

Abstract
In mammalian cells, bulky DNA adducts located in the template but not the coding strand of genes block elongation by RNA polymerase II (RNAPII). The blocked RNAPII targets these transcription-blocking adducts to undergo more rapid excision repair than adducts located elsewhere in the genome. In excision repair, coupled incisions are made in the damaged DNA strand on both sides of the adduct. The fate of RNAPII in the course of this transcription-coupled repair (TCR) pathway is unclear. To address the fate of RNAPII, we used methods that control transcription to initiate a discrete “wave” of elongation complexes. Analyzing genome-wide transcription and repair by next-generation sequencing, we identified locations of elongation complexes and transcription-repair coupling events in genes throughout the genome. Using UV-exposed human skin fibroblasts, we found that, at the dose used, a single wave of elongation complexes was blocked within the first 25 kb of genes. TCR occurred where the elongation complexes were blocked, and repair was associated with the dissociation of these complexes. These results indicate that individual elongation complexes do not engage in multiple rounds of TCR with successive lesions. Our results are consistent with a model in which RNAPII is dissociated after the dual incision of the transcription-blocking lesion, perhaps by Cockayne syndrome group B translocase, or during the synthesis of a repair patch.
Keywords: gene transcription, nucleotide excision repair, RNA polymerase II, DNA damage, DNA repair
Introduction
Bulky adducts in DNA are repaired exclusively by nucleotide excision repair in mammalian cells. Excision repair in eukaryotes requires six factors, (RPA, XPA, XPC, TFIIH, XPG, and XPF-ERCC1) and entails damage recognition, melting of the duplex to form a “repair bubble,” and dual incision of the damaged DNA strand. The excised, adduct-containing 26–27–nucleotide (nt)2 product dissociates, and DNA polymerase and DNA ligase synthesize a repair patch (1–3).
The genome is not repaired uniformly. DNA-binding proteins including histones inhibit repair, likely by blocking access of repair factors to the damage (4–6). Also, there is an enhanced rate of repair of the template strand of actively transcribed genes, called transcription-coupled repair (TCR) (7–10).
An emerging consensus is that the enhanced rate of TCR compared with global repair results from RNAPII revealing to repair factors transcription-blocking adducts that would otherwise be blocked by components of chromatin and actively participating in recruiting repair factors to damage sites. RNAPII is blocked by template but not coding strand lesions and remains stably bound at the damage site (11–13). Evidence for efficient excision where RNAPII is stalled has been reported (11, 13, 14). To access a transcription-blocking adduct, the transcription-repair factor TFIIH likely has a central role, because TFIIH interacts with RNAPII during transcription initiation and because during global repair, TFIIH is involved in forming the repair bubble, it interacts with repair factors, and following repair, excision products released from chromatin are found bound to TFIIH (15–17). In addition, Cockayne syndrome complementation group B (CSB) protein, which is required for TCR (18) and which induces the forward translocation of RNAPII (14, 19), likely positions stalled RNAPII so as to allow excision of the transcription-blocking adduct in the presence of RNAPII. The presence of the transcription bubble likely facilitates repair as well. Excision repair occurs more rapidly in vitro when the adduct is located in a region of nonhomology resembling a transcription or repair bubble (20).
In an alternative model of TCR, it was suggested that TFIIS acts as a transcription-repair coupling factor by causing RNAPII stalled at an adduct to “back up,” thereby revealing the adduct to the repair enzyme assembly. It was suggested that following repair, the backed-up RNAPII may resume elongation (12). The backing-up effect of TFIIS has been demonstrated; however, it is reversed by CSB in vitro (14, 19). Furthermore, TFIIS is not required for TCR in yeast (21) or human cells (22).
An interesting aspect of TCR is the fate of RNAPII stalled at a template strand lesion. We have addressed the fate of RNAPII in vivo using methods to control transcription, to locate elongating and stalled transcription complexes genome-wide (native elongation transcript sequencing (NET-seq)) (23), and to locate excision repair sites genome-wide (XR-seq) (8). Our results suggest that RNAPII that has participated in transcription-coupled repair does not continue transcribing the template and does not engage in additional rounds of TCR. This is consistent with the dissociation of RNAPII following incision of a transcription-blocking lesion.
Results
Control of transcription with DRB
Our experiments utilized the transcription inhibitor 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside (DRB) to control RNAPII elongation. Under physiological conditions, transcription proceeds through the formation of a paused, promoter-proximal RNAPII complex before entering into a uniform elongation mode. DRB prevents the conversion of this paused complex to a fully elongating complex by inhibiting the positive transcription elongation factor complex (24). The DRB effect is reversible, and to create a discrete “wave” of RNAPs transcribing along the template, we used a pulse–chase–pulse scheme termed double DRB (DRB2) treatment illustrated in Fig. 1A, in which cells were first incubated in DRB for 2 h, to prevent new, actively elongating complexes and to permit RNAPII complexes already elongating during treatment with DRB to complete and terminate transcription. After 2 h, which is sufficient for the completion of even the longest transcripts, the DRB was washed off, and the cells were incubated without DRB for 10 min. Then DRB was added again. During the 10-min DRB-free period, RNAPII elongation begins from promoter proximal pause sites in a synchronized manner. Fig. 1B illustrates RNA levels produced in unirradiated XPC mutant cells at various times during the DRB2 procedure. RNA levels were measured by RT-qPCR using primers located at +2, +10, +50, and +100 kb in the OPA1 gene, which is 200 kb in length. The increase in progressively distal RNA levels with time illustrates a discrete wave of RNAPII transcribing along the gene at ∼1 kb/min during the DRB2 treatment, in agreement with reported rates of RNAPII elongation (24).
Figure 1.
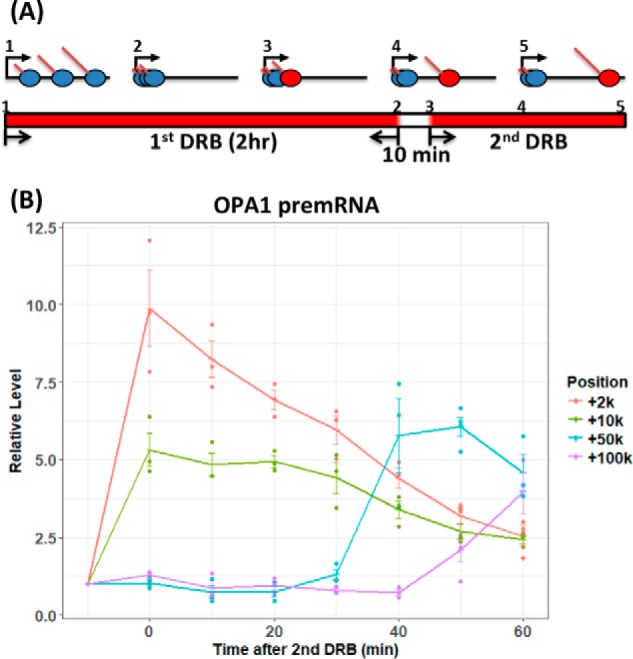
Wave of transcription elongation during the DRB2 treatment regimen. A, scheme of DRB2 treatments. Prior to treatment (step 1), elongating RNAPII (blue ovals) was distributed at random locations throughout the gene body. During the 2-h initial treatment with DRB, actively elongating RNAPII completed transcription. Initially transcribing complexes remained at the promoter-proximal pause site (step 2), unable to elongate because of the inhibition of the positive transcription factor elongation complex by DRB. After the 2-h DRB treatment, DRB medium was washed off, and the cells were incubated without DRB for 10 min. During this incubation, a wave of RNAPII complexes (red ovals in step 3–5) was “released” and initiates elongation. DRB was then added after 10 min to prevent additional RNAPII complexes from initiating elongation. The red sections of the bar at the bottom of A indicate periods of incubation with DRB. B, quantitative RT-PCR analysis of OPA1 nascent RNA during the DRB2 regimen. Total RNA was harvested from XP-C cells at different times (x axis). The end of the initial 2-h DRB treatment period is denoted −10 min, and the end of the DRB-free treatment period is denoted 0 min. RNA was harvested up to 60 min following the second addition of DRB. Introns in pre-mRNA from different positions (+2, +10, +50, and +100 kb) were quantified by RT-qPCR at each time point. RT-qPCR signals were normalized to the 18S rRNA signal and then were expressed on the y axis relative to the signal before RNAPII release at −10 min. The experiment was done three times. Individual data points for each replicate are plotted. Trend lines connect mean values, and S.E. values are shown.
Overall effects of controlled transcription on repair
To measure repair under various transcription conditions, we used the in vivo excision assay, described previously (25). In this assay, the 26–27–nt excision products are isolated from UV-irradiated cells, radiolabeled, and resolved on sequencing gels as shown in Fig. 2B. The irradiation produces two bulky adducts in DNA, cyclobutane pyrimidine dimers (CPDs), and pyrimidine-pyrimidone (6-4) photoproducts. Both are substrates for excision repair. However, because the (6-4) photoproducts are efficiently recognized by the core excision repair factors, their rates of repair are only modestly affected by transcription, and we analyzed only the repair of CPDs to gain insight into TCR (8). This was done by selectively immunoprecipitating the excision products with an anti–CPD-DNA antibody prior to radiolabeling. Excision products generated within the cell are vulnerable to degradation in vivo, and the repair signal at any time point reflects the combination of formation and degradation of the excision product. XPC mutant cells were used in all of our experiments; these cells are incapable of “global” excision repair, and thus the repair signal from these cells arises solely from TCR (26, 27).
Figure 2.

Effect of DRB on DNA repair of CPDs in XPC mutant cells. A, three UV/DRB (red) regimens were used: UV only (top), DRB after UV (middle), and the DRB2 regimen (DRB pulse, no DRB chase, DRB pulse) with UV (bottom). B, excision repair assays under the three conditions were conducted. C, quantitative results of repair experiments. The primary (non-degraded) excision products indicated with brackets in B were quantitated and normalized to the 60-min −DRB treatment repair signal, and means of three or more experiments are plotted as bars in C. Individual data points for each of the experiments are plotted in addition to S.E.
Repair was measured following the three treatment regimens shown in Fig. 2A. In the DMSO control, the cells were irradiated and then incubated in culture medium. In the DRB regimen, the cells were irradiated and then cultured in DRB. In the DRB2 regimen, the cells were incubated in a DRB-containing medium for 2 h, after which the medium was replaced with DRB-free medium for 10 min to enable elongation to begin from the 5′ end of genes. After 10 min, the cells were irradiated with UV, and DRB was added to the medium. The raw repair data in Fig. 2B and the quantitative values in Fig. 2C show that compared with the DMSO control, the DRB and DRB2 treatments changed the excision kinetics. These observed changes in transcription-coupled repair levels are consistent with decreased repair associated with decreased transcription caused by DRB. In the case of single DRB after UV treatment, the amount of RNAPII elongating at the time of irradiation is sufficient to initiate nearly normal levels of TCR at early time points, but repair at 60 min is lower than control because of a lack of newly initiated transcription. During the DRB2 regimen, the low repair at all time points reflects the single wave of transcription. Importantly, the relatively high repair in control cells at 60 min presumably results from continued formation of newly elongating transcripts at the 5′ end of genes.
Global analysis of individual elongation complexes and repair events
XR-seq experiments were performed with DRB2-treated cells to locate specific sites of repair throughout the genome. In XR-seq, excision products with CPD adducts are isolated, sequenced, and mapped to the genome (8). In addition, NET-seq (23) was performed to identify where elongating RNAP is located during the experiments. In NET-seq, nascent RNA that is bound to RNAPII and thus protected from RNase digestion is isolated, sequenced, and mapped to the genome. Fig. 3 shows a scatter plot in which each gene is represented by a dot, plotted along the x axis based upon its relative transcription level (NET-seq value) and along the y axis based upon its repair level (XR-seq value). The extent of repair and transcription are expressed as excision products or nascent RNA sequencing reads per kilobase per million total reads (RPKM). The results are shown for both coding (Fig. 3, NTS, left panels) and template (Fig. 3, TS, right panels) strands. Overall, the positive association between transcription and repair of the TS exhibited in Fig. 3 by these DRB2-treated cells indicates that the greater the likelihood of a gene being transcribed, the greater the likelihood that it will undergo transcription-coupled repair.
Figure 3.

Transcription-coupled repair in XPC mutant cells as a function of transcription. XR-seq results are plotted as repair reads per kilobase per million reads (RPKM, y axis, log10) versus NET-seq results, which are plotted as nascent transcript reads per kilobase per million reads (RPKM, x axis, log10). The right panels illustrate transcription and repair of the template strand at 30-min (top) and 60-min (bottom), and the left panels illustrate antisense transcription and coding strand repair at the 30-min (top) and 60-min (bottom) time points.
The left panels of Fig. 3 illustrate TCR of the NTS of genes. Compared with the signals from the TS (right panels), the dots are more concentrated in the lower left quandrants of the NTS panels, which indicates reduced transcription and repair levels. Earlier studies demonstrated the existence of transcription-coupled repair of the coding strand (NTS) of genes in the vicinity of the promoter, and it was attributed to antisense transcription from bidirectional promoters that occurs in this region (8–10). Our results demonstrate that this antisense transcription and transcription-repair coupling are produced by the DRB2 procedure and indicate that levels of antisense transcription and TCR are uniformly low among genes.
To further assess the distribution of repair along the template, the scatter plots in Fig. 4 were generated to illustrate the relative number of reads per gene (y axis) as a function of gene length (x axis). The top two rows illustrate repair of the template strand (TS), and repair of the coding strand is shown in the bottom two rows (NTS). Plots of template strand repair clearly illustrate two major populations (high RPKM and low RPKM) in all conditions, representing genes with high and low transcription-coupled repair levels. As in Fig. 3, predominantly low TCR is seen from the NTS, which is due to bidirectional transcription from the promoter/enhancer regions.
Figure 4.

Transcription-coupled repair in XPC mutant cells as a function of gene length. The cells were treated with the three treatment regimens (DMSO, left column; DRB, middle column; DRB2, right column) and allowed to repair for 30 or 60 min. The data were analyzed by scatter plot. The length of each gene in bp is plotted on the x axis (log10) versus XR-seq repair reads per kilobase per million total reads (RPKM, y axis, log10). The top two rows and the bottom two rows illustrate results of the template (TS) and coding (NTS) strands, respectively, and the 30- and 60-min repair times are indicated on the right side of the panels. In the TS (top two rows), TCR was clustered in high RPKM and low RPKM populations. To illustrate trends in TCR versus gene length among the high RPKM populations, linear regression lines and slope value are shown.
Interestingly, the distributions of high RPKM template strand repair groups as a function of gene length (Fig. 4, top two rows) were different between control, DRB, and DRB2 conditions. Linear regression lines drawn on the plot and slope values illustrate trends in TCR versus gene length. In the control, at 30 min (top left panel) there were fewer repair reads per kb in long genes compared with short genes, and this negative correlation was stronger at 60 min (Fig. 4, second row, left panel). Relevant to this observation, earlier studies (8) (also see below) showed an elevated level of transcription-repair coupling at the 5′ end of genes compared with repair within gene bodies. In the control (DMSO) cells in Fig. 4, it is likely that this higher level of repair at the 5′ end more strongly influences the overall repair level of short genes compared with long genes and is responsible for the negative correlation. Consistent with this interpretation, the negative correlation between gene size and number of reads is absent in the single DRB-treated cells (middle panels), in which there is no new transcription after UV and thus reduced repair at the 5′ end of genes. In the DRB2 condition (Fig. 4, right column), there was no obvious difference between 30 and 60 min, but there was a negative correlation between RPKM and gene length. This is because, during the 10-min DRB-free incubation before UV, the likelihood that a discrete wave of elongating RNAPII complexes is generated depends upon promoter activity (Fig. 3), not gene length. When the level of transcription-coupled repair is then expressed as a function of gene length, longer genes exhibit less repair per kb than short genes (Fig. 4, right column).
Analysis of a discrete wave of repair
To address whether a single elongating RNAPII could induce multiple transcription coupled events on one template, we selected for further analysis 1646 genes based upon length over 100 kb and exhibiting higher repair levels (RPKM over 0.1). These criteria increase the likelihood that multiple repair events per gene would be detected if they occurred. If one RNAPII could induce multiple repair events (transcription resumes following TCR), we expect to see the repair signal shift to the 3′ end of the template with time following DRB treatment. We plotted the repair signals from the first 100 kb of the template strand of these genes in Fig. 5A after normalizing to the sequencing depth and the relative repair levels we observed from the excision assay (Fig. 2). Repair of the non-transcribed coding strands (NTS) are plotted for reference.
Figure 5.

Location of DNA repair in genes larger than 100 kb. A, analysis was limited to 1646 genes with relatively high repair signals (RPKM > 0.1, see Fig. 3) and genes over 100 kb long. Repair in the first 100 kb from the TSS was analyzed (bin size, 250 nt). Repair signals were first normalized to the relative quantity of primary excision product (Fig. 2) and are plotted in aggregate view (top row), gene-by-gene heat map (middle two rows), and as heat maps representing the ratio of signals at 60 or 30 min (bottom row). In the aggregate view (top row), the dots illustrate the results of each bin, and lines illustrate the smoothed data. Also, the aggregate view illustrates repair in both the transcribed strand (TS) and the non-transcribed coding strand (NTS). The NTS 30-min (red) and 60-min (blue) data are largely superimposed, and the blue curve predominates. B, repair distributions are plotted for cells incubated in DRB-free medium for either 10 or 5 min in the DRB2 regimen. The data are plotted in aggregate view (top row), gene-by-gene heat maps (middle two rows), and as the ratio of 10- to 5-min incubations (bottom row). Repair was for 60 min.
In control DMSO-treated cells (Fig. 5A, left column), most of the signal is distributed evenly on the gene body, and there is an elevated signal close to the transcription start sites (TSSs) at 30 min after UV. At 60 min, the signal from the gene body overall decreased, and the signal near the TSS increased. The increased repair near the TSS is consistent with earlier studies (8) as mentioned above and consistent with repair associated with transcription initiated after UV. The decreased repair in the gene bodies at 60 min is consistent with a single round of TCR, initiated by RNAPII complexes transcribing at the time of irradiation, and largely complete by ∼30 min. These results are also consistent with the results in Fig. 4. The stronger negative correlation between repair and gene length at 60 min compared with 30 min in Fig. 4 reflects the reduced repair in gene bodies, which, for longer genes, had a more profound effect than the increased repair near the TSS.
When DRB was added after UV (Fig. 5A, middle column), repair signals again were relatively evenly distributed on the gene bodies but were in this circumstance reduced near the TSS. The reduced repair levels near the TSS support the conclusion that in control cells (left column), the elevated repair near the TSS is due to transcription elongation that began following UV. As in control cells, repair in the gene bodies was lower at 60 min compared with 30 min, again suggesting that each transcription-coupled repair event is not followed by elongation and additional TCR.
In the DRB2 regimen (Fig. 5A, right column), the cells exhibited very little repair in the gene bodies, as expected from the depletion of elongating RNAPII complexes during the 2-h pretreatment with DRB. The cells incubated for 30 min following UV exhibited repair as a peak within 10 kb of the TSS, resulting from the wave of elongating RNAPII that was produced during the 10 min of DRB-free incubation before UV. Most importantly, the peak of repair near the TSS decreased at 60 min but did not shift dramatically in the 3′ direction. Some repair was seen up to approximately +25 kb; however, this distance is rather small considering the elongation rate of RNAPII (24, 28), and this modest extension of repair in the 3′ direction may reflect the elongation of multiple RNAPII complexes during the 10-min DRB-free incubation, with the first polymerase repairing the first lesion encountered and dissociating and then the second polymerase repairing the next lesion downstream following repair of the first. In addition, the lesion distribution is expected to effect the observed repair pattern. In these experiments, the 20 J/m2 dose is expected to produce ∼1 lesion/strand per 10 kb, and ∼90% of the photoproducts are CPDs.
To examine whether multiple RNAPII complexes begin elongation during the 10-min DRB-free incubation, we compared the XR-seq patterns following 5- and 10-min DRB-free incubations, under the DRB2 condition and with 60-min repair. The results in Fig. 5B show that the shorter 5-min RNAPII release condition produced a slightly narrower repair peak. The broader repair peak following the 10-min release is exclusively due to more repair in the 3′ direction and is consistent with release of multiple RNAPII complexes during the DRB-free incubation time only in a fraction of the templates. Thus the results in Fig. 5A (right column), in which the peak of repair decreases from 30 to 60 min, indicate that following transcription-coupled repair, the RNAPII that targeted the transcription-blocking adduct for repair does not proceed to initiate repair at the next lesion downstream in the template strand.
Analysis of a discrete wave of transcription
To further characterize the disposition of RNAPII in transcription-repair coupling, we used NET-seq to map the location of RNA-bound elongating RNAPII under the DRB2 condition, 30 and 60 min after UV (UV) or no UV (Ctl). RNAPII was located mainly in the first bin (within 250 nt of the TSS) under all conditions, indicating that most of the RNAPII remained at the proximal pause site, and only a limited amount was released during the 10-min DRB-free incubation (data not shown). Therefore, the analysis focused on regions between 1 and 100 kb of the genes examined. The plots in Fig. 6 show the smoothed, normalized reads per million (top panels), the reads per gene (middle panels), the 60-min/30-min ratios of reads per gene (bottom panels), and the UV/control ratio of reads per gene (right column). The top panels show transcription from the transcribed strand (TS) and antisense transcription of the coding strand (NTS).
Figure 6.

Distribution of elongating RNAPII. This figure shows the NET-seq data for the same genes as in Fig. 5. RNAP-associated RNA was isolated from XPC mutant cells treated with the DRB2 regimen either in the absence of UV (Ctl) or with UV (UV) treatment at the end of the DRB-free incubation. Mammalian NET-seq signals of the first 1 kb to 100 kb from the TSS were analyzed (bin size, 250 nt) as nascent transcript reads per kb per million total reads (RPM). Relative transcription was plotted in an aggregate view (top panel) or as gene-by-gene heat maps (middle panels). In the aggregate view, the dots illustrate the data for each bin, and lines show the smoothed data trend. The aggregate view also shows elongation sites in both the TS and the NTS (coding strand). The difference between control and +UV is shown as the UV/control (Ctl) ratio at 30- or 60-min repair time in the two panels on the right. The difference between the 30- and 60-min repair times is shown as the 60/30-min ratios for +UV and −UV treatments in the bottom two panels. To calculate the ratio between any two different conditions, signals of individual genes were smoothed (80 bins).
The NET-seq results in Fig. 6 (top and middle panels, left column) show that in the minus UV control (Ctl) at 30 min there was a discrete concentration of RNAPII located within 25 kb of the TSS, and the distribution of RNAPII was similar among genes. After 60 min, RNAPII elongated, as indicated by the loss of the concentrated elongation complexes near the TSS, and the distance traversed in the 3′ direction was gene-specific. Following UV and 30-min repair (Fig. 6, top and middle panels, right column), RNAPII was concentrated within 10 kb of the TSS, and this distribution is similar to the distribution of repair under the same condition (Fig. 5A, right column). Importantly, following 60 min of repair (Fig. 6, top and middle panels, right column), the density of RNAPII near the TSS decreased but was not associated with an increase of RNAPII downstream (Fig. 6, compare bottom left and right panels). These data are consistent with dissociation of RNAPII from the template after it participates in a single round of TCR.
Transcription and TCR in the OPA1 gene
The effects of UV damage on transcription elongation and repair kinetics are visually illustrated in Fig. 7 with screenshots showing locations of both RNAPII (NET-seq, blue) and excision repair (XR-seq, red) within the OPA1 gene (108 kb) under the DRB2 condition. Quantitative values for transcription shown on the plot were obtained by calculating total NET-seq reads from 1 to 20 kb and from 40 to 60 kb (red horizontal bars at top). In the absence of UV, the nascent transcripts, representing elongation complexes, are concentrated near the TSS 30 min after release from the promoter proximal pause site. By 60 min, they are seen to have elongated further into the gene body. Following UV, elongation complexes are more highly concentrated near the TSS at 30 min, and at 60 min, there is a striking loss of elongation complexes and failure to elongate into the gene body. Repair events are also concentrated near the TSS at 30 min. After 60 min, there is a striking reduction in repair near the TSS, and there is only a modest increase in repair in the 3′ direction because of limited release of more than one elongating RNAPII during the 10-min DRB-free incubation period. Thus the sites of repair coincide with the sites of elongation complexes. The distribution of elongation complexes and repair events as a function of time indicates that individual RNAPII complexes are unable to continuously progress downstream and engage in multiple rounds of TCR and are apparently dissociated from the template in association with TCR.
Figure 7.
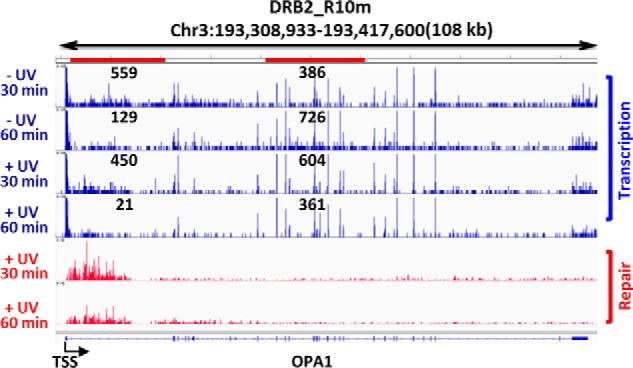
Screenshot illustrating XR-seq and NET-seq results for the OPA1 gene in XPC mutant cells. NET-seq (blue) and XR-seq (red) results from XP-C cells following the DRB2 regimen are illustrated together with a representation of the OPA1 gene (bottom). We note that a number of transcription elongation sites are seen throughout the OPA1 gene in both the presence and the absence of UV, including many of the strongest peak signals. These signals are likely splice sites, because this method is known to detect RNAPII complexes stalled at splice sites (23). Total reads from 1 to 20 kb and 40 to 60 kb (red horizontal bars at top) were calculated and are shown on the plot. Signals from splice sites contributed significantly to these read numbers; nevertheless, the values observed remain consistent with progression of RNAPII along the template from 30 to 60 min in the absence of UV and loss of elongating RNAPII from 30 to 60 min following UV. The repair signal peak near the promoter decreased from 30 to 60 min following UV as the wave of transcription and TCR was completed and the repair products were degraded.
Discussion
Our findings support the concept that RNAPII dissociates from the template during transcription-coupled repair. Our methods appear reliable because they showed that DRB did reduce transcription in a controllable manner, and reduction of transcription with DRB produced an expected overall reduction in TCR as measured in XPC mutant cells. Our data show blockage of elongation where elongating RNAPII is expected to encounter the first CPD in the template. We detect, under a controlled, discrete wave of transcription, essentially no additional repair or transcription further downstream following repair of the first adduct encountered. Our findings extend those of Andrade-Lima et al. (29) obtained by different methods that focused on recovery of transcription following UV.
In light of our findings combined with numerous reports on the fates of RNAPII and the transcript upon encountering a photoproduct, we propose the model shown in Fig. 8. In this illustration, the three template strand adducts that undergo TCR require three different RNAPIIs that each initiate transcription at the upstream promoter. The polymerases each undergo transcription blockage, TCR, and RNAPII dissociation, with the successive removal of lesions from the template in the 5′ to 3′ direction and eventual clearance of adducts from the template enabling synthesis of full-length transcripts.
Figure 8.
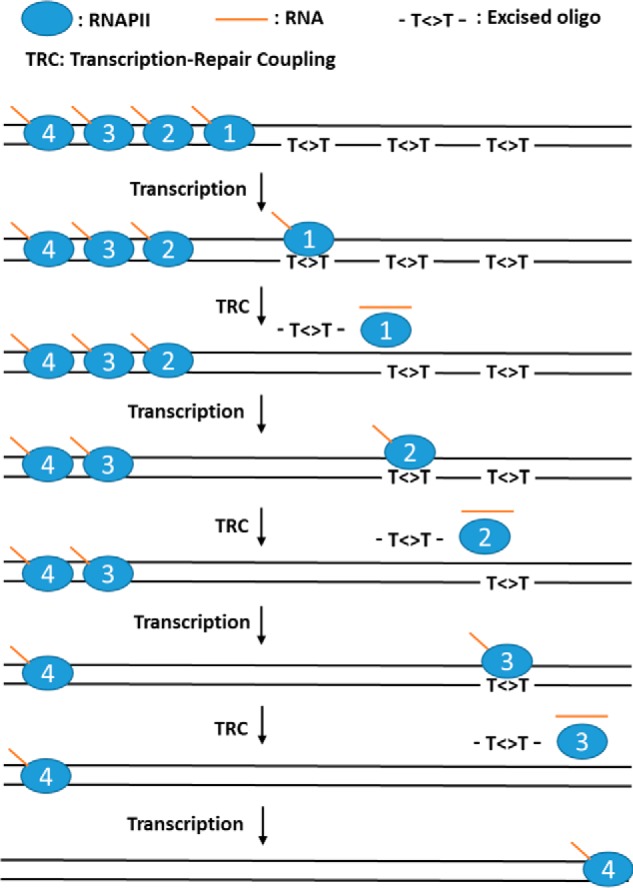
Model for TCR in mammalian cells. For simplicity we show a gene with four RNAPIIs in poised state at the 5′ region of the gene and three CPDs in the transcribed strand. After each RNAPII-CPD encounter, the damage is repaired, and the RNAPII behind continues transcription until it encounters the next CPD, resulting in repair and dissociation of RNAPII and continuation of repair by the following RNAPIIs until the track is completely cleared of damage. In our experiments, the DRB2 (pulse–chase–pulse) regimen permitted only limited release of RNAPII during the 10-min DRB-free chase period, allowing repair of only the initial damage site at the 5′ end of the gene.
The point at which RNAP is dissociated during TCR remains unclear. Regarding the possibility of dissociation before repair, it is difficult to envisage how RNAPII could target a transcription-blocking adduct for repair after it dissociates. Dissociation of RNAPII independently of repair assisted by RNAP-interacting factors has been observed in vitro with cell extracts (11, 13, 16, 30) but may be artifactual for various reasons, such as loss of compartmentalization in preparation of cell extracts. In our experiments, TCR-independent RNAPII dissociation may have occurred in the irradiated cells but been undetected by our methods. However, this cannot be a major pathway of RNAPII transcriptional termination because in that case there would not have been TCR targeted to the template strand.
In light of these findings and considerations, we propose that RNAPII dissociation occurs in some way in conjunction with repair. One mechanism for RNAP dissociation in conjunction with repair has been described in Escherichia coli. Mfd, the transcription-repair coupling factor in E. coli (31, 32), binds simultaneously to the upstream face of stalled RNAP and to the upstream template. The translocation activity of Mfd is activated by this interaction, and the translocation action of Mfd dissociates RNAP from the template (31–33). The dissociated RNAP remains tethered to the template via the template-bound Mfd in a semi-stable complex that has a half-life of ∼10 min in isolation. Mfd in this conformation reveals its UvrA-binding site and exhibits a high affinity for UvrA, the damage recognition subunit of the E. coli excision repair enzyme. Binding of UvrA induces dissociation of Mfd and RNAP from the DNA and delivery of the repair enzyme to the damage, with formation of a damaged DNA–UvrA–UvrB preincision complex, which makes dual incisions in the presence of UvrC (31, 32).
The mammalian CSB protein has been considered analogous to Mfd because CSB is required for transcription-coupled repair (18, 34) and because, like Mfd, CSB is a large protein with a centrally located helicase motifs region (35, 36). However, unlike Mfd, CSB alone does not remove RNAP stalled on a DNA template, although similar to Mfd, CSB induces forward translocation of RNAP (14, 16, 19). It is conceivable that, by analogy with Mfd, removal of RNAPII by CSB would occur in the presence of repair factors; however, this was not observed (14).
Other relevant differences exist between excision repair in humans and E. coli. During repair in E. coli, there is no melting of the adducted DNA substrate duplex, no repair bubble is formed, and after dual incisions are made, the excision product remains annealed to the chromosome together with the UvrB-UvrC repair factors. These are dissociated by the action of DNA helicase II (UvrD gene product) and DNA polymerase I (37, 38). Furthermore, in E. coli, in the absence of Mfd, RNA polymerase stalled at an adduct inhibits repair of the adduct (39). In contrast, in humans, the dual incisions are made in a repair bubble (1–3) or, in the case of coupled repair, in a transcription-repair bubble, apparently in the presence of the stalled RNAPII (11, 13, 14). Thus, whereas both prokaryotes and eukaryotes perform dual incision-based excision repair and both perform global and transcription-coupled repair, the corresponding repair proteins in prokaryotes and eukaryotes are not homologous, their reaction mechanisms are different, and the mechanism for dissociation of mammalian RNAPII during TCR must be different from the corresponding mechanism in E. coli.
The scenario most consistent with the presently available information is that in mammalian cells, dissociation of adduct-blocked RNAPII occurs following dual incision. Eukaryotic dual incision sites are the same in global and transcription-coupled repair (25), and in both repair pathways, the excision product is released and degraded, although before degradation, the released product can be temporarily isolated either complexed with TFIIH or in partially degraded form complexed with RPA (25). Removal of the damaged section of template within the transcription/repair bubble may precede or occur together with dissociation of RNAPII. Dissociation of RNAPII from the template may be facilitated by the forward translocation action of CSB (19) and/or by repair synthesis. RNAPII in elongation mode is known to remain bound to the template in active form even under harsh conditions, and CSB translocase or proteins associated with repair synthesis may provide the force needed to dissociate RNAPII from the template following dual incision. The results presented in this paper are not consistent with RNAPII resuming transcription either before repair synthesis, by “jumping the gap” in the template, or after repair synthesis, by remaining in a paused, transcription-competent, template-bound state during repair synthesis.
Experimental procedures
Cells culture and DRB treatment
XP-C (GM15983) human skin fibroblast cells were purchased from Coriell Institute. All of the cells were grown in DMEM-high glucose (ThermoFisher) supplemented with 10% (v/v) fetal bovine serum (Sigma) and maintained at 37 °C in 5% CO2. DRB or DMSO was added to the medium when cells were ∼80% confluent. The final concentrations of DRB and DMSO were 100 μm and 0.1% (v/v). For post-UV DRB treatment, medium was collected, and DRB was added to the medium just before irradiation. The cells were irradiated with UVC (20 J/m2). Then the DRB containing medium was added back, and the cells were incubated for 30 or 60 min at 37 °C in 5% CO2. For DRB2 regimen, the cells were first incubated with DRB at 37 °C in 5% CO2 for 2 h. After collecting the medium with DRB, cells were wash two times with PBS and then incubated in fresh medium for 10 min. Then medium was removed, and the cells were irradiated with UVC. After irradiation, medium with DRB was added back, and the cells were incubated for 30 or 60 min at 37 °C in 5% CO2.
Real-time quantitative PCR analysis of OPA1 transcription
Total RNA of XP-C cells was purified using TRIzol reagent (Ambion). First-strand synthesis was performed using an iScript cDNA synthesis kit (Bio-Rad). Real-time PCR was performed using iTaqTM Universal SYBR®s Green Supermix (Bio-Rad). Primers to detect different regions of the OPA1 gene and 18S rRNA were based on the published sequences (24, 40).
Excision assay and excision repair sequencing (XR-seq) library preparation
The cells were irradiated with 20 J/m2 UVC and were incubated at 37 °C in 5% CO2 for 30 or 60 min. Excision assays were performed according to the published procedures (25). In brief, excision products were first extracted by the Hirt procedure. After phenol-chloroform extraction and ethanol precipitation, CPD-containing excision products were purified by immunoprecipitation. Purified excision products were radiolabeled with 32P-cordycepin and resolved on 10% urea-polyacrylamide sequencing gels. The signals were detected by phosphorimaging and quantified by normalizing to the internal control 50-mer “spike” DNA added to each reaction before the labeling reaction. Signals for treated cells were normalized to the signals from control cells (DMSO) and repaired for 60 min in each experiment.
Library generation for XR-seq was based on the published procedure (8). The cells were lysed in hypotonic buffer with Nonidet P-40. Then excision products were purified using antibodies against TFIIH components (anti-p89, SC-293 and anti-p62, SC-292 from Santa Cruz). After adaptor ligation, CPD excision products were purified again by immunoprecipitation using an anti-CPD-DNA antibody (Cosmo Bio USA). After repair by CPD photolyase, the fragments were amplified by PCR, and the libraries were sequenced by Hiseq 2500 (1 × 50).
XR-seq experiments were performed two times, and the data presented in text figures are from averaging of the two experiments. To illustrate the concordance of the two experiments, Fig. S1 shows individually the results of the two experiments that were averaged to obtain the values plotted in the critical XR-seq experiment in Fig. 5.
Mammalian NET-seq library preparation
Library generation for NET-seq followed the published procedure with some modification (23). The cells were lysed, and nuclei were purified. The nuclei were resuspended in NUN1 buffer (20 mm Tris-Cl, pH 7.9, 75 mm NaCl, 0.5 mm EDTA, 50% (v/v) glycerol) and then were lysed with NUN2 buffer (20 mm HEPES-KOH, pH 7.6, 300 mm NaCl, 0.2 mm EDTA, 7.5 mm MgCl2, 1% (v/v) Nonidet P-40, and 1 m urea). After centrifugation, the chromatin fraction containing elongating RNAPII was collected. RNAPII–RNA–DNA complexes were released by Micrococcal nuclease digestion and were enriched by the immunoprecipitation with the antibody recognizing the C-terminal domain of RNAPII (MABI0601 from Medical & Biological Laboratories). Phosphorylation at the 5′ end of fragments was performed by T4-PNK. Fragments were used for the generation of libraries using a kit for small RNA library generation (NEBNext Small RNA Prep Set for Illumina), and the libraries were sequenced by Hiseq 2500 (2 × 50).
NET-seq experiments were done twice, and the data presented in text figures represent the average of the two experiments. To illustrate the concordance of the two experiments, Fig. S2 shows individually the results of the two experiments that were averaged to obtain the values plotted in the critical NET-seq experiment in Fig. 6.
Analysis of data from high-throughput sequencing
For XR-seq, adaptor sequences were first removed using BBDuk tool of BBMap with the options: ktrim = r k = 21 hdist = 2 minlen = 1. Duplicated sequences were then removed using Collapser tool of FASTX. After removing 50-mers (contaminant), unique reads were mapped to the human genome (hg19) using bowtie2 with the option “very sensitive.” Gene structure annotations were downloaded from University of California, Santa Cruz Table Browser. Annotations were modified for genes with multiple isoforms or overlapping regions to generate longest non-overlapped genes annotation (25,839 genes). Aligned reads were strand-specifically assigned to the genes using Intersect tool of Bedtools with the option “c.” For intragene distribution, genes with RPKM ≥ 0.1 in all conditions and the length ≥100 kb were selected (1646 genes). Genes were separated into the 250 bases/bin, and the aligned reads were assigned to each bin using Intersect tool of Bedtools with the options “c” and “F 0.5.” The number of reads was first normalized to the number of total aligned reads and then was normalized to the relative CPD repair levels from Fig. 2 (DMSO 30 min: 1.07, DMSO 60 min: 1, DRB 30 min: 1.06, DRB 60 min: 0.56, DRB2 30 min: 0.52, DRB2 60 min: 0.31).
For NET-seq, adaptor sequences were first removed using BBDuk tool of BBMap with the options: ktrim = r k = 21 hdist = 2 minlen = 1 tpe tbo. Reads were mapped to the human genome (hg19) using BBMap with the options: maxindel = 100,000 intronlen = 10 ambig = random qin = 33. After mapping, reads with proper pair were selected, and the locations of RNAPII were defined as the positon of first base from the 3′ end of the isolated RNA. Positions of RNAPII were assigned to each bin of the same gene sets used in XR-seq in the same manner that XR-seq sites were assigned. The number of reads was normalized to total reads and was aligned to the 1–100-kb region of the gene sets. For comparing the ratio between different conditions, data from each gene was smoothed by loess methods to decrease variations caused by bins without any reads.
Author contributions
Y.-Y. C. and J. H. data curation; Y.-Y. C. formal analysis; Y.-Y. C. and J. H. investigation; Y.-Y. C. visualization; Y.-Y. C. writing-review and editing; J. H., A. S., and C. P. S. conceptualization; A. S. and C. P. S. supervision; A. S. funding acquisition; C. P. S. writing-original draft.
Supplementary Material
Acknowledgment
We thank Michael G. Kemp (Wright State University) for useful comments.
This work was supported by National Institutes of Health Grants GM118102 and ES027255. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- nt
- nucleotide(s)
- RNAPII
- RNA polymerase II
- TCR
- transcription-coupled repair
- CSB
- Cockayne syndrome group B
- NET-seq
- native elongation transcript sequencing
- XR-seq
- excision repair sequencing
- DRB
- 5,6-dichlorobenzimidazole 1-β-d-ribofuranoside
- DRB2
- double DRB
- RT-qPCR
- real-time quantitative PCR
- CPD
- cyclobutane pyrimidine dimer
- RPKM
- reads per kilobase per million total reads
- TSS
- transcription start site.
References
- 1. Wood R. D. (1997) Nucleotide excision repair in mammalian cells. J. Biol. Chem. 272, 23465–23468 10.1074/jbc.272.38.23465 [DOI] [PubMed] [Google Scholar]
- 2. Reardon J. T., and Sancar A. (2005) Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol. 79, 183–235 10.1016/S0079-6603(04)79004-2 [DOI] [PubMed] [Google Scholar]
- 3. Sancar A. (2016) Mechanisms of DNA repair by photolyase and excision nuclease (Nobel lecture). Angew. Chem. Int. Ed. Engl. 55, 8502–8527 10.1002/anie.201601524 [DOI] [PubMed] [Google Scholar]
- 4. Thoma F. (1999) Light and dark in chromatin repair: repair of UV-induced DNA lesions by photolyase and nucleotide excision repair. EMBO J. 18, 6585–6598 10.1093/emboj/18.23.6585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smerdon M. J., and Conconi A. (1999) Modulation of DNA damage and DNA repair in chromatin. Prog. Nucleic Acid Res. Mol. Biol. 62, 227–255 [DOI] [PubMed] [Google Scholar]
- 6. Hara R., Mo J., and Sancar A. (2000) DNA damage in the nucleosome core is refractory to repair by human excision nuclease. Mol. Cell. Biol. 20, 9173–9181 10.1128/MCB.20.24.9173-9181.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mellon I., Spivak G., and Hanawalt P. C. (1987) Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 51, 241–249 10.1016/0092-8674(87)90151-6 [DOI] [PubMed] [Google Scholar]
- 8. Hu J., Adar S., Selby C. P., Lieb J. D., and Sancar A. (2015) Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 29, 948–960 10.1101/gad.261271.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu J., Adebali O., Adar S., and Sancar A. (2017) Dynamic maps of UV damage formation and repair for the human genome. Proc. Natl. Acad. Sci. U.S.A. 114, 6758–6763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adar S., Hu J., Lieb J. D., and Sancar A. (2016) Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 113, E2124–E2133 10.1073/pnas.1603388113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Selby C. P., Drapkin R., Reinberg D., and Sancar A. (1997) RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res. 25, 787–793 10.1093/nar/25.4.787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donahue B. A., Yin S., Taylor J. S., Reines D., and Hanawalt P. C. (1994) Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc. Natl. Acad. Sci. U.S.A. 91, 8502–8506 10.1073/pnas.91.18.8502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tremeau-Bravard A., Riedl T., Egly J. M., and Dahmus M. E. (2004) Fate of RNA polymerase II stalled at a cisplatin lesion. J. Biol. Chem. 279, 7751–7759 10.1074/jbc.M309853200 [DOI] [PubMed] [Google Scholar]
- 14. Lainé J. P., and Egly J. M. (2006) Initiation of DNA repair mediated by a stalled RNA polymerase IIO. EMBO J. 25, 387–397 10.1038/sj.emboj.7600933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kemp M. G., Gaddameedhi S., Choi J. H., Hu J., and Sancar A. (2014) DNA repair synthesis and ligation affect the processing of excised oligonucleotides generated by human nucleotide excision repair. J. Biol. Chem. 289, 26574–26583 10.1074/jbc.M114.597088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Selby C. P., and Sancar A. (1997) Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem. 272, 1885–1890 10.1074/jbc.272.3.1885 [DOI] [PubMed] [Google Scholar]
- 17. Kemp M. G., Reardon J. T., Lindsey-Boltz L. A., and Sancar A. (2012) Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 287, 22889–22899 10.1074/jbc.M112.374447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Hoffen A., Natarajan A. T., Mayne L. V., van Zeeland A. A., Mullenders L. H., and Venema J. (1993) Deficient repair of the transcribed strand of active genes in Cockayne's syndrome cells. Nucleic Acids Res. 21, 5890–5895 10.1093/nar/21.25.5890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Selby C. P., and Sancar A. (1997) Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. U.S.A. 94, 11205–11209 10.1073/pnas.94.21.11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mu D., and Sancar A. (1997) Model for XPC-independent transcription-coupled repair of pyrimidine dimers in humans. J. Biol. Chem. 272, 7570–7573 10.1074/jbc.272.12.7570 [DOI] [PubMed] [Google Scholar]
- 21. Verhage R. A., Heyn J., van de Putte P., and Brouwer J. (1997) Transcription elongation factor S-II is not required for transcription-coupled repair in yeast. Mol. Gen. Genet. 254, 284–290 10.1007/s004380050417 [DOI] [PubMed] [Google Scholar]
- 22. Mackinnon-Roy C., Stubbert L. J., and McKay B. C. (2011) RNA interference against transcription elongation factor SII does not support its role in transcription-coupled nucleotide excision repair. Mutat. Res. 706, 53–58 10.1016/j.mrfmmm.2010.10.010 [DOI] [PubMed] [Google Scholar]
- 23. Nojima T., Gomes T., Carmo-Fonseca M., and Proudfoot N. J. (2016) Mammalian NET-seq analysis defines nascent RNA profiles and associated RNA processing genome-wide. Nat. Protoc. 11, 413–428 10.1038/nprot.2016.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Veloso A., Kirkconnell K. S., Magnuson B., Biewen B., Paulsen M. T., Wilson T. E., and Ljungman M. (2014) Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res. 24, 896–905 10.1101/gr.171405.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu J., Choi J. H., Gaddameedhi S., Kemp M. G., Reardon J. T., and Sancar A. (2013) Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem. 288, 20918–20926 10.1074/jbc.M113.482257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Venema J., van Hoffen A., Karcagi V., Natarajan A. T., van Zeeland A. A., and Mullenders L. H. (1991) Xeroderma pigmentosum complementation group C cells remove pyrimidine dimers selectively from the transcribed strand of active genes. Mol. Cell. Biol. 11, 4128–4134 10.1128/MCB.11.8.4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Hoffen A., Venema J., Meschini R., van Zeeland A. A., and Mullenders L. H. (1995) Transcription-coupled repair removes both cyclobutane pyrimidine dimers and 6-4 photoproducts with equal efficiency and in a sequential way from transcribed DNA in xeroderma pigmentosum group C fibroblasts. EMBO J. 14, 360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fuchs G., Voichek Y., Rabani M., Benjamin S., Gilad S., Amit I., and Oren M. (2015) Simultaneous measurement of genome-wide transcription elongation speeds and rates of RNA polymerase II transition into active elongation with 4sUDRB-seq. Nat. Protoc. 10, 605–618 10.1038/nprot.2015.035 [DOI] [PubMed] [Google Scholar]
- 29. Andrade-Lima L. C., Veloso A., Paulsen M. T., Menck C. F., and Ljungman M. (2015) DNA repair and recovery of RNA synthesis following exposure to ultraviolet light are delayed in long genes. Nucleic Acids Res. 43, 2744–2756 10.1093/nar/gkv148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hara R., Selby C. P., Liu M., Price D. H., and Sancar A. (1999) Human transcription release factor 2 dissociates RNA polymerases I and II stalled at a cyclobutane thymine dimer. J. Biol. Chem. 274, 24779–24786 10.1074/jbc.274.35.24779 [DOI] [PubMed] [Google Scholar]
- 31. Fan J., Leroux-Coyau M., Savery N. J., and Strick T. R. (2016) Reconstruction of bacterial transcription-coupled repair at single-molecule resolution. Nature 536, 234–237 10.1038/nature19080 [DOI] [PubMed] [Google Scholar]
- 32. Selby C. P. (2017) Mfd protein and transcription-repair coupling in Escherichia coli. Photochem. Photobiol. 93, 280–295 10.1111/php.12675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park J. S., Marr M. T., and Roberts J. W. (2002) E. coli transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell 109, 757–767 10.1016/S0092-8674(02)00769-9 [DOI] [PubMed] [Google Scholar]
- 34. Venema J., Mullenders L. H., Natarajan A. T., van Zeeland A. A., and Mayne L. V. (1990) The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl. Acad. Sci. U.S.A. 87, 4707–4711 10.1073/pnas.87.12.4707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Troelstra C., van Gool A., de Wit J., Vermeulen W., Bootsma D., and Hoeijmakers J. H. (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell 71, 939–953 10.1016/0092-8674(92)90390-X [DOI] [PubMed] [Google Scholar]
- 36. Xu J., Lahiri I., Wang W., Wier A., Cianfrocco M. A., Chong J., Hare A. A., Dervan P. B., DiMaio F., Leschziner A. E., and Wang D. (2017) Structural basis for the initiation of eukaryotic transcription-coupled DNA repair. Nature 551, 653–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Husain I., Van Houten B., Thomas D. C., Abdel-Monem M., and Sancar A. (1985) Effect of DNA polymerase I and DNA helicase II on the turnover rate of UvrABC excision nuclease. Proc. Natl. Acad. Sci. U.S.A. 82, 6774–6778 10.1073/pnas.82.20.6774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adebali O., Chiou Y. Y., Hu J., Sancar A., and Selby C. P. (2017) Genome-wide transcription-coupled repair in Escherichia coli is mediated by the Mfd translocase. Proc. Natl. Acad. Sci. U.S.A. 114, E2116–E2125 10.1073/pnas.1700230114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Selby C. P., and Sancar A. (1990) Transcription preferentially inhibits nucleotide excision repair of the template DNA strand in vitro. J. Biol. Chem. 265, 21330–21336 [PubMed] [Google Scholar]
- 40. Singh J., and Padgett R. A. (2009) Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 16, 1128–1133 10.1038/nsmb.1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.