously, additional structural elements are required for efficient HemN activity. Consequently, HemW is not an inactivated potential coproporphyrinogen III dehydrogenase.
Figure 1.
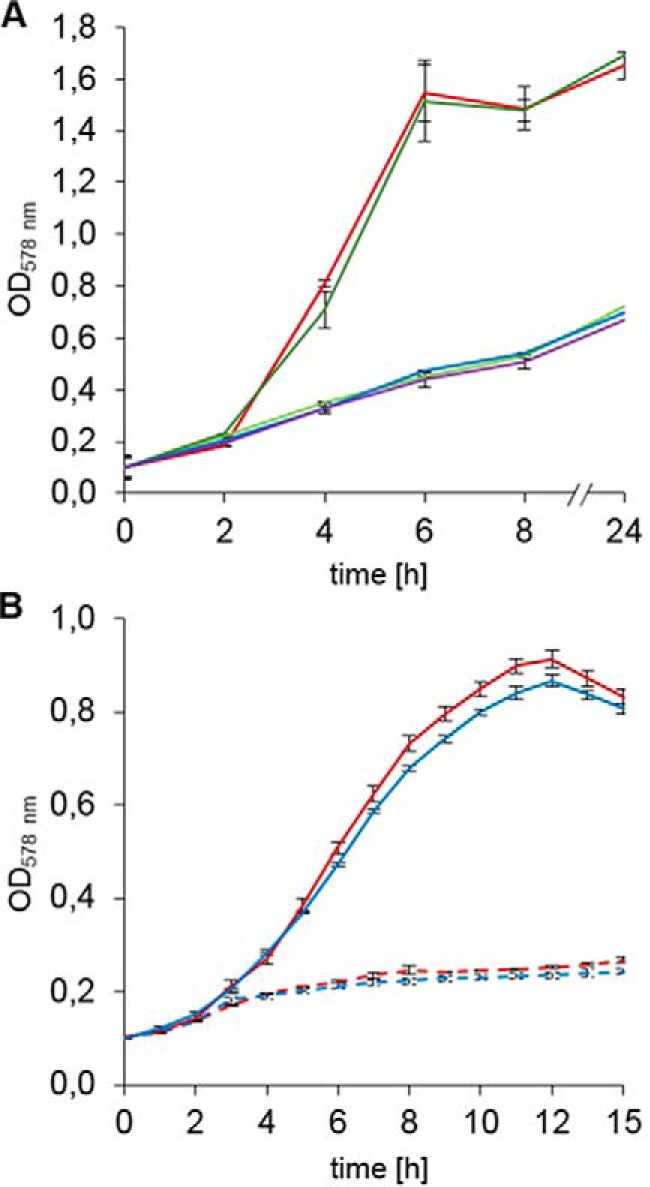
Growth behavior of E. coli hemN and hemW mutants. A, E. coli hemW does not encode a coproporphyrinogen III dehydrogenase. Cells were grown anaerobically at 37 °C in LB medium containing 10 mm NaNO3 and no antibiotics in tightly sealed anaerobic flasks. Changes in optical density at A578 were followed spectroscopically. Values for each strain are averages of three independent experiments with three parallel cultures. The growth of wildtype BW25113 (red), E. coli ΔhemN (green), ΔhemN with pET-3a-hemN (E. coli hemN) (orange), ΔhemN with pGEXhemWF25C (blue), and ΔhemN with pGEXhemWF25C+46N-term (purple) were compared. B, slight growth phenotype of the E. coli hemW mutant. The anaerobic growth in M9 minimal medium supplemented with 5 mm KNO3 and glycerol as non-fermentable carbon source was compared. Without KNO3, almost no growth was observed. Growth with KNO3 of wildtype E. coli BW25113 (red solid line) and E. coli JW2922 (ΔhemW) (blue solid line) and growth without KNO3 of E. coli BW25113 (red dashed line) and E. coli JW2922 (ΔhemW) (blue dashed line) were compared. Changes in optical density at A578 were followed spectroscopically. Values for each strain are averages of six independent experiments with three parallel cultures. Error bars represent S.D.
E. coli HemW binds a [4Fe–4S] cluster
For the biochemical and biophysical characterization, E. coli HemW was produced as a glutathione S-transferase (GST) fusion protein in E. coli BL21 (DE3). After anaerobic chromatographic purification and removal of the GST tag by PreScission protease cleavage, an apparent homogenous protein was obtained. SDS-PAGE analysis revealed a single protein band after staining with Coomassie Blue (Fig. 2A). The protein had a relative molecular weight of ∼45,000 ± 5,000, which nicely corresponds to the calculated molecular mass for the HemW monomer of 42,584 Da. Approximately 12.5 mg of purified HemW were obtained per liter of culture. To elucidate whether E. coli HemW coordinates an iron–sulfur cluster, the iron and sulfur contents of the protein were determined. For native, purified HemW, no obvious absorption around A410–A425 was detectable in the UV/visible absorption spectrum. Purified HemW exhibited 0.5 mol of iron and 0 mol of sulfur/mol of HemW, and an A420:A280 ratio of 0.04. Consequently, we carried out reconstitution of the obviously labile [Fe–S] cluster via treatment of the protein with iron ammonium citrate and lithium sulfide. After reconstitution, the iron and sulfur content of HemW increased to 3.8 mol of iron and 2.5 mol of sulfur/mol of HemW and an A420:A280 ratio of 0.19. As a consequence, the typical absorbance for [Fe–S] clusters at 420 nm became clearly visible (Fig. 2B). To further characterize the cluster type of HemW, the iron–sulfur cluster of HemW was reconstituted with ammonium [57Fe]ferric citrate, and Mössbauer spectroscopy of 57Fe-reconstituted HemW was performed. Mössbauer spectra were recorded for samples containing HemW. Spectra without further addition revealed one dominant quadrupole doublet (83% of the total intensity) with an isomer shift (δ) of 0.49 mm/s and a quadrupole splitting parameter (ΔEQ) of 1.00 mm/s, which are typical of [4Fe–4S]2+ clusters (Fig. 2C, dashed line). Moreover, a second quadrupole doublet (17% of the total intensity) with an isomer shift (δ) of 1.48 mm/s and a quadrupole splitting parameter (ΔEQ) of 3.30 mm/s was detected (Fig. 2C, dotted line). The solid line in Fig. 2 represents the superposition of the two quadrupole doublets. This spectrum is consistent with a coordination of the iron–sulfur cluster by three cysteine ligands and one potential nitrogen/oxygen ligand. The three cysteine residues are likely the Cys16, Cys20, and Cys23 of the CX3CX2C motif at the N terminus of the protein sequence. The high isomer shift of the second quadrupole doublet excludes an origin from [Fe–S] clusters but reveals high-spin Fe(II) sites with six hard oxygen or nitrogen ligands; the component is therefore assigned to adventitiously bound Fe(II) in the protein, presumably remaining from the reconstitution procedure. However, various attempts to reduce the [4Fe–4S]2+ cluster with different electron donor systems (e.g. sodium dithionite and titanium III citrate with redox mediators) for subsequent EPR analysis failed. In contrast, the [4Fe–4S] cluster of the related E. coli HemN could be reduced at such conditions (18) and used for successful EPR measurements. Surprisingly, cyclic voltammetry measurements clearly indicated a redox transition of the iron–sulfur cluster of HemW at around −410 mV (Fig. 3). The potential of −410 mV is in the range of values found for other radical SAM enzymes (20). At this redox potential, both dithionite and titanium III citrate should serve as efficient electron donors for HemW. Obviously, electron donor compounds are prevented from accessing the [4Fe–4S] cluster for reduction; consequently, no radical reaction can be initiated. A similar explanation has been suggested for the [Fe–S] cluster in succinate dehydrogenase subunit B, which appears to be inaccessible for oxidants and toxins (21).
Figure 2.

Spectroscopic analyses of the HemW-bound [4Fe–4S]2+ cluster. A, SDS-PAGE analysis of the purification of recombinant HemW from E. coli. Recombinant HemW in E. coli cell-free lysates was loaded onto glutathione-Sepharose. The immobilized GST-HemW fusion was digested by PreScission protease, and released HemW was eluted. Resulting fractions were separated via SDS-PAGE, and proteins were visualized using Coomassie Blue staining. In lane 1, total cellular extract was loaded; lanes 2 and 3 show the washing steps; lanes 4–7 show the purified HemW protein; and lane 8 shows the eluate of the cleaved GST tag. A marker mixture with proteins of known relative molecular weights is shown in lane M. B, UV-visible spectra of 10 μm HemW before (solid line) and after (dashed line) reconstitution of the iron–sulfur cluster. C, zero-field Mössbauer spectrum of purified HemW (0.55 mm) recorded at 80.00 K. The solid line represents a fit of the experimental data (dots) with two Lorentzian quadrupole doublets (dashed and dotted lines). The analysis revealed a dominant component (83% of the total intensity) with an isomer shift (δ) of 0.49 mm/s and a quadrupole splitting parameter (ΔEQ) of 1.00 mm/s and second quadrupole doublet (17% of the total intensity) with δ = 1.48 mm/s and ΔEQ = 3.30 mm/s (dotted line). The high isomer shift of the latter excludes an origin from [Fe–S] clusters but reveals high-spin Fe(II) sites with six hard oxygen or nitrogen ligands; the component is therefore assigned to adventitiously bound Fe(II) in the protein, presumably remaining from the reconstitution procedure.
Figure 3.
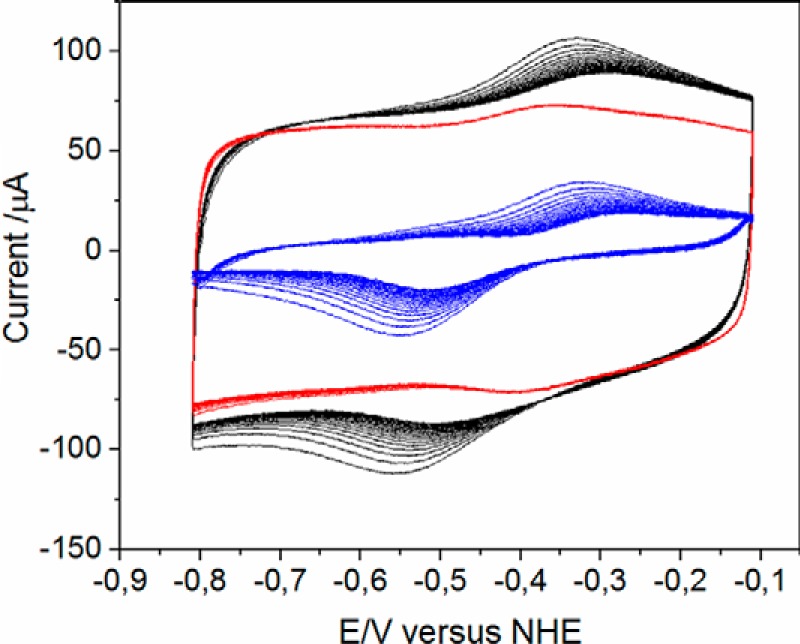
Redox potential of the HemW-bound [4Fe–4S] cluster. Cyclic voltammograms of HemW (black), buffer (red), and the resulting difference of HemW and buffer (blue) are shown. A redox potential of ∼410 mV was deduced. NHE, normal hydrogen electrode.
The HemW [4Fe–4S]2+ cluster promotes protein dimerization
To study the influence of the iron–sulfur cluster on the oligomerization state of HemW, experiments using size-exclusion chromatography of anaerobically purified and reconstituted HemW and of a HemW variant (C16S/C20S/C23S) lacking the [4Fe–4S] cluster were performed (Fig. 4). For the [4Fe–4S] cluster–containing HemW, two fractions corresponding to monomeric (fraction 16) and dimeric protein (fraction 14) were detected (Fig. 4A). Calibration of the column revealed that fraction 16 for the monomeric protein corresponded to an Mr of 45,000 ± 5000 and fraction 14 for the dimeric protein corresponded to an Mr of 87,000 ± 6000. Interestingly, an increased amount of iron–sulfur cluster was spectroscopically detected at 420 nm for the dimeric HemW species compared with the monomeric form (Fig. 4A, dashed line). Nevertheless, iron–sulfur clusters were also detected in monomeric HemW, which indicated a dynamic transition between monomeric and dimeric HemW. This transition between monomeric and dimeric proteins was tested by rechromatography of the collected separated monomeric and dimeric HemWs in fraction 14 and fraction 16 on the gel filtration column. We observed that rechromatography of the dimeric HemW in fraction 14 resulted again in two equal-sized protein absorption peaks in fractions 14 and 16 (Fig. 4C). However, most iron–sulfur cluster absorption was detected for the dimeric protein in fraction 14. Analogously, rechromatography of monomeric protein in fraction 16 also generated two absorption maxima in fractions 14 and 16; however, the bigger peak in fraction 16 represented the monomeric protein (Fig. 4D). An analytical gel filtration analysis of HemW without [Fe–S] cluster, caused by the replacement of cysteines 16, 20, and 23 with serines in the iron–sulfur cluster binding motif, revealed only one single peak in fraction 16 corresponding to a monomeric protein (Fig. 4B). The HemW-C16S/C20S/C23S variant was subjected to iron–sulfur cluster reconstitution experiment analogously to the wildtype protein prior to these experiments but remained iron–sulfur cluster-free as determined spectroscopically and by iron and sulfur determinations. Obviously, an equilibrium exists between the monomeric and dimeric protein; however, formation of a dimeric HemW is favored by the incorporation of the iron–sulfur cluster.
Figure 4.
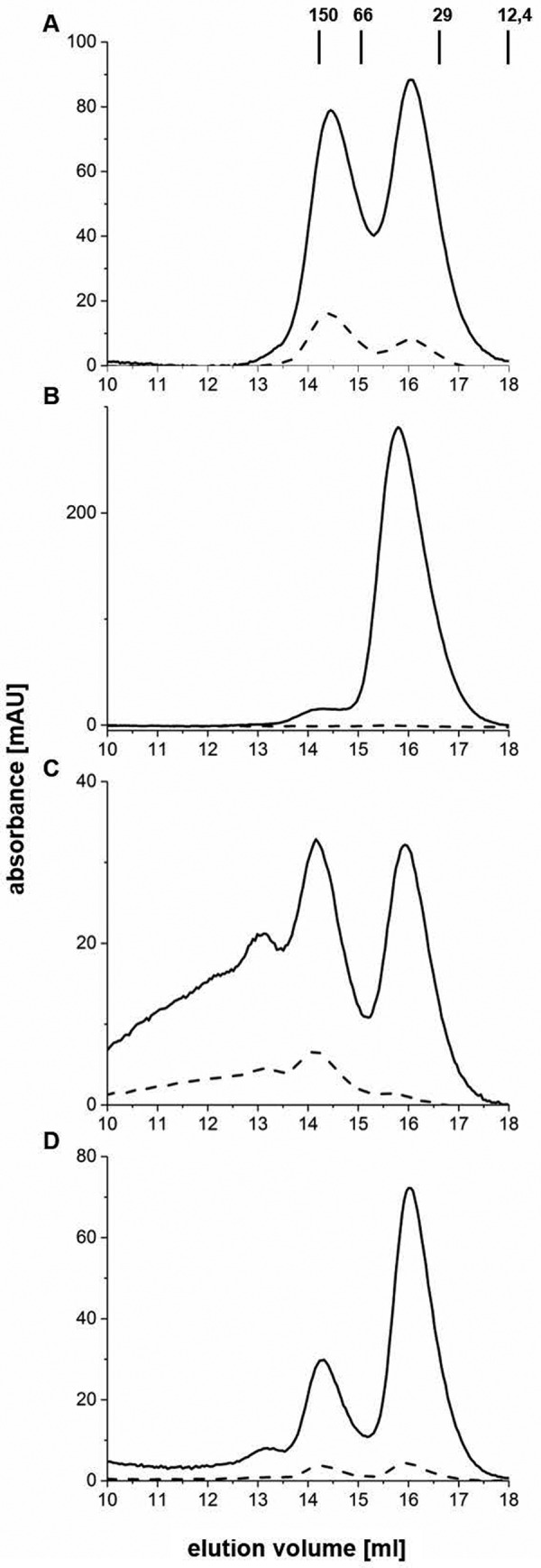
Influence of the iron–sulfur cluster on the oligomeric state of HemW. Analytical gel permeation chromatography analyses of HemW (A, C, and D) and the HemW-C16S/C20S/C23S variant (B) were performed using a Superdex 200 10/300 GL column on an ÄKTA purifier system (GE Healthcare) with a flow rate of 0.5 ml/min. Protein absorption was followed at 280 nm (solid line), and iron–sulfur cluster absorption was followed at 420 nm (dashed line). 40 μm concentrations of anaerobically prepared and iron–sulfur-reconstituted HemW and of the triple mutant were chromatographed. The separated protein peaks of the HemW (A) in fractions 14 and 16 were individually collected and rechromatographed. The rerun of fraction 14 is shown in C, and that of fraction 16 is shown in D. Apart from monomeric (fraction 16) and dimeric HemW (fraction 14) detected during the rechromatography shown in C and D, an additional shoulder peak was detected in fraction 13, most likely due to protein aggregation resulting from the concentration of HemW prior to the second chromatography. mAU, milliabsorbance units.
E. coli HemW binds SAM
The amino acid sequence analysis of E. coli HemW clearly revealed two binding sites for SAM similar to the radical SAM enzyme HemN from E. coli. Overall, in E. coli HemN, 19 amino acid residues are known from the crystal structure of the protein to coordinate two SAM molecules and one [4Fe–4S] cluster. Of the involved 19 HemN amino residues, 11 (Arg184, Gly113, Thr114, Cys66, Cys62, Cys69, Gln172, Asp209, Tyr56, Gly112, and Glu145, clockwise around the binding site (Fig. S2)) were found to be identical in HemW (Arg138, Gly67, Thr68, Cys20, Cys16, Cys23, Gln126, Asp163, Tyr10, Gly66, and Glu95) and three were found to be homologous (Ile211 in HemN and Met165 in HemW, Phe240 in HemN and Tyr194 in HemW, and Phe68 in HemN and Tyr22 in HemW) when the amino acid sequences of the proteins were aligned. Due to the low amino acid sequence conservation at the C terminus of both proteins, an alignment of four amino acid residues of HemN (Ala243, Ala242, Phe310, and Ile329) did not match the corresponding residues of HemW. This region of HemN is involved in coproporphyrinogen III coordination and most likely represents the heme-binding region of HemW. Only the cysteine (Cys71) and the following residue (Gly70) used to clearly differentiate HemN from HemW proteins were clearly different (Phe25 and Asp24). For experimentally studying SAM binding of HemW, the purified reconstituted protein was incubated with [14C]SAM, and the mixture was passed over a desalting column for the removal of non-incorporated free SAM. Protein-bound [14C]SAM was subsequently quantified using liquid scintillation counting. The control experiment was carried out using BSA and [14C]SAM. The HemW-[14C]SAM complex was eluted in the protein-containing fractions (Fig. 5A, solid line, fractions 1–4). Some free [14C]SAM eluted in the later, small-molecule fractions. In contrast, all [14C]SAM incubated with BSA eluted in the small-molecule fraction (Fig. 5A, dashed line, fractions 6–14). Consequently, SAM binding to HemW was clearly demonstrated. The highly conserved structure of the SAM and [4Fe–4S]-cluster binding site suggested the presence of two SAM molecules. However, due to the unknown amount of already bound SAM in the tested HemW proteins and the unknown exchange rate between bound and unbound SAM, it was not possible to determine the stoichiometry of SAM binding to HemW.
Figure 5.

SAM binding and SAM cleavage by HemW. A, SAM binding assay. 100 μm HemW was incubated with 0.5 μCi of [14C]SAM and fractionated via a desalting column. The radioactive fractions were analyzed using liquid scintillation counting. Solid line, HemW + [14C]SAM; dashed line, BSA + [14C]SAM. B, the SAM cleavage assays were performed for 25 μm E. coli HemW (solid line) supplemented with heme, 25 μm E. coli HemN without substrate (dashed line), and 25 μm E. coli HemN with its substrate coproporphyrinogen III (dotted line). After addition of 0.6 mm dithionite as a potential electron donor, 0.6 mm SAM was added, and the mixture was incubated. The reaction was stopped with formic acid. Samples were chromatographically separated on a Hypercarb column with appropriate marker substances. SAM (indicated with a dot) and formed 5′-deoxyadenosine (indicated with a star) were detected at 254 nm. Background controls without protein or BSA did not yield the 5′-deoxyadenosine–specific peak. HemN in the presence of substrate revealed full SAM cleavage. Without substrate, 5% residual SAM cleavage was observed for HemN. HemW with heme revealed comparable residual SAM cleavage activity.
Analysis of the SAM cleavage capacity of HemW
The classical radical SAM enzyme chemistry requires the reduction of the [4Fe–4S] cluster and homolytic cleavage of the S–C(5′) bond of SAM with the generation of the 5′-deoxyadenosyl radical. HemN usually requires its substrate coproporphyrinogen III for radical formation (10). However, in the absence of the substrate, residual enzymatic SAM cleavage of usually less than 15% of the reaction with substrate was observed. To test HemW for full or residual SAM cleavage activity, reconstituted HemW protein was incubated with SAM with and without heme as a potential substrate under reducing conditions. The disappearance of SAM with the parallel formation of deoxyadenosine was monitored by HPLC analysis (Fig. 5B, solid line). The same experiment was performed as a control with purified HemN in the absence and presence of substrate (Fig. 5B, dashed and dotted line). In this case, under tested conditions E. coli HemN revealed full SAM cleavage activity in the presence of the substrate coproporphyrinogen III (Fig. 5B, dotted line) and less than 5% of its SAM cleavage activity without substrate (Fig. 5B, dashed line). Comparable residual SAM cleavage capacity was observed for HemW with the addition (Fig. 5B, solid line) and without the addition of heme (not shown). Clearly, E. coli HemW revealed only the residual SAM cleavage activity comparable with HemN without substrate (Fig. 5B).
HemW is a heme-binding protein
Previous studies with the HemW homolog from L. lactis revealed heme binding to the protein (13). To test for heme binding of E. coli HemW and determine its specificity, the purified HemW protein and heme were incubated anaerobically overnight and analyzed spectrophotometrically (Fig. 6A, dotted line). As a control, the protein solution used and free heme were analyzed in parallel. HemW showed only the typical protein absorption at A280 and little absorption for the iron–sulfur cluster (Fig. 6A, solid line). Free heme showed the typical spectrum with peaks around A400 and A580 (Fig. 6A, dashed line). The HemW-heme complex revealed a broad absorption peak between A380 and A420 besides the protein absorbance at A280 (Fig. 6A, dotted line). To determine the specificity of heme binding, all three samples (HemW, heme, and HemW-heme complex) were subjected to extensive dialysis overnight and subsequent spectroscopic analyses. Although the spectrum for HemW did not change (Fig. 6B, solid line) and the spectrum for the HemW-heme complex only lost its small increase around A400 (Fig. 6B, dotted line), all free heme was gone (Fig. 6B, dashed line). Identical results were obtained for the dialyzed and Superdex® 200 gel-filtrated HemW-heme complex (data not shown). Interestingly, reduction ot the HemW-heme complex resulted in an increase of absorption at A424, generating a Soret band, and further absorption peaks at A531 and A559 (Fig. 6C, dashed line). In contrast, free reduced heme shows an absorption peak at around A400. These results demonstrate the specificity of HemW-heme interaction.
Figure 6.

Spectroscopic characterization of HemW heme binding. Shown are UV-visible spectra from A250 to A700. A, spectra of 20 μm HemW (solid line) and 20 μm HemW after binding of 20 μm heme (dotted line). The dashed line shows the spectrum of 20 μm free heme. B, spectra of 20 μm HemW (solid line), HemW plus bound heme (dotted line), and free heme all after extensive dialysis (dashed line). Free heme was completely removed after dialysis, whereas HemW and the stable HemW-heme complex remained. C, spectra of the 20 μm HemW-heme complex after dialysis (solid line) and after subsequent addition of 1 mm DTT (dotted line). To emphasize the changes at wavelengths from A500 to A650, these parts of the spectra were enlarged. mAU, milliabsorbance units.
For further analysis of the nature and stoichiometry of HemW-heme interaction, complexes were analyzed via SDS-PAGE with heme staining and acidified butanone extraction. HemW and equimolar amounts of heme were incubated overnight and subjected in duplicate to SDS-PAGE analyses. Subsequently, one half of the gel was stained with Coomassie Brilliant Blue for detection of separated proteins (Fig. 7A, lane 1), whereas the proteins on the second half of the gel were blotted onto a nitrocellulose membrane for heme staining. The detection of the HemW-bound heme was based on its intrinsic peroxidase activity by incubation with the ECL reagent (Fig. 7A, lane 2). The observed heme staining of E. coli HemW indicated stably bound heme. To obtain further evidence for the possible covalent nature of heme binding, butanone extraction experiments, which can result either in release of non-covalently linked heme in the organic phase or in still bound, covalently linked heme in the aqueous phase (22), were performed. Cytochrome c was used as a positive control, clearly indicating the presence of covalently bound heme in the aqueous phase (Fig. 7B, middle tube). Hemoglobin with non-covalently bound heme was used as a negative control. Here, almost all heme was extracted in the upper organic phase (Fig. 7B, right tube). Butanone extraction of HemW incubated with heme revealed a completely clear upper phase and a slightly brownish lower phase (Fig. 7B, left tube). The presence of HemW-derived heme in the lower aqueous phase indicated covalently bound heme. Although our experiment pointed toward covalently bound heme, strong binding of the heme in a tight hydrophobic pocket resistant to SDS and butanone treatment cannot be excluded. Subsequently, the heme staining assay was used to identify the binding stoichiometry of HemW and heme. For this purpose, a solution of 10 μm HemW (Fig. 7C, lanes 1–5) was titrated with heme in increasing concentrations from 5 to 25 μm (Fig. 7C, lanes 6–10). The subsequent heme staining revealed an increase of the heme bound to HemW up to a heme concentration of 10 μm, indicating that apparent saturation of the signal occurred after addition of equimolar amounts of heme (Fig. 7C, lanes 6–10). Alternatively, the stoichiometry of heme binding by HemW was determined spectroscopically by using 20 μm native HemW titrated with increasing amounts of heme. Measurements of the optical density at 416 nm revealed heme binding saturation at 20 μm (Fig. 7D). These results clearly indicate a specific binding with a stoichiometry of one molecule of heme per HemW monomer.
Figure 7.

One molecule of HemW covalently binds one molecule of heme. A, 25 μm E. coli HemW was incubated with an equimolar amount of heme. After SDS-PAGE, separated proteins were either stained with Coomassie Brilliant Blue (A, lane 1) or blotted onto a PVDF membrane. The peroxidase activity of heme is detectable based on the reaction with ECL reagent (A, lane 2). M, marker with proteins of known relative molecular masses. B, for butanone extraction, a 25 μm concentration of either HemW, cytochrome c, or hemoglobin was incubated with 15 μm heme overnight. Afterward, the pH was adjusted to pH 1.5 with 10% HCl. Ice-cold 2-butanone was added and carefully mixed. The heme bound to protein found in the lower aqueous phase indicated the covalent binding of heme to HemW (B, left tube). Butanone extraction from cytochrome c with covalently bound heme (horse heart; 1 mg/ml) served as a positive control (B, middle tube), and hemoglobin with non-covalently bound heme served as a negative control (B, right tube). C, heme stoichiometry of HemW binding. For heme staining, purified E. coli HemW (10 μm) was titrated with increasing amounts of heme (5, 10, 15, 20, and 25 μm. Following SDS-PAGE, the gel was stained with Coomassie Brilliant Blue for protein visualization (lanes 1–5), and the blotted membrane was stained with ECL reagent for bound heme (lanes 5–10). M, marker with proteins of known relative molecular masses. For spectroscopic determination of heme binding, 20 μm HemW was incubated with different amounts of heme. D, titration curve for the determination of the stoichiometry of HemW and heme. Absorption was measured at A416 and plotted against increasing concentrations of heme. The intersection of the two linear slopes indicates the saturation of heme at ∼20 m heme. Binding of 1 mol of heme/mol of HemW monomer was concluded.
Heme binding is independent of the presence of the iron–sulfur cluster
Aerobically prepared HemW without iron–sulfur cluster bound heme as efficiently as anaerobically prepared HemW with the cluster. Similarly, the HemW triple mutant (HemW-C16S/C20S/C23S) also bound heme with high efficiency. In agreement, the Mössbauer spectrum of HemW supplemented with non-enriched heme (natural isotope distribution, only 2.2% 57Fe) showed the same spectrum and identical fit parameters as HemW without further additions. The presence of heme only slightly changed the observed redox potential of the iron–sulfur cluster to around −410 mV (Fig. 3). These results demonstrate that the iron–sulfur cluster was not affected by heme binding. Vice versa, the iron–sulfur cluster did not influence heme binding by HemW. Moreover, the presence of SAM did not change heme binding of HemW.
Respiratory nitrate reductase and bacterioferritin are interaction partners of HemW
To determine specific targets for the potential heme chaperone HemW, multiple interaction partners were tested using the bacterial adenylate cyclase two-hybrid (BACTH) system in P. aeruginosa. The P. aeruginosa system was used because high background noise levels, which obscured the results, were observed for similar experiments in E. coli. In this study, the hemoenzymes bacterial ferritin BfrA; bacterioferritin BfrB; catalase KatA; the last enzyme of heme biosynthesis ferrochelatase, HemH; and the heme-containing subunit of the respiratory nitrate reductase, NarI, were analyzed for their interaction with HemW. The reasons for testing BfrA and -B as well KatA and ferrochelatase HemH are obvious because all proteins are heme-binding/storing enzymes. The consecutive β-galactosidase assays revealed the highest Miller units for the combination of HemW with NarI, indicating their strong affinity. Furthermore, HemW interacted with BfrA and BfrB but not with HemH or KatA. In agreement, in the inverse experiment, neither HemH nor KatA interacted with HemW (Fig. 8).
Figure 8.
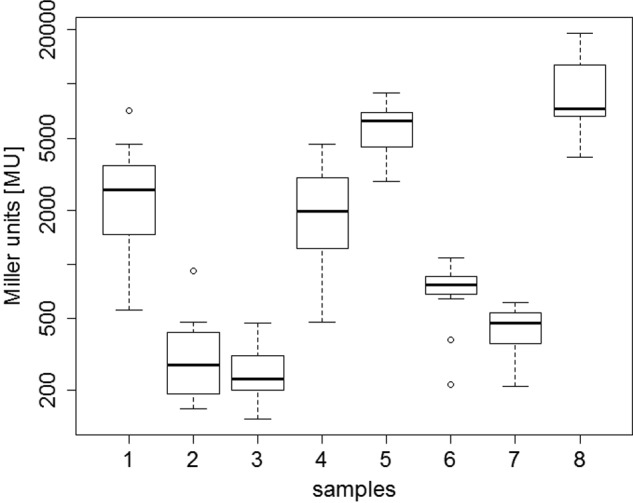
Identification of HemW protein interaction partner using a bacterial two-hybrid system. Shown are the resulting β-galactosidase activities in Miller units (MU) for the testing of the following bait-prey pairs: HemW-BfrA with pUT18C-hemW/pKT25-bfrA (1), HemW-HemH with pUT18C-hemW/pKT25-hemH (2), HemW-KatA with pUT18C-hemW/pKT25-katA (3), HemW-BfrB with pKT25-hemW/pUT18C-bfrB (4), HemW-NarI with pUT18C-hemW/pKNT25-narI (5), and HemH-BfrB with pUT18C-hemH/pKT25-bfrB (6). pKNT25/pUT18 served as a negative control (7), and pKT25-zip/pUT18C-zip served as a positive control (8). Error bars represent S.D.
HemW transfers heme to heme-depleted quinol-nitrate oxidoreductase NarGHI
The strongest interaction of HemW was found with the heme-containing subunit NarI of the respiratory nitrate oxidoreductase NarGHI. Under anaerobic conditions and the presence of nitrate, E. coli utilizes this enzyme for energy generation by replacing oxygen with nitrate as the terminal electron acceptor. This respiratory complex has the ability to use all three natural quinones for energy generation (23, 24). For our approach, ubiquinol served as electron donor, and the membrane-anchored subunit NarI provides the quinol-binding and oxidation site. Two low-spin hemes (bH and bL) involved in the electron transfer from quinols to the subunit NarG are coordinated by NarI (25). The presence of both hemes was demonstrated to be essential for the oxidation of quinols and the overall activity of NarGHI as deduced from analysis of NarI variants having lost either heme bH or bL (26). To unambiguously identify HemW as a true heme chaperone, heme transfer analyses from HemW to the quinol-nitrate oxidoreductase NarGHI from E. coli were performed. Thus, Nar-enriched membrane vesicles from E. coli wildtype MC4100 and the heme-deficient E. coli ΔhemA were prepared. The absence of heme in the heme-depleted membrane vesicles due to the ΔhemA gene mutation was obvious by the visible change in color of the membrane preparation (Fig. 9B) and the corresponding UV-visible spectra (Fig. 9A). The enzymatic activity of heme-depleted nitrate oxidoreductase was first tested spectrophotometrically using a quinol analog as electron donor (27). A classical spectrophotometric activity assay for the nitrate reductase NarGHI was used. This assay is based on the absorption changes of the artificial electron donor, the menaquinol analog 2-methyl-1,4-naphtoquinol (menadiol), used. The oxidation from menadiol to menadione during the reduction of nitrate to nitrite was followed spectroscopically at 260 nm. As mentioned above, only heme-containing NarGHI can catalyze menadiol oxidation in the presence of nitrate. Results of the heme transfer to the NarGHI are summarized in box plots in Fig. 9C. Membrane vesicles prepared from E. coli wildtype MC4100 served as a positive control in this experiment (Fig. 9C, lane A). Activity assays of membrane vesicles containing heme-depleted quinol-nitrate oxidoreductase revealed only low residual enzymatic activity (Fig. 9C, lane B). However, addition of HemW preincubated with heme led to a significant nitrate reductase activity (Fig. 9C, lane C). Addition of NADH further increased the observed enzyme activity (Fig. 9C, lane D). Over 50% of the potential nitrate reductase activity was restored. Full restoration might be hampered by the possible instability of the heme-free apoenzyme compared with the nitrate reductase holoenzyme. In the negative controls, E. coli ΔhemA mutant membrane vesicles with the addition of solely heme or apo-HemW exhibited residual background activity identical to the results of the heme-free nitrate reductase (Fig. 9C, lanes E and F). Similarly, the combination of heme, HemW, and NADH did not react with the nitrate reductase substrate used (Fig. 9C, lane H). SAM did not influence the heme transfer reaction. To analyze the influence of the [4Fe–4S] cluster on heme transfer, a HemW triple mutant (HemW-C16S/C20S/C23S) lacking the cluster was tested. The activity of NarGHI was not restored by the triple-mutant HemW (Fig. 9C, lane G), whereas the heme binding behavior of the mutant enzyme was comparable with wildtype HemW. The observed catalytic activity of the nitrate oxidoreductase in the membrane vesicles prepared from the heme-deficient E. coli ΔhemA mutant after addition of heme-loaded HemW clearly indicates successful heme transfer from HemW to the heme-requiring NarI subunit. These results support the function of HemW as a heme chaperone.
Figure 9.

HemW-mediated heme transfer to the nitrate oxidoreductase NarGHI. A, comparative spectroscopic analysis of prepared membrane vesicles from E. coli wildtype MC4100 and the corresponding E. coli ΔhemA mutant. The recorded absorption spectrum at around A425 indicated the absence of bound heme cofactor in NarGHI produced by E. coli ΔhemA mutant (dashed line) in contrast to the spectrum recorded for the identical membrane vesicle preparation from wildtype E. coli. B, the decolorization of the prepared membrane vesicles (left tube, wildtype; right tube, ΔhemA mutant) due to the depletion of heme is optically visible. C, enzyme assays were performed with membrane vesicles isolated from E. coli MC4100/pVA700 overexpressing narGHJI (labeled MC4100) and membrane vesicles with overproduced heme-depleted nitrate oxidoreductase isolated from E. coli ΔhemA/pVA700 (labeled ΔhemA SHSP18). The heme-depleted nitrate oxidoreductase was incubated with HemW-heme (C), HemW-heme + NADH (D), and [4Fe–4S] clusterless HemW-C16S/C20S/C23S–heme (G) and as negative controls solely with heme (E), HemW (F), or HemW and NADH (H), respectively. 20 mm 2-methyl-1,4-naphtoquinol (menadiol) served as electron donor, 2 mm nitrate served as electron acceptor, and 5 mm NADH and 1.5 mm free heme were used where indicated. The range between −1 and 18 μmol/min × ml is shown. Error bars represent S.D.
Slight growth phenotype of the E. coli hemW mutant
In light of the heme chaperone function of HemW for the respiratory nitrate reductase NarGHI, growth experiments with wildtype and a hemW mutant under anaerobic, nitrate respiratory conditions with the non-fermentable carbon source glycerol were performed. Under the tested growth conditions, the hemW mutant showed a slight, but highly reproducible growth phenotype (Fig. 1B, blue line). In the absence of nitrate, almost no growth was observed. The observed growth of the hemW mutant indicated the presence of intact nitrate reductase and a second heme-inserting system supplementing for the inactivated HemW. Backup systems for essential functions were observed in E. coli for catalases, ribonucleotide reductases, and pyruvate kinases to name a few.
Human HemW homologue RSAD1 binds heme
RSAD1 from Homo sapiens (30% amino acid sequence identity, 50% homology) was analyzed for heme binding via its recombinant production in E. coli, affinity purification, and heme staining (Fig. 10A). Clearly, heme bound strongly to human RSAD1. Additionally, a typical absorption spectrum was also recorded for the H. sapiens RSAD1-heme complex (Fig. 10B). Altogether, the experiments for human RSAD1 confirmed the results for bacterial HemW, suggesting the ubiquitous function of HemW/RSAD1 as a heme chaperone.
Figure 10.
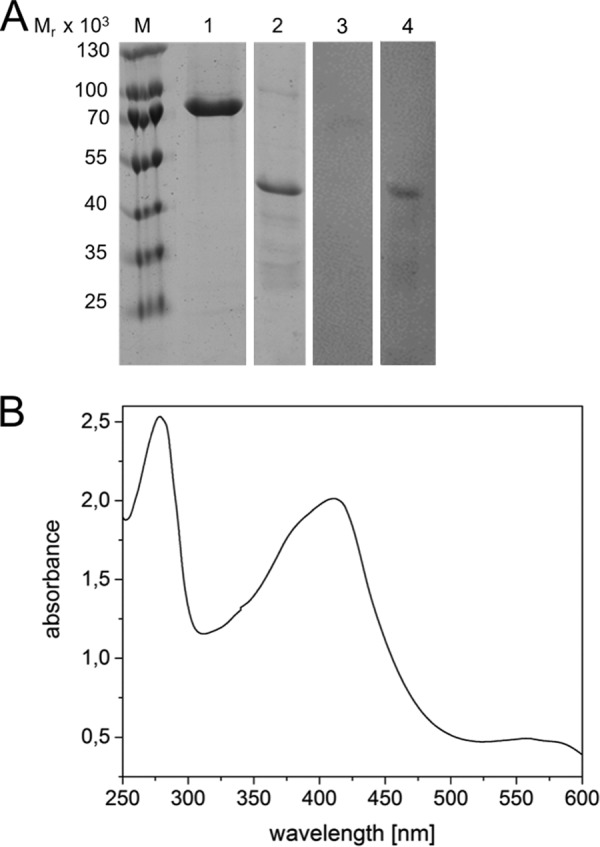
Heme binding of HemW (RSAD1) from humans. A, for heme staining, purified RSAD1/HemW proteins of H. sapiens (lanes 1 and 3) and E. coli (lanes 2 and 4) were incubated with heme overnight and separated via SDS-PAGE. The proteins were stained with InstantBlueTM (Expedeon Inc., San Diego, CA) (lanes 1 and 2) or blotted onto nitrocellulose membrane with subsequent ECL treatment for heme staining (lanes 3 and 4). A marker indicates the relative molecular mass of the separated proteins. B, UV-visible spectra of H. sapiens RSAD1 incubated with heme overnight. An absorption maximum at 410 nm indicated heme binding. Peaks at 531 and 556 nm are typical for incorporated heme.
Discussion
HemW protein inserts heme into proteins with different functions in respiration. Genes encoding HemW/RSAD1 are found in the genomes of almost all organisms (bacteria, archaea, plants, and animals) with the noticeable exception of fungi. The presence of hemW genes nicely correlates with the utilization of heme-dependent aerobic and anaerobic respiration. Consequently, hemW is even found in organisms that utilize heme taken up from the environment for this process such as Lactococcus (13). In contrast, strict fermentative organisms such as Clostridium, deficient in heme biosynthesis and heme uptake due to the absence of classical heme-dependent respiratory processes, also lack HemW. How does this radical SAM protein-based heme chaperone work? As shown in the model in Fig. 11, heme can be derived from biosynthesis and heme import. Interestingly, no stable complex formation between HemW and ferrochelatase (HemH), the last enzyme of the heme biosynthesis, was observed (28). However, heme from HemW was found to interact with BfrA and BfrB. In 1999, Hassett and co-workers (29) reported that P. aeruginosa catalase A (KatA) requires bacterial ferritin A (BfrA) for full activity. They proposed that BfrA not only stores iron for incorporation into heme but also the necessary prosthetic heme group of KatA. Consequently, BfrA could function as a heme transporter between HemH, the last enzyme of heme biosynthesis, and the heme-accepting protein KatA.
Figure 11.

Working model of E. coli HemW. Heme from heme biosynthesis is transferred via the bacterioferritin Bfr (gray) as a carrier to the heme chaperone HemW (orange). Dimerized, the [4Fe–4S] cluster–containing HemW localizes to the membrane where it interacts with its target protein, NarI (yellow), a subunit of the respiratory nitrate reductase NarGHI. After heme incorporation into apo-NarI, the holo-NarGHI catalyzes the reduction of nitrate to nitrite. PPO is protoporphyrinogen IX oxidase; FC (HemH) is ferrochelatase.
Recently, a radical SAM protein (ChuW) with heme-degrading activity from E. coli was described. ChuW utilizes a radical-based mechanism for heme ring opening and methylation of the resulting open-chain tetrapyrrole (30). E. coli ChuW has an amino acid sequence identity of 28% to E. coli HemW. However, almost all identical amino acid residues are part of the N-terminal radical SAM element of both proteins. No common heme-binding domain was detected.
Based on these observations and other data, a model for HemW (depicted in Fig. 11) was deduced. Heme produced in heme biosynthesis is transferred via bacterioferritin to HemW where it is covalently bound. In the presence of the [4Fe–4S] cluster, HemW dimerizes, becomes localized at the membrane, and interacts with its target NarI. Transfer of the heme requires an intact [4Fe–4S] cluster and might involve radical chemistry. The exact mechanism of the NADH stimulation of heme transfer remains to be determined Finally, the human RSAD1 protein was found to bind heme tightly, indicating the general importance of the HemW/RSAD1 enzyme family for the heme insertion into cellular proteins. Future experiments will focus on the biochemistry of the heme release from HemW to target proteins and the role of the radical chemistry in that process.
Experimental procedures
Primers, strains, and plasmids
The primers, strains, and plasmids used in this study are listed in Tables S1–S3.
Cloning, expression, and purification of E. coli hemW
The hemW gene was PCR-amplified from E. coli genomic DNA using the primers hemWE.c.-pGEX-for and hemWE.c.-pGEX-rev harboring a BamHI and a XhoI restriction site (underlined in Table S1), respectively. The 1137-bp PCR product was cloned into the respectively restricted vector pGEX-6P-1 according to the manufacturer's instructions, yielding the plasmid pGEX-HemWE.c.. The vector encoded HemW with an N-terminal GST tag and a cleavage site for PreScission protease (GE Healthcare). GST-HemW was produced in E. coli BL21 (DE3) with the help of the plasmid pGEX-HemWE.c.. Two-liter cultures were grown aerobically in LB medium and 100 mg/ml ampicillin at 200 rpm at 37 °C. hemW gene expression was induced at an attenuance at A578 of 0.6 by addition of 0.5 mm isopropyl β-d-thiogalactoside, and cultivation was continued overnight at 17 °C at 200 rpm. Cells were harvested by centrifugation at 4000 × g for 15 min at 4 °C. All following steps occurred under strict anaerobic conditions at 20 °C. For HemW purification, cells were resuspended in 10 ml of buffer 1 (140 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, 5% glycerol, 1 mm DTT, pH 7.4), and cells were disrupted by a single passage through a French press at 19,200 p.s.i. Cell debris and insoluble proteins were removed by centrifugation for 60 min at 25,000 × g at 4 °C. The soluble protein fraction was loaded onto a glutathione-Sepharose column (Macherey-Nagel, Düren, Germany). HemW was liberated from the column by cleavage of the GST tag overnight with PreScission protease (GE Healthcare) according to the manufacturer's instructions. HemW-containing fractions were eluted, pooled, and concentrated by ultrafiltration using an Amicon membrane with a 30-kDa molecular mass cutoff (Merck Millipore, Billerica, MA). Protein concentrations were determined with a colorimetric assay using Bradford reagent with bovine serum albumin as standard according to the manufacturer's instructions. (Sigma-Aldrich). The triple-mutant hemWC16S/C20S/C23S was constructed using the Q5® site-directed mutagenesis kit (New England Biolabs, Frankfurt, Germany) according to the manufacturer's instructions. Successful construction of mutations was confirmed by DNA sequencing of the complete hemW gene variant. Production and purification of the HemW variant were performed analogously to wildtype HemW.
Absorption spectroscopy
UV-visible absorption spectra of HemW and HemW-heme complexes were recorded on a Jasco V-650 spectrophotometer in buffer 1 using the same buffer as a blank. The recording wavelengths were from A250 to A600. Heme (10 mg) was dissolved in 1 ml of 0.1 m NaOH and incubated at room temperature for 1 h. After addition of 1 ml of Tris-HCl (1 m, pH 7.6) the solution was centrifuged at 12,100 × g at room temperature for 10 min. The supernatant was filtered to remove insoluble residues, and the concentration was determined as A385 X/58.44 = x × 500 = x mmol/liter.
In vitro iron–sulfur cluster analysis
The in vitro reconstitution of [Fe–S] clusters was performed as described previously (31). After reconstitution of the [Fe–S] cluster, the excess iron and sulfide was removed by centrifugation at 12,100 × g at 4 °C and subsequent passage of the protein solution through a NAP-25 column (GE Healthcare) according to the manufacturer's instructions. The iron content of purified HemW was determined according to a protocol described elsewhere (32). After denaturation of the protein with 1 m perchloric acid, bathophenanthroline was used as the chelating reagent. The sulfur content was determined as described previously (33).
Mössbauer spectroscopy
The final HemW concentration used for Mössbauer spectroscopy analysis of E. coli HemW was 350 μm. Sample preparation was performed under strict anaerobic conditions. The iron–sulfur cluster of HemW was reconstituted with ammonium [57Fe]ferric citrate. For the sample containing HemW supplemented with heme, an equimolar ratio of heme to HemW was added, and the mixture was incubated overnight at 20 °C. The solutions were transferred to 350-μl Mössbauer cups and frozen in liquid nitrogen. Mössbauer spectra were recorded on a spectrometer with alternating constant acceleration of the γ-source. The minimum experimental line width was 0.24 mm/s (full width at half-height). The sample temperature was maintained constant in an Oxford Instruments Variox cryostat, whereas the 57Co/Rh source (1.8 GBq) was kept at room temperature. Isomer shifts are quoted relative to iron metal at 300 K.
Cyclic voltammetry
Cyclic voltammetry measurements were performed using an Ametek Versastat 3. The measurements were carried out in a self-made anaerobic three-electrode electrochemical cell flushed with nitrogen. As the reference, a silver/silver chloride electrode was used (3 mol liter−1 KCl). All potentials in the text and figures are given versus normal hydrogen electrode (+210 mV). A platinum wire was used as the counterelectrode with glassy carbon as the working electrode. Before each measurement, the platinum wire was annealed in a natural gas flame, and the glassy carbon electrode was pretreated in nitric acid, neutralized, polished with 0.05-μm alumina, and annealed again in a natural gas flame. For each experiment, 20 cycles were recorded. The potential slightly drifted only over the first 10 cycles and stabilized thereafter. In this work, only the stabilized potential is discussed. The cycles were recorded with a scan rate of 1 V s−1. All electrochemical experiments were carried out at ambient temperature in a 100-μl drop. Samples contained 120 μm HemW, 120 μm heme, or 500 μm S-adenosylmethionine (Sigma-Aldrich) in diverse combinations. Samples were prepared under anaerobic conditions in a glove box (Coy Laboratories) and transferred into HPLC vials before injecting into the cyclic voltammetry chamber directly on the glassy carbon electrode.
SAM binding and cleavage analyses
The SAM binding assays were performed as described previously (34). For this purpose, 100 μm purified HemW or BSA was incubated with 0.5 μCi of S-[carboxyl-14C]SAM (1.48–2.22 GBq/mmol, 0.1 mCi/ml) at 25 °C for 1 h. Mixtures were separated via chromatography through an illustraTM NAPTM-5 desalting column (GE Healthcare). Fractions (200 μl) were collected and analyzed by liquid scintillation counting (PerkinElmer Life Sciences).
For SAM cleavage, 25 μm purified HemW (free or loaded with heme) was incubated with 0.6 mm sodium dithionite as potential electron donor and 0.6 mm SAM overnight at 17 °C under anaerobic conditions. Reactions were stopped by adding 5% formic acid. For HPLC analysis, the samples were centrifuged at 16,100 × g for 10 min. HPLC analysis was performed as described previously (20). In detail, for the separation of 5′-deoxyadenosine from SAM, a Hypercarb column (Thermo Fisher Scientific, Waltham, MA) in a Jasco 2000 system with a flow rate of 0.2 ml/min was used at room temperature. A 5-ml gradient of 0.1% TFA in H2O and 0.08% TFA in acetonitrile was applied. SAM and 5′-deoxyadenosine were detected at A254. Appropriate markers were used to calibrate the column.
Determination of the native molecular mass
An ÄKTA purifier system for gel permeation chromatography with a Superdex 200 HR 10/300 column was used (GE Healthcare). The column was equilibrated using buffer 1 and calibrated using carbonic anhydrase (Mr = 9,000), bovine serum albumin (Mr = 66,200), yeast alcohol dehydrogenase (Mr = 150,000), and β-amylase (Mr = 200,000) as marker proteins. A sample containing purified recombinant HemW was chromatographed under identical conditions with a flow rate of 0.25 ml/min under anaerobic conditions.
Heme binding assays
Heme (1 mg) was dissolved in 100 μl of 100 mm NaOH and thoroughly mixed (34). After 30 min, 100 μl of 1 m Tris, pH 7.4, was added. The solution was centrifuged at 4 °C for 10 min at 12,100 × g. The concentration was determined using ϵ385 = 58.44 (mm cm)−1. HemW was incubated with an equimolar concentration of heme under anaerobic conditions at 20 °C overnight, and the formed complex was used for further analyses including spectroscopy, heme staining, and the in vitro transfer reaction to NarGHI.
For detection of the stable HemW-heme complex, the heme-complexed protein was separated via SDS-PAGE followed by electrophoretic transfer to an Amersham Biosciences Hybond-ECL nitrocellulose membrane (GE Healthcare). After three washing steps with buffer 2 (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4), the nitrocellulose membrane was incubated for 5 min with Amersham Biosciences ECLTM Prime Western Blotting Detection Reagent (GE Healthcare). Heme was detected by its intrinsic peroxidase activity (35) with a charge-coupled device camera. Acidic butanone extraction was performed as described elsewhere (22, 36).
Protein-protein interaction studies using BACTH
The BACTH System kit (Euromedex, Souffelweyersheim, France) was used to analyze the interaction between P. aeruginosa HemW and selected partner proteins from the same bacterium (BfrA, KatA, HemH, BfrB, and NarI). Corresponding genes were amplified by PCR using P. aeruginosa genomic DNA as template DNA. The primers used are listed in Table S1. Genes were integrated into plasmids pKT25, pKNT25, pUT18, and pUT18C, respectively (Euromedex). For the detection of the in vivo interaction, selected plasmids, encoding the genes for the proteins of interest, were cotransformed into the reporter strain E. coli BTH101 and washed after transformation twice with M63 buffer (2 g of (NH4)2SO4, 13.6 g of KH2PO4, 0.5 mg of FeSO4·7H2O, 1 ml of 1 m MgSO4·7H2O, 10 ml of 20% maltose, 2 ml of 0.05% thiamin, pH 7.0). Colonies were selected on M63 agar plates containing ampicillin (100 μg/ml), kanamycin (50 μg/ml), or streptomycin (100 μg/ml); 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) (40 μg/ml); and isopropyl β-d-thiogalactoside (0,5 mm). Incubation was for 4–8 days at 30 °C. β-Galactosidase assays were performed using the Miller method according to Griffith and Wolf (37). For this purpose, blue E. coli BTH101 colonies were used to inoculate LB medium supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin. One hundred microliters of bacterial suspension were centrifuged at 4400 × g for 5 min at 4 °C. The resulting pellet was resuspended in 900 μl of Z buffer (60 mm Na2PO4·7H2O, 40 mm NaH2PO4·H2O, 10 mm KCl, 1 mm MgSO4·7H2O, 50 mm β-mercaptoethanol), and the cell density was measured at A600. Afterward, one drop of 0.1% SDS and chloroform were added. The sample was incubated for 5 min at 30 °C at 300 rpm. The reaction was started by adding 200 μl of o-nitrophenyl β-d-galactopyranoside (4 mg/ml) dissolved in 100 mm phosphate buffer (60 mm Na2HPO4·7H2O, 40 mm NaH2PO4·H2O, pH 7.0). The suspension was mixed and incubated at 30 °C. The reaction was terminated by adding 500 μl of 1 m Na2CO3. A centrifugation step for 5 min at 12,700 × g removed cell debris and chloroform. Optical densities were recorded at A420 and A550. Miller units (MU) were calculated using the equation MU = 1000 × (A420 − (1.75 × A550)/(TR × V × A600) where TR is time of the reaction in minutes and V is the volume of culture used in the assay in ml. The units indicate the change in A420/min/ml of cells/A600.
Heme transfer to heme-free quinol-nitrate oxidoreductase NarGHI
The heme auxotroph hemA strain SHSP18 (38) was transformed with the pVA700 plasmid (39), allowing overproduction of the NarGHI complex. Isolation of the transformants was performed on an LB agar plate supplemented with 40 mm glucose and 150 μm δ-aminolevulinic acid. The following steps were performed simultaneously for the hemA strain SHSP18 and the wildtype strain E. coli MC4100 (40) both transformed with the pVA700 plasmid. An LB overnight culture supplemented with glucose (40 mm), sodium formate (12.5 mm), sodium selenite (2 μm), sodium molybdate (2 μm), and phosphate buffer (100 mm, pH 6.8) was inoculated with a single colony of the corresponding strains. The production culture was then inoculated with this overnight culture to an initial A600 of 0.05 in the identical medium and incubated for 24 h at 37 °C. The grown cells were pelleted by centrifugation and kept at −20 °C until use. Cells were resuspended in 50 mm MOPS buffer, 1 mm MgCl2, pH 7.2, and broken by two passages through a French press at 1100 p.s.i. Intact cells and cell debris were removed by a centrifugation at 14,000 × g. Membrane vesicles were obtained after ultracentrifugation at 40,000 × g for 90 min and kept at −80 °C until use. The amount of NarGHI in the various preparations was determined via immunoelectrophoresis. The various membrane vesicle preparations from the wildtype strain had 85–95 mg/ml total protein with 5.6–7.9 mg/ml (6.5–8.3%) NarGHI protein, whereas the preparation from the hemA mutant yielded 60–106 mg/ml total protein with 3.6–5.8 mg/ml (5.5–6.0%) NarGHI protein. Standard deviations between 3 and 5% were observed. All measurements were performed with equal amounts of NarGHI (4 μg) and 1.5 μm purified HemW/HemW-C16S/C20S/C23S. For the assay, 20 mm 2-methyl-1,4-naphtoquinol (menadiol) as electron donor, 5 mm NADH, 1.5 mm free heme, and 2 mm nitrate as electron acceptor were added where indicated. A quartz cell with 1.4-ml volume was used. The assay was performed under strict anaerobic conditions at 30 °C. The activity of E. coli quinol-nitrate oxidoreductase was measured spectrophotometrically as outlined before (27). The changes in absorption of the ubiquinol analog 2-ethyl-4-naphtoquinol (menadiol) caused by oxidation were measured at A260. A quartz cell with 1.4-ml volume was used. The assay was performed under strict anaerobic conditions at 30 °C. One unit of quinol-nitrate oxidoreductase activity is the amount of nitrate oxidoreductase catalyzing the production of 1 μmol of menadione/min.
Author contributions
V.H., S.K., T.M., S.B., F.A., K.M., E.B., W.L., K.K., P.S., and M.J. formal analysis; V.H., S.K., T.M., S.B., F.A., K.M., E.B., K.K., P.S., M.B., and M.J. investigation; V.H., S.K., T.M., S.B., F.A., and A.M. methodology; V.H. writing-original draft; S.K., T.M., S.B., F.A., W.L., and P.S. data curation; A.M. and D.J. conceptualization; A.M., W.L., P.S., M.B., M.J., and D.J. project administration; M.B. and D.J. supervision; M.J. and D.J. writing-review and editing; D.J. funding acquisition.
Supplementary Material
Acknowledgments
We especially thank Léa Sylvi for excellent technical assistance and Gunhild Layer for critical reading of the manuscript.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Forschergruppe FOR1220 PROTRAIN). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2 and Tables S1–S3.
- SAM
- S-adenosylmethionine
- RSAD1
- radical SAM domain–containing 1
- CPDH
- coproporphyrinogen III dehydrogenase
- BACTH
- bacterial adenylate cyclase two-hybrid
- E.c.
- E. coli.
References
- 1. Frey P. A., Hegeman A. D., and Ruzicka F. J. (2008) The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 43, 63–88 10.1080/10409230701829169 [DOI] [PubMed] [Google Scholar]
- 2. Landgraf B. J., McCarthy E. L., and Booker S. J. (2016) Radical S-adenosylmethionine enzymes in human health and disease. Annu. Rev. Biochem. 85, 485–514 10.1146/annurev-biochem-060713-035504 [DOI] [PubMed] [Google Scholar]
- 3. Wang J., Woldring R. P., Román-Meléndez G. D., McClain A. M., Alzua B. R., and Marsh E. N. (2014) Recent advances in radical SAM enzymology: new structures and mechanisms. ACS Chem. Biol. 9, 1929–1938 10.1021/cb5004674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byer A. S., Shepard E. M., Peters J. W., and Broderick J. B. (2015) Radical S-adenosyl-l-methionine chemistry in the synthesis of hydrogenase and nitrogenase metal cofactors. J. Biol. Chem. 290, 3987–3994 10.1074/jbc.R114.578161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frey P. A., and Reed G. H. (2011) Pyridoxal-5′-phosphate as the catalyst for radical isomerization in reactions of PLP-dependent aminomutases. Biochim. Biophys. Acta 1814, 1548–1557 10.1016/j.bbapap.2011.03.005 [DOI] [PubMed] [Google Scholar]
- 6. Mehta A. P., Abdelwahed S. H., Mahanta N., Fedoseyenko D., Philmus B., Cooper L. E., Liu Y., Jhulki I., Ealick S. E., and Begley T. P. (2015) Radical S-adenosylmethionine (SAM) enzymes in cofactor biosynthesis: a treasure trove of complex organic radical rearrangement reactions. J. Biol. Chem. 290, 3980–3986 10.1074/jbc.R114.623793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sanyal I., Cohen G., and Flint D. H. (1994) Biotin synthase: purification, characterization as a [2Fe-2S]cluster protein, and in vitro activity of the Escherichia coli bioB gene product. Biochemistry 33, 3625–3631 10.1021/bi00178a020 [DOI] [PubMed] [Google Scholar]
- 8. Helbig K. J., and Beard M. R. (2014) The role of viperin in the innate antiviral response. J. Mol. Biol. 426, 1210–1219 10.1016/j.jmb.2013.10.019 [DOI] [PubMed] [Google Scholar]
- 9. Layer G., Moser J., Heinz D. W., Jahn D., and Schubert W. D. (2003) Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of radical SAM enzymes. EMBO J. 22, 6214–6224 10.1093/emboj/cdg598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Layer G., Verfürth K., Mahlitz E., and Jahn D. (2002) Oxygen-independent coproporphyrinogen-III oxidase HemN from Escherichia coli. J. Biol. Chem. 277, 34136–34142 10.1074/jbc.M205247200 [DOI] [PubMed] [Google Scholar]
- 11. Layer G., Grage K., Teschner T., Schünemann V., Breckau D., Masoumi A., Jahn M., Heathcote P., Trautwein A. X., and Jahn D. (2005) Radical S-adenosylmethionine enzyme coproporphyrinogen III oxidase HemN: functional features of the [4Fe-4S] cluster and the two bound S-adenosyl-l-methionines. J. Biol. Chem. 280, 29038–29046 10.1074/jbc.M501275200 [DOI] [PubMed] [Google Scholar]
- 12. Dailey H. A., Gerdes S., Dailey T. A., Burch J. S., and Phillips J. D. (2015) Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin. Proc. Natl. Acad. Sci. U.S.A. 112, 2210–2215 10.1073/pnas.1416285112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abicht H. K., Martinez J., Layer G., Jahn D., and Solioz M. (2012) Lactococcus lactis HemW (HemN) is a haem-binding protein with a putative role in haem trafficking. Biochem. J. 442, 335–343 10.1042/BJ20111618 [DOI] [PubMed] [Google Scholar]
- 14. Kranz R. G., Richard-Fogal C., Taylor J. S., and Frawley E. R. (2009) Cytochrome c biogenesis: mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol. Mol. Biol. Rev. 73, 510–528 10.1128/MMBR.00001-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shepherd M., Heath M. D., and Poole R. K. (2007) NikA binds heme: a new role for an Escherichia coli periplasmic nickel-binding protein. Biochemistry 46, 5030–5037 10.1021/bi700183u [DOI] [PubMed] [Google Scholar]
- 16. Hannibal L., Collins D., Brassard J., Chakravarti R., Vempati R., Dorlet P., Santolini J., Dawson J. H., and Stuehr D. J. (2012) Heme binding properties of glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 51, 8514–8529 10.1021/bi300863a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hannappel A., Bundschuh F. A., and Ludwig B. (2012) Role of Surf1 in heme recruitment for bacterial COX biogenesis. Biochim. Biophys. Acta 1817, 928–937 10.1016/j.bbabio.2011.09.007 [DOI] [PubMed] [Google Scholar]
- 18. Layer G., Pierik A. J., Trost M., Rigby S. E., Leech H. K., Grage K., Breckau D., Astner I., Jänsch L., Heathcote P., Warren M. J., Heinz D. W., and Jahn D. (2006) The substrate radical of Escherichia coli oxygen-independent coproporphyrinogen III oxidase HemN. J. Biol. Chem. 281, 15727–15734 10.1074/jbc.M512628200 [DOI] [PubMed] [Google Scholar]
- 19. Xu K., Delling J., and Elliott T. (1992) The genes required for heme synthesis in Salmonella typhimurium include those encoding alternative functions for aerobic and anaerobic coproporphyrinogen oxidation. J. Bacteriol. 174, 3953–3963 10.1128/jb.174.12.3953-3963.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuehner M., Schweyen P., Hoffmann M., Ramos J. V., Reijerse E. J., Lubitz W., Broering M., and Layer G. (2016) The auxiliary [4Fe-4S] cluster of the radical SAM heme synthase from Methanosarcina barkeri is involved in electron transfer. Chem. Sci. 7, 4633–4643 10.1039/C6SC01140C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rouault T. A. (2015) Mammalian iron-sulphur proteins: novel insights into biogenesis and function. Nat. Rev. Mol. Cell Biol. 16, 45–55 10.1038/nrm3909 [DOI] [PubMed] [Google Scholar]
- 22. Teale F. W. (1959) Cleavage of the haem-protein link by acid methylethylketone. Biochim. Biophys. Acta 35, 543 10.1016/0006-3002(59)90407-X [DOI] [PubMed] [Google Scholar]
- 23. Rendon J., Pilet E., Fahs Z., Seduk F., Sylvi L., Hajj Chehade M., Pierrel F., Guigliarelli B., Magalon A., and Grimaldi S. (2015) Demethylmenaquinol is a substrate of Escherichia coli nitrate reductase A (NarGHI) and forms a stable semiquinone intermediate at the NarGHI quinol oxidation site. Biochim. Biophys. Acta 1847, 739–747 10.1016/j.bbabio.2015.05.001 [DOI] [PubMed] [Google Scholar]
- 24. Unden G., Steinmetz P. A., and Degreif-Dunnwald P. (2014) The aerobic and anaerobic respiratory chain of Escherichia coli and Salmonella enterica: enzymes and energetics. EcoSal Plus 6 10.1128/ecosalplus.ESP-0005-2013 [DOI] [PubMed] [Google Scholar]
- 25. Bertero M. G., Rothery R. A., Palak M., Hou C., Lim D., Blasco F., Weiner J. H., and Strynadka N. C. (2003) Insights into the respiratory electron transfer pathway from the structure of nitrate reductase A. Nat. Struct. Biol. 10, 681–687 10.1038/nsb969 [DOI] [PubMed] [Google Scholar]
- 26. Magalon A., Lemesle-Meunier D., Rothery R. A., Frixon C., Weiner J. H., and Blasco F. (1997) Heme axial ligation by the highly conserved His residues in helix II of cytochrome b (NarI) of Escherichia coli nitrate reductase A. J. Biol. Chem. 272, 25652–25658 10.1074/jbc.272.41.25652 [DOI] [PubMed] [Google Scholar]
- 27. Lanciano P., Magalon A., Bertrand P., Guigliarelli B., and Grimaldi S. (2007) High-stability semiquinone intermediate in nitrate reductase A (NarGHI) from Escherichia coli is located in a quinol oxidation site close to heme bD. Biochemistry 46, 5323–5329 10.1021/bi700074y [DOI] [PubMed] [Google Scholar]
- 28. Dailey H. A., Dailey T. A., Gerdes S., Jahn D., Jahn M., O'Brian M. R., and Warren M. J. (2017) Prokaryotic heme biosynthesis: multiple pathways to a common essential product. Microbiol. Mol. Biol. Rev. 81, e00048–16 10.1128/MMBR.00048-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ma J. F., Ochsner U. A., Klotz M. G., Nanayakkara V. K., Howell M. L., Johnson Z., Posey J. E., Vasil M. L., Monaco J. J., and Hassett D. J. (1999) Bacterioferritin A modulates catalase A (KatA) activity and resistanceto hydrogen peroxide in Pseudomonas aeruginosa. J. Bacteriol. 181, 3730–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. LaMattina J. W., Nix D. B., and Lanzilotta W. N. (2016) Radical new paradigm for heme degradation in Escherichia coli O157:H7. Proc. Natl. Acad. Sci. U.S.A. 113, 12138–12143 10.1073/pnas.1603209113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fluhe L., Knappe T. A., Gattner M. J., Schäfer A., Burghaus O., Linne U., and Marahiel M. A. (2012) The radical SAM enzyme AlbA catalyzes thioether bond formation in subtilosin A. Nat. Chem. Biol. 8, 350–357 10.1038/nchembio.798 [DOI] [PubMed] [Google Scholar]
- 32. Fish W. W. (1988) Rapid colorimetric micromethod for the quantitation of complexed iron in biological samples. Methods Enzymol. 158, 357–364 10.1016/0076-6879(88)58067-9 [DOI] [PubMed] [Google Scholar]
- 33. Beinert H. (1983) Semi-micro methods for analysis of labile sulfide and of labile sulfide plus sulfane sulfur in unusually stable iron-sulfur proteins. Anal. Biochem. 131, 373–378 10.1016/0003-2697(83)90186-0 [DOI] [PubMed] [Google Scholar]
- 34. Storbeck S., Walther J., Müller J., Parmar V., Schiebel H. M., Kemken D., Dülcks T., Warren M. J., and Layer G. (2009) The Pseudomonas aeruginosa nirE gene encodes the S-adenosyl-L-methionine-dependent uroporphyrinogen III methyltransferase required for heme d1 biosynthesis. FEBS J. 276, 5973–5982 10.1111/j.1742-4658.2009.07306.x [DOI] [PubMed] [Google Scholar]
- 35. Owens C. P., Du J., Dawson J. H., and Goulding C. W. (2012) Characterization of heme ligation properties of Rv0203, a secreted heme binding protein involved in Mycobacterium tuberculosis heme uptake. Biochemistry 51, 1518–1531 10.1021/bi2018305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vargas C., McEwan A. G., and Downie J. A. (1993) Detection of c-type cytochromes using enhanced chemiluminescence. Anal. Biochem. 209, 323–326 10.1006/abio.1993.1127 [DOI] [PubMed] [Google Scholar]
- 37. Griffith K. L., and Wolf R. E. Jr. (2002) Measuring β-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem. Biophys. Res. Commun. 290, 397–402 10.1006/bbrc.2001.6152 [DOI] [PubMed] [Google Scholar]
- 38. Săsărman A., Surdeanu M., Szégli G., Horodniceanu T., Greceanu V., and Dumitrescu A. (1968) Hemin-deficient mutants of Escherichia coli K-12. J. Bacteriol. 96, 570–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guigliarelli B., Magalon A., Asso M., Bertrand P., Frixon C., Giordano G., and Blasco F. (1996) Complete coordination of the four Fe-S centers of the β subunit from Escherichia coli nitrate reductase. Physiological, biochemical, and EPR characterization of site-directed mutants lacking the highest or lowest potential [4Fe-4S] clusters. Biochemistry 35, 4828–4836 10.1021/bi952459p [DOI] [PubMed] [Google Scholar]
- 40. Peters J. E., Thate T. E., and Craig N. L. (2003) Definition of the Escherichia coli MC4100 genome by use of a DNA array. J. Bacteriol. 185, 2017–2021 10.1128/JB.185.6.2017-2021.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.