

Abstract
The links between oncogenic drivers and cancer cell metabolism have emerged over the past several decades, indicating that constitutive oncogenic growth signaling can render cancers susceptible to metabolic interventions. While significant progress has been achieved in identifying metabolic vulnerabilities of cancer cells, the complexity of the tumor microenvironment (TME) and the dynamic nature of organismal circadian metabolism challenge the precision of targeting cancer metabolism. Herein, current progress in the areas of cancer metabolism and TME metabolism is reviewed, highlighting how cancer metabolism can be accurately and precisely targeted.
Introduction
The “Precision Medicine” revolution in cancer has focused on the identification of compounds or biologic agents with high levels of molecular specificity and the tailored use of these agents in subsets of patients whose tumors have unique molecular characteristics. The use of such “precise” therapies can protect from off-target side effects, but not from on-target side effects. Such on-target effects can impact remote tissues, but also non-cancerous cells within the tumor microenvironment. The complex interplay between stromal elements, immune cells, and blood vessels can drastically impact the response to therapy and the development of resistance. Hence, through on-target effects on non-cancerous tissues both within and outside of the tumor microenvironment, “precise” therapies can fail to “accurately” treat a patient's cancer.
This issue is particularly notable with metabolic therapies that target pathways that are ubiquitous in both malignant and normal host cells. In this review, we will summarize alteration in tumor and TME metabolism, current therapies targeting these alterations, and strategies for improving the accuracy of metabolic therapies.
Basic concepts in tumor metabolism
Simplicity for the sake of understanding cancer metabolism has provided key conceptual frameworks over the past decades. The first of these frameworks is that cancer cell metabolism is altered to support biomass accumulation rather than efficient energy production. This was built on the observations of Louis Pasteur and Otto Warburg. In the 1880s, Pasteur showed that oxygen could suppress glycolysis or fermentation in yeast. Warburg then demonstrated in the 1920s that animal and human cancer tissues continued to display high glycolytic rates, even in the presence of oxygen[1, 2]. The former phenomenon is called the Pasteur effect, while the latter is coined the Warburg effect. Warburg's initial interpretation, that tumor mitochondria were dysfunctional, turned out to be incorrect. Rather, oxidative metabolism is held at sub-maximal levels so that alternative pathways may use glucose carbon to support the metabolic demands of constitutive cell growth and division.
The second major framework in tumor metabolism, based on work in the 1990s, is that the metabolic changes are driven by changes in oncogenes and tumor suppressors, as well as the hypoxia inducible factors (HIFs), which rewire metabolic pathways [3-6]. Human oncogenes, such as MYC, PI3K, and RAS, drive neoplastic cell proliferation by stimulating cancer cell metabolism through transcriptional, translational, and post-translational alterations of metabolic enzymes, hence promoting biosynthesis [6].
The constitutive oncogenic drive for cancer cells to grow and proliferate renders cancer cells dependent on a continuous nutrient supply to meet the constant demand of uncontrolled proliferative metabolism, which is distinct from normal maintenance metabolism that occurs in the vast majority of non-proliferative adult mammalian cells and provides a therapeutic window [7]. This window is limited by three major factors, 1) the complexity and heterogeneity of tumor metabolism itself, the cellular complexity of the TME and the role of metabolic pathways in the non-cancerous cells in the TME and in interactions between the tumor and the TME, and the use of tumor-like metabolism by a small subset of normal tissues, largely stem cells.
Complexity of tumor metabolism
The central metabolic pathways are conserved and evolved over billions of years. Specifically, glycolysis is the most conserved pathway that converts glucose to pyruvate (Figure 1), which is further catabolized to lactate, alanine, alcohol, or transported into the mitochondrion depending on the metabolic wiring of the cell [5, 6]. The conversion of glucose to lactate via glycolysis and lactate dehydrogenase A (LDHA) is known as the Warburg effect. The mitochondrial pyruvate carrier transports pyruvate into the mitochondrion, where it is converted by pyruvate dehydrogenase (PDH) to acetyl-CoA for further catabolism through the tricarboxylic acid (TCA) cycle. Hypoxia, through HIF-1, reverses the Pasteur effect and activates glycolytic genes as well as activating pyruvate dehydrogenase kinase to inhibit the conversion of pyruvate to acetyl-CoA while favoring its conversion to lactate via LDHA.
Figure 1.
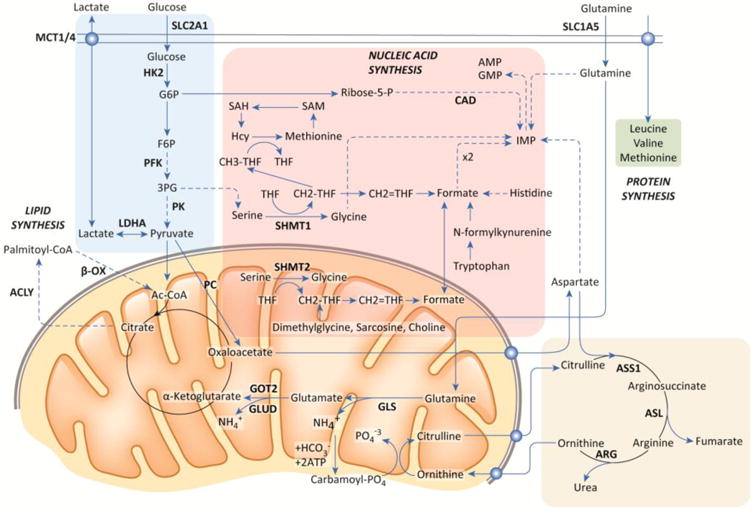
Overview of Cellular Metabolic Pathways, Central metabolic pathways and their connections are outlined, including glycolysis, the TCA cycle, nucleic acid synthesis, lipid synthesis, and the urea cycle. Cancer cells utilize these pathways to varying extents depending on the genetics and tissue of origin of the tumor, as discussed in the main text. Glucose and glutamine can both converted into substrates that can be oxidized via the TCA cycle, but intermediates of these pathways can be diverted to provide substrates for nucleic acid and amino acid synthesis or to replenish TCA cycle intermediates. Glucose can also be converted to lactate and exported (the Warburg Effect). Alternatively, lactate can also be taken up and oxidized as fuel.
There are exit and entry points into the mitochondrion and the TCA cycle that are involved in catabolism or biosynthesis [5]. Citrate produced from glucose in the mitochondrion (or glutamine through reductive carboxylation) is transported into the cytosol and converted by acetyl-CoA lyase (ACLY) to acetyl-CoA, providing a precursor for fatty acid synthesis or acetylation reactions. Succinyl-CoA, downstream of citrate, provides a precursor for heme biosynthesis. Oxaloacetate is converted by glutamate oxaloacetate transaminase (GOT2) to aspartate, which is exported into the cytosol for nucleotide biosynthesis. Glutamine is converted by glutaminase to ammonia and glutamate, which is further transformed to α-ketoglutarate for catabolism in the TCA cycle [8]. Under specific conditions, such as hypoxia or a truncated TCA cycle (eg., fumarate hydratase (FH) mutations in certain cancers), α-ketoglutarate can undergo reductive carboxylation to produce citrate in support of fatty acid synthesis [9]. Branched chain amino acids (BCAA; Leu, Ile, Val) are converted to ketoacids and ultimately to TCA cycle intermediates for catabolism [10]. Lipids transported into mitochondria can be catabolized through fatty acid oxidation (FAO) [11]. Ketone bodies (acetoacetate, β-hydroxybutyrate, and acetone) can also be converted to acetyl-CoA and catabolized by the mitochondrion. Further, pyruvate can be converted by pyruvate carboxylase (PC) to oxaloacetate to enter the TCA cycle, in addition to its conversion to acetyl-CoA by PDH [12].
These pathways are rewired in cancer cells under specific conditions. For example, studies with human lung cancers reveal the role of PC in the use of glucose [13], whereas BCAA are utilized in a genetically engineered mouse model (GEMM) of pancreatic cancer [10], FH mutant tumors undergo glutamine reductive carboxylation [14], and human liver cancers can catabolize ketone bodies [15]. It should also be noted that cancer cells can also turn on themselves and digest their own constituents via autophagy to generate metabolic intermediates for survival, they can eat albumin from the environment through macropinocytosis, and they can even eat other nearby cells through entosis [16, 17].
While engaging the mitochondria to catabolize nutrients is essential for the survival of cancer cells, the generation of byproducts can also be toxic and expose additional vulnerabilities such as the induction of autophagy by ammonia, the inhibition of NOS by nitric oxide, and oxidative stress generated by reactive oxygen species [18]. Cancer cells can therefore require increased activation of anti-oxidant defense mechanisms. One example of this is the NRF2 transcriptional axis, which is activated through the ROS-induced inactivation of KEAP to release active NRF2, which enters the nucleus and transactivates genes involved in redox homeostasis to reduce oxidants [19, 20]. In a fraction of human lung cancers, KEAP1 is mutated, allowing for the constitutive activity of NRF2, which increases the anti-oxidant programs for cell survival. Another example is the increased synthesis and use of long chain polyunsaturated fatty acids, particularly in high mesenchymal cell state tumors, which leads to increased dependence on glutathione and the lipid peroxidase GPX4 to avoid ferroptosis [21]. Hence, such rewiring exposes new vulnerabilities to oxidant stress in these tumors.
While different metabolic pathways have been shown to be important in different tumor subtypes, there can also be heterogeneity in metabolic characteristics within an individual tumor. Certain regions of a tumor are more hypoxic than others that are more proximal to blood vessels [22, 23]. In fact, it is documented that in some settings hypoxic cancer cells generate lactate that is converted to pyruvate for mitochondrial catabolism in more oxygenated cancer cells [24, 25]. This was further supported by a recent study demonstrating that poorly perfused regions of human non-small cell lung tumors predominantly fuel the TCA cycle with glucose, while well-perfused regions preferentially use non-glucose alternatives[26].
The work summarized above has allowed for a deep understanding of cancer cell intrinsic metabolic changes. However, with a richer appreciation for the cellular complexity of the tumor microenvironment (TME), this understanding is insufficient for a strategy to target tumor metabolism, noting that targets can be precisely hit without accuracy in the context of a complex TME that changes spatially and temporally.
Metabolism in the TME
The tumor tissue is highly complex and variable depending on the tissue of origin [27] (Figure 2). For example, while leukemias and liver cancers tend to be composed primarily of malignant cells, cancers involving the pancreas display highly variable composition with remarkable heterogeneity of the stromal versus cancer cellular content. In addition to the genomically altered cancer cell, the tumor microenvironment can contain stromal fibroblasts, glial cells, macrophages, myeloid derived tumor suppressor cells, and tumor tolerant T and B cells, which are all fed by neo-vasculature and drained by highly variable lymphatics. These tumor-associated cells operate within the same metabolic milieu as the cancer and can be involved in tumor formation, maintenance, and therapy response and resistance. As such, the understanding of the complexity of the community of cells that comprises the tumor tissue is essential for accurate and precise targeting of tumor metabolism.
Figure 2.
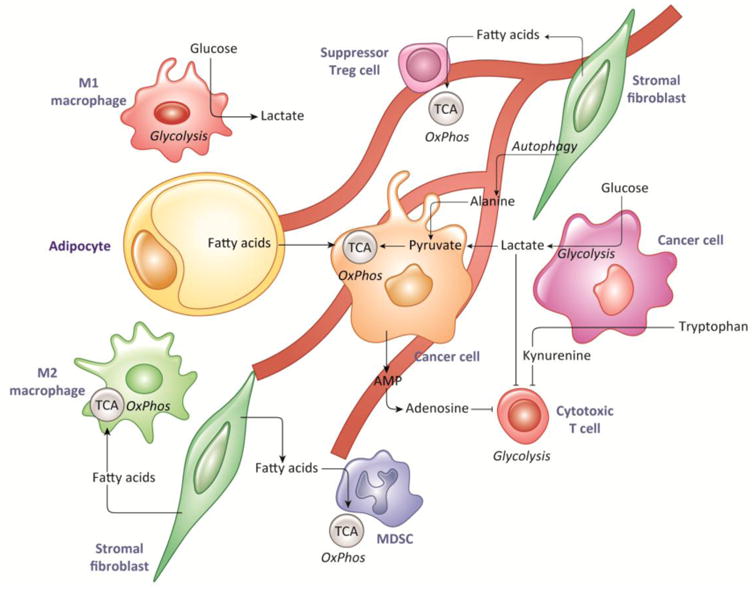
Metabolism in the Tumor Microenvironment, In addition to tumor cells, the tumor microenvironment is composed of fibroblasts, macrophages, myeloid-derived suppressor cells, regulatory and cytotoxic T cells, and endothelial cells, among others. These cells depend on specific metabolic substrates for appropriate function, which can be disrupted in the TME or with therapy. Cancer cells can reprogram these cells to produce fuels such as lactate, alanine, and fatty acids that are then consumed by the cancer cells themselves. They can also produce immunosuppressive metabolites, including lactate, adenosine, and kynurenine.
Stromal cells can undergo metabolic changes in response to signals from the tumor cells as well as in response to the hypoxic and acidic microenvironment [28]. These metabolic changes can then feed-back to support tumor growth. For examples, pancreatic stellate cells can produce alanine to support the metabolism of pancreatic cancer cells [29]. Prostate cancer-associated fibroblasts can produce exosomes that supply amino acids to tumor cells and reprogram tumor metabolism away from oxidative energy production [30]. Tumor derived hydrogen peroxide can induce cancer-associated fibroblasts to consume glucose into lactate, which can then be used as an oxidative metabolite by the cancer cells [31]. Ovarian cancer cells have been shown to promote lipolysis in adipocytes and transfer of free fatty acids to tumor cells for use in beta-oxidation [32]. More broadly, the community of cancer and stromal cells co-exists in an abnormal metabolic environment that selects for synergistic commensal interactions, which manifest as clinically detectable cancers.
The tumor neo-vasculature is required for oxygen and nutrient delivery, for removal of waste products, and for delivery of chemotherapeutic agents. Endothelial cells are thus essential to the metabolism of the tumor, but they also have their own unique metabolic features [33]. Endothelial cells in general are highly glycolytic, even in the presence of oxygen, reminiscent of the Warburg effect [34]. There are also roles for glutamine and lipids in endothelial metabolism [33]. Tumor endothelial cells are abnormal and have even higher rates of glycolysis than non-tumor endothelial cells, and blocking glycolysis can lead to normalization of the tumor vasculature and improved delivery of chemotherapeutic agents [35]. In addition to improving drug delivery, normalization of tumor blood vessels can alleviate hypoxia, which reverse tumor metabolic changes and can alleviate selective pressure for tumor evolution and metastasis [36]. In addition to hypoxia, lactate produced from tumors can signal to endothelial cells to promote angiogenesis [37], thus meeting the metabolic demands of the tumor.
The dynamism of the immune system further complicates the TME through the contributions of macrophages, myeloid derived cells, and lymphocytes, which have metabolic profiles and needs that change in different states of activation [38, 39]. T cells provide an instructive example. Quiescent T cells oxidize glucose and fatty acids to produce ATP, but then switch to aerobic glycolysis upon activation. Memory T cells return to a resting state-like metabolic program, but have increased mitochondrial mass and spare respiratory capacity, which provides a proliferative advantage upon re-stimulation with antigen [40]. In a tumor, the metabolic dynamics of T cell activation and persistence are then overlaid with the complexity of tumor cell metabolism and the TME. While these immune metabolism/tumor metabolism interactions pose challenges, they also provide an opportunity to identify cancer-cell dependent pathways that produce immunosuppressive metabolites such as lactate, adenosine, and kynurenine [41].
The challenge in metabolic anti-tumor therapy is therefore not only to identify and collect metabolic inhibitors with appropriate drug properties, but to apply them with an understanding of their effect on the tumor, the TME, and the interaction between them in order to achieve both therapeutic precision and accuracy.
Cancer metabolic inhibitors and drugs
It is notable that the first rationally designed anti-cancer drug, developed by Sidney Farber and colleagues in the 1940's, targeted 1C metabolism by blocking the folate pathway [42]. This success in targeting cancer metabolism triggered studies using 2-deoxyglucose in human studies in the 1950's, which were met with side effects [43, 44]. Interest in targeting cancer metabolism waned and was replaced by a focus on kinases. However, the resurgence of interest in cancer metabolism has revived metabolic inhibitors and led to their use in preclinical studies, and in some cases, clinical trials. For example, metformin, which inhibits mitochondrial Complex I and is used clinically to treat diabetes, and hydroxychloroquine, which inhibits autophagy and is used clinically to treat malaria as well as rheumatic diseases, have been exhumed and placed into cancer clinical trials (clinicaltrials.gov). Increased interest in cancer metabolism has also driven the identification of novel chemical entities that target metabolic pathways for cancer therapy.
Because many, but not all, human cancers display avid uptake of radio-labeled 2-deoxyglucose as determined by positron emission tomography, it stands to reason that targeting glucose metabolism could be of therapeutic importance. Compounds targeting glucose metabolism include inhibitors of glucose import, of glucose phosphorylation by hexokinase 2 (HK2), of conversion of pyruvate into lactate, and of export of lactate out of the cell [45-48]. Pathways involving NAD+ synthesis, glutamine metabolism, fatty acid synthesis and mitochondrial function have been targeted by small molecules [24, 48-52]. Table 1 summarizes metabolic enzymes and inhibitors that have been studied in in vitro or in pre-clinical or clinic settings.
Table 1. Cancer metabolic inhibitors tested in selected studies.
| Protein | Compound | Study | Finding | Ref |
|---|---|---|---|---|
| GLUT1 | STF-31 | pre-clinical | Xenograft activity | [46] |
| Xct (SLC7A11) | Sulfasalazine | pre-clinical | [89] | |
| MCT1 (SLC16A1) | AZD3965 | clinical | ongoing | [53] ClinicalTrials.gov Identifier: NCT01791595 |
| HK2 | Substituted glucosamine | In vitro | [90] | |
| GAPDH | 3-bromo pyruvate | pre-clinical in vivo | [64] | |
| PKM2 | Activator: TEPP-46 | pre-clinical in vivo | Xenograft activity | [57] |
| LDHA | GNE-140 | In vitro | nM, poor PK | [91] |
| LDHA | 2-((3-cyanopyridin-2-yl)thio)acetamides | In vitro | nM, poor PK | [92] |
| LDHA | Quinoline-3-sulfonamides | In vitro | [93] | |
| LDHA | Pyrazole-based | In vitro | nM, cell active | [94] |
| LDHA | Fragment-based | nM in vitro | [95] | |
| LDHA | FX-11 | pre-clinical in vivo | off-target effects | [47] |
| NAMPT | APO-866 | clinical | Phase I: thrombo-cytopenia | [96] |
| GLS | BPTES | Pre-clinical | Survival GEMM model liver cancer, kidney | [58, 97] |
| GLS | CB-839 | clinical | Phase I/II Solid tumor Plus nivolumab | [98] ClinicalTrials.gov Identifier: NCT02771626 |
| FASN | TVB-2640 | clinical | PD study resectable colon cancer | ClinicalTrials.gov Identifier: NCT02980029 |
| ACC | ND-646 | Pre-clinical | GEMM model lung cancer | [99] |
| Mitochondrial Complex I | phenformin | clinical | Phase I combination in melanoma | ClinicalTrials.gov Identifier: NCT03026517 |
| Mitochondrial Complex 1 | metformin | clinical | Interventional with standard therapy; metastatic breast cancer | ClinicalTrials.gov Identifier: NCT01310231 |
| PDH, OGDH | CPI-613 | clinical | Phase I Metastatic pancreas cancer; CRs | [100] |
| IDH1 | AG-120 | clinical | Interventional plus azacytidine; AML | ClinicalTrials.gov Identifier: NCT02677922 |
| IDH2 | Enasidenib (IDHIFA) | clinical | FDA approved 2017; AML | [101] |
Notes: GLUT1, glucose transporter; Xct amino acid antiporter; MCT1, monocarboxylate transporter 1; HK2, hexokinase 2; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PKM2, pyruvate kinase M2; LDH, lactate dehydrogenase; NAMPT, nicotinamide phosphoribosyltransferase; GLS, glutaminase; FASN, fatty acid synthase; ACC, acetyl-CoA carboxylase; PDH, pyruvate dehydrogenase; OGDH, oxoglutarate dehydrogenase; IDH, isocitrate dehydrogenase; nM, nanomolar IC50; PK, pharmacokinetics; GEMM, genetically engineered mouse model; CRs, complete remissions; AML, acute myelogenous leukemia; FDA, Food and Drug Administration.
A number of lessons have been learned from studies summarized in Table 1 as well as other related studies. First is that there is a heterogeneity of responses to specific inhibitors depending on the genetics and tissue origin of the model tested, as well as combination therapies used. For example, an inhibitor of the lactate transporter MCT1 appears to have variable responses in different models, with MCT1 high/MCT4 low tumor responding better [53, 54]. The concomitant use of complex I inhibitors (metformin or phenformin), rewire cells toward a further dependency on glycolysis and sensitize to the effects of MCT1 inhibition [55, 56]. Activators of PKM2 also seems variable depending on whether specific tumors rely heavily on PKM2 for survival, and the role of PKM2 in tumorigenesis appears much more complicated than originally surmised [57]. Glutaminase inhibition in preclinical models appears to depend on specific models and on potential synergies with other inhibitors. For example, while glutaminase (Gls) genetic loss or pharmacological inhibition in a MYC-inducible mouse liver cancer model seems effective in delaying tumorigenesis or prolonging survival, inhibition of glutaminase does not seem to affect lung or pancreas cancer in GEMM models [58-60]. However, under oxidative stress, GLS inhibition appears more effective in pancreas or a subtype of lung cancer that depends on the anti-oxidant activity of activated Nrf2 [61]. The study by Yuneva et al. was prescient in pointing out that both the genetic basis and tissue origin of a cancer could determine its metabolic re-wiring in vivo [62]. Specifically, the Yuneva study documented that in mouse liver cancer, MET oncogene-driven tumors were glycolytic while MYC oncogene-driven tumors were glutaminolytic [62]. This observation suggested that MET oncogene-driven liver cancers would be more sensitive to glycolytic but not glutaminolytic inhibitors. Moreover, they went on to show that a MYC oncogene driven lung cancer model, contrary to the MYC-driven liver cancer, was able to use both glucose and glutamine, but these tumors could also generate glutamine through glutamine synthetase, which is induced in lung tissue.
The second lesson learned from Table I and related studies is that in vitro activity does not portend in vivo activity due to off-target effects, suboptimal pharmacokinetic properties of the compounds in question, or profound differences between in vitro versus in vivo tumor metabolism. For example, many chemical entities have been generated against LDHA and some are quite effective in in vitro assays but are ineffective in vivo due to poor pharmacodynamic properties. Other tool compounds against LDHA, such as FX11, are effective in vivo, but have off-target effects at high concentrations and hence their anti-tumorigenic activity may not be due entirely to LDHA inhibition [47, 63]. In addition to inhibiting GAPDH, 3-bromopyruvate is a highly active alkylating agent that is likely to have pleiotropic effects [64]. Exploitation of increased transporter (MCT1) expression in tumor cells, however, could increase 3-bromopyruvate uptake and enhance target cellular accuracy [65]. Cancer dependency on specific metabolic pathways, and hence sensitivity to pathway inhibition, can be very context-dependent. Since even alteration in composition of culture media can have significant effects on cellular metabolite levels [66], it is perhaps not surprising that there can be profound differences between in vitro and in vivo metabolic dependencies. In non-small cell lung cancer, for example, cultured cells have high glutaminolytic flux and are sensitive to glutaminase inhibitors. However, in vivo tumors utilize less glutamine and are insensitive to glutaminase inhibition [59]. The other consideration, given lessons learned from LDHA, is the importance of cellular concentrations of target enzymes. In this regard, it is intriguing to note from global cellular proteomics studies of NIH3T3 fibroblasts and human Hela cells that the concentrations of glycolytic enzymes vary dramatically by orders of magnitude from HK2 to LDHA [67, 68]. Copies of glycolytic enzymes involved in the catabolism of hexoses tend to be low (∼50,000-200,000 copies/cell) relative to the much higher (50 to over 100 million copies/cell) protein copy numbers per cell of glycolytic enzymes involved in the metabolism of trioses, such as LDHA, estimated to have a concentration of up to 16 μM. Hence, it may be more fruitful to focus on enzymes with low copy number such as HK2, particularly since HK2 is induced in proliferating cells, while HK1 tends to be involved in maintenance metabolism of non-proliferative cells.
The third lesson learned is that metabolic synthetic essentiality can be exploited to enhance tumor selectivity. As alluded to above, oxidative stress can synergize with glutaminase inhibition in a lung cancer model that depends on Nrf2 [61] and inhibitors of oxidative phosphorylation can increase dependence on glycolysis and sensitivity to MCT1 inhibition [45]. mTOR inhibition can also synergize with glutaminase inhibition [69, 70]. The re-wiring of metabolism by targeted kinase inhibition can lead to new vulnerabilities, such as sensitivity to phenformin after BRAF inhibition in melanoma [71]. Loss of isoforms of metabolic genes adjacent to tumor suppressor genes can drive sensitivity to inhibition or loss of the remaining isoforms, a concept dubbed “collateral lethality” [72, 73]. The combination of glycolytic, glutaminolytic and oxidative metabolic inhibitors could hypothetically uncover new vulnerabilities that are likely tumor type specific. The effects of these combinations on non-cancer cells in the TME should be consider for full understanding of their effects in vivo.
The fourth lesson, one still at early stages, is the potential for metabolic inhibitors to affect elements of the TME and therefore antagonize or synergize therapeutic response. Metabolic inhibitors can alter the tumor vasculature, either by changing tumor endothelial cell metabolism directly or by altering metabolic signals in the TME sensed by endothelial cells, thus altering angiogenesis, metabolite and oxygen supply, and therapeutic access [33, 35]. The success of checkpoint inhibition and chimeric antigen receptor T cell therapy in specific clinical settings has put particular focus on metabolic interactions with the anti-tumor function of cytolytic T cells, tumor suppressive macrophages, or myeloid and B suppressor cells [38, 41, 74, 75]. As described above, T cells have specific metabolic requirements at different stages of activation and persistence. The use of metabolic therapies may alter this process when used in combination with immunotherapies. Intriguingly and further demonstrating the complexity of these interactions, one study suggests that phenformin can inhibit myeloid-derived suppressor cells and increase the anti-tumor effect of PD-1 blockade in melanoma [74]. How glycolytic and glutaminase inhibitors alter T cell function or fates will be important areas to explore, particularly given the observation that mTOR inhibition could skew T cell differentiation away from the tumor permissive Treg lymphocytes toward more cytolytic T cells. [76, 77].
Concluding Remarks
While significant advances have been achieved in understanding cancer metabolism and developing metabolic therapies, several issues remain to be addressed (see Outstanding Questions Box). We anticipate that a richer understanding of tumor, organismal, and TME metabolism will allow for application of such therapies with both precision and accuracy.
Outstanding Questions.
How can metabolic enzymes be targeted precisely with potent inhibitors without causing an immune-suppressive microenvironment?
Could some metabolic drugs promote a tumor-permissive microenvironment and therefore lose efficacy, despite diminishing tumor cell intrinsic growth?
What are the synthetic essentialities with specific metabolic drugs that could be exploited to improve safety and efficacy?
Could new technologies be developed to assess or visualize the effects of metabolic drugs on components of the tumor microenvironment in vivo?
With regard to tumor intrinsic metabolism, our knowledge of synthetic essentialities between metabolic pathways as well as with other pathways is still rudimentary. It is notable that passenger genomic deletions, such as loss of ENO1 or ME2 found in specific cancers could uncover new druggable targets with the remaining isoenzymes, ENO2 and ME3, respectively [73, 78]. Additional studies to uncover synthetic lethal vulnerabilities will help tremendously in streamlining experimental combination metabolic therapies. It is also expected that blockage of key metabolic pathways would trigger other survival mechanisms such as autophagy and macropinocytosis. As such, better definition of the interplay of these cellular pathways will be essential. Lastly, better definition is needed of links between the genomic alterations of human cancers and their metabolic phenotypic manifestations (by metabolic nuclear medicine imaging approaches).
Improvements in the use of genomic data to define the TME composition [79] are needed to take advantage of this expanding resource. In addition, the use of advanced technologies to directly define the tumor immune microenvironment by flow cytometry will greatly enhance to strategic use of metabolic drugs. Beyond T cells, better understanding of how metabolic inhibitors influence macrophages, myeloid suppressor, and B suppressor cells will aid in determining responses versus resistance due to cell extrinsic mechanisms in immune-competent hosts.
The proliferation of committed normal tissue stem cells in less than 1% of adult mammalian cells relies on biosynthetic pathways similar to those used by cancer cells [80]. However, the normal stem cell cycle oscillator tends to couple with the circadian clock to orchestrate tissue repair and regeneration in synchrony with the solar cycle of fasting and feeding coupled with sleep and wake periods that are linked to cellular metabolism [81, 82]. These normal pathways are under tight control of feed-back loops that return cells to a resting state when nutrients and growth signals are insufficient. Emerging evidence has shown oncogenic disruption of the clock in cancer cells [83-85]. This presents the possibility that metabolic toxicity to normal tissues could be spared and efficacy against cancer cells increased by taking the circadian clock into consideration. For example, a large clinical trial showed that the effectiveness of 5-FU in colorectal cancer depends both on the time of treatment and gender of the patient [86, 87]. Further studies are needed to determine the role of the circadian clock in controlling the metabolism of cells in the TME. Such refinements based on time of administration could allow for the more accurate use of targeted therapies, improving therapeutic outcomes [88].
Trends Box.
Oncogenes rewire cellular metabolism to meet the energetic and substrate demands of the tumor, but this rewiring also create new opportunities for therapy
Significant advances have been made in understanding cancer cell intrinsic metabolism, the metabolism of non-tumor cells in the tumor microenvironment, and the metabolic interactions between them.
Rising interest in the metabolic vulnerabilities of cancer has led to the development of novel therapies targeting diverse aspects of nutrient transport and utilization
Application of metabolic therapies has been hindered by context-dependentvariations in substrate utilization and by effects on normal cells both within and outsideof the tumor microenvironment.
Acknowledgments
Our original work is supported by NCI grants CA051497 and CA057341, and the Ludwig Institute for Cancer Research. AJW is supported by the Children's Hospital of Philadelphia Department of Pediatrics training grant, 5T32GM007367.
Footnotes
Conflicts of Interest. CVD is a consultant for Rafael Pharmaceutical, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Warburg O, et al. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8(6):519–30. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pasteur L. The Physiological Theory of Fermentation. Kessinger Publishing; LLC: 1879. [Google Scholar]
- 3.Koppenol WH, et al. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–37. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 4.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134(5):703–7. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 5.Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2(10):881–98. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26(9):877–90. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altman BJ, et al. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–34. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buescher JM, et al. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol. 2015;34:189–201. doi: 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayers JR, et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science. 2016;353(6304):1161–5. doi: 10.1126/science.aaf5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Camarda R, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22(4):427–32. doi: 10.1038/nm.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng T, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108(21):8674–9. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sellers K, et al. Pyruvate carboxylase is critical for non-small-cell lungcancer proliferation. J Clin Invest. 2015;125(2):687–98. doi: 10.1172/JCI72873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullen AR, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7(5):1679–1690. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang, et al. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res. 2016;26(10):1112–1130. doi: 10.1038/cr.2016.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Commisso C, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633–7. doi: 10.1038/nature12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Overholtzer M, et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131(5):966–79. doi: 10.1016/j.cell.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 18.Gorrini C, et al. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12(12):931–47. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 19.Pandey P, et al. The see-saw of Keap1-Nrf2 pathway in cancer. Crit Rev Oncol Hematol. 2017;116:89–98. doi: 10.1016/j.critrevonc.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1signaling pathway in cancer. Genes Dev. 2013;27(20):2179–91. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viswanathan VS, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mucaj V, et al. Effects of hypoxia and HIFs on cancer metabolism. Int J Hematol. 2012;95(5):464–70. doi: 10.1007/s12185-012-1070-5. [DOI] [PubMed] [Google Scholar]
- 23.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Outschoorn UE, et al. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14(1):11–31. doi: 10.1038/nrclinonc.2016.60. [DOI] [PubMed] [Google Scholar]
- 25.Danhier P, et al. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim Biophys Acta. 2017;1858(8):556–572. doi: 10.1016/j.bbabio.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 26.Hensley CT, et al. Metabolic Heterogeneity in Human Lung Tumors. Cell. 2016;164(4):681–94. doi: 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maley CC, et al. Classifying the evolutionary and ecological features of neoplasms. Nat Rev Cancer. 2017;17(10):605–619. doi: 10.1038/nrc.2017.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strickaert A, et al. Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene. 2017;36(19):2637–2642. doi: 10.1038/onc.2016.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sousa CM, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536(7617):479–83. doi: 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao H, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. 2016;5:e10250. doi: 10.7554/eLife.10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez-Outschoorn UE, et al. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. 2014;25:47–60. doi: 10.1016/j.semcancer.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Nieman KM, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17(11):1498–503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong BW, et al. Endothelial cell metabolism in health and disease: impact of hypoxia. EMBO J. 2017;36(15):2187–2203. doi: 10.15252/embj.201696150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Bock K, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154(3):651–63. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 35.Cantelmo AR, et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell. 2016;30(6):968–985. doi: 10.1016/j.ccell.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–22. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee DC, et al. A lactate-induced response to hypoxia. Cell. 2015;161(3):595–609. doi: 10.1016/j.cell.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 38.Murray PJ, et al. SnapShot: Immunometabolism. Cell Metab. 2015;22(1):190–190 e1. doi: 10.1016/j.cmet.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 39.O'Neill LA, et al. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–65. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pearce EL, et al. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buck MD, et al. Metabolic Instruction of Immunity. Cell. 2017;169(4):570–586. doi: 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Farber S. Some observations on the effect of folic acid antagonists on acute leukemia and other forms of incurable cancer. Blood. 1949;4(2):160–7. [PubMed] [Google Scholar]
- 43.Landau BR, Lubs HA. Animal responses to 2-deoxy-D-glucoseadministration. Proc Soc Exp Biol Med. 1958;99(1):124–7. doi: 10.3181/00379727-99-24268. [DOI] [PubMed] [Google Scholar]
- 44.Landau BR, et al. Certain metabolic and pharmacologic effects in cancer patients given infusions of 2-deoxy-D-glucose. J Natl Cancer Inst. 1958;21(3):485–94. [PubMed] [Google Scholar]
- 45.Doherty JR, et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014;74(3):908–20. doi: 10.1158/0008-5472.CAN-13-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan DA, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3(94):94ra70. doi: 10.1126/scitranslmed.3002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Le A, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037–42. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491(7424):364–73. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 49.Tennant DA, et al. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10(4):267–77. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 50.Galluzzi L, et al. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12(11):829–46. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- 51.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10(9):671–84. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 52.Jones NP, Schulze A. Targeting cancer metabolism--aiming at a tumour's sweet-spot. Drug Discov Today. 2012;17(5-6):232–41. doi: 10.1016/j.drudis.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 53.Polanski R, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res. 2014;20(4):926–937. doi: 10.1158/1078-0432.CCR-13-2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Noble RA, et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica. 2017;102(7):1247–1257. doi: 10.3324/haematol.2016.163030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013;123(9):3685–92. doi: 10.1172/JCI69741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marchiq I, et al. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015;75(1):171–80. doi: 10.1158/0008-5472.CAN-14-2260. [DOI] [PubMed] [Google Scholar]
- 57.Anastasiou D, et al. Pyruvate kinase M2 activators promote tetramerformation and suppress tumorigenesis. Nat Chem Biol. 2012;8(10):839–47. doi: 10.1038/nchembio.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiang Y, et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest. 2015;125(6):2293–306. doi: 10.1172/JCI75836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davidson SM, et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016;23(3):517–28. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Son J, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–5. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Romero R, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017 doi: 10.1038/nm.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuneva MO, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15(2):157–70. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen CY, et al. Identification of a potent inhibitor targeting human lactate dehydrogenase A and its metabolic modulation for cancer cell line. Bioorg Med Chem Lett. 2016;26(1):72–5. doi: 10.1016/j.bmcl.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 64.Ganapathy-Kanniappan S, et al. Anticancer efficacy of the metabolic blocker 3-bromopyruvate: specific molecular targeting. Anticancer Res. 2013;33(1):13–20. [PubMed] [Google Scholar]
- 65.Birsoy K, et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat Genet. 2013;45(1):104–8. doi: 10.1038/ng.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cantor JR, et al. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell. 2017;169(2):258–272 e17. doi: 10.1016/j.cell.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwanhausser B, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–42. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 68.Beck M, et al. The quantitative proteome of a human cell line. Mol Syst Biol. 2011;7:549. doi: 10.1038/msb.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tanaka K, et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest. 2015;125(4):1591–602. doi: 10.1172/JCI78239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lampa M, et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS One. 2017;12(9):e0185092. doi: 10.1371/journal.pone.0185092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan P, et al. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc Natl Acad Sci U S A. 2013;110(45):18226–31. doi: 10.1073/pnas.1317577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Muller FL, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488(7411):337–42. doi: 10.1038/nature11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dey P, et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature. 2017;542(7639):119–123. doi: 10.1038/nature21052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim SH, et al. Phenformin Inhibits Myeloid-Derived Suppressor Cells and Enhances the Anti-Tumor Activity of PD-1 Blockade in Melanoma. J Invest Dermatol. 2017;137(8):1740–1748. doi: 10.1016/j.jid.2017.03.033. [DOI] [PubMed] [Google Scholar]
- 75.O'Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15–23. doi: 10.1084/jem.20151570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.He WL, et al. CD155T/TIGIT Signaling Regulates CD8+ T Cell Metabolism and Promotes Tumor Progression in Human Gastric Cancer. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-17-0381. [DOI] [PubMed] [Google Scholar]
- 77.Kishton RJ, et al. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017;26(1):94–109. doi: 10.1016/j.cmet.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leonard PG, et al. SF2312 is a natural phosphonate inhibitor of enolase. Nat Chem Biol. 2016;12(12):1053–1058. doi: 10.1038/nchembio.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gentles AJ, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weger M, et al. Stem cells and the circadian clock. Dev Biol. 2017 doi: 10.1016/j.ydbio.2017.09.012. [DOI] [PubMed] [Google Scholar]
- 81.Bass J, Lazar MA. Circadian time signatures of fitness and disease. Science. 2016;354(6315):994–999. doi: 10.1126/science.aah4965. [DOI] [PubMed] [Google Scholar]
- 82.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330(6009):1349–54. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Altman BJ, et al. Correspondence: Oncogenic MYC persistently upregulates the molecular clock component REV-ERBalpha. Nat Commun. 2017;8:14862. doi: 10.1038/ncomms14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Altman BJ, et al. MYC Disrupts the Circadian Clock and Metabolism in Cancer Cells. Cell Metab. 2015;22(6):1009–19. doi: 10.1016/j.cmet.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Papagiannakopoulos T, et al. Circadian Rhythm Disruption Promotes Lung Tumorigenesis. Cell Metab. 2016;24(2):324–31. doi: 10.1016/j.cmet.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Levi F, et al. Randomised multicentre trial of chronotherapy with oxaliplatin, fluorouracil, and folinic acid in metastatic colorectal cancer. International Organization for Cancer Chronotherapy. Lancet. 1997;350(9079):681–6. doi: 10.1016/s0140-6736(97)03358-8. [DOI] [PubMed] [Google Scholar]
- 87.Innominato PF, et al. The circadian timing system in clinical oncology. Ann Med. 2014;46(4):191–207. doi: 10.3109/07853890.2014.916990. [DOI] [PubMed] [Google Scholar]
- 88.Ballesta A, et al. Systems Chronotherapeutics. Pharmacol Rev. 2017;69(2):161–199. doi: 10.1124/pr.116.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Timmerman LA, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24(4):450–65. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin H, et al. Discovery of a Novel 2,6-Disubstituted Glucosamine Series of Potent and Selective Hexokinase 2 Inhibitors. ACS Med Chem Lett. 2016;7(3):217–22. doi: 10.1021/acsmedchemlett.5b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boudreau A, et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat Chem Biol. 2016;12(10):779–86. doi: 10.1038/nchembio.2143. [DOI] [PubMed] [Google Scholar]
- 92.Cui W, et al. Discovery of 2-((3-cyanopyridin-2-yl)thio)acetamides as human lactate dehydrogenase A inhibitors to reduce the growth of MG-63 osteosarcoma cells: Virtual screening and biological validation. Bioorg Med Chem Lett. 2016;26(16):3984–7. doi: 10.1016/j.bmcl.2016.06.083. [DOI] [PubMed] [Google Scholar]
- 93.Billiard J, et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013;1(1):19. doi: 10.1186/2049-3002-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rai G, et al. Discovery and Optimization of Potent, Cell-Active Pyrazole-Based Inhibitors of Lactate Dehydrogenase (LDH) J Med Chem. 2017 doi: 10.1021/acs.jmedchem.7b00941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ward RA, et al. Design and synthesis of novel lactate dehydrogenase A inhibitors by fragment-based lead generation. J Med Chem. 2012;55(7):3285–306. doi: 10.1021/jm201734r. [DOI] [PubMed] [Google Scholar]
- 96.Sampath D, et al. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol Ther. 2015;151:16–31. doi: 10.1016/j.pharmthera.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 97.Shroff EH, et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A. 2015;112(21):6539–44. doi: 10.1073/pnas.1507228112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gross MI, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870. [DOI] [PubMed] [Google Scholar]
- 99.Svensson RU, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22(10):1108–1119. doi: 10.1038/nm.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Alistar A, et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: a single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017;18(6):770–778. doi: 10.1016/S1470-2045(17)30314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bose P, et al. Treatment of Relapsed/Refractory Acute Myeloid Leukemia. Curr Treat Options Oncol. 2017;18(3):17. doi: 10.1007/s11864-017-0456-2. [DOI] [PubMed] [Google Scholar]