

Abstract
Pyoverdines are important siderophores that guarantee iron supply to important pathogenic and non-pathogenic pseudomonads in host habitats. A key characteristic of all pyoverdines is the fluorescent dihydroxyquinoline group that contributes two ligands to the iron complexes. Pyoverdines are derived from the non-ribosomally synthesized peptide ferribactin, and their fluorophore is generated by periplasmic oxidation and cyclization reactions of d-tyrosine and l-diaminobutyric acid. The formation of the fluorophore is known to be driven by the periplasmic tyrosinase PvdP. Here we report that the putative periplasmic oxidoreductase PvdO of Pseudomonas fluorescens A506 is required for the final oxidation of dihydropyoverdine to pyoverdine, which completes the fluorophore. The pvdO deletion mutant accumulates dihydropyoverdine, and this phenotype is fully complemented by recombinant PvdO. The autoxidation of dihydropyoverdine at alkaline pH and the presence of high copper concentrations can mask this phenotype. Mutagenesis of conserved residues with potential catalytic function identified Glu-260 as an essential residue whose mutation abolished function without affecting stability or transport. Glu-260 of PvdO is at the exact position of the active-site cysteine in the structurally related formylglycine-generating enzyme. Evolution thus used the same protein fold for two distinct functionalities. As purified PvdO was inactive, additional factors are required for catalysis.
Keywords: fluorophore formation, periplasmic maturation, Pseudomonas fluorescens, pyoverdines, siderophore, Pseudomonas, iron, oxidation-reduction (redox), biosynthesis
Introduction
The acquisition of iron is often a limiting factor for bacteria that live in host habitats, including pathogenic as well as mutualistic species (1). Fluorescent pseudomonads warrant their iron supply by pyoverdines, fluorescent siderophores that are derived from non-ribosomally synthesized peptides that are called ferribactins (2) (Fig. 1). Ferribactins are transported into the periplasm in acylated form and deacylated thereafter by PvdQ (3–5). In the periplasm, a 5,6-dihydroxyquinoline fluorophore is generated from ferribactin, and the conserved glutamic acid residue at the first position of the peptide is modified (6–8). Iron can then be chelated by the two hydroxyl groups of the fluorophore and four further ligands from side chains of the peptide moiety (9, 10). Only one enzyme, PvdP, is currently believed to catalyze the complete oxidation of the fluorophore (6). However, the often found dihydropyoverdine had been postulated in the past to be an obligate intermediate in fluorophore biogenesis (11, 12), and a mutant strain was known that produced dihydropyoverdine (13), suggesting two successive oxidation steps for fluorophore biogenesis. The pvdP gene is usually encoded adjacent to the pvdMNO operon, which encodes three further periplasmic proteins that might act as enzymes (Fig. 2). We recently have shown that PvdN is responsible for one of the glutamic acid modifications, the conversion of this residue to succinamide (7). The functions of PvdM and PvdO have not been resolved yet, but based on interposon mutagenesis, both proteins are postulated to be essential for pyoverdine formation (14, 15). The structure of PvdO has been solved recently (16). It has been shown to be structurally related to the formylglycine-generating enzyme (FGE),2 but because the catalytic residues of FGE are not conserved in PvdO (16), no function could be assigned to this important protein so far. Being a periplasmic protein, PvdO can be expected to play a role in periplasmic pyoverdine maturation processes, which are fluorophore formation and pyoverdine modification (Fig. 1).
Figure 1.
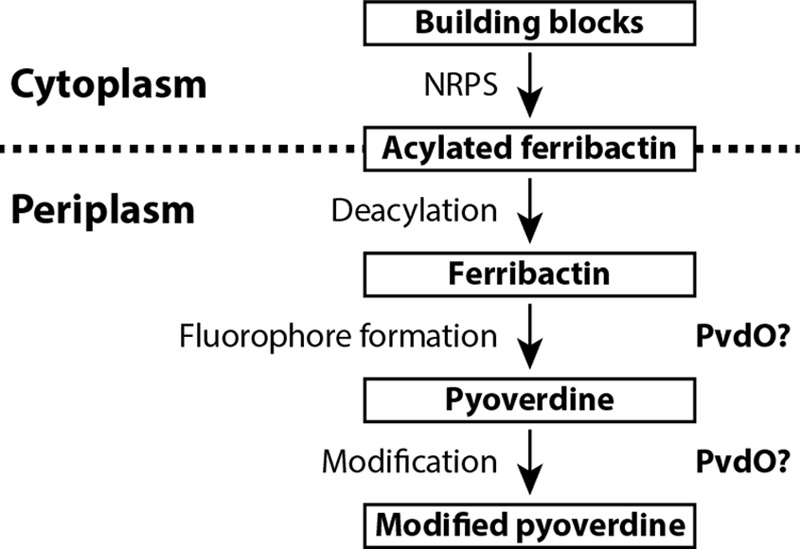
Schematic overview of pyoverdine biosynthesis stages. PvdO is a putative oxidoreductase and, being a periplasmic protein, could be involved in fluorophore formation or pyoverdine modification. NRPS, non-ribosomal peptide synthetase.
Figure 2.

Comparison of genomic environments of pvdO in representative Pseudomonas genomes.
Here we describe that PvdO from Pseudomonas fluorescens A506 is required for the last oxidation step during fluorophore formation. We found that the dihydro-form of the fluorophore was the final product of a mutant strain with a scar-less in-frame pvdO deletion, and this phenotype was fully complemented in trans by pvdO. The requirement of PvdO can be masked by a PvdO-independent oxidation under non-physiological conditions. Purified PvdO from the periplasmic fraction did not contain potentially catalytic metal ions. However, the protein as purified was inactive, and catalysis therefore requires additional factors that are absent in the in vitro system.
Results
A pvdO deletion strain produces a non-fluorescent but active siderophore
In the course of our functional analyses of the periplasmic enzymes that are involved in pyoverdine maturation, we deleted the gene encoding PvdO in our model strain P. fluorescens A506. Because pvdO is organized with two other genes in the pvdMNO operon, we applied scar-less in-frame deletion to avoid polar effects. An essential phenotype was previously postulated for all three genes in the operon, but we could recently demonstrate that this is not the case for pvdN (7). In-frame deletions of the other two genes of the operon, pvdM and pvdO, resulted in differential phenotypes; the ΔpvdM strain could not grow under iron limitation as induced by EDDHA, but the ΔpvdO strain was indeed able to grow under this condition, indicating that PvdO might be dispensable for functional pyoverdine formation (Fig. 3, top). However, we noted that the pvdO deletion strain did not secrete the fluorescent pyoverdine into the medium (Fig. 3, bottom). These initial observations already suggested that PvdO might be somehow involved in a late step of pyoverdine maturation, as apparently an active siderophore was formed that did not show fluorescence. In addition, pvdM was found to be indeed essential for the formation of an active siderophore, and this explains the former polar effects of the interposon mutagenesis of pvdN as well as of pvdO (14).
Figure 3.

Evidence for a non-fluorescent pyoverdine generated in the pvdO deletion strain. Shown is the growth of strain A506 (WT) and its derivatives deleted in pvdM (ΔpvdM), pvdO (ΔpvdO), pvdP (ΔpvdP), or pvdN and pvdO (ΔpvdNO) on pyoverdine-inducing solid casamino acid (CAA) medium without or with EDDHA for additional iron depletion. Indicated complementation strains are included in the analysis. Growth on EDDHA-containing plates correlates with the formation of pyoverdines and (in case of the ΔpvdO strain) with dihydropyoverdine, as monitored by fluorescence (bottom).
PvdO oxidizes dihydropyoverdine and thereby completes the formation of the pyoverdine fluorophore
We extracted pyoverdines from culture supernatant of wildtype P. fluorescens A506 and the corresponding pvdO deletion strain and examined them by mass spectrometry (Fig. 4A). Elution profiles of the respective masses are shown on the left, and the corresponding mass spectra are depicted on the right. The assigned structures are illustrated in Fig. 4 (B and D). While the wildtype strain produced the well-known pyoverdines PVD-α-ketoglutaric acid (2), PVD-succinamide (3), and PVD-succinic acid (4), the data clearly showed that the pyoverdines of the pvdO deletion mutant were the known dihydro-intermediates of the pyoverdines with the different modifications known for the first residue, namely glutamic acid (5), α-ketoglutaric acid (6), succinamide (7), and succinic acid (8). The succinic acid form (8) was very low-abundant, and some ferribactin (9) was detected in both preparations, especially in the pvdO deletion strain. The glutamic acid variant (1) was not detected in the wildtype strain, which is due to the known quantitative conversion of 1 to 2 and 3 by PvdN and PtaA (7, 8). The identity of the dihydropyoverdine compounds was verified by high-resolution mass spectrometry (calculated exact masses are denoted in parentheses; see Ref. 8 for the other pyoverdine masses): 5, 1192.560 atomic mass units (a.m.u.) (1192.560 a.m.u.); 6, 1191.528 a.m.u. (1191.528 a.m.u.); 7, 1162.550 a.m.u. (1162.549 a.m.u.). The data demonstrated not only that PvdO is responsible for the final oxidation of dihydropyoverdine, but also that the side chain–modifying enzymes accept the dihydro-substrate.
Figure 4.

PvdO catalyzes the oxidation of dihydropyoverdine. A, analysis of pyoverdines produced by wildtype P. fluorescens A506 (top) and the corresponding dihydropyoverdines (DH-PVD) produced by the pvdO deletion strain (bottom). Chromatograms on the left depict the elution profiles of the numbered compounds that are assigned in B and D. C, detection of fluorescence (left) or iron-binding capacity in a CAS overlay assay (right) after isoelectric focusing of the pyoverdines of indicated strains. WT, wildtype A506; ΔpvdO, strains with pvdO deletion; ΔpvdO/ΔpvdN, strain with an additional pvdN deletion; ΔpvdO/pvdO, pvdO-complemented pvdO deletion strain.
The pyoverdines of the wildtype and the ΔpvdO-, ΔpvdN/ΔpvdO-, and pvdO-complemented ΔpvdO strains were analyzed by isoelectric focusing, followed by fluorescence detection (Fig. 4C, left) or the CAS overlay assay (Fig. 4C, right). The fluorescence detection confirmed the pyoverdines detected by mass spectrometry, with the succinic acid and α-ketoglutaric acid forms migrating at the lower part of the gel and the succinamide and glutamic acid forms migrating at the upper part. Note that the epifluorescence was recorded without a filter, as the dihydropyoverdines (middle two lanes) exhibit only a rather weak blue fluorescence that can be thereby detected. The CAS overlay assay demonstrated iron binding of all identified siderophores, including the dihydropyoverdines, which agrees with former reports about a complementation of pyoverdine deficiency by dihydropyoverdine as well as with affinity measurements (13).
Non-enzymatic oxidation of dihydropyoverdine masks PvdO activity
The finding that PvdO was required for the final oxidation of dihydropyoverdine in the described cultivations raised the question of why PvdO was not required for the reported PvdP-dependent fluorophore formation from ferribactin in previous in vitro experiments (6). In that study, it had been clearly shown that the mature fluorophore was generated in assays that contained PvdP as the only enzyme (6). It thus was very likely that the previously applied in vitro conditions had somehow triggered a PvdO-independent completion of fluorophore oxidation. Obvious unusual conditions that had been used in these assays were (i) the alkaline pH 9.0, which is non-physiological as the periplasmic compartment is slightly acidic, and (ii) the inclusion of 250 μm CuSO4, which is a copper stress condition. These conditions had been found to be the optimum conditions for the PvdP-catalyzed reaction (6). As fluorescence is quenched by copper ions, we used the oxidation-dependent increase of chromophore absorbance at 405 nm to examine the potential influence of pH and copper on dihydropyoverdine oxidation (Fig. 5A). Non-catalytic oxidation was very rapid at pH 9.0, and this was significantly slowed down by the addition of 250 μm CuSO4. At pH 8, there was essentially the same effect of copper, but autoxidation proceeded at a much slower rate. At pH 7, there was hardly any autoxidation observed. In conclusion, alkaline autoxidation occurs rapidly without involvement of copper as catalyst. The alkaline autoxidation had already been recognized in early studies on the structure of dihydropyoverdine (17). In that study, it was also shown that chelation of iron inhibits the autoxidation (17), and our data indicate that chelation of copper has the same effect.
Figure 5.
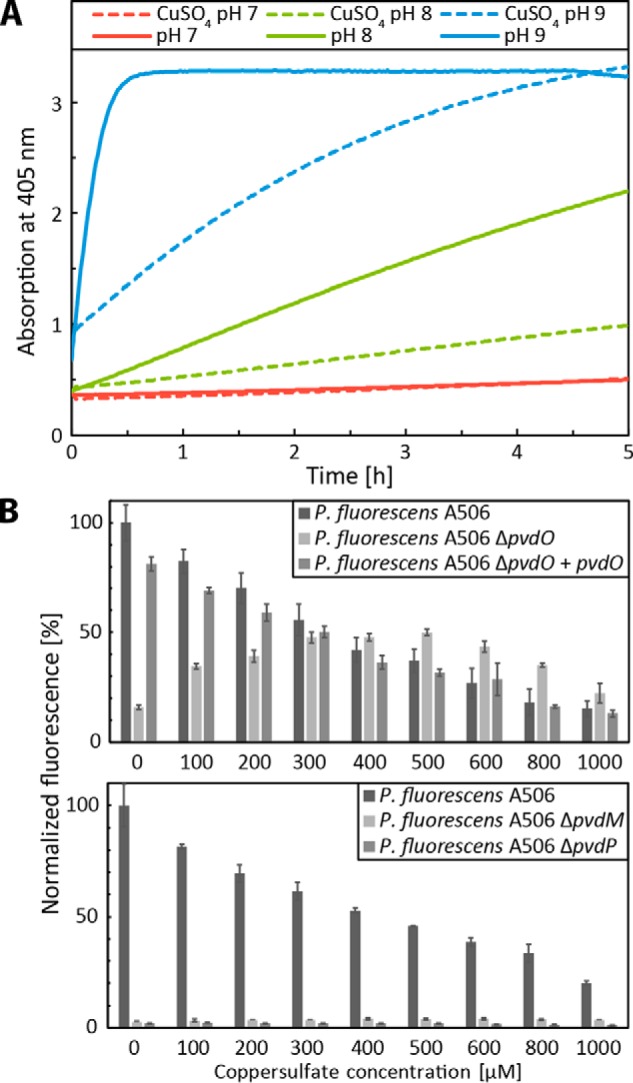
Non-physiological oxidation of dihydropyoverdines by alkaline autoxidation in vitro and copper-induced oxidation in growing cultures. A, pH-dependent autoxidation of dihydropyoverdine as monitored by absorbance increase at 405 nm. Kinetics of assays containing 250 μm CuSO4 (dashed lines) are directly compared with kinetics in the absence of copper. Copper was added after 1 min of incubation. The data of the copper-containing assays were corrected for copper absorption. See “Experimental procedures” for details. B, pyoverdine fluorescence of culture supernatants of wildtype A506, its ΔpvdO derivative, and the pvdO-complemented ΔpvdO strain after 24 h of growth in CAS medium supplemented with the indicated CuSO4 concentrations (top). Control measurements are shown, indicating the absence of fluorescence with strains deleted in pvdM (ΔpvdM) or pvdP (ΔpvdP) after a 48-h incubation (bottom). Error bars are derived from triplicate assays of independent cultures. Fluorescence was normalized to the wildtype levels in the absence of copper. The decrease of fluorescence at higher copper concentrations is due to the quenching of pyoverdine fluorescence by copper ions.
The assay system for the PvdP-dependent formation of completely oxidized pyoverdine from ferribactin did not monitor the potential formation of a dihydropyoverdine intermediate (6). The rapid autoxidation of this intermediate at the applied non-physiological pH 9 masked the PvdO requirement in these assays.
However, the effect of copper is more complex. When we analyzed the impact of copper on the formation of fluorescent pyoverdines in growing cultures, we noted differential effects (Fig. 5B, top). As expected, in the absence of copper, the wildtype strain produced fluorescent pyoverdine, and the medium of the pvdO deletion strain contained only very little fluorescence due to the formation of dihydropyoverdine. However, abundant copper in the bacterial cultures promoted the oxidation of dihydropyoverdine in the ΔpvdO background, and fluorescence reached the level of the wildtype strain at ∼300 μm copper. At higher copper concentrations, fluorescence was gradually quenched by copper. The positive influence of copper on dihydropyoverdine oxidation thus had a maximum effect in the range of 200–500 μm copper, which coincides with the reported optimum of PvdP activity at 250 μm copper (6). Therefore, although copper binding to dihydropyoverdine partially inhibits alkaline autoxidation (Fig. 5A), the same high copper concentrations can artificially trigger a dihydropyoverdine-oxidizing activity in the presence of whole cells (see “Discussion”). In control experiments, strains lacking PvdM or PvdP did not produce any fluorescent pyoverdine in the presence of added copper even after prolonged incubations, excluding alternative explanations for fluorescence increase (Fig. 5B, bottom).
PvdO and PvdP generally co-occur in fluorescent pseudomonads
As shown above, PvdO is required for the oxidation of dihydropyoverdine under physiological conditions in P. fluorescens A506. It can therefore be expected that PvdO must always co-occur with PvdP, because PvdP is essential for the preceding ring formation. We thus examined the presence of pvdO and pvdP in all genomes of Pseudomonas strains available in the Pseudomonas Genome Database (18) in August 2017. In full agreement with the prediction, these analyses demonstrated a complete co-occurrence of the two genes (Fig. 6). Individual mismatches (in 13 of 3347 analyzed genomes) could all be traced back to unfinished genome assemblies or gaps.
Figure 6.

Co-occurrence of PvdO and PvdP homologs within the genus Pseudomonas. Numbers to the right of the bar graph indicate the total number of genomes analyzed for the respective species. Note that the two genes co-occurred in 3334 of 3347 genomes, and the 13 exceptions were attributed to unfinished genomes.
Mutational analysis of PvdO
As PvdO is somehow required for the oxidation of dihydropyoverdine in living cells under physiological conditions, it was interesting to investigate its structural and biochemical properties.
We homology-modeled the structure of PvdO from P. fluorescens A506 (PvdOA506) to the known structures of PvdO from Pseudomonas aeruginosa PAO1 (PvdOPAO1; 73% identity; PDB 5HHA) and the structurally related FGE (25% identity; PDB 2Q17) from Streptomyces coelicolor. Neither PvdOPAO1 nor FGE contain a redox active cofactor associated with the purified proteins used in crystallographic analyses. However, FGE is suggested to employ a copper cofactor to oxidize cysteine to formylglycine (19), and according to a structural analysis study on PvdOPAO1, FGE is the only so-far known protein that is structurally related to PvdO, although active-site cysteines of FGE are not conserved in PvdO (16). The PvdOPAO1- and FGE-based structural models of PvdOA506 are in most parts highly similar with respect to the backbone structure and differ mainly in the conformation of surface loop structures (Video S1). The FGE-derived model was suggestive of a potential His/His/Glu non-heme iron-binding site that resembled known sites (20, 21). These residues (His-188, His-218, and Glu-260) are highly conserved (Fig. 7D), as are several other potentially important candidate residues (Glu-179, Asn-210, Tyr-215, Asp-216, Met-217, and Asp-257) that were included in a mutational analysis. Also, several non-conserved residues with potential metal-chelating propensity were included as negative controls (Gln-187 and Met-214). As in the case of PvdO from P. aeruginosa (16), all six cysteines of P. fluorescens A506 PvdO form disulfides and thus are unlikely to be involved in metal binding. Exchanged residues are indicated in the sequence of Fig. 7D as well as in the structural model in Fig. 7E.
Figure 7.

Effects of alanine exchanges of residues with potential catalytic function. A, effect of the indicated single-alanine exchanges on the formation of fluorescent pyoverdine and control of the presence of the respective PvdO variants in the periplasm by SDS-PAGE/Western blotting. B, subcellular fractionation of all analyzed PvdO variants. Cytoplasmic (C), membrane (M), and periplasmic (P) fractions were analyzed for the presence of PvdO by SDS-PAGE/Western blotting. Note than in case of stable PvdO variants, the protein is exclusively detectable in the periplasm, as expected for Sec transport. C, Coomassie-stained PvdO preparations that had been used for metal detections. D, sequence logo of the mutated regions (see “Experimental procedures” for details). Exchanged residues in PvdOA506 are colored red, and distinct regions are highlighted in yellow and blue. The residue numbers on top of the sequence logo indicate the residue position in PvdOA506. E, homology model of the PvdOA506 structure based on the known structure of PvdO from P. aeruginosa (PDB code 5HHA). Exchanged residues are highlighted in red. Left, overview of the structure; right, close-up view. The model was visualized using Chimera (36). Masses of marker proteins (in kDa) are indicated on the left side of blots.
Using our functional complementation system described above, we examined for all of these individual PvdO variants their ability to complement the ΔpvdO phenotype in terms of pyoverdine production (Fig. 7A, top), their stability (Fig. 7A, bottom), and their transport into the periplasm (Fig. 7B). Fortunately, while a number of exchanges caused an instability of the protein and consequently a loss of functionality, several of the exchanges, namely PvdO(D257A) and PvdO(E260A), abolished complementation of the mutant phenotype without compromising protein stability. The transport of these variants into the periplasm was unaffected, indicating that the non-functionality was not caused by a mislocalization. As expected from the stability analysis shown in Fig. 7A, the other PvdO variants were not detected in any subcellular fraction (Fig. 7B), indicating degradation to non-detectable levels. From all of these biochemical data, it can be concluded that the residues Asp-257 and Glu-260 are essential for dihydropyoverdine oxidation in living P. fluorescens A506 cells, without being required for protein integrity or subcellular targeting.
As a complementary analysis, we also evaluated pyoverdine formation during growth on iron-depleted or iron-limited solid media (Fig. 8). As expected, pvdO-deleted strains or strains containing inactivated or destabilized PvdO variants grew with reduced rate under iron-depleted conditions (i.e. in the presence of the chelator EDDHA; Fig. 8, A and C) and could not form fluorescent pyoverdine halos (Fig. 8, C and D). Some fluorescence surrounding the strain with the D257A variant indicates a residual activity that had not been detected with planctonic growth. In all other cases, the results of this assay were only confirmative. Together, the in vivo data described above indicated that the two acidic residues Asp-257 and Glu-260 of PvdO were important for the oxidation of dihydropyoverdine in living cells.
Figure 8.
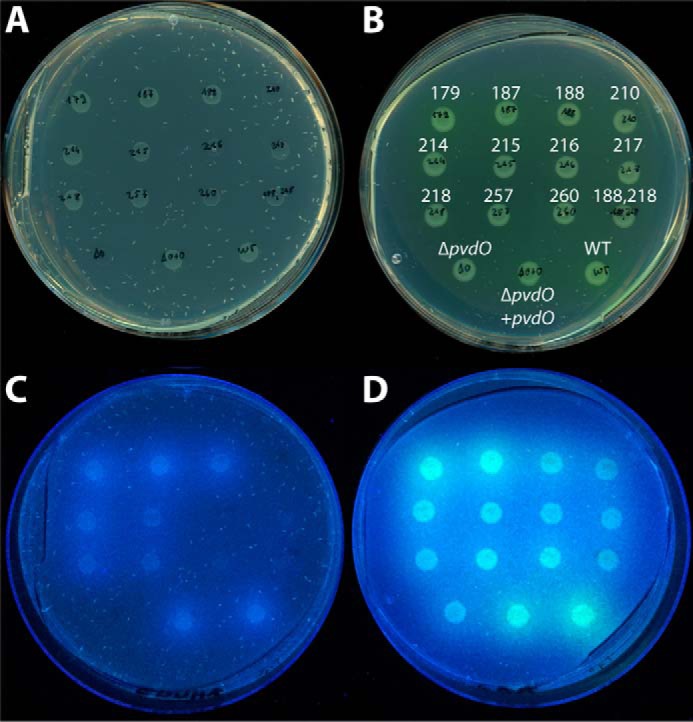
Analysis of physiological functionality of PvdO variants with single-amino acid exchanges. A and C, CAA plates containing iron-depleting EDDHA; B and D, CAA plates without EDDHA. A and B show the growth of spotted cultures on the respective media, whereas B and D show the fluorescence caused by produced pyoverdines. The amino acid exchanges and control strains are denoted in B. All plates contain the same organization of spotted strains.
Based on this knowledge, we then carried out analyses in vitro. We purified wildtype PvdO and the E260A variant from the periplasmic fraction of E. coli harboring a pvdO expression system (Fig. 7C). The protein migrated near 35 kDa, which is slightly above the calculated mass of 30 kDa for the Strep-tagged protein. Using ICP-MS, we did not detect any potentially redox-active iron, copper, cobalt, or manganese ions or zinc ions as constituents of the purified proteins. Therefore, as discussed in more detail below, either a cofactor was not stably assembled or catalysis does not depend on metal ions. The activity of purified PvdO and its variant PvdO(E260A) was not detectable in vitro (Fig. S1), which can be due to various reasons (see “Discussion”).
Discussion
PvdO is required for dihydropyoverdine oxidation
The functional role of PvdO has been matter of speculation since its importance had been recognized by Ochsner et al. in 2002 (15). It was known that no fluorescent pyoverdine could be formed in a pvdO mutant strain that had been generated by interposon mutagenesis (15, 14). However, the deletion strains were not complemented, and the fact that pvdO is organized in the pvdMNO operon made the interpretations difficult due to possible polar effects. PvdN, which is encoded by the same operon and had also been believed to be essential for pyoverdine formation, is in fact not essential and catalyzes the conversion of the fluorophore-attached glutamic acid to succinamide (7). To clarify this issue for PvdM and PvdO, we carried out in-frame scar-less deletions of these two genes, analyzed their phenotypes, and successfully complemented them. While PvdM turned out to be indeed essential for pyoverdine formation, the ΔpvdO phenotype was more complex; there was still some growth possible on iron-depleted medium, albeit the typical yellow-greenish pyoverdine fluorescence was absent from this mutant (Fig. 3). This prompted us to investigate the spectrum of potential pyoverdine precursors produced by that strain, and we discovered that PvdO is required for the oxidation of dihydropyoverdine in vivo under physiological conditions (Fig. 4). The pvdO gene fully complemented the ΔpvdO phenotype and produced wildtype levels of the oxidized pyoverdine fluorophore. Thus, no secondary mutations had caused the phenotype of the gene deletion. The role in fluorophore biogenesis was unexpected, because the early postulated dihydropyoverdine intermediate (11, 12) had not been considered as relevant anymore when PvdP had been shown to catalyze the complete conversion of ferribactin to fluorescent pyoverdine (6). In that in vitro study, however, the full oxidation had been demonstrated in assays that contained 250 μm CuSO4 at pH 9.0. As we observed that dihydropyoverdine is rapidly oxidized at pH 9.0 even in the presence of 250 μm copper (Fig. 5A), and as PvdO is responsible for the oxidation of dihydropyoverdine under physiological conditions, it is likely that dihydropyoverdine rather than pyoverdine is the product of PvdP. However, it is noteworthy that a growing ΔpvdO strain could generate fluorescent pyoverdine in the presence of 200–500 μm CuSO4. This copper effect most likely relates to living cell constituents, and it is intriguing that the optimal concentration in the published PvdP assays is the same as in our in vivo assays (Fig. 5B). It thus might be speculated that an occupancy of low-affinity copper sites on cellular proteins such as PvdP can contribute, besides alkaline autoxidation, to an artificial full oxidation to pyoverdine under these conditions. As PvdO strictly co-occurs with PvdP in fluorescent pseudomonads (Fig. 6), it is clear that the oxidation of dihydropyoverdine must generally be catalyzed in a PvdO-dependent manner under physiological conditions not only in P. fluorescens but also in the other species.
Although required, the exact mode by which PvdO contributes to the oxidation of dihydropyoverdine remains unclear
We initially expected a transition metal cofactor as a constituent of PvdO and thus homology-modeled the structure with the related S. coelicolor FGE structure and later also with the recently published P. aeruginosa PvdO structure as templates to identify potential metal-binding sites. Several residues were possibly involved in an iron-binding site, but there was no good evidence for any copper site. The mutational analysis therefore included residues with potential metal-binding properties in the environment of the suspected iron site, and we examined the influence of the mutations on fluorescent pyoverdine formation as well as on stability (Figs. 7 and 8). The exchange of the presumed histidine ligands strongly affected PvdO stability, which argued against a primarily catalytic function. Only the mutation of two acidic residues, Asp-257 and Glu-260, resulted in complete (E260A) or strongly reduced (D257A) functionality without any significant effect on protein stability. We had analyzed not only wildtype PvdO but also the E260A variant, as we suspected Glu-260 to be an essential ligand for the supposed metal ion. To our surprise, neither the wildtype nor the mutated protein contained any detectable metal ions, although PvdO had been purified under mild conditions by affinity chromatography. The absence of metals pointed to a mechanism similar to the metal-independent alkaline autoxidation (17) in which the deprotonation of the catechol is the initializing step for the oxidation. It is proposed that unpaired electrons that are generated during the oxidation are stabilized between the neighboring catechol hydroxylates, which promotes the oxidation (17). To this mechanism fits the essential role of Glu-260, and also the significant contribution of a second carboxylic acid, Asp-257. If PvdO directly catalyzes the oxidation of dihydropyoverdine, it probably enables the deprotonations at pH 6.5 in the periplasm, stabilizes the oxyanions, and catalyzes the electron transfer to an unknown electron acceptor (Fig. 9A). Once the ring system is oxidized, tautomerizations would readily generate the 5,6-dihydroxyquinoline fluorophore. However, because we could not reconstitute the activity in vitro, PvdO most likely needs to interact with another component in the periplasm to perform its function. Obviously, this could be a specific electron acceptor that channels the electrons into the respiratory electron transport chain. The inactivity of purified PvdO can be taken as an argument against a more direct electron transfer to oxygen, as implied by an autoxidation-like mechanism. The possibility certainly also cannot be excluded that an unknown cofactor is required that is lost during purification or that PvdO is a protein that enables another enzyme to oxidize dihydropyoverdine, although the active-site conservation argues for a direct enzymatic involvement (see below).
Figure 9.

Role of PvdO in fluorophore formation of pyoverdines and identification of functionally important residues in a potential active-site cavity of PvdO. A, biosynthetic reactions leading from ferribactin to pyoverdine via the intermediate dihydropyoverdine. PvdP is required for the initial oxidative cyclization reaction that results in the catechol system. PvdO is required for the final oxidation, resulting in the mature fluorophore. B, the two active-site residues Asp-257 and Glu-260 (red) and their position on the surface of PvdO relative to the other residues mutated in this study (blue). C (left), active-site cavity shown by the APBS (37)-calculated electrostatic potential-colored surface (scale from −10 (red) through 0 (white) to +10 (blue)). Surface-exposed side-chain regions of Asp-257 and Glu-260 are colored purple. Middle and right, superposition of the structure from FGE and PvdOA506 structural model, highlighting the respective active-site residues at overlapping positions. The active-site Cys-272 of FGE is at exactly the same position as Glu-260 in PvdO.
The active-site cavity appears to be conserved between FGE and PvdO
When we examined the position of the identified key acidic residues Glu-260 and Asp-257 in the homology-modeled protein, we readily recognized that these residues were located in a pocket-like cavity that was likely to represent the substrate-binding site (Fig. 9, B and C). Interestingly, we found that the supposed binding pocket was exactly at the position reported to be the active site of the structurally related FGEs (PDB code 2Q17). One of the two active-site cysteines in FGE is in fact superimposable with Glu-260, which we had identified as an essential residue of PvdO (Fig. 9C). Glu-254 of PvdOPAO1, which corresponds to Glu-260 of PvdOA506, had already been recognized to be at the position of the active-site cysteine in human FGE (16). Although no function had been assigned to this residue in that study, the corresponding surface cleft was proposed to represent a binding site for an unknown substrate. Therefore, nature apparently used the same protein fold and the same active-site region to generate two distinct functionalities for distinct subcellular compartments. While the cytoplasmic FGEs can use cysteines for active-site metal coordination (22), periplasmic PvdO does not have this cysteine and therefore does not bind copper at that position. Instead, PvdO function requires a glutamic acid that has been placed at the exact position of the copper ligands in FGEs. This finding strongly argues for a direct enzymatic function of PvdO, which remains to be demonstrated in the future. This will help to ultimately clarify the mechanism by which dihydropyoverdine is oxidized.
Experimental procedures
Strains and growth conditions
The strain P. fluorescens A506 was utilized for all physiological assays. Escherichia coli DH5α λ pir+ was employed for cloning and E. coli Rosetta 2 (DE3) pLysSRARE2 for protein production. The E. coli strains were cultivated at 37 °C and P. fluorescens strains at 30 °C unless noted otherwise. LB-Medium (1% (w/v) tryptone, 1% (w/v) NaCl, 0.5% (w/v) yeast extract) was used for standard cultivations. Appropriate antibiotics were supplemented at the following final concentrations: 100 μg/ml ampicillin, 25 μg/ml chloramphenicol, 50 μg/ml kanamycin, 20 μg/ml tetracycline. The pyoverdine-plate assay and the relative pyoverdine quantification assay were performed as described previously (7, 8).
Genetic methods and plasmids
Construction of all plasmids and scar- and marker-less deletions were performed as described previously (7, 8). All primers used for this study are listed in Table 1.
Table 1.
Primers used in this study
| Name | Sequence | Restriction site | Purpose |
|---|---|---|---|
| pEXH5-RBS-F-MR | GGCGCGGGATCCGTTTAACTTTAAGAAGGAGATATAC | BamHI | Forward primer for subcloning from pEXH5 into pME6010 |
| pEXH5-strep-term-R-MR | CCCCTTGAATTCAAAAAAAACCCCGCCCTGTCAGGGGCGGGGTTTTTTTTTTTACTTTTCGAACTGCGGGTGGCTCC | EcoRI | Reverse primer for subcloning from pEXH5 into pME6010 |
| pEXPT7-HA-term-R-MR | CCCCTTGAATTCAAAAAAAACCCCGCCCTGTCAGGGGCGGGGTTTTTTTTTTTAGGCGTAGTCCGGCACGTCGTACGGG | EcoRI | Reverse primer for subcloning from pEXH5 into pME6010 with HA-tag |
| PfA506-dpvdNO-F2-MR | CTAACCACCACGAAGTAAGTCCATGACCCGCGAACTCTAATATCTTGTAGG | Forward primer for pvdMNO right flanking region | |
| PfA506-matPvdO-strep-R-MR | CCGATTAAGCTTTTACTTTTCGAACTGCGGGTGGCTCCAGAGTTCGCGTACTACCCGGAACCCAATCCAATC | HindIII | Reverse primer for cloning PvdO-coding region into pEXH5 |
| PfA506-PvdM-DF-MR | ACCGTCGTGGTTGTCGAAGTAG | pvdM genomic deletion control primer | |
| PfA506-PvdM-DR-MR | GGGCCAGCAGTTGGTCCTTGAGTTC | pvdM genomic deletion control primer | |
| PfA506-PvdM-F1-MR | CGCCTTAAGCTTTATGGCGGTGCATGAAGAAGAAGTCG | HindIII | Forward primer for pvdM left flanking region |
| PfA506-PvdM-F2-MR | CCTCAACTGACTGAGATGGCAATGACAAACCGCTAACCACCACGAAGTAAGTCC | Forward primer for pvdM right flanking region | |
| PfA506-PvdM-F-MR | TCAGCGCATATGACAAAATCACGTTCGAAAAAGGCGCTGTATATC | NdeI | Forward primer for cloning PvdM-coding region into pEXH5 |
| PfA506-PvdM-HA-R-MR | GGCTAAAAGCTTTTAGGCGTAGTCCGGCACGTCGTACGGGTAGCGGTTGGCCAAGGGTTTGGCGGCTTTTTGGAC | HindIII | Reverse primer for cloning PvdM-coding region into pEXH5 |
| PfA506-PvdM-R1-MR | TGTCATTGCCATCTCAGTCAGTTGAGG | Reverse primer for pvdM left flanking region | |
| PfA506-PvdM-R2-MR | ATCTGCGGATCCTTGCAGGTGCAACTTGAAGGCTTCG | BamHI | Reverse primer for pvdM right flanking region |
| PfA506-pvdO_D216A-F-MR | CTTTGGCATGTATGCCATGCACGGCAATGT | Primer for D216A exchange by oePCR | |
| PfA506-pvdO_D216A-R-MR | CCGTGCATGGCATACATGCCAAAGGCATTC | Primer for D216A exchange by oePCR | |
| PfA506-PvdO_D257A-F-MR | GCAATGCCTGGGGCGAGGCGCCGGTGTTTT | Primer for D257A exchange by oePCR | |
| PfA506-PvdO_D257A-R-MR | CGCCCCAGGCATTGCCGCGAATCTGGTAGG | Primer for D257A exchange by oePCR | |
| PfA506-PvdO_E179A-F-MR | CCCGTTCGACGCCGGCAAGGGCTACAGCAT | Primer for E179A exchange by oePCR | |
| PfA506-PvdO_E179A-R-MR | CCCTTGCCGGCGTCGAACGGGAAGGGGAAC | Primer for E179A exchange by oePCR | |
| PfA506-pvdO_E260A-F-MR | GCAATGACTGGGGCGCCGCGCCGGTGTTTT | Primer for E260A exchange by oePCR | |
| PfA506-pvdO_E260A-R-MR | GAAAACACCGGCGCGGCGCCCCAGTCATTG | Primer for E260A exchange by oePCR | |
| PfA506-PvdO_H188A-F-MR | ACAGCATCGCCCAGGCCGCCAACACCTACG | Primer for H188A exchange by oePCR | |
| PfA506-PvdO_H188A-R-MR | GTAGGTGTTGGCGGCCTGGGCGATGCTGTA | Primer for H188A exchange by oePCR | |
| PfA506-PvdO_H218A-F-MR | ACCGAATGCCTTTGGCATGTATGACATGGCCGGCAATGTG | Primer for H218A exchange by oePCR | |
| PfA506-PvdO_H218A-R-MR | ATACACATTGCCGGCCATGTCATACATGCCAAAGGCATTC | Primer for H218A exchange by oePCR | |
| PfA506-pvdO_M214A-F-MR | TACCCACCGAATGCCTTTGGCGCCTATGAC | Primer for M214A exchange by oePCR | |
| PfA506-pvdO_M214A-R-MR | GTGCATGTCATAGGCGCCAAAGGCATTCGG | Primer for M214A exchange by oePCR | |
| PfA506-pvdO_M217A-F-MR | CTTTGGCATGTATGACGCCCACGGCAATGT | Primer for M217A exchange by oePCR | |
| PfA506-pvdO_M217A-R-MR | GCCGTGGGCGTCATACATGCCAAAGGCATT | Primer for M217A exchange by oePCR | |
| PfA506-pvdO_N210A-F-MR | AGCTACCCACCGGCCGCCTTTGGCATGTAT | Primer for N210A exchange by oePCR | |
| PfA506-pvdO_N210A-R2-MR | TCATACATGCCAAAGGCGGCCGGTGGGTAG | Primer for N210A exchange by oePCR | |
| PfA506-pvdO_Q187A-F-MR | ACAGCATCGCCGCCCACGCCAACACCTACG | Primer for Q187A exchange by oePCR | |
| PfA506-pvdO_Q187A-R-MR | GTAGGTGTTGGCGTGGGCGGCGATGCTGTA | Primer for Q187A exchange by oePCR | |
| PfA506-pvdO_Y215A-F-MR | TGGCATGGCCGACATGCACGGCAATGTGTA | Primer for Y215A exchange by oePCR | |
| PfA506-pvdO_Y215A-R-MR | GTGCATGTCGGCCATGCCAAAGGCATTCGG | Primer for Y215A exchange by oePCR | |
| PfA506-PvdO-DF-MR | CTGCAGGAAACCTGGAAGCGTGAG | pvdO genomic deletion control primer | |
| PfA506-PvdO-DR-MR | CATGACAAACGAAGCTGTGGCGTGAC | pvdO genomic deletion control primer | |
| PfA506-PvdO-F1-MR | TTAGCCGGATCCCCGAAGGGCTGGCGATGATCTATG | BamHI | Forward primer for pvdO left flanking region |
| PfA506-PvdO-F2-MR | TCCCTTTGAACGAGACGCCTCCATGCGCGAACTCTAATATCTTGTAGG | Forward primer for pvdO right flanking region | |
| PfA506-PvdO-F-MR | TTACCGCATATGACGCCATCCCGACTCAAACCGCTCAC | NdeI | Forward primer for cloning PvdO-coding region into pEXH5 |
| PfA506-PvdO-R1-MR | CATGGAGGCGTCTCGTTCAAAGGG | Reverse primer for pvdO left flanking region | |
| PfA506-PvdO-R2-MR | GCACGCAAGCTTCCTGGTTCGTTCTTGTTCTGTAGG | HindIII | Reverse primer for pvdO right flanking region |
| PfA506-PvdP-DF-MR | GTTGCAGTTCCTTGTCGCTGAGGTTG | pvdP genomic deletion control primer | |
| PfA506-PvdP-DR-MR | CCGGCCAGCATTTCTGGCTATTC | pvdP genomic deletion control primer | |
| PfA506-PvdP-F1-MR | TGCTCTAAGCTTTTGGTCGACATCTGCGACACGTCGATGATCAC | HindIII | Forward primer for pvdP left flanking region |
| PfA506-PvdP-F2-MR | ACGCTTTAAAACGTGTTGCAGGCGCTCTAGG | Forward primer for pvdP right flanking region | |
| PfA506-PvdP-R1-MR | GCGCCTGCAACACGTTTTAAAGCGTTGTCATTGGCTACCTTAGGAAACGGC | Reverse primer for pvdP left flanking region | |
| PfA506-PvdP-R2-MR | TTAAACCCCGGGAGGTACTGGGTAGCCTGTCCAGGCGTATCGACGAG | XmaI | Reverse primer for pvdP right flanking region |
Biochemical methods
SDS-PAGE, successive Western blotting, and in-gel colloidal Coomassie staining for protein analysis were performed according to standard protocols (23–26). Development of Western blots was performed according to the manufacturer's instructions using a StrepMAB-Classic monoclonal antibody (IBA, Göttingen, Germany) for detection of Strep-tagged proteins. As secondary antibody, anti-mouse-horseradish peroxidase conjugate (Carl Roth, Karlsruhe, Germany) was employed. Images were recorded using the MF-ChemiBIS version 4.2 imaging system (DNR Bio-Imaging Systems, Jerusalem, Israel).
Subcellular fractionation of P. fluorescens A506 was performed as reported previously (27) with minor modifications; cultures (50 ml) were grown to OD600 of 1, and a 1-ml fraction volume was used throughout. The samples were not further concentrated by precipitation for further analysis.
The subcellular fractionation of E. coli was carried out as described earlier, with minor modifications (28). Briefly, 10 100-ml cultures in 0.5-liter Erlenmeyer flasks with four baffles each were inoculated with 4 ml of the appropriate overnight culture and incubated at 30 °C and ∼180 rpm until the OD600 reached 0.6. Then the expression was induced with 1 mm isopropyl β-d-1-thiogalactopyranoside for 3 h, maintaining the incubation conditions. Subsequently, cells were sedimented in 250-ml centrifugation buckets for 10 min at 3857 × g and 4 °C. The pellets were resuspended in 40 ml of TES buffer (20% (w/v) sucrose, 10 mm Tris buffer, pH 8.0, 1 mm EDTA) and incubated at room temperature for 10 min. The cells were transferred to 45-ml centrifugation buckets and sedimented for 10 min at 2990 × g and 4 °C. Pellets were then resuspended in 1.5 ml of ice-cold 5 mm MgSO4 each and transferred to 2-ml plastic tubes. Resuspended cells were then incubated for 20 min on ice and subsequently centrifuged for 10 min at 4 °C and 16,060 × g. The supernatants (periplasmic fraction) were collected and immediately sterile filtered through a 0.2-μm PES filter. The protein was purified utilizing one 1.5-ml Strep-Tactin® Superflow® column (IBA) according to the manufacturer's instructions using 100 mm Tris-HCl buffer, pH 8.0, with 150 mm NaCl as running buffer. After elution, the elution fractions E3 and E4 were combined and diluted with 100 mm Tris-HCl, pH 8.0, to lower the NaCl concentration to 50 mm for subsequent ICP-MS analysis. The protein was then concentrated using a Vivaspin® 6 concentrator (cutoff 10 kDa; Sartorius, Göttingen, Germany). The protein concentration was estimated via absorption at 280 nm using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Samples were subsequently analyzed by ICP-MS (SpurenAnalytisches Laboratorium Dr. Baumann; Pirkensee, Germany).
A Jasco V-650 spectrophotometer (Jasco, Gross-Umstadt, Germany) was used for in vitro dihydropyoverdine oxidation assays with the following settings: wavelength, 405 nm; bandwidth, 2 nm; measurement interval, 1 min; total measurement time, 5 h; temperature, 20 °C; stirrer, 800 rpm. As buffers for the assay, 50 mm HEPES, pH 7, 50 mm HEPES, pH 8, and 50 mm CHES, pH 9, were used. The assays started with the addition of 10 μl of freshly thawed dihydropyoverdine stock solution to a final assay volume of 2 ml in the respective buffer. After 1 min of incubation time, CuSO4 was added from a 100 mm stock solution to a final concentration of 250 μm. The absorptivity caused by CuSO4 was subtracted from the respective kinetics measurement from the time point of the addition on.
In vitro activity assays were carried out in 50 mm PIPES, pH 6.5, in a final total volume of 2 ml. For each measurement, 10 μl of the dihydropyoverdine stock solution were mixed with reaction buffer, and the assay was started after 1 min of pre-equilibration time by adding 100 μl of the protein (0.4 mg/ml in elution buffer) to a final concentration of 20 μg/ml. As blank, the same volume of elution buffer was used, and blank data were subtracted. The settings for the Jasco V-650 spectrophotometer were the same as for the above-described dihydropyoverdine oxidation assays.
Pyoverdine isolation, the isoelectric focusing assay in conjunction with the chrome azurol S overlay assay, and the ultraperformance liquid chromatography–high-resolution MS analysis were carried out as reported previously (7, 8).
Bioinformatic methods
The initial classification of PvdO was performed employing the InterPro web service (29, 30). To generate the sequence logo, PvdO homologs were identified by searching through all amino acid sequences of all complete and draft genomic sequences deposited in the Pseudomonas Genome Database (18) with phmmer version 3.1b (31). The redundancy was subsequently reduced by using the CD-HIT algorithm from the CD-HIT Suite (32), utilizing a similarity threshold of 0.95. Sequences were aligned with MAFFT version 7.310 (33) using the G-INS-i setting. The resulting sequence alignment was visualized using WebLogo 3 (34), plotting the probability of the occurrence of each amino acid at each position.
The distribution analysis was performed by searching the already downloaded sequences from the Pseudomonas Genome Database (18) with phmmer version 3.1b (31). Utilizing an in-house developed software, a taxonomy table reduced to species level was constructed, and the identified homologs were classified accordingly. Subsequently, the number of found homologs in one species was divided by the total number of organisms attributed to the same species, giving the percentage of occurrence. To model the structure of PvdO, the SWISS-MODEL web service (35) was used and in conjunction with UCSF Chimera (36) and APBS (37), and the models were visualized. Sequence identities were calculated from alignments generated using Clustal Ω (38).
To visualize the genomic contexts of pvdO in multiple organisms, the MGcV web service (39) was used.
Author contributions
M. T. R. performed the experiments, prepared the figures, and analyzed the data together with T. B., and G. D. performed the MS analyses. T. B. conceived and coordinated the study. T. B. and M. T. R. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Sybille Traupe for technical support.
This work was supported by German Science Foundation (DFG) Grant GRK1798 “Signaling at the Plant-Soil Interface” and Project BR 2285/7-1. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Video S1 and Fig. S1.
- FGE
- formylglycine-generating enzyme
- CAS
- chrome azurol S
- CHES
- N-cyclohexyl-2-aminoethanesulfonic acid
- EDDHA
- ethylenediamine di(o-hydroxy)phenylacetic acid
- ICP-MS
- inductively coupled plasma mass spectrometry
- PVD
- pyoverdine
- PDB
- Protein Data Bank
- CAA
- casamino acid
- a.m.u.
- atomic mass units
- oePCR
- overlap extension PCR.
References
- 1. Ellermann M., and Arthur J. C. (2017) Siderophore-mediated iron acquisition and modulation of host-bacterial interactions. Free Radic. Biol. Med. 105, 68–78 10.1016/j.freeradbiomed.2016.10.489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cézard C., Farvacques N., and Sonnet P. (2015) Chemistry and biology of pyoverdines, Pseudomonas primary siderophores. Curr. Med. Chem. 22, 165–186 [DOI] [PubMed] [Google Scholar]
- 3. Yeterian E., Martin L. W., Guillon L., Journet L., Lamont I. L., and Schalk I. J. (2010) Synthesis of the siderophore pyoverdine in Pseudomonas aeruginosa involves a periplasmic maturation. Amino Acids 38, 1447–1459 10.1007/s00726-009-0358-0 [DOI] [PubMed] [Google Scholar]
- 4. Drake E. J., and Gulick A. M. (2011) Structural characterization and high-throughput screening of inhibitors of PvdQ, an NTN hydrolase involved in pyoverdine synthesis. ACS Chem. Biol. 6, 1277–1286 10.1021/cb2002973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hannauer M., Schäfer M., Hoegy F., Gizzi P., Wehrung P., Mislin G. L. A., Budzikiewicz H., and Schalk I. J. (2012) Biosynthesis of the pyoverdine siderophore of Pseudomonas aeruginosa involves precursors with a myristic or a myristoleic acid chain. FEBS Lett. 586, 96–101 10.1016/j.febslet.2011.12.004 [DOI] [PubMed] [Google Scholar]
- 6. Nadal-Jimenez P., Koch G., Reis C. R., Muntendam R., Raj H., Jeronimus-Stratingh C. M., Cool R. H., and Quax W. J. (2014) PvdP is a tyrosinase that drives maturation of the pyoverdine chromophore in Pseudomonas aeruginosa. J. Bacteriol. 196, 2681–2690 10.1128/JB.01376-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ringel M. T., Dräger G., and Brüser T. (2016) PvdN enzyme catalyzes a periplasmic pyoverdine modification. J. Biol. Chem. 291, 23929–23938 10.1074/jbc.M116.755611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ringel M. T., Dräger G., and Brüser T. (2017) The periplasmic transaminase PtaA of Pseudomonas fluorescens converts the glutamic acid residue at the pyoverdine fluorophore to α-ketoglutaric acid. J. Biol. Chem. 292, 18660–18671 10.1074/jbc.M117.812545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Albrecht-Gary A.-M., Blanc S., Rochel N., Ocaktan A. Z., and Abdallah M. A. (1994) Bacterial iron transport: coordination properties of pyoverdin PaA, a peptidic siderophore of Pseudomonas aeruginosa. Inorg. Chem. 33, 6391–6402 10.1021/ic00104a059 [DOI] [Google Scholar]
- 10. Teintze M., Hossain M. B., Barnes C. L., Leong J., and van der Helm D. (1981) Structure of ferric pseudobactin: a siderophore from a plant growth promoting Pseudomonas. Biochemistry 20, 6446–6457 10.1021/bi00525a025 [DOI] [PubMed] [Google Scholar]
- 11. Baysse C., Budzikiewicz H., Uría Fernández D., and Cornelis P. (2002) Impaired maturation of the siderophore pyoverdine chromophore in Pseudomonas fluorescens ATCC 17400 deficient for the cytochrome c biogenesis protein CcmC. FEBS Lett. 523, 23–28 10.1016/S0014-5793(02)02915-0 [DOI] [PubMed] [Google Scholar]
- 12. Jacques P., Ongena M., Bernard F., Fuchs R., Budzikiewicz H., and Thonart P. (2003) Fluorescent Pseudomonas mainly produce the dihydroform of pyoverdine at low specific growth rate. Lett. Appl. Microbiol. 36, 259–262 10.1046/j.1472-765X.2003.01304.x [DOI] [PubMed] [Google Scholar]
- 13. Bultreys A., Gheysen I., Maraite H., and de Hoffmann E. (2001) Characterization of fluorescent and nonfluorescent peptide siderophores produced by Pseudomonas syringae strains and their potential use in strain identification. Appl. Environ. Microbiol. 67, 1718–1727 10.1128/AEM.67.4.1718-1727.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lamont I. L., and Martin L. W. (2003) Identification and characterization of novel pyoverdine synthesis genes in Pseudomonas aeruginosa. Microbiology 149, 833–842 10.1099/mic.0.26085-0 [DOI] [PubMed] [Google Scholar]
- 15. Ochsner U. A., Wilderman P. J., Vasil A. I., and Vasil M. L. (2002) GeneChip® expression analysis of the iron starvation response in Pseudomonas aeruginosa: identification of novel pyoverdine biosynthesis genes. Mol. Microbiol. 45, 1277–1287 10.1046/j.1365-2958.2002.03084.x [DOI] [PubMed] [Google Scholar]
- 16. Yuan Z., Gao F., Bai G., Xia H., Gu L., and Xu S. (2017) Crystal structure of PvdO from Pseudomonas aeruginosa. Biochem. Biophys. Res. Commun. 484, 195–201 10.1016/j.bbrc.2016.12.181 [DOI] [PubMed] [Google Scholar]
- 17. Teintze M., and Leong J. (1981) Structure of pseudobactin A, a second siderophore from plant growth promoting Pseudomonas B10. Biochemistry 20, 6457–6462 10.1021/bi00525a026 [DOI] [PubMed] [Google Scholar]
- 18. Winsor G. L., Griffiths E. J., Lo R., Dhillon B. K., Shay J. A., and Brinkman F. S. L. (2016) Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas Genome Database. Nucleic Acids Res. 44, D646–D653 10.1093/nar/gkv1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meury M., Knop M., and Seebeck F. P. (2017) Structural basis for copper-oxygen mediated C–H bond activation by the formylglycine-generating enzyme. Angew. Chem. Int. Ed. Engl. 56, 8115–8119 10.1002/anie.201702901 [DOI] [PubMed] [Google Scholar]
- 20. Buongiorno D., and Straganz G. D. (2013) Structure and function of atypically coordinated enzymatic mononuclear non-heme-Fe(II) centers. Coord. Chem. Rev. 257, 541–563 10.1016/j.ccr.2012.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kovaleva E. G., and Lipscomb J. D. (2008) Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat. Chem. Biol. 4, 186–193 10.1038/nchembio.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holder P. G., Jones L. C., Drake P. M., Barfield R. M., Bañas S., de Hart G. W., Baker J., and Rabuka D. (2015) Reconstitution of formylglycine-generating enzyme with copper(II) for aldehyde tag conversion. J. Biol. Chem. 290, 15730–15745 10.1074/jbc.M115.652669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 24. Burnette W. N. (1981) “Western Blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 112, 195–203 10.1016/0003-2697(81)90281-5 [DOI] [PubMed] [Google Scholar]
- 25. Towbin H., Staehelin T., and Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U.S.A. 76, 4350–4354 10.1073/pnas.76.9.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neuhoff V., Arold N., Taube D., and Ehrhardt W. (1988) Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 9, 255–262 10.1002/elps.1150090603 [DOI] [PubMed] [Google Scholar]
- 27. Ize B., Viarre V., and Voulhoux R. (2014) Cell fractionation. Methods Mol. Biol. 1149, 185–191 10.1007/978-1-4939-0473-0_15 [DOI] [PubMed] [Google Scholar]
- 28. Taubert J., Hou B., Risselada H. J., Mehner D., Lünsdorf H., Grubmüller H., and Brüser T. (2015) TatBC-independent TatA/Tat substrate interactions contribute to transport efficiency. PLoS One 10, e0119761 10.1371/journal.pone.0119761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., Pesseat S., Quinn A. F., Sangrador-Vegas A., Scheremetjew M., Yong S.-Y., Lopez R., and Hunter S. (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 10.1093/bioinformatics/btu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finn R. D., Attwood T. K., Babbitt P. C., Bateman A., Bork P., Bridge A. J., Chang H.-Y., Dosztányi Z., El-Gebali S., Fraser M., Gough J., Haft D., Holliday G. L., Huang H., Huang X., et al. (2017) InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 10.1093/nar/gkw1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eddy S. R. (2011) Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 10.1371/journal.pcbi.1002195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang Y., Niu B., Gao Y., Fu L., and Li W. (2010) CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682 10.1093/bioinformatics/btq003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Katoh K., and Standley D. M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crooks G. E., Hon G., Chandonia J.-M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L., and Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258 10.1093/nar/gku340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 37. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Overmars L., Kerkhoven R., Siezen R. J., and Francke C. (2013) MGcV: the microbial genomic context viewer for comparative genome analysis. BMC Genomics 14, 209 10.1186/1471-2164-14-209 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.