

Abstract
The tricarboxylic acid (TCA) cycle is a central route for oxidative phosphorylation in cells, and fulfills their bioenergetic, biosynthetic, and redox balance requirements. Despite early dogma that cancer cells bypass the TCA cycle and primarily utilize aerobic glycolysis, emerging evidence demonstrates that certain cancer cells, especially those with deregulated oncogene and tumor suppressor expression, rely heavily on the TCA cycle for energy production and macromolecule synthesis. As the field progresses, the importance of aberrant TCA cycle function in tumorigenesis and the potentials of applying small molecule inhibitors to perturb the enhanced cycle function for cancer treatment start to evolve. In this review, we summarize current knowledge about the fuels feeding the cycle, effects of oncogenes and tumor suppressors on fuel and cycle usage, common genetic alterations and deregulation of cycle enzymes, and potential therapeutic opportunities for targeting the TCA cycle in cancer cells. With the application of advanced technology and in vivo model organism studies, it is our hope that studies of this previously overlooked biochemical hub will provide fresh insights into cancer metabolism and tumorigenesis, subsequently revealing vulnerabilities for therapeutic interventions in various cancer types.
KEYWORDS: glutaminolysis, the TCA cycle, cancer metabolism, glycolysis
Introduction
Cancer is a disease characterized by the accumulation of genetic alterations and gene deregulations, resulting in uncontrolled cell proliferation that demands both increased energy production and macromolecule synthesis. To cope with increased metabolic stress, malignant cells often reprogram their biochemical pathways to enable rapid uptake and breakdown of nutrients, thus contributing to disease transformation, maintenance, and progression (Hanahan and Weinberg, 2011; Ward and Thompson, 2012). The birth of cancer metabolism research extends back to the early 20th century, when Otto Warburg noted the heavy dependence of cancer cells on glycolysis for growth (Warburg et al., 1927). Indeed, various types of cancer cells increase their glucose uptake and preferentially utilize glucose through aerobic glycolysis (Gillies and Gatenby, 2007; Pavlova and Thompson, 2016). This effect was subsequently applied in the clinic for tumor imaging and detection through positron emission tomography scans of radiolabeled glucose analogs (Papathanassiou et al., 2009). These early findings laid the groundwork for a recent revival of interest in cancer metabolism research, which has lead to discoveries showing overactivation and/or rewiring of multiple metabolic pathways in cancer cells. In just the last ten years, the significance of metabolic reprogramming has led to its inclusion with the classic hallmarks of cancer (Hanahan and Weinberg, 2011). Accumulating evidence indicates that exploiting the unique metabolic dependencies of tumor cells represents an exciting new direction of targeted therapy (Pathania et al., 2009; Kishton and Rathmell, 2015).
The tricarboxylic acid (TCA) cycle is a central hub for energy metabolism and macromolecule synthesis and redox balance. The cycle is composed of a series of biochemical reactions occurring in the mitochondrial matrix, which allow aerobic organisms to oxidize fuel sources and provide energy, macromolecules, and redox balance to the cell. Aberrant TCA cycle function is implicated in a wide variety of pathological processes. Genetic diseases with compromised TCA cycle function due to inherited cycle enzyme mutations, such as fumarase (FH) deficiency, are rare but severe (Rustin et al., 1997). Moreover, several TCA cycle enzymes are deregulated in obesity, including citrate synthase, which exhibits reduced activity in obese mice (Cummins et al., 2014). Multiple neurodegenerative disorders such as Alzheimer’s disease are associated with reduced activity of the α-ketoglutarate dehydrogenase complex (KGDHC) (Gibson et al., 2010). In light of the widely accepted belief that cancer cells primarily utilize aerobic glycolysis, the role of the TCA cycle in cancer metabolism and tumorigenesis has been overlooked until recently.
With the application of contemporary technology, such as unbiased and targeted metabolomics, as well as genetic and biochemical studies using animal models, many recent advances have been made in the field of cancer metabolism. Studies have demonstrated that tumor cells can indeed uncouple glycolysis from the TCA cycle, allowing the use of additional fuel sources such as glutamine to meet their heightened metabolic needs (Chen and Russo, 2012) (Pavlova and Thompson, 2016). Importantly, glutamine is now established as an important nutrient source across numerous cancer types, especially for MYC-driven cancers (DeBerardinis and Cheng, 2010). The role of lipid metabolism in tumorigenesis has also received increased attention in recent years. Altogether, these studies have provided convincing evidence to establish the role of the TCA cycle in cancer metabolism and tumorigenesis (Sajnani et al., 2017). Importantly, various oncogenes and tumor suppressors regulate both the uptake and breakdown of fuel sources in the TCA cycle by regulating the expression of fuel transporters and/or activity of cycle enzymes in cancer cells (Chen and Russo, 2012). Multiple cycle enzymes, including aconitase (also known as aconitate hydratase, AH), isocitrate dehydrogenase (IDH), FH, succinate dehydrogenase (SDH) and KGDHC, are frequently mutated or deregulated in human cancers (Eng et al., 2003; Juang, 2004; Yan et al., 2009). Recent results from clinical testing suggest that targeting reprogrammed metabolic pathways, including the TCA cycle, could provide a new and promising therapeutic avenue for the treatment of a broad spectrum of cancers.
Fuels feeding the TCA cycle
The TCA cycle serves as a convergence point in the cellular respiration machinery, which integrates multiple fuel sources derived from the diet including glucose, glutamine, and fatty acids. Through various biochemical reactions, the cycle produces intermediates for use as building blocks in macromolecule synthesis, as well as energy and electron acceptors that are utilized in downstream cellular processes such as the electron transport chain (ETC) reactions. Although both normal and tumor cells can catabolize all major types of fuels, they differ in the rate of uptake and catabolism of each fuel. While glucose provides the main source of pyruvate entering the TCA cycle in normal cells, cancer cells often shunt glucose away from the TCA cycle for catabolism through anaerobic glycolysis, and thus are more dependent on glutamine and fatty acids to replenish TCA cycle intermediates (Eagle, 1955).
Glucose
Glucose is imported into the cell by glucose transporters (GLUT) and serves as the most common fuel source in mammalian cells (Fig. 1). In normal cells, most cellular glucose enters the TCA cycle in the form of pyruvate, although glucose can also be utilized for lactate production or macromolecular synthesis through the pentose phosphate pathway (Fig. 1). Through glycolysis, one glucose molecule is converted into two pyruvate molecules, which are primarily oxidized to produce acetyl-CoA feeding the TCA cycle. Alternatively, under hypoxic conditions, pyruvate may be converted to lactate as well. Progression through the TCA cycle occurs when heightened energetic needs arise (Fig. 1). Glucose can also be synthesized through gluconeogenesis, a process reciprocally regulated compared to glycolysis in order to keep the metabolism of the cell efficient (Berg JM, 2002).
Figure 1.
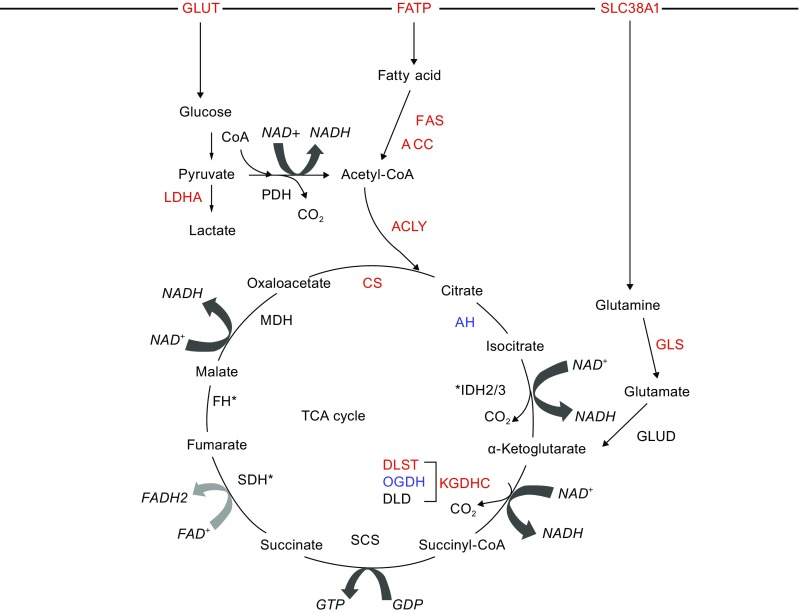
Transporters, fuels, enzymes, and biochemical reactions driving the TCA cycle. The typical input for the TCA cycle is acetyl-CoA, which is derived from pyruvate, the end product of glycolysis. Through a series of redox reactions, chemical bond energy from acetyl-CoA is harvested to produce high-energy electrons, which are carried to the electron transport chain by nicotinamide adenine dinucleotide (NAD+) and flavin adenine dinucleotide (FAD). Subsequent oxidative phosphorylation results in the production of adenosine triphosphate (ATP) from each acetyl-CoA. Because oxygen is required to regenerate NAD+ and FAD, the TCA cycle only proceeds in aerobic environments. There are a total of 8 steps in the TCA cycle, three of which are irreversible; the generation of citrate from oxaloacetate and acetyl-CoA by CS; the conversion of isocitrate to α-KG by IDH3; and the formation of succinyl-CoA from α-KG by KGDHC (Berg JM, 2002; Akram, 2014). The biochemical reactions in the TCA cycle are regulated by several means including substrate availability, product inhibition, and allosteric regulation, allowing the cell to control energy production based on its energy status (NADH/NAD+ ratio, ATP availability) and nutrient availability (Berg JM, 2002). Intermediates in the cycle can be derived from outside sources, such as the production of acetyl-CoA from β-oxidation of fatty acids or the production of α-KG from protein catabolism, particularly glutaminolysis (Houten and Wanders, 2010; Akram, 2014). Importantly, deregulation of TCA cycle enzymes, such as mutations and gene deregulations, or aberrant accumulation of TCA intermediates can have disease-relevant consequences. Proteins that are upregulated in cancer are highlighted as red and downregulated as blue, while enzymes mutated are marked with an asterisk. Abbreviations: CS: citrate synthase, AH: aconitase, IDH: isocitrate dehydrogenase, KGDHC: α-ketoglutarate dehydrogenase complex, OGDH: α-KG dehydrogenase, DLST: dihydrolipoamide S-succinyltransferase, DLD: dihydrolipoamide dehydrogenase, SCS: succinyl-CoA synthase, SDH: succinate dehydrogenase, FH: fumarate hydratase, MDH: malate dehydrogenase, PDH: pyruvate dehydrogenase, GLUT: glucose transporter, FATP: fatty acid transporter, SCL38A: sodium-coupled neutral amino acid transporter, ACLY: adenosine triphosphate citrate lyase, ACC: acetyl-CoA carboxylase, FAS: fatty acid synthase, GLS: glutaminase, GDH: glutamate dehydrogenase
Cancer cells markedly increase their glucose usage, as noted by Otto Warburg nearly 100 years ago. Tumors acquire additional glucose by upregulating the high-affinity glucose transporters GLUT1 and GLUT3, while simultaneously downregulating lower affinity transporters (Birnbaum et al., 1987; Baron-Delage et al., 1996). Not only do cancer cells increase the rate of glucose uptake and utilization, but the fate of imported glucose differs from that in normal cells as well. While normal cells and some cancer cells, such as lung cancer stem cells and leukemic cells, oxidize glucose in the mitochondria (Gatenby and Gillies, 2004; Gao et al., 2016; Kishton et al., 2016), most cancer cells preferentially break down glucose to produce lactate even in normoxic conditions (Kim et al., 2006), The process of aerobic glycolysis only generates 2 ATP per glucose molecule, a drastic reduction from 38 ATP when glucose is oxidized through the TCA cycle. To meet their heightened energetic needs, cancer cells turns to other fuel sources, such as glutamine, to feed the TCA cycle.
Glutamine
In addition to glucose, amino acids can also fuel the TCA cycle. Amino acids enter the cycle after being converted to either acetyl-CoA or α-keto acid intermediates: pyruvate, oxaloacetate, and succinyl-CoA (Berg JM, 2002). Glutamine is the most abundant amino acid in the human body, serving to transport nitrogen in the plasma for biosynthesis of non-essential amino acids, such as purines and pyrimidines, as well as fatty acids, or entering the TCA cycle in the form of α-ketoglutarate (α-KG) (Reitzer et al., 1979; Brosnan, 2003; Wang et al., 2017). Glutaminolysis, the breakdown of glutamine, is critical in replenishing cycle intermediates in proliferating cells. Glutamine is first hydrolyzed by glutaminase (GLS) to yield glutamate, which subsequently is either dehydrogenated by glutamate dehydrogenase (GLUD) to form α-KG or functions as a co-substrate for the transaminases, glutamate oxaloacetate transaminase and glutamate pyruvate transaminase to form alanine and aspartate respectively. α-KG is a substrate for oxidative decarboxylation by KGDHC or for reductive carboxylation by IDH2 (Mullen et al., 2011). Thus, glutaminolysis serves as a common pathway for both anaplerotic and cataplerotic processes.
The importance of glutaminolysis in cancer cell proliferation was noted decades ago by Harry Eagle, who found that HeLa cells preferred a molar excess of 10- to 100- fold of glutamine for maximum growth (Eagle, 1955). This metabolic dependence is partially driven by the glycolytic phenotype seen in certain types of cancer cells. Due to the excessive conversion of glucose to lactate, tumor cells use anaplerotic reactions to replenish TCA cycle intermediates, which is largely achieved through increased glutaminolysis (DeBerardinis et al., 2007). To do so, cancer cells upregulate both glutamine transporters and enzymes catalyzing glutaminolysis, thus uncoupling this pathway from growth factor-mediated stimuli (Fig. 1) (Pavlova and Thompson, 2016). The proto-oncogene MYC is a critical regulator of glutaminolysis and upregulates both glutamine transporters and GLS (Wise et al., 2008; Gao et al., 2009). Elevated levels of GLS and glutamine transporters enable tumor cells to derive large portions of their energy and macromolecules through glutamine catabolism, leading to glutamine addiction in numerous cancer types including myeloma and glioma (Bolzoni et al., 2016; Márquez et al., 2017).
Fatty acids
The third type of fuel source in cancer cells is fatty acids, which enter the TCA cycle after undergoing β-oxidation to generate acetyl-CoA. Acetyl-CoA is the substrate for both the fatty acid synthesis pathway and the TCA cycle, making lipogenesis an important convergence point for TCA cycle flux and cellular biosynthesis (Migita et al., 2008). In the process of β-oxidation, the acyl chain undergoes oxidation, introducing a double bond, followed by hydration to alcohol and oxidation to ketone. Finally, co-enzyme A cleaves the acyl tail to yield an acetyl-CoA and reduces the fatty acid chain length by two carbons. This process generates more acetyl-CoA per molecule than does either glucose or glutamine (Berg JM, 2002). De novo synthesis of fatty acids is critical to supply lipids for cell membrane formation in rapidly proliferating cells, and is regulated by fatty acid biosynthetic enzymes: adenosine triphosphate citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FAS). ACLY converts citrate to oxaloacetate and cytosolic acetyl-CoA. This cytosolic acetyl-CoA is carboxylated by ACC to form malonyl-CoA, which is then combined with additional acetyl-CoA until the 16-carbon unsaturated fatty acid palmitate is formed. Palmitate can then be modified to form additional required components of cell membrane.
While enzymes regulating lipid synthesis are often expressed in low levels in most normal tissue (Clarke, 1993), they are overexpressed in multiple types of cancers. ACLY is overexpressed in non-small cell lung cancer, breast cancer, and cervical cancer among others (Migita et al., 2008; Xin et al., 2016; Wang et al., 2017). ACC is upregulated in non-small cell lung cancer and hepatocellular carcinoma (Wang et al., 2016; Svensson and Shaw, 2017). FAS is overexpressed in prostate and breast cancers (Swinnen et al., 2002; Menendez et al., 2004). In tumor cells where the demand is much greater, lipogenesis occurs via these overexpressed enzymes. The increased activation and overexpression of these enzymes in tumors correlates with disease progression, poor prognosis, and is being investigated as a potential biomarker of metastasis (Xin et al., 2016).
Oncogenes and tumor suppressors impinging on the TCA cycle
Genetic alterations and/or deregulations of tumor suppressors or oncogenes often drive metabolic reprograming in cancers, although this effect can differ based on specific alterations or deregulations, and is often context-dependent. Several oncogenes, including MYC, HIF, P53, and RAS, are known to regulate the metabolic phenotype of tumors and play a critical role in determining how the TCA cycle is utilized in these cancer cells.
MYC
The proto-oncogene MYC controls a wide range of cellular processes, including cell proliferation, metabolism, cellular differentiation and genomic instability, and is a dominant driver of tumor transformation and progression (Meyer and Penn, 2008). Aberrant MYC activity, resulting from chromosomal translocations, gene amplifications or increased mRNA/protein stability, is found in over half of all human cancers (Gabay et al., 2014). Importantly, MYC is a central regulator of cellular metabolism, and can promote a broad range of metabolic pathways, such as aerobic glycolysis, glutaminolysis, mitochondrial biogenesis, oxidative phosphorylation, and nucleotide and amino acid biosynthesis (Adhikary and Eilers, 2005; Gabay et al., 2014; Wahlstrom and Henriksson, 2015). As stated early in this review article, MYC transcriptionally activates key genes and enzymes regulating glutaminolysis, and serves as the principal driver of glutamine metabolism through the TCA cycle (i.e., glutamine anaplerosis). Specifically, to promote the import of glutamine into the cell, MYC transcriptionally upregulates glutamine transporters ASC amino acid transporter 2 (ASCT2) and system N transporter (SN2). Additionally, Gao et al. demonstrated that MYC controls the conversion of glutamine to glutamate by activating glutaminase 1 (GLS1) through transcriptional suppression of its negative regulator miR-23a/b (Wise et al., 2008; Gao et al., 2009). There are two independent pathways that control the conversion of glutamate to α-KG entering the TCA cycle: one controlled by GLUD and another by aminotransferases. MYC-dependent cancer cells can utilize either GLUD or aminotransferases to convert glutamine to α-KG for the TCA cycle (Wise et al., 2008; Wang et al., 2011). MYC may also play a role in directing fatty acid oxidation and directing its metabolites into the TCA cycle by way of acetyl-CoA. Specifically, MYC expression leads to the upregulation of fatty acid transporters (e.g., fatty acid-binding protein 4) and fatty acid oxidation genes such as hydroxyacyl-CoA dehydrogenase (Wang et al., 2011; Edmunds et al., 2015).
HIF
Hypoxia-inducible factors (HIFs) are transcription factors that respond to reduced oxygen availability. HIFs are heterodimers composed of an oxygen-dependent α-subunit and a constitutively expressed β-subunit. Under normoxia, the α-subunit is targeted for degradation upon hydroxylation by prolyl hydroxylases (PHD) and subsequent ubiquitination by von Hippel-Lindau (VHL) tumor suppressor. Tumors activate HIFα either in the face of hypoxia resulting from poor vascularization or due to genetic abrogation such as VHL loss (Gordan and Simon, 2007). HIF activation orchestrates a metabolic program that promotes the catabolism of glucose through aerobic glycolysis and thus shifts glucose away from the TCA cycle (Semenza, 2012). HIF promotes glycolysis and lactate production through transcriptional upregulation of glucose transporters (SLC2A1 and SLC2A3), glycolytic enzymes (e.g., hexokinase (HK) and pyruvate kinase (PK)), and lactate dehydrogenase A (LDHA) (Kim et al., 2006). Kim et al. demonstrated that HIF1 suppresses glucose metabolism through the TCA cycle (i.e., glucose anaplerosis) by directly activating pyruvate dehydrogenase kinase 1 (PDK-1), a negative regulator of cycle enzyme pyruvate dehydrogenase (PDH) (Kim et al., 2006). To compensate for the reduction of glucose feeding the TCA cycle, tumor cells with HIF activation often increase the usage of glutamine (Le et al., 2012). Under hypoxia conditions, glutamine largely fuels the TCA cycle in the form of α-KG to promote reductive carboxylation that produces citrate for lipogenesis (Wise et al., 2008; Metallo et al., 2011; Gameiro et al., 2013).
P53
P53 is a transcription factor and known tumor suppressor that regulates many important cellular pathways, including cell survival, DNA repair, apoptosis, and senescence (Bensaad et al., 2009). Wild-type P53 plays an important role in metabolism by striking a balance between bioenergetics and biosynthesis. One of the ways it does so is by lowering rates of glycolysis and promoting oxidative phosphorylation. P53 acts to suppress glycolysis by directly downregulating glucose transporters (GLUT1 and GLUT4) and indirectly inhibiting the activity of glycolytic enzymes, phosphofructokinase 1 (PFK1) and phosphoglycerate mutase (Kondoh et al., 2005; Bensaad et al., 2006, 2009; Zhang et al., 2013). To promote oxidative phosphorylation, P53 ensures availability of anapleurotic substrates, glucose, and glutamine, to the TCA cycle. As an activator of PDH, P53 downregulates PDH’s negative regulator PDK2 and indirectly activates PDHA1 (PDH A1 subunit). Additionally, P53 promotes glutamine incorporation into the TCA through direct transcriptional upregulation of glutaminase 2 (GLS2) (Zhang et al., 2011; Contractor and Harris, 2012). In solid tumors, P53 is commonly mutated and somatic mutations of P53 occur in more than 50% of human malignancies (Kruiswijk et al., 2015). Subsequently, loss of wild-type P53 function has a significant impact on cellular metabolism, leading to enhanced glycolysis and repressed oxidative phosphorylation in these tumor cells.
RAS
The most frequently mutated RAS subfamily genes in cancer are KRAS, NRAS, and HRAS, which serve as intercellular signaling molecules to transduce extracellular signaling from receptor tyrosine kinase to downstream effectors (Pylayeva-Gupta et al., 2011; Stephen et al., 2014). RAS plays a critical role in activating scavenging pathways in certain types of tumors and promotes nutrient uptake through both the extracellular and intracellular sources (Pylayeva-Gupta et al., 2011; Stephen et al., 2014). For example, Kamphorst et al. demonstrated that KRAS-driven pancreatic cells scavenge proteins, such as glutamine, from the extracellular space and utilize them to fuel the TCA cycle (Kamphorst et al., 2015). Additionally, it has been shown that KRAS-driven non-small cell lung cancer cells utilize autophagy to access intracellular supplies of glutamine to promote TCA cycle function (Guo et al., 2011; Strohecker and White, 2014). Moreover, KRAS-driven cancer cells can scavenge branch chain amino acids (i.e., isoleucine, valine, and leucine) and convert them into acetyl-CoA to fuel the TCA cycle (Mayers et al., 2014). A recent study by Kerr et al. demonstrated that copy number gain of mutant KRAS associated with tumor progression can promote glucose anaplerosis to fuel the TCA cycle (Kerr et al., 2016).
Cycle enzyme alterations in cancer
The biochemical reactions in the TCA cycle are catalyzed by a number of enzymes. Recent findings show that multiple cycle enzymes are either mutated or deregulated in a broad spectrum of cancer, resulting in characteristic metabolic and epigenetic changes that are correlated with disease transformation and progression.
SDH
Succinate dehydrogenase (SDH), also known as complex II, has roles in the TCA cycle and the ETC. SDH is a heterotetrameric enzyme complex composed of 4 subunits (SDHA, SDHB, SDHC, and SDHD), which catalyzes the oxidation of succinate to fumarate in the TCA cycle, while simultaneously reducing ubiquinone to ubiquinol in the ETC (Chandel, 2015). Mutations in SDHA, SDHB, SDHC, SDHD and SDH assembly factor 2 (SDHAF2) have been identified in hereditary paragangliomas (hPGLs) and pheochromocytomas (PCCs) (Table 1) (Baysal et al., 2000; Niemann and Muller, 2000; Astuti et al., 2001; Baysal et al., 2002; Hao et al., 2009; Bayley et al., 2010; Burnichon et al., 2010). Heterozygous mutations in SDH predispose patients to hPGL and PCC. Loss of heterozygosity as a result of a second mutation in the wild-type SDH allele triggers neoplastic transformation; thus, SDH is classified as a tumor suppressor gene (Gottlieb and Tomlinson, 2005). Additionally, mutations in SDH have also been identified in gastrointestinal stromal tumors, renal tumors, thyroid tumors, neuroblastoma, and testicular seminoma, implicating its importance in a wide range of cancer (Bardella et al., 2011).
Table 1.
Summary of cycle enzyme genetic alterations in cancer
| Gene | Genetic alterations | Tumor context | Consequence of alterations |
References |
|---|---|---|---|---|
| SDHA | c.91C>T c.1765C>T c.212G>A c.674C>T, c.818C>T c.341A>G c.367C>A c.441delG c.725_736del c.989_9990insTA c.1753C>T c.1865G>A c.1873C>T c.1886A>T |
Paragangliomas Pheochromocytomas |
Leads to reduction or loss of enzymatic activity of the SDH catalytic subunit and defective function of mitochondrial complex II | (Burnichon et al., 2010; Bardella et al., 2011; Korpershoek et al., 2011; Dwight et al., 2013; Evenepoel et al., 2015; Pillai et al., 2017) |
| c.2T>C c.91C>T c.113A>T c.160C>T c. 206C>T c.224G>A c.244A>T c.T273I c.457-3 457-1delCAG c.457-2 c457delCAG c.511C>T c.553C>T c.562C>T c.688delG c.767C>T c.778G>A c.800C>T c.818C>T c.985C>T c.1043-1055del c.1046 147delTG c.1151C>G c.1255G>A c.1334C>T c.1357G>A c.1361C>A c.1471G>T c.1534C>T c.1690G>A c.1765C>T c.1766G>A c.1794G>C c.1795-1G>T c.1873C>T c.1969G>A |
Gastrointestinal stromal tumors | (Pantaleo et al., 2011; Italiano et al., 2012; Belinsky et al., 2013a; Belinsky et al., 2013b; Miettinen et al., 2013; Oudijk et al., 2013; Miettinen and Lasota, 2014; Evenepoel et al., 2015; Jiang et al., 2015) | ||
| c.2T>C | Renal cell carcinoma | (Jiang et al., 2015) | ||
| SDHB | c.-1- ?_72+ ?del c.-1- ?_765+ ?del c.3G>A c.21delC c.49delA c.72+1G>A c.73_76delGCCT c.79C>A c.136C>G c.137G>A c.141G>A c.155delC c.166-170delCCTCA c.203G>A c.213C>T c.221insCCAG c.238A>G c.268C>T c.269G>A c.270C>G c.271G>A c.277T>C c.287-2A>G c.287- ?_540+ ?del c.291G>A c.293G>A c.299C>T c.300-4delCCTCA c.300T>C c.312insCACTGCA c.328C>T c.394T>C c.402C>T c.416T>C c.421-2A>G c.423+1G>A c.438G>A c.540G>A c.541-2A>G c.549_552delTACinsATACAG c.557G>A c.558-3C>G c.566G>A c.589C>T c.649C>T c.650G>T c.653G>A c.688C>T c.689G>A c.689G>T c.708T>C c.718_719delCT c.721G>A c.724C>G c.724C>A c.724C>T c.725G>A c.736A>T c.761C>T c.765+1G>A c.778G>C c.780delG c.847delTCTC c.859G>A c.881C>A c.889+1G>A |
Paraganglioma Pheochromocytoma |
Reduces SDH catalytic activity and causes defects in enzymatic activity in mitochondrial complex II | (Neumann et al., 2004, 2009; Bardella et al., 2011; Sjursen et al., 2013; Evenepoel et al., 2015; Bennedbaek et al., 2016) |
| c.32G>A c.88delC c.136C>T c.137G>A c.847-50delTCTC |
Renal cell carcinoma | (Vanharanta et al., 2004; Ricketts et al., 2008; Paik et al., 2014) | ||
| c.392delC | Thyroid carcinoma | (Zantour et al., 2004) | ||
| IVS1+1G>T c.17_dup26GTCG{dup26}GCCA c.17 42dup c.43+1C>T c.45_46insCC c.72+1G>T c.137G>A c.274T>A c.380T>G c.423+1G>C c.423+1G>A c.423+20T>A c.600G>T c.725G>A |
Gastrointestinal stromal tumors | (McWhinney et al., 2007; Pasini et al., 2008; Janeway et al., 2011; Miettinen et al., 2013; Miettinen and Lasota, 2014) | ||
| c.418G>T | Neuroblastoma | (Schimke et al., 2010) | ||
| c.587G>A | Pituitary carcinoma | (Tufton et al., 2017) | ||
| c.136C>T | T-cell acute leukemia | (Baysal, 2007) | ||
| SDHC | c.1A>G c.2T>A c.3G>A c.23dupA c.39C>A c.43C>T c.77 + 4760A>G c.78-2A>G c.78-19C>T c.112A>G c.126G>A c.140-5527C>A c.148C>T c.166A>T c.173T>C c.191_207del17 c.210C>G c.214C>T c.218insA c.224G>A c.242G>T c.242-5580C>A, c.212C>A c.253_255dupTTT c.397C>T c.405+1G>T c.439C>T c.496C>G IVS4+1G>A |
Paraganglioma Pheochromocytoma |
Leads to reduced SDH enzymatic activity and defective function in mitochondrial complex II | (Douwes Dekker et al., 2003; Mannelli et al., 2007; Peczkowska et al., 2008; Neumann et al., 2009; Bennedbaek et al., 2016; Pillai et al., 2017) |
| IVS5+1G>A c.1A>G c.6delT c.43C>T c.57delG c.224G>A c.301delT c.380A>G c.397C>T c.405+1G>A c.455G>C |
Gastrointestinal stromal tumors | (McWhinney et al., 2007; Pasini et al., 2008; Janeway et al., 2011; Miettinen et al., 2013; Miettinen and Lasota, 2014) | ||
| SDHD | c.2T>A c.3G>C c.14G>A c.33C>A c.33C>T c.34G>A c.36_37delTG c.49C>T c.50G>T c.52+2T>G c.53-2A>G c.53+2T>G c.55dupT c.64C>T c.106C>T c.112C>T c.118A>G c.120_ 127delCCCAGAAT c.129G>A c.149A>G c.168_169delTT c.169 + 5G>A, c.53-889G>A c.170-1G>T c.184_185insTC c.191_192delTC c.204-216del13bp c.206_218del13bp c.208A>G c.230T>G c.233_242del10bp c.242C>T c.252T>G c.274G>T c.276_278delCTA c.284T>C c.302T>C c.314+1G>C c.317delG c.325C>T c.334_337delACTG c.337_340delGACT c.341A>G c.341_342delAT c.361C>T c.367G>A c.370delG c.386_387insT c.408delT c.416T>C c.441delG c.443G>T IVS1+2T>G |
Paraganglioma Pheochromocytoma |
Reduces efficacy of SDH and impairs mitochondrial complex II activity | (Gimm et al., 2000; Taschner et al., 2001; Dannenberg et al., 2002; Douwes Dekker et al., 2003; Lee et al., 2003; Neumann et al., 2004; Simi et al., 2005; Galera-Ruiz et al., 2008; Neumann et al., 2009; Evenepoel et al., 2015; Bennedbaek et al., 2016; Pillai et al., 2017) |
| c.34G>A c.57delG c.352delG c.416T>C |
Gastrointestinal stromal tumors | (Pasini et al., 2008; Janeway et al., 2011; Miettinen et al., 2013; Oudijk et al., 2013; Miettinen and Lasota, 2014) | ||
| c.129G>A | Testicular seminoma | (Galera-Ruiz et al., 2008; Evenepoel et al., 2015) | ||
| SDHAF2 | c.68C>T c.139A>G c.232 G>A |
Paraganglioma Pheochromocytoma |
Leads to loss of flavination of SDH, reducing stability and activity of the enzyme complex | (Hao et al., 2009; Bayley et al., 2010; Pillai et al., 2017) |
| FH | p.Gln4X 1-bp del. In codon 17 p.Arg58X p.Asn64Thr p.Ala74Pro p.His137Arg p.Gln142Arg 2-bp del. In codon 181 Lys187Arg Lys del. In codon 187 Arg190His -15 splice site p.Gly239Val p.Arg300X 1-bp del. In codon 507 |
Multiple leiomyomatosis | Leads to loss of FH enzymatic activity and accumulation of fumarate in the cell | (Tomlinson et al., 2002) |
| c.1?_c.*100del c.1?_404+?del c.111insA c.127_128delGA c.138+1_138+10del10 c.147delT c.157G>T c.172C>T c.191A>C c.220G>C c.233del c.247_249+1delGAGGinsA c.250-2A>G c.266T>C c.298delA c.305C>G c.349A>G c.410A>G c.425A>G c.431C>T c.434A>G c.455T>C c.503T>C c.560A>G c.568C>T c.568delAC c.569G>A c.569G>T c.575A>G c.632A>G c.666delC c.698G>A c.780delGC c.782-788 7-bp del. c.806T>C c.808G>T c.810delA c.815T>C c.821C>T c.823C>T c.824G>A c.836T>A c.869G>A c.875T>C c.891T>A c.898C>T c.952C>T c.964A>G c.968G>A c.989A>G c.1002T>G 2-bp ins @1004 c.1020T>A c.1025C>A c.1028A>G c.1060G>A c.1083-1086delTGAA c.1108-2A>G c.1121-1123 del TAC c.1123delA c.1126T>C c.1138insA c.1144A>G c.1162delA c.1187A>C c.1189G>A c.1210G>T c.1234del c.1265A>G 8-bp dup @ 1300-1307 c.1339delG c.1349-1352delATGA c.1371G>A c.1431insAAA |
Hereditary leiomymatosis and renal cell carcinoma | (Toro et al., 2003; Wei et al., 2006; Pfaffenroth and Linehan, 2008; Gardie et al., 2011; Smit et al., 2011; Chen et al., 2014; Wong et al., 2014; Arenas Valencia et al., 2017) | ||
| c.220G>C c.426+1G>A c.988A>G c.994delA |
Type 2 papillary renal cell carcinoma | (Gardie et al., 2011) | ||
| c.1394G>A c.352A>C |
Leydig cell tumors (Carvajal-Carmona et al.) | (Carvajal-Carmona et al., 2006) | ||
| 435insAAA 691G>A |
Ovarian mucinous cystadenoma | (Ylisaukko-oja et al., 2006) | ||
| IDH1 | p.Arg100Gln p.Arg132His p.Arg132Cys p.Arg132Ser p.Arg132Leu p.Arg132Gly |
Gliomas/Glioblastomas | Increases affinity for NADPH/α-KG; reduces affinity for isocitrate; increases production of 2-HG | (Parsons et al., 2008; Dang et al., 2009; Yan et al., 2009; Pusch et al., 2011) |
| p.Arg132His p.Arg132Cys p.Arg132Ser p.Arg132Gly p.Arg132Leu |
Acute myeloid leukemia | (Mardis et al., 2009; Abbas et al., 2010; Bayley et al., 2010) | ||
| p.Arg132Cys p.Arg132Leu p.Arg132Gly p.Arg132Ser |
Myelodysplastic syndromes/ Myeloproliferative neoplasms | (Kosmider et al., 2010; Pardanani et al., 2010) | ||
| p.Arg132Cys p.Arg132His p.Arg132Leu p.Arg132Ser |
Chondrosarcoma | (Amary et al., 2011) | ||
| p.Arg132His p.Arg132Gly p.Arg132Ser p.Arg132Cys |
Acute lymphoblastic leukemia | (Kang et al., 2009; Zhang et al., 2012) | ||
| p.Gly70Asp p.Val71Ile p.Gly105Gly; p.Val1781Ile p.Gly123Arg p.Ile130Met p.His133Gln p.Ala134Asp |
Thyroid carcinoma | (Hemerly et al., 2010; Murugan et al., 2010) | ||
| p.Arg132Cys p.Arg132His |
Prostate carcinoma | (Kang et al., 2009; Ghiam et al., 2012) | ||
| IDH2 | p.Arg172Gly p.Arg172Met p.Arg172Lys |
Gliomas/Glioblastomas | Increases affinity for NADPH/α-KG; reduces affinity for isocitrate; increases production of 2-HG | (Yan et al., 2009) |
| p.Arg140Gln p.Arg172Lys p.Arg172Gln p.Arg172Thr p.Arg172Gly |
Angioimmunoblastic T-cell lymphoma | (Cairns et al., 2012; Lemonnier et al., 2016) | ||
| p.Arg140Gln p.Arg140Trp p.Arg172Lys p.Arg172Met |
Acute myeloid leukemia | (Abbas et al., 2010; Gross et al., 2010; Pardanani et al., 2010) | ||
| p.Arg140Gln p.Arg140Leu |
Myelodysplastic syndromes/ Myeloproliferative neoplasms | (Kosmider et al., 2010; Pardanani et al., 2010) | ||
| p.Arg172Ser | Chondrosarcoma | (Amary et al., 2011) |
FH
Fumarate hydratase (FH) is a homotetrameric cycle enzyme that catalyzes the stereospecific and reversible hydration of fumarate to L-malate. Beyond its mitochondrial role, FH is also expressed in the cytoplasm where it participates in the urea cycle as well as nucleotide and amino acid metabolism (Adam, 2014 #160). Heterozygous mutations in FH predispose patients to multiple cutaneous and uterine leiomyomas (MCUL), as well as hereditary leiomyomatosis and renal cell cancer (HLRCC) (Table 1) (Launonen et al., 2001; Tomlinson et al., 2002). Additionally, mutations in FH have been identified in bladder, breast and testicular cancer (Table 1) (Carvajal-Carmona et al., 2006; Ylisaukko-oja et al., 2006). Mutations predisposing patients to MCUL or HLRCC occur across the gene and include missense, frameshift, nonsense and large deletions at the FH locus (Table 1) (Bensaad et al., 2006). Similar to SDH, the enzymatic activity of FH is completely absent in HLRCC patients due to a loss of the remaining wild-type allele (Wei et al., 2006).
IDH
The IDH family is comprised of three isoforms (IDH1, IDH2, and IDH3) that convert isocitrate to α-KG. Only IDH2 and 3 are expressed in the mitochondria, while IDH1 is expressed in the cytoplasm. IDH1 and IDH2 function as homodimers that catalyze the conversion of α-KG to isocitrate and require NADP+ as a co-factor, whereas IDH3 is a heterodimer (IDH3A, IDH3B, and IDH3G) that can only oxidize isocitrate to α-KG and requires NAD+ as a co-factor (Chandel, 2015). Unlike FH and SDH, mutations in IDH1 and 2 are somatic heterozygous missense mutations that occur primarily at the active arginine residues that are critical for isocitrate binding (IDH1: R132; IDH2: R172, R140; Table 1) (Parsons et al., 2008; Dang et al., 2009; Mardis et al., 2009; Yan et al., 2009). No mutations in IDH3 have been reported so far. IDH1/2 mutations occur frequently in low-grade glioma and secondary glioblastoma (~80%), but can also occur in acute myeloid leukemia (20%), angioimmunoblastic T-cell lymphomas (20%), and rarely in other malignancies such as thyroid, colorectal, and prostate cancer (Table 1) (Kang et al., 2009; Yen et al., 2010; Ghiam et al., 2012; Ohgaki and Kleihues, 2013; Yen et al., 2017). These neomorphic mutations result in the gained function of converting α-KG to 2-hydroxyglutarate (2-HG), an oncometabolite.
Deregulation of other cycle enzymes
Beyond mutations detected for cycle enzymes, several studies have demonstrated that other cycle enzymes, CS, AH, and KGDHC, are deregulated in cancer. CS catalyzes a rate-limiting step in the TCA cycle and is either overexpressed or has increased enzymatic activity in pancreatic, ovarian, and renal cancer (Schlichtholz et al., 2005; Lin et al., 2012; Chen et al., 2014). AH is a reversible enzyme that catalyzes the conversion of citrate to isocitrate and its expression is downregulated in both gastric and prostate cancer (Singh et al., 2006; Wang et al., 2013). KGDHC is a rate-limiting enzyme of the TCA cycle and has three components including α-KG dehydrogenase (OGDH), dihydrolipoamide S-succinyltransferase (DLST), and dihydrolipoamide dehydrogenase (DLD). OGDH is downregulated in colorectal cancer as the result of promoter hypermethylation and similar promoter hypermethylation has been documented in breast, lung, esophageal, cervical, and pancreatic cancer (Hoque et al., 2008; Ostrow et al., 2009; Fedorova et al., 2015). Interestingly, Snezhkina and colleagues have demonstrated that an alternative splice variant of OGDH that is tumor specific is overexpressed in colorectal cancer (Snezhkina et al., 2016). OGDH is regulated by Ca2+, adenine nucleotides, and NADH, and the tumor-specific isoform lacks three regions of the protein and exhibits reduced sensitivity to Ca2+. Additionally, Anderson et al. found that the E2 component of KGDHC, DLST, is upregulated in T-cell acute lymphoblastic leukemia (T-ALL) (Anderson et al., 2016).
Disease mechanisms underlying cycle enzyme alterations
Genetic alterations can occur in multiple cycle enzymes; however, their mechanisms of action in tumorigenesis differ. Both SDH and FH are classical tumor suppressor genes, and predispose individuals with heritable mutated genes to cancer when the second wild-type allele is lost (Chandel, 2015). Inactivating mutations in FH result in a build-up of fumarate and metabolic reprograming (Pollard et al., 2005), which includes an increased dependence on glycolysis and glutamine anaplerosis (Aspuria et al., 2014). In tumor cells harboring mutant FH, an accumulation of fumarate results in succination of cysteine-modifying proteins such as kelch-like ECH-associated protein 1 (KEAP1) and mitochondrial aconitase (ACO2) (Yang et al., 2014). Loss-of-function mutations of SDH result in the accumulation of millimolar concentrations of succinate and reduced levels of fumarate and malate (Pollard et al., 2005), which lead to disruption of multiple metabolic pathways including central carbon metabolism (Yang et al., 2013; Aspuria et al., 2014). On the other hand, IDH1/2 missense mutations render the enzymes acquiring neomorphic activity that can convert α-KG to 2-HG (Dang et al., 2009). 2-HG is an oncometabolite that acts as a competitive inhibitor to α-KG-dependent dioxygenases, such as hypoxia-inducible factor (HIF), prolyl hydroxylases (PDHs), JmjC domain-containing histone demethylases, and ten-eleven translocation (TET) family of 5mC DNA hydroxylases (Chowdhury et al., 2011; Xu et al., 2011; Koivunen et al., 2012). The inhibition of these dioxygenases results in broad epigenomic alterations that both suppress differentiation and promote proliferation. Mutations in IDH2, FH, and SDH share a common mechanism of inhibiting α-KG-dependent dioxygenases through 2-HG, fumarate, or succinate, respectively (Hoekstra et al., 2015). Both FH and SDH mutations induce a state of pseudohypoxia, where 2-HG, fumarate or succinate can inhibit PHDs, resulting in stabilization of HIF. Additionally, mutations of FH, SDH, and IDH1/2 cause increased production of reactive oxygen species (ROS), either directly by mutated SDH or indirectly in tumor cells with mutant IDH1/2 and FH (Hoekstra et al., 2015). For example, glioma cells with IDH mutations have increased ROS and reduced GSH levels due to insufficient NADPH pools (Shi et al., 2015). In cancer cells with FH mutations, the accumulation of fumarate results in elevated levels of succinic-glutathione (GSF), which acts as an alternative substrate for GSH reductase, ultimately leading to decreased levels of NADPH and GSH (Sullivan et al., 2013).
Potential approaches to target the TCA cycle
Therapeutically targeting the TCA cycle function in cancer is an attractive strategy to treat cancer and two strategies are currently being tested in the clinic. Many tumors utilize glutamine as a fuel source for the TCA cycle, thus suppression of glutaminolysis through small molecule inhibitors is an attractive approach to therapeutically target these tumors (Seltzer et al., 2010; Cheng et al., 2011; Le et al., 2012; Yuneva et al., 2012; Gameiro et al., 2013). An initial strategy utilized glutamine analogues, such as 6-diazo-5-oxo-L-norleucine, to target glutaminolysis (Ovejera et al., 1979; Ahluwalia et al., 1990; Griffiths et al., 1993). While these compounds highlight the potential of targeting glutamine anaplerosis, they ultimately failed to enter clinics due to high tissue toxicities. Additional studies have demonstrated that glutamine limitation, through either depletion of glutamine in the plasma (L-aspariginase) or blocking glutamine transport (sulfasalazine), can provide therapeutic benefit (Oettgen et al., 1967; Lo et al., 2008; Chan et al., 2014; Parmentier et al., 2015; Rodman et al., 2016; Roh et al., 2016; Shitara et al., 2017). Recently, GLS inhibitors, such as CB-839, an orally available, potent, and specific inhibitor of GLS, have shown anti-tumor efficacy. CB-839 disrupts the conversion of glutamine to glutamate and alters a number of downstream pathways, including the TCA cycle, glutathione production, and amino acid synthesis (Gross et al., 2010; Jacque et al., 2015). Phase I clinical trials are currently underway for CB-839, and examine its effectiveness for the treatment of both hematological malignancies and solid tumors (NCT02071927 and NCT02071888).
Besides targeting glutaminolysis outside the TCA cycle through GLS inhibition, several recent studies indicate that KGDHC represents a striking vulnerability for numerous cancers, and is a promising therapeutic target. Utilizing a MYC-driven model of T-ALL, Anderson and colleagues demonstrated that heterozygous inactivation of DLST (the E2 enzyme of KGDHC) was sufficient to significantly delay tumor onset without impacting normal animal development (Anderson et al., 2016). Additionally, they show that DLST inactivation in T-ALL cells disrupts the TCA cycle, while slowing cell growth and inducing apoptosis (Anderson et al., 2016). Allen et al. conducted a focused siRNA screen on TCA cycle enzymes, and found that many cancer cells highly depend on OGDH (the E1 component of KGDHC) for growth and survival (Allen et al., 2016). A recent study by Ilic et al. demonstrated that cancer cells harboring oncogenic PI3K mutations require all three components of KGDHC, OGDH in particular, for proliferation (Ilic et al., 2017). These findings support the rationale to target KGDHC for cancer treatment. CPI-613 is a lipoate analog that can simultaneously inhibit both PDH and KGDHC, as lipoate is a co-factor for both enzyme complexes. While CPI-613 stimulates PDK to phosphorylate and inactivate PDH (Zachar et al., 2011), CPI-613 can also induce a burst of mitochondria ROS through acting on DLD (the E3 component of KGDHC) and suppression of the E2 subunit of KGDHC, DLST (Stuart et al., 2014). Currently, CPI-613 is being tested in phase I and II clinical trials, as a single agent or in combination with standard chemotherapy, to treat cancers (NCT02168140, NCT01902381, NCT02232152, and NCT01766219). Limited data published from these trials have already shown that CPI-613 is generally well tolerated with minimal toxicity (Pardee et al., 2014; Lycan et al., 2016). While a phase I trial indicated that CPI-613 may be effective as a single agent for treating hematological malignancies, a phase II trial for small cell lung carcinoma show no efficacy as a single agent (Pardee et al., 2014; Lycan et al., 2016).
Finally, mutations in TCA cycle gene IDH2 provide a unique opportunity for therapeutic intervention. Not only can mutant IDH serve as a biomarker, but also their neomorphic enzymatic activity can be targeted through small molecule inhibition. Currently, there are several small molecule inhibitors of mutant IDH2 in clinical development, including enasidenib (AG-221) that inhibits mutant IDH2 and AG-881 that targets both mutant IDH1 and IDH2 (Dang, 2016 #135). These compounds act by binding to the active catalytic site of mIDH1/2 enzymes and blocking the conformational change required to convert α-KG into 2-HG. AG-221 is an orally available inhibitor of mutant IDH2-R140 and IDH2-R172 (Yen et al., 2017), and is currently undergoing phase I/II clinical trials as a single agent for the treatment of AML and solid tumors (e.g., glioma and angioimmunoblastic T-cell lymphoma; NCT01915498 and NCT02273739, respectively). Preclinical data demonstrate that AG-221 can dramatically reduce 2-HG levels. Additionally, AG-221 results in cellular differentiation of tumor cells in murine xenograft models (Yen et al., 2017). Preliminary data from the AML clinical trial demonstrate that AG-221 alone led to a 41% object response rate and a 28% complete response rate. New phase I and III clinical trials will soon start and will examine the effectiveness of AG-221 alone in comparison to conventional therapy, as well as AG-221 in combination with standard induction and consolidation therapy (NCT02577406 and NCT02632708). The dual target inhibitor of mutant IDH1 and mutant IDH2, AG-881, is an orally available inhibitor that can pass the blood-brain barrier and may serve as a better option for glioma patients (Medeiros et al., 2017). Currently, AG-881 is in phase I clinical trial for AML patients with mutant IDH1/2, and a clinical trial for patients with glioma will begin soon (NCT02492737 and NCT02481154).
Future perspectives
The TCA cycle is a critical metabolic pathway that allows mammalian cells to utilize glucose, amino acids, and fatty acids. The entry of these fuels into the cycle is carefully regulated to efficiently fulfill the cell’s bioenergetic, biosynthetic, and redox balance requirements. Multiple types of cancer are marked by drastic changes to TCA cycle enzymes, which result in characteristic metabolic and epigenetic changes that are correlated with disease transformation and progression. As a result, several components of the TCA cycle may be exploited therapeutically for the treatment of disease. However, due to the importance of the TCA cycle in normal cell development, high toxicity is a concern of this approach. Interestingly, although decreased KGDHC activity is associated with neurodegenerative diseases (Gibson et al., 2010), inhibiting KGDHC through CPI-613 is well tolerated in clinical testing (Pardee et al., 2014; Lycan et al., 2016). Additionally, 50% reduction of DLST, the E2 component of KGDHC, in zebrafish significantly delays MYC-driven leukemogenesis, without causing any detectable abnormalities (Anderson et al., 2016). Importantly, others show that cancer cells with IDH mutations become insensitive to treatment with mutant IDH inhibitors in vivo, owing to the metabolic rewiring and enhanced usage of the TCA cycle (Grassian et al., 2014; Tateishi et al., 2015). Emerging studies demonstrate that cancer cells utilize the TCA cycle differently from those of normal cells, making it likely that cancer cells will be more sensitive to inhibitors targeting the reprogrammed metabolic pathways in the TCA cycle (Kishton et al., 2016). These observations support the notion that targeting the TCA cycle by small molecule inhibitors of cycle enzymes and/or enzymes regulating the cycle could serve as a productive approach for cancer treatment.
Cancer cells often escape treatment through compensatory pathways (Obre and Rossignol, 2015; Zugazagoitia et al., 2016), and the metabolic properties of cancer cells are often context-dependent (Yuneva et al., 2012; Kishton et al., 2016; Martinez-Outschoorn et al., 2017). Hence, the key for successful metabolism-based therapies against cancer relies on both the identification of the “oncometabolic” enzyme(s) responsible for metabolic reprogramming and an in-depth understanding of the activity and flexibility of the altered pathways in the context of each specific cancer type. Despite the established role of the TCA cycle in tumorigenesis, its involvement in cancer metabolism remains incompletely understood. Additionally, how the TCA cycle interacts with other biochemical and cell signaling pathways is yet to be characterized. Owing to the impact of microenvironment on cellular metabolism and oncogenic signaling, it is critically important to study the contribution of the TCA cycle to cancer metabolism and tumorigenesis in vivo. Importantly, researchers started to successfully apply untargeted/targeted metabolomics and respiratory analyses to animal model organisms. The intensive research effort in the coming years will undoubtedly deepen our understanding of the role of this central metabolic hub that was once overlooked in tumorigenesis, reveal vulnerabilities for therapeutic intervention, and eventually bring this targeted approach from infancy up to maturity.
ACKNOWLEDGEMENTS
This work was supported by a young investigator award from the Leukemia Research Foundation, a Ralph Edwards Career Development Professorship from Boston University, a Scholar grant from the St. Baldrick’s Foundation, an ignition award from Boston University, a clinical translational institute pilot from the National Institute of Health (1UL1TR001430), an institutional grant (IRG–72-001-36-IRG) from the American Cancer Society, and a grant from the Mary Kay Ash Foundation to H.F., a young investigator award from the Alex Lemonade Stand to N.M.A., P.M. acknowledges training support through T32GM008541 from the National Institutes of Health. The content of this research is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
ABBREVIATIONS
α-KG, α-ketoglutarate; ACC, acetyl-CoA carboxylase; ACLY, adenosine triphosphate citrate lyase; AH, aconitate hydratase; DLD, dihydrolipoamide dehydrogenase; DLST, dihydrolipoamide S-succinyltransferase; ETC, electron transport chain; FAS, fatty acid synthase; FH, fumarase; GLS, glutaminase; GLUD, glutamate dehydrogenase; GLUT, glucose transporters; HIFs, hypoxia-inducible factors; HK, hexokinase; HLRCC, hereditary leiomyomatosis and renal cell cancer; IDH, isocitrate dehydrogenase; KGDHC, α-ketoglutarate dehydrogenase complex; LDHA, lactate dehydrogenase A; MCUL, multiple cutaneous and uterine leiomyomas; OGDH, α-KG dehydrogenase; PDH, pyruvate dehydrogenase; PDK-1, pyruvate dehydrogenase kinase 1; PFK1, phosphofructokinase 1; PHD, prolyl hydroxylases; PK, pyruvate kinase; SDH, succinate dehydrogenase; TCA, tricarboxylic acid; TET, ten-eleven translocation; VHL, von Hippel-Lindau
COMPLIANCE WITH ETHICS GUIDELINES
The authors declare no conflicts of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Footnotes
Nicole M. Anderson and Patrick Mucka equally contributed to this study.
References
- Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, van Putten WJ, Rijneveld AW, Lowenberg B, Valk PJ. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116:2122–2126. doi: 10.1182/blood-2009-11-250878. [DOI] [PubMed] [Google Scholar]
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- Ahluwalia GS, Grem JL, Hao Z, Cooney DA. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther. 1990;46:243–271. doi: 10.1016/0163-7258(90)90094-I. [DOI] [PubMed] [Google Scholar]
- Akram M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem Biophys. 2014;68:475–478. doi: 10.1007/s12013-013-9750-1. [DOI] [PubMed] [Google Scholar]
- Allen EL, Ulanet DB, Pirman D, Mahoney CE, Coco J, Si Y, Chen Y, Huang L, Ren J, Choe S, et al. Differential aspartate usage identifies a subset of cancer cells particularly dependent on OGDH. Cell Rep. 2016;17:876–890. doi: 10.1016/j.celrep.2016.09.052. [DOI] [PubMed] [Google Scholar]
- Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- Anderson NM, Li D, Peng HL, Laroche FJ, Mansour MR, Gjini E, Aioub M, Helman DJ, Roderick JE, Cheng T, et al. The TCA cycle transferase DLST is important for MYC-mediated leukemogenesis. Leukemia. 2016;30:1365–1374. doi: 10.1038/leu.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas Valencia C, Rodriguez Lopez ML, Cardona Barreto AY, Garavito Rodriguez E, Arteaga Diaz CE. Hereditary leiomyomatosis and renal cell cancer syndrome: identification and clinical characterization of a novel mutation in the FH gene in a Colombian family. Fam Cancer. 2017;16:117–122. doi: 10.1007/s10689-016-9922-4. [DOI] [PubMed] [Google Scholar]
- Aspuria PJ, Lunt SY, Varemo L, Vergnes L, Gozo M, Beach JA, Salumbides B, Reue K, Wiedemeyer WR, Nielsen J, et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014;2:21. doi: 10.1186/2049-3002-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011;1807:1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Baron-Delage S, Mahraoui L, Cadoret A, Veissiere D, Taillemite JL, Chastre E, Gespach C, Zweibaum A, Capeau J, Brot-Laroche E, et al. Deregulation of hexose transporter expression in Caco-2 cells by ras and polyoma middle T oncogenes. Am J Physiol. 1996;270:G314–G323. doi: 10.1152/ajpgi.1996.270.2.G314. [DOI] [PubMed] [Google Scholar]
- Bayley JP, Kunst HP, Cascon A, Sampietro ML, Gaal J, Korpershoek E, Hinojar-Gutierrez A, Timmers HJ, Hoefsloot LH, Hermsen MA, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11:366–372. doi: 10.1016/S1470-2045(10)70007-3. [DOI] [PubMed] [Google Scholar]
- Baysal BE. A recurrent stop-codon mutation in succinate dehydrogenase subunit B gene in normal peripheral blood and childhood T-cell acute leukemia. PLoS ONE. 2007;2:e436. doi: 10.1371/journal.pone.0000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, Yee HA, Brackmann DE, Slattery WH, 3rd, Myers EN, et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet. 2002;39:178–183. doi: 10.1136/jmg.39.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinsky MG, Rink L, Flieder DB, Jahromi MS, Schiffman JD, Godwin AK, Mehren M. Overexpression of insulin-like growth factor 1 receptor and frequent mutational inactivation of SDHA in wild-type SDHB-negative gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2013;52:214–224. doi: 10.1002/gcc.22023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinsky MG, Rink L, von Mehren M. Succinate dehydrogenase deficiency in pediatric and adult gastrointestinal stromal tumors. Front Oncol. 2013;3:117. doi: 10.3389/fonc.2013.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennedbaek M, Rossing M, Rasmussen AK, Gerdes AM, Skytte AB, Jensen UB, Nielsen FC, Hansen TV. Identification of eight novel SDHB, SDHC, SDHD germline variants in Danish pheochromocytoma/paraganglioma patients. Hered Cancer Clin Pract. 2016;14:13. doi: 10.1186/s13053-016-0053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28:3015–3026. doi: 10.1038/emboj.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5. New York: W. H. Freeman and Company; 2002. [Google Scholar]
- Birnbaum MJ, Haspel HC, Rosen OM. Transformation of rat fibroblasts by FSV rapidly increases glucose transporter gene transcription. Science (New York, NY) 1987;235:1495–1498. doi: 10.1126/science.3029870. [DOI] [PubMed] [Google Scholar]
- Bolzoni M, Chiu M, Accardi F, Vescovini R, Airoldi I, Storti P, Todoerti K, Agnelli L, Missale G, Andreoli R, et al. Dependence on glutamine uptake and glutamine addiction characterize myeloma cells: a new attractive target. Blood. 2016;128:667–679. doi: 10.1182/blood-2016-01-690743. [DOI] [PubMed] [Google Scholar]
- Brosnan JT. Interorgan amino acid transport and its regulation. J Nutr. 2003;133:2068S–2072S. doi: 10.1093/jn/133.6.2068S. [DOI] [PubMed] [Google Scholar]
- Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, Jouanno E, Jeunemaitre X, Benit P, Tzagoloff A, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset P, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119:1901–1903. doi: 10.1182/blood-2011-11-391748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Carmona LG, Alam NA, Pollard PJ, Jones AM, Barclay E, Wortham N, Pignatelli M, Freeman A, Pomplun S, Ellis I, et al. Adult leydig cell tumors of the testis caused by germline fumarate hydratase mutations. J Clin Endocrinol Metab. 2006;91:3071–3075. doi: 10.1210/jc.2006-0183. [DOI] [PubMed] [Google Scholar]
- Chan WK, Lorenzi PL, Anishkin A, Purwaha P, Rogers DM, Sukharev S, Rempe SB, Weinstein JN. The glutaminase activity of L-asparaginase is not required for anticancer activity against ASNS-negative cells. Blood. 2014;123:3596–3606. doi: 10.1182/blood-2013-10-535112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS. Navigating metabolism. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2015. [Google Scholar]
- Chen JQ, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim Biophys Acta. 2012;1826:370–384. doi: 10.1016/j.bbcan.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Chen L, Liu T, Zhou J, Wang Y, Wang X, Di W, Zhang S. Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS ONE. 2014;9:e115708. doi: 10.1371/journal.pone.0115708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci USA. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SD. Regulation of fatty acid synthase gene expression: an approach for reducing fat accumulation. J Anim Sci. 1993;71:1957–1965. doi: 10.2527/1993.7171957x. [DOI] [PubMed] [Google Scholar]
- Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012;72:560–567. doi: 10.1158/0008-5472.CAN-11-1215. [DOI] [PubMed] [Google Scholar]
- Cummins TD, Holden CR, Sansbury BE, Gibb AA, Shah J, Zafar N, Tang Y, Hellmann J, Rai SN, Spite M, et al. Metabolic remodeling of white adipose tissue in obesity. Am J Physiol Endocrinol Metab. 2014;307:E262–E277. doi: 10.1152/ajpendo.00271.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg H, Dinjens WN, Abbou M, Van Urk H, Pauw BK, Mouwen D, Mooi WJ, de Krijger RR. Frequent germ-line succinate dehydrogenase subunit D gene mutations in patients with apparently sporadic parasympathetic paraganglioma. Clin Cancer Res. 2002;8:2061–2066. [PubMed] [Google Scholar]
- DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douwes Dekker PB, Hogendoorn PC, Kuipers-Dijkshoorn N, Prins FA, van Duinen SG, Taschner PE, van der Mey AG, Cornelisse CJ. SDHD mutations in head and neck paragangliomas result in destabilization of complex II in the mitochondrial respiratory chain with loss of enzymatic activity and abnormal mitochondrial morphology. J Pathol. 2003;201:480–486. doi: 10.1002/path.1461. [DOI] [PubMed] [Google Scholar]
- Dwight T, Mann K, Benn DE, Robinson BG, McKelvie P, Gill AJ, Winship I, Clifton-Bligh RJ. Familial SDHA mutation associated with pituitary adenoma and pheochromocytoma/paraganglioma. J Clin Endocrinol Metab. 2013;98:E1103–E1108. doi: 10.1210/jc.2013-1400. [DOI] [PubMed] [Google Scholar]
- Eagle H. The minimum vitamin requirements of the L and HeLa cells in tissue culture, the production of specific vitamin deficiencies, and their cure. J Exp Med. 1955;102:595–600. doi: 10.1084/jem.102.5.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds LR, Sharma L, Kang A, Lu J, Vockley J, Basu S, Uppala R, Goetzman ES, Beck ME, Scott D, et al. c-Myc programs fatty acid metabolism and dictates acetyl-CoA abundance and fate. J Biol Chem. 2015;290:20100. doi: 10.1074/jbc.A114.580662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193–202. doi: 10.1038/nrc1013. [DOI] [PubMed] [Google Scholar]
- Evenepoel L, Papathomas TG, Krol N, Korpershoek E, de Krijger RR, Persu A, Dinjens WN. Toward an improved definition of the genetic and tumor spectrum associated with SDH germ-line mutations. Genet Med. 2015;17:610–620. doi: 10.1038/gim.2014.162. [DOI] [PubMed] [Google Scholar]
- Fedorova MS, Kudryavtseva AV, Lakunina VA, Snezhkina AV, Volchenko NN, Slavnova EN, Danilova TV, Sadritdinova AF, Melnikova NV, Belova AA, et al. Downregulation of OGDHL expression is associated with promoter hypermethylation in colorectal cancer. Mol Biol (Mosk) 2015;49:678–688. doi: 10.1134/S0026893315040044. [DOI] [PubMed] [Google Scholar]
- Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014 doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galera-Ruiz H, Gonzalez-Campora R, Rey-Barrera M, Rollon-Mayordomo A, Garcia-Escudero A, Fernandez-Santos JM, DeMiguel M, Galera-Davidson H. W43X SDHD mutation in sporadic head and neck paraganglioma. Anal Quant Cytol Histol. 2008;30:119–123. [PubMed] [Google Scholar]
- Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, Lopez-Larrubia P, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17:372–385. doi: 10.1016/j.cmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Shen Y, Jin F, Miao Y, Qiu X. Cancer stem cells in small cell lung cancer cell line H446: higher dependency on oxidative phosphorylation and mitochondrial substrate-level phosphorylation than non-stem cancer cells. PLoS ONE. 2016;11:e0154576. doi: 10.1371/journal.pone.0154576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardie B, Remenieras A, Kattygnarath D, Bombled J, Lefevre S, Perrier-Trudova V, Rustin P, Barrois M, Slama A, Avril MF, et al. Novel FH mutations in families with hereditary leiomyomatosis and renal cell cancer (HLRCC) and patients with isolated type 2 papillary renal cell carcinoma. J Med Genet. 2011;48:226–234. doi: 10.1136/jmg.2010.085068. [DOI] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Ghiam AF, Cairns RA, Thoms J, Dal Pra A, Ahmed O, Meng A, Mak TW, Bristow RG. IDH mutation status in prostate cancer. Oncogene. 2012;31:3826. doi: 10.1038/onc.2011.546. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta. 2010;1802:122–134. doi: 10.1016/j.bbadis.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies RJ, Gatenby RA. Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? J Bioenerg Biomembr. 2007;39:251–257. doi: 10.1007/s10863-007-9085-y. [DOI] [PubMed] [Google Scholar]
- Gimm O, Armanios M, Dziema H, Neumann HP, Eng C. Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer Res. 2000;60:6822–6825. [PubMed] [Google Scholar]
- Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev. 2007;17:71–77. doi: 10.1016/j.gde.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5:857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, Slocum KL, Pu M, Lin F, Vickers C, et al. IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 2014;74:3317–3331. doi: 10.1158/0008-5472.CAN-14-0772-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths M, Keast D, Patrick G, Crawford M, Palmer TN. The role of glutamine and glucose analogues in metabolic inhibition of human myeloid leukaemia in vitro. Int J Biochem. 1993;25:1749–1755. doi: 10.1016/0020-711X(88)90303-5. [DOI] [PubMed] [Google Scholar]
- Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemerly JP, Bastos AU, Cerutti JM. Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur J Endocrinol. 2010;163:747–755. doi: 10.1530/EJE-10-0473. [DOI] [PubMed] [Google Scholar]
- Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, Ras C, Seifar RM, van Minderhout I, Cornelisse CJ, Hogendoorn PC, Breuning MH, Suijker J, et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget. 2015;6:38777–38788. doi: 10.18632/oncotarget.6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque MO, Kim MS, Ostrow KL, Liu J, Wisman GB, Park HL, Poeta ML, Jeronimo C, Henrique R, Lendvai A, et al. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008;68:2661–2670. doi: 10.1158/0008-5472.CAN-07-5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic N, Birsoy K, Aguirre AJ, Kory N, Pacold ME, Singh S, Moody SE, DeAngelo JD, Spardy NA, Freinkman E, et al. PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc Natl Acad Sci USA. 2017;114:E3434–E3443. doi: 10.1073/pnas.1617922114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiano A, Chen CL, Sung YS, Singer S, DeMatteo RP, LaQuaglia MP, Besmer P, Socci N, Antonescu CR. SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer. 2012;12:408. doi: 10.1186/1471-2407-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015;126:1346–1356. doi: 10.1182/blood-2015-01-621870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Zhang Y, Zhou YH, Hou YY, Wang JY, Li JL, Li M, Tong HX, Lu WQ. A novel germline mutation in SDHA identified in a rare case of gastrointestinal stromal tumor complicated with renal cell carcinoma. Int J Clin Exp Pathol. 2015;8:12188–12197. [PMC free article] [PubMed] [Google Scholar]
- Juang HH. Modulation of mitochondrial aconitase on the bioenergy of human prostate carcinoma cells. Mol Genet Metab. 2004;81:244–252. doi: 10.1016/j.ymgme.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75:544–553. doi: 10.1158/0008-5472.CAN-14-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, Lee JY, Yoo NJ, Lee SH. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009;125:353–355. doi: 10.1002/ijc.24379. [DOI] [PubMed] [Google Scholar]
- Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531(7592):110–113. doi: 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kishton RJ, Rathmell JC. Novel therapeutic targets of tumor metabolism. Cancer J. 2015;21:62–69. doi: 10.1097/PPO.0000000000000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, Macintyre AN, Goraksha-Hicks P, de Cubas AA, Liu T, et al. AMPK is essential to balance glycolysis and mitochondrial metabolism to control T-ALL cell stress and survival. Cell Metab. 2016;23:649–662. doi: 10.1016/j.cmet.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]
- Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, Badoual C, Gadessaud N, Venisse A, Bayley JP, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96:E1472–E1476. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, Hunault-Berger M, Slama B, Vey N, Lacombe C, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia. 2010;24:1094–1096. doi: 10.1038/leu.2010.52. [DOI] [PubMed] [Google Scholar]
- Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, Sistonen P, Herva R, Aaltonen LA. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001;98:3387–3392. doi: 10.1073/pnas.051633798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Chionh SB, Chong SM, Taschner PE. Hereditary paraganglioma due to the SDHD M1I mutation in a second Chinese family: a founder effect? Laryngoscope. 2003;113:1055–1058. doi: 10.1097/00005537-200306000-00026. [DOI] [PubMed] [Google Scholar]
- Lemonnier F, Cairns RA, Inoue S, Li WY, Dupuy A, Broutin S, Martin N, Fataccioli V, Pelletier R, Wakeham A, et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development. Proc Natl Acad Sci U S A. 2016;113:15084–15089. doi: 10.1073/pnas.1617929114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Cheng TL, Tsai WH, Tsai HJ, Hu KH, Chang HC, Yeh CW, Chen YC, Liao CC, Chang WT. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep. 2012;2:785. doi: 10.1038/srep00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo M, Wang YZ, Gout PW. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol. 2008;215:593–602. doi: 10.1002/jcp.21366. [DOI] [PubMed] [Google Scholar]
- Lycan TW, Pardee TS, Petty WJ, Bonomi M, Alistar A, Lamar ZS, Isom S, Chan MD, Miller AA, Ruiz J. A phase II clinical trial of CPI-613 in patients with relapsed or refractory small cell lung carcinoma. PLoS ONE. 2016;11:e0164244. doi: 10.1371/journal.pone.0164244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannelli M, Ercolino T, Giache V, Simi L, Cirami C, Parenti G. Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J Med Genet. 2007;44:586–587. doi: 10.1136/jmg.2007.051045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Márquez J, Alonso FJ, Matés JM, Segura JA, Martín-Rufián M, Campos-Sandoval JA. Glutamine addiction in gliomas. Neurochem Res. 2017;42(6):1735–1746. doi: 10.1007/s11064-017-2212-1. [DOI] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11–31. doi: 10.1038/nrclinonc.2016.60. [DOI] [PubMed] [Google Scholar]
- Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, Yuan C, Bao Y, Townsend MK, Tworoger SS, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med. 2014;20:1193–1198. doi: 10.1038/nm.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhinney SR, Pasini B, Stratakis CA. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357:1054–1056. doi: 10.1056/NEJMc071191. [DOI] [PubMed] [Google Scholar]
- Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31:272–281. doi: 10.1038/leu.2016.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Ropero S, Mehmi I, Atlas E, Colomer R, Lupu R. Overexpression and hyperactivity of breast cancer-associated fatty acid synthase (oncogenic antigen-519) is insensitive to normal arachidonic fatty acid-induced suppression in lipogenic tissues but it is selectively inhibited by tumoricidal alpha-linolenic and gamma-linolenic fatty acids: a novel mechanism by which dietary fat can alter mammary tumorigenesis. Int J Oncol. 2004;24:1369–1383. [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- Miettinen M, Lasota J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs)—a review. Int J Biochem Cell Biol. 2014;53:514–519. doi: 10.1016/j.biocel.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen M, Killian JK, Wang ZF, Lasota J, Lau C, Jones L, Walker R, Pineda M, Zhu YJ, Kim SY, et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am J Surg Pathol. 2013;37:234–240. doi: 10.1097/PAS.0b013e3182671178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, et al. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68:8547–8554. doi: 10.1158/0008-5472.CAN-08-1235. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugan AK, Bojdani E, Xing M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem Biophys Res Commun. 2010;393:555–559. doi: 10.1016/j.bbrc.2010.02.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- Neumann HP, Erlic Z, Boedeker CC, Rybicki LA, Robledo M, Hermsen M, Schiavi F, Falcioni M, Kwok P, Bauters C, et al. Clinical predictors for germline mutations in head and neck paraganglioma patients: cost reduction strategy in genetic diagnostic process as fall-out. Cancer Res. 2009;69:3650–3656. doi: 10.1158/0008-5472.CAN-08-4057. [DOI] [PubMed] [Google Scholar]
- Niemann S, Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- Obre E, Rossignol R. Emerging concepts in bioenergetics and cancer research: metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int J Biochem Cell Biol. 2015;59:167–181. doi: 10.1016/j.biocel.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Oettgen HF, Old LJ, Boyse EA, Campbell HA, Philips FS, Clarkson BD, Tallal L, Leeper RD, Schwartz MK, Kim JH. Inhibition of leukemias in man by L-asparaginase. Cancer Res. 1967;27:2619–2631. [PubMed] [Google Scholar]
- Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19:764–772. doi: 10.1158/1078-0432.CCR-12-3002. [DOI] [PubMed] [Google Scholar]
- Ostrow KL, Park HL, Hoque MO, Kim MS, Liu J, Argani P, Westra W, Van Criekinge W, Sidransky D. Pharmacologic unmasking of epigenetically silenced genes in breast cancer. Clin Cancer Res. 2009;15:1184–1191. doi: 10.1158/1078-0432.CCR-08-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudijk L, Gaal J, Korpershoek E, van Nederveen FH, Kelly L, Schiavon G, Verweij J, Mathijssen RH, den Bakker MA, Oldenburg RA, et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol. 2013;26:456–463. doi: 10.1038/modpathol.2012.186. [DOI] [PubMed] [Google Scholar]
- Ovejera AA, Houchens DP, Catane R, Sheridan MA, Muggia FM. Efficacy of 6-diazo-5-oxo-L-norleucine and N-[N-gamma-glutamyl-6-diazo-5-oxo-norleucinyl]-6-diazo-5-oxo-norleucine against experimental tumors in conventional and nude mice. Cancer Res. 1979;39:3220–3224. [PubMed] [Google Scholar]
- Paik JY, Toon CW, Benn DE, High H, Hasovitz C, Pavlakis N, Clifton-Bligh RJ, Gill AJ. Renal carcinoma associated with succinate dehydrogenase B mutation: a new and unique subtype of renal carcinoma. J Clin Oncol. 2014;32:e10–e13. doi: 10.1200/JCO.2012.47.2647. [DOI] [PubMed] [Google Scholar]
- Pantaleo MA, Astolfi A, Indio V, Moore R, Thiessen N, Heinrich MC, Gnocchi C, Santini D, Catena F, Formica S, et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst. 2011;103:983–987. doi: 10.1093/jnci/djr130. [DOI] [PubMed] [Google Scholar]
- Papathanassiou D, Bruna-Muraille C, Jouannaud C, Gagneux-Lemoussu L, Eschard JP, Liehn JC. Single-photon emission computed tomography combined with computed tomography (SPECT/CT) in bone diseases. Joint Bone Spine. 2009;76:474–480. doi: 10.1016/j.jbspin.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Pardanani A, Lasho TL, Finke CM, Mai M, McClure RF, Tefferi A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia. 2010;24:1146–1151. doi: 10.1038/leu.2010.77. [DOI] [PubMed] [Google Scholar]
- Pardee TS, Lee K, Luddy J, Maturo C, Rodriguez R, Isom S, Miller LD, Stadelman KM, Levitan D, Hurd D, et al. A phase I study of the first-in-class antimitochondrial metabolism agent, CPI-613, in patients with advanced hematologic malignancies. Clin Cancer Res. 2014;20:5255–5264. doi: 10.1158/1078-0432.CCR-14-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmentier JH, Maggi M, Tarasco E, Scotti C, Avramis VI, Mittelman SD. Glutaminase activity determines cytotoxicity of L-asparaginases on most leukemia cell lines. Leuk Res. 2015;39:757–762. doi: 10.1016/j.leukres.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie G, et al. Clinical and molecular genetics of patients with the Carney–Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- Pathania D, Millard M, Neamati N. Opportunities in discovery and delivery of anticancer drugs targeting mitochondria and cancer cell metabolism. Adv Drug Deliv Rev. 2009;61:1250–1275. doi: 10.1016/j.addr.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peczkowska M, Cascon A, Prejbisz A, Kubaszek A, Cwikla BJ, Furmanek M, Erlic Z, Eng C, Januszewicz A, Neumann HP. Extra-adrenal and adrenal pheochromocytomas associated with a germline SDHC mutation. Nat Clin Pract Endocrinol Metab. 2008;4:111–115. doi: 10.1038/ncpendmet0726. [DOI] [PubMed] [Google Scholar]
- Pfaffenroth EC, Linehan WM. Genetic basis for kidney cancer: opportunity for disease-specific approaches to therapy. Expert Opin Biol Ther. 2008;8:779–790. doi: 10.1517/14712598.8.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S, Gopalan V, Lo CY, Liew V, Smith RA, Lam AK. Silent genetic alterations identified by targeted next-generation sequencing in pheochromocytoma/paraganglioma: a clinicopathological correlations. Exp Mol Pathol. 2017;102:41–46. doi: 10.1016/j.yexmp.2016.12.007. [DOI] [PubMed] [Google Scholar]
- Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- Pusch S, Sahm F, Meyer J, Mittelbronn M, Hartmann C, von Deimling A. Glioma IDH1 mutation patterns off the beaten track. Neuropathol Appl Neurobiol. 2011;37:428–430. doi: 10.1111/j.1365-2990.2010.01127.x. [DOI] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979;254:2669–2676. [PubMed] [Google Scholar]
- Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, Maher ER. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260–1262. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- Rodman SN, Spence JM, Ronnfeldt TJ, Zhu Y, Solst SR, O’Neill RA, Allen BG, Guan X, Spitz DR, Fath MA. Enhancement of radiation response in breast cancer stem cells by inhibition of thioredoxin- and glutathione-dependent metabolism. Radiat Res. 2016;186:385–395. doi: 10.1667/RR14463.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016;381:96–103. doi: 10.1016/j.canlet.2016.07.035. [DOI] [PubMed] [Google Scholar]
- Rustin P, Bourgeron T, Parfait B, Chretien D, Munnich A, Rötig A. Inborn errors of the Krebs cycle: a group of unusual mitochondrial diseases in human. Biochim Biophys Acta. 1997;1361:185–197. doi: 10.1016/S0925-4439(97)00035-5. [DOI] [PubMed] [Google Scholar]
- Sajnani K, Islam F, Smith RA, Gopalan V, Lam AK. Genetic alterations in Krebs cycle and its impact on cancer pathogenesis. Biochimie. 2017;135:164–172. doi: 10.1016/j.biochi.2017.02.008. [DOI] [PubMed] [Google Scholar]
- Schimke RN, Collins DL, Stolle CA. Paraganglioma, neuroblastoma, and a SDHB mutation: resolution of a 30-year-old mystery. Am J Med Genet A. 2010;152A:1531–1535. doi: 10.1002/ajmg.a.33384. [DOI] [PubMed] [Google Scholar]
- Schlichtholz B, Turyn J, Goyke E, Biernacki M, Jaskiewicz K, Sledzinski Z, Swierczynski J. Enhanced citrate synthase activity in human pancreatic cancer. Pancreas. 2005;30:99–104. doi: 10.1097/01.mpa.0000153326.69816.7d. [DOI] [PubMed] [Google Scholar]
- Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, Tsukamoto T, Rojas CJ, Slusher BS, Rabinowitz JD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Sun B, Shi W, Zuo H, Cui D, Ni L, Chen J. Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biol. 2015;36:655–662. doi: 10.1007/s13277-014-2644-z. [DOI] [PubMed] [Google Scholar]
- Shitara K, Doi T, Nagano O, Fukutani M, Hasegawa H, Nomura S, Sato A, Kuwata T, Asai K, Einaga Y, et al. Phase 1 study of sulfasalazine and cisplatin for patients with CD44v-positive gastric cancer refractory to cisplatin (EPOC1407) Gastric Cancer. 2017 doi: 10.1007/s10120-017-0720-y. [DOI] [PubMed] [Google Scholar]
- Simi L, Sestini R, Ferruzzi P, Gagliano MS, Gensini F, Mascalchi M, Guerrini L, Pratesi C, Pinzani P, Nesi G, et al. Phenotype variability of neural crest derived tumours in six Italian families segregating the same founder SDHD mutation Q109X. J Med Genet. 2005;42:e52. doi: 10.1136/jmg.2004.030353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK, Desouki MM, Franklin RB, Costello LC. Mitochondrial aconitase and citrate metabolism in malignant and nonmalignant human prostate tissues. Mol Cancer. 2006;5:14. doi: 10.1186/1476-4598-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjursen W, Halvorsen H, Hofsli E, Bachke S, Berge A, Engebretsen LF, Falkmer SE, Falkmer UG, Varhaug JE. Mutation screening in a Norwegian cohort with pheochromocytoma. Fam Cancer. 2013;12:529–535. doi: 10.1007/s10689-013-9608-0. [DOI] [PubMed] [Google Scholar]
- Smit DL, Mensenkamp AR, Badeloe S, Breuning MH, Simon ME, van Spaendonck KY, Aalfs CM, Post JG, Shanley S, Krapels IP, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 2011;79:49–59. doi: 10.1111/j.1399-0004.2010.01486.x. [DOI] [PubMed] [Google Scholar]
- Snezhkina AV, Krasnov GS, Zaretsky AR, Zhavoronkov A, Nyushko KM, Moskalev AA, Karpova IY, Afremova AI, Lipatova AV, Kochetkov DV, et al. Differential expression of alternatively spliced transcripts related to energy metabolism in colorectal cancer. BMC Genomics. 2016;17:1011. doi: 10.1186/s12864-016-3351-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Strohecker AM, White E. Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy. 2014;10:384–385. doi: 10.4161/auto.27320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart SD, Schauble A, Gupta S, Kennedy AD, Keppler BR, Bingham PM, Zachar Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014;2:4. doi: 10.1186/2049-3002-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51:236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson RU, Shaw RJ (2017) Lipid synthesis is a metabolic liability of non-small cell lung cancer. In: Cold Spring Harbor symposia on quantitative biology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor [DOI] [PubMed]
- Swinnen JV, Roskams T, Joniau S, Van Poppel H, Oyen R, Baert L, Heyns W, Verhoeven G. Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. Int J Cancer. 2002;98:19–22. doi: 10.1002/ijc.10127. [DOI] [PubMed] [Google Scholar]
- Taschner PE, Jansen JC, Baysal BE, Bosch A, Rosenberg EH, Brocker-Vriends AH, van Der Mey AG, van Ommen GJ, Cornelisse CJ, Devilee P. Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer. 2001;31:274–281. doi: 10.1002/gcc.1144. [DOI] [PubMed] [Google Scholar]
- Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, Wiederschain D, Bedel O, Deng G, Zhang B, et al. Extreme vulnerability of IDH1 mutant cancers to NAD+ depletion. Cancer Cell. 2015;28:773–784. doi: 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, Turner ML, Stewart L, Duray P, Tourre O, Sharma N, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95–106. doi: 10.1086/376435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufton N, Roncaroli F, Hadjidemetriou I, Dang MN, Denes J, Guasti L, Thom M, Powell M, Baldeweg SE, Fersht N, et al. Pituitary carcinoma in a patient with an SDHB mutation. Endocr Pathol. 2017 doi: 10.1007/s12022-017-9474-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, Lehtonen R, Januszewicz A, Jarvinen H, Juhola M, et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. 2004;74:153–159. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstrom T, Henriksson MA. Impact of MYC in regulation of tumor cell metabolism. Biochim Biophys Acta. 2015;1849:563–569. doi: 10.1016/j.bbagrm.2014.07.004. [DOI] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Mai C, Wei YL, Zhao JJ, Hu YM, Zeng ZL, Yang J, Lu WH, Xu RH, Huang P. Decreased expression of the mitochondrial metabolic enzyme aconitase (ACO2) is associated with poor prognosis in gastric cancer. Med Oncol. 2013;30:552. doi: 10.1007/s12032-013-0552-5. [DOI] [PubMed] [Google Scholar]
- Wang MD, Wu H, Fu GB, Zhang HL, Zhou X, Tang L, Dong LW, Qin CJ, Huang S, Zhao LH, et al. Acetyl-coenzyme A carboxylase alpha promotion of glucose-mediated fatty acid synthesis enhances survival of hepatocellular carcinoma in mice and patients. Hepatology. 2016;63:1272–1286. doi: 10.1002/hep.28415. [DOI] [PubMed] [Google Scholar]
- Wang D, Yin L, Wei J, Yang Z, Jiang G. ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumour Biol. 2017;39:1010428317698338. doi: 10.1177/1010428317698338. [DOI] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol. 2012;4:a006783. doi: 10.1101/cshperspect.a006783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MH, Toure O, Glenn GM, Pithukpakorn M, Neckers L, Stolle C, Choyke P, Grubb R, Middelton L, Turner ML, et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet. 2006;43:18–27. doi: 10.1136/jmg.2005.033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MH, Tan CS, Lee SC, Yong Y, Ooi AS, Ngeow J, Tan MH. Potential genetic anticipation in hereditary leiomyomatosis-renal cell cancer (HLRCC) Fam Cancer. 2014;13:281–289. doi: 10.1007/s10689-014-9703-x. [DOI] [PubMed] [Google Scholar]
- Xin M, Qiao Z, Li J, Liu J, Song S, Zhao X, Miao P, Tang T, Wang L, Liu W, et al. miR-22 inhibits tumor growth and metastasis by targeting ATP citrate lyase: evidence in osteosarcoma, prostate cancer, cervical cancer and lung cancer. Oncotarget. 2016;7:44252–44265. doi: 10.18632/oncotarget.10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lane AN, Ricketts CJ, Sourbier C, Wei MH, Shuch B, Pike L, Wu M, Rouault TA, Boros LG, et al. Metabolic reprogramming for producing energy and reducing power in fumarate hydratase null cells from hereditary leiomyomatosis renal cell carcinoma. PLoS ONE. 2013;8:e72179. doi: 10.1371/journal.pone.0072179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Ternette N, Su H, Dabiri R, Kessler BM, Adam J, Teh BT, Pollard PJ. The succinated proteome of FH-mutant tumours. Metabolites. 2014;4:640–654. doi: 10.3390/metabo4030640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen KE, Bittinger MA, Su SM, Fantin VR. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene. 2010;29:6409–6417. doi: 10.1038/onc.2010.444. [DOI] [PubMed] [Google Scholar]
- Yen K, Travins J, Wang F, David MD, Artin E, Straley K, Padyana A, Gross S, DeLaBarre B, Tobin E, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7(5):478–493. doi: 10.1158/2159-8290.CD-16-1034. [DOI] [PubMed] [Google Scholar]
- Ylisaukko-oja SK, Cybulski C, Lehtonen R, Kiuru M, Matyjasik J, Szymanska A, Szymanska-Pasternak J, Dyrskjot L, Butzow R, Orntoft TF, et al. Germline fumarate hydratase mutations in patients with ovarian mucinous cystadenoma. Eur J Hum Genet. 2006;14:880–883. doi: 10.1038/sj.ejhg.5201630. [DOI] [PubMed] [Google Scholar]
- Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15:157–170. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachar Z, Marecek J, Maturo C, Gupta S, Stuart SD, Howell K, Schauble A, Lem J, Piramzadian A, Karnik S, et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents in vivo. J Mol Med (Berl) 2011;89:1137–1148. doi: 10.1007/s00109-011-0785-8. [DOI] [PubMed] [Google Scholar]
- Zantour B, Guilhaume B, Tissier F, Louvel A, Jeunemaitre X, Gimenez-Roqueplo AP, Bertagna X. A thyroid nodule revealing a paraganglioma in a patient with a new germline mutation in the succinate dehydrogenase B gene. Eur J Endocrinol. 2004;151:433–438. doi: 10.1530/eje.0.1510433. [DOI] [PubMed] [Google Scholar]
- Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci USA. 2011;108:16259–16264. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wei H, Tang K, Lin D, Zhang C, Mi Y, Wang L, Wang C, Wang M, Wang J. Mutation analysis of isocitrate dehydrogenase in acute lymphoblastic leukemia. Genet Test Mol Biomark. 2012;16:991–995. doi: 10.1089/gtmb.2011.0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38:1551–1566. doi: 10.1016/j.clinthera.2016.03.026. [DOI] [PubMed] [Google Scholar]