

Abstract
The Plasmodium falciparum variant surface antigen PfEMP1 expressed on the surface of infected erythrocytes is thought to play a major role in the pathology of severe malaria. As the sequence pool of the var genes encoding PfEMP1 expands there are opportunities, despite the high degree of sequence diversity demonstrated by this gene family, to reconstruct full-length var genes from small sequence tags generated from patient isolates. To test whether this is possible we have used a set of recently laboratory adapted ICAM-1-binding parasite isolates to generate sequence tags and, from these, to identify the full-length PfEMP1 being expressed by them. In a subset of the strains available we were able to produce validated, full-length var gene sequences and use these to conduct biophysical analyses of the ICAM-1 binding regions.
Introduction
The pathology of severe Plasmodium falciparum malaria is not simple and is thought to involve the ability of erythrocytes infected with this parasite to bind to host endothelial cells lining the microvasculature1. The protein responsible for most of this adhesion is the variant surface antigen Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1). This is encoded by a family of approximately 60 var genes per parasite genome with limited overlap of the var gene repertoires between isolates2–4. As PfEMP1 can be targeted by the host immune system, it undergoes a complex process of switching transcription between var gene family members5 resulting in (mostly) mutually exclusive expression of a single member of the family on the surface of the infected erythrocyte. Consistent with their role in avoiding immune clearance, the extracellular PfEMP1 sequences are highly variable consisting of DBL and CIDR regions that vary in their number, position and composition. Several studies have sought to relate motifs from PfEMP1s to aspects of malaria disease using var gene upstream sequences6, the relatively conserved N-terminal DBL1 domain7 and a more complex categorization of the combination of specific DBL and CIDR domain types in ‘domain cassettes’4. All of these have shown associations between classes of PfEMP1s and disease, most notably the upsA types and CIDR1α8,9.
Linked to the variable nature of PfEMP1 has been the expectation that the diversity in sequence and DBL/ CIDR composition would translate into variable adhesion characteristics of erythrocytes infected with parasites expressing different var genes. This is seen in several laboratory-adapted isolates for which the full-length sequences of the expressed PfEMP1s are known and regions of PfEMP1s have been mapped to specific binding behaviours1,10, revealing a wide range of potential receptors on host cells. However, it has been more challenging to link binding to a specific receptor with disease, with quite diverse results being reported, except for placental malaria and CSA11, and in the more recent case of binding to Endothelial Protein C Receptor (EPCR)12. Despite these difficulties, we are beginning to understand more about the functions of specific regions of PfEMP1, in particular the role of defined domains in binding to specific host receptors (e.g. CIDRα1 to EPCR; some DBLβ to ICAM-1) and thereby developing algorithms to understand the potential binding characteristics of a PfEMP1 variant from its sequence alone13. The challenge running in parallel with this is how to obtain the full-length PfEMP1 sequences from patient samples that may be limited in quantity.
There are molecular techniques to identify and sequence var genes but these are labour intensive and not suited to anything beyond limited throughput14. However, as more var gene sequences have become available in genome databases, while the diversity of the gene family is, as expected, very high, there are significant overlaps in var gene sequences coming from divergent parasite isolates. This latter finding suggests that it may be possible to look for existing var gene sequences for almost any PfEMP1 in the sequence databases and to use this information to provide a framework to generate de novo PfEMP1 full-length data from small sequence tags produced by RT-PCR from patient samples. To test whether this approach is feasible, we used a set of recently laboratory-adapted ICAM-1-binding isolates to see whether we could identify the full-length var gene sequences being expressed and then to use these to characterise their respective PfEMP1 binding characteristics.
Results
Identification of dominantly expressed var genes of culture-adapted patient isolates
In order to identify novel var genes that mediate binding to ICAM-1, three ICAM-1-selected, culture-adapted patient isolates – BC12, J1 and PCM7 – were re-selected on ICAM-1 and returned to culture for expansion before RNA extraction and cDNA synthesis. Expressed var genes were identified by PCR amplification of homology regions in the DBLα domain using universal primers15 to produce short sequence tags known as DBLα tags. Twenty-four DBLα tags from each culture-adapted patient isolate were cloned, sequenced and named alphabetically in order of cloning frequency. The number of unique sequences varied between isolates but all contained a prominent DBLα tag that was cloned at a high frequency, namely BC12a, J1a and PCM7a in isolates BC12, J1 and PCM7, respectively (Fig. 1A). Reverse transcriptase quantitative PCR (RT-qPCR), carried out with tag-specific primers designed against regions of variable sequence within the DBLα tag, confirmed that the most frequently cloned DBLα tag was in fact the most highly expressed tag for each isolate (Fig. 1B). DBLα tags J1b and J1d, from isolate J1, and PCM7d, from isolate PCM7, were also expressed at a relatively high level (Fig. 1B), suggesting they may also have a role in mediating ICAM-1-binding.
Figure 1.
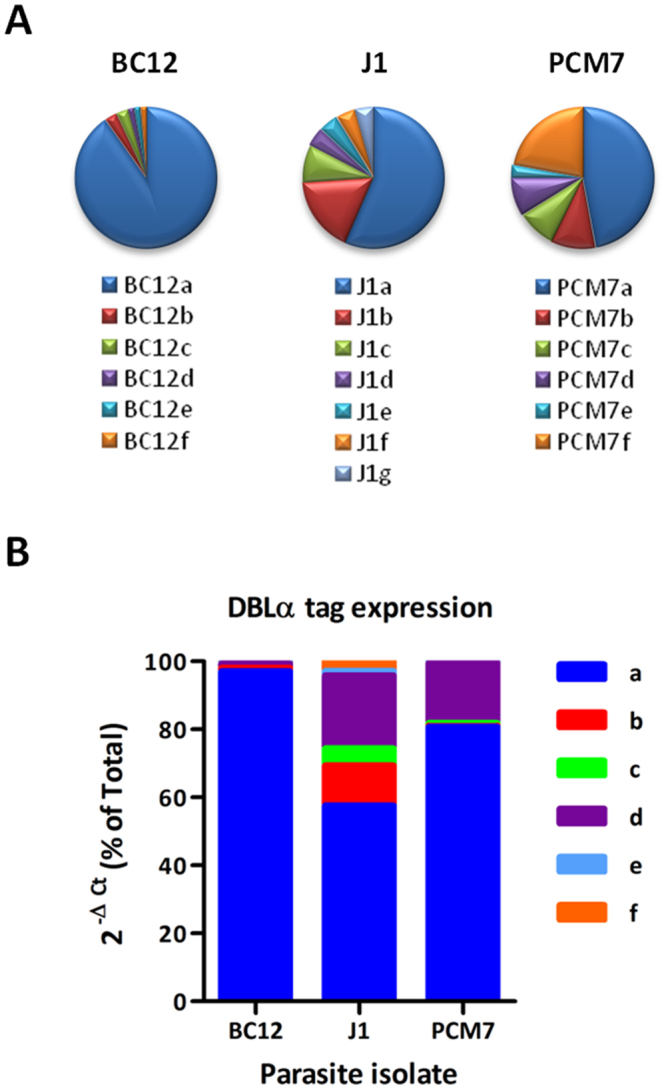
Identification of expressed DBLα tags of three ICAM-1 selected culture-adapted patient isolates. (A) Expressed DBLα tags were identified by RT-PCR of ICAM-1 selected parasite isolates BC12, J1 and PCM7, cloned and 24 tags from each sequenced by Sanger sequencing. Unique tags were named a, b, c, etc for each strain. PCM7f refers to a set of unique DBLα tags found only once in the cloning experiments, for which qPCR was not performed. (B) RT-qPCR was carried out with primers specific to each DBLα tag identified in A. Ct values were normalised against the ASL internal control gene and are shown as percentage of total for each cDNA. Results from three different RNA extractions for BC12 and PCM7 and a single RNA extraction for J1 are shown.
Reconstruction of full length var genes using the Pf3k var gene database
Sequencing var genes de novo is challenging due to their size (4.5–10 kb) and sequence composition which is highly variable with short regions of homology3,4. Here, we tested whether searching the only comprehensive var gene database for a match to a short DBLα tag can predict the full length var gene sequence. The highly expressed DBLα tags of BC12, J1 and PCM7 coming from the qRT-PCR analysis were BLAST searched against the Pf3k var gene database with parameters of at least 99% identity and 95% overlap. Five of the six DBLα tags resulted in ≤10 hit sequences each, whereas J1a resulted in 153 hits (Table 1). The sequences obtained from the tags have been submitted to Genbank (# MG220391 – MG220396 – Table 1).
Table 1.
Number of hits to each DBLα tag in the Pf3k var gene database detected by BLAST search.
| DBLα tag | Accession number | # database hits |
|---|---|---|
| BC12a | MG220391 | 2 |
| J1a | MG220392 | 153 |
| J1b | MG220393 | 1 |
| J1d | MG220394 | 3 |
| PCM7a | MG220395 | 4 |
| PCM7d | MG220396 | 10 |
The accession numbers refer to the extended var gene sequence for each tag.
The two returned sequences to BC12a share 99% sequence identity (based on measuring similarity across the whole length of the returned sequences and not restricted to the DBLα tag) and are therefore essentially the same gene. Similarly, the respective returned sequences to J1d and PCM7a, share 99% sequence identity within each group. Sequence hits to PCM7d (N = 10) shared 97–99% identity, with one hit containing an additional ~200 bp insertion. The 153 hits to J1a share sequence similarity over ~3 kb at the 5′ end but are variable downstream. Therefore, it appears that the more sequence hits that are returned from the DBLα tag search, the more likely it is that they will be of varying full-length sequence, at least in our small sample set.
Returned sequence hits to each DBLα tag were chosen as reference sequences for primer design based on length and consensus sequence. Primers were designed every ~1 kb to produce overlapping products which were cloned and sequenced. In addition, we performed 5′ upstream (UPS) typing using primers specific for UPS types A, B and C6 which revealed BC12a, J1d, PCM7a and PCM7d to be UPS B type and J1a UPS C type. We were unable to UPS type J1b. BC12a, J1a and J1b fragments shared ≥99% identity with their respective reference sequences (Fig. 2A,B and C). J1d fragments downstream of the DBLα tag shared ≥99% identity with its reference sequence but the UPS fragment (J1d_1-567) had no significant similarity in the coding region (Fig. 2D). PCM7a fragments amplified across three quarters of the length of the reference gene with ≥99% identity but the remaining fragments failed to amplify (Fig. 2E). PCM7d fragments also shared ≥99% identity with their reference sequence with the exception of two 3′ fragments whose sequences were highly AT rich, suggesting that priming occurred within the intron (Fig. 2F, denoted by question marks). A conserved primer to exon 216 was used to fully sequence PCM7a and J1b. However, we were unable to gain the complete exon 1 sequence of genes BC12a, J1a, J1d and PCM7d due to either multiple products of the exon 2 PCR or cloning difficulty (data not shown).
Figure 2.
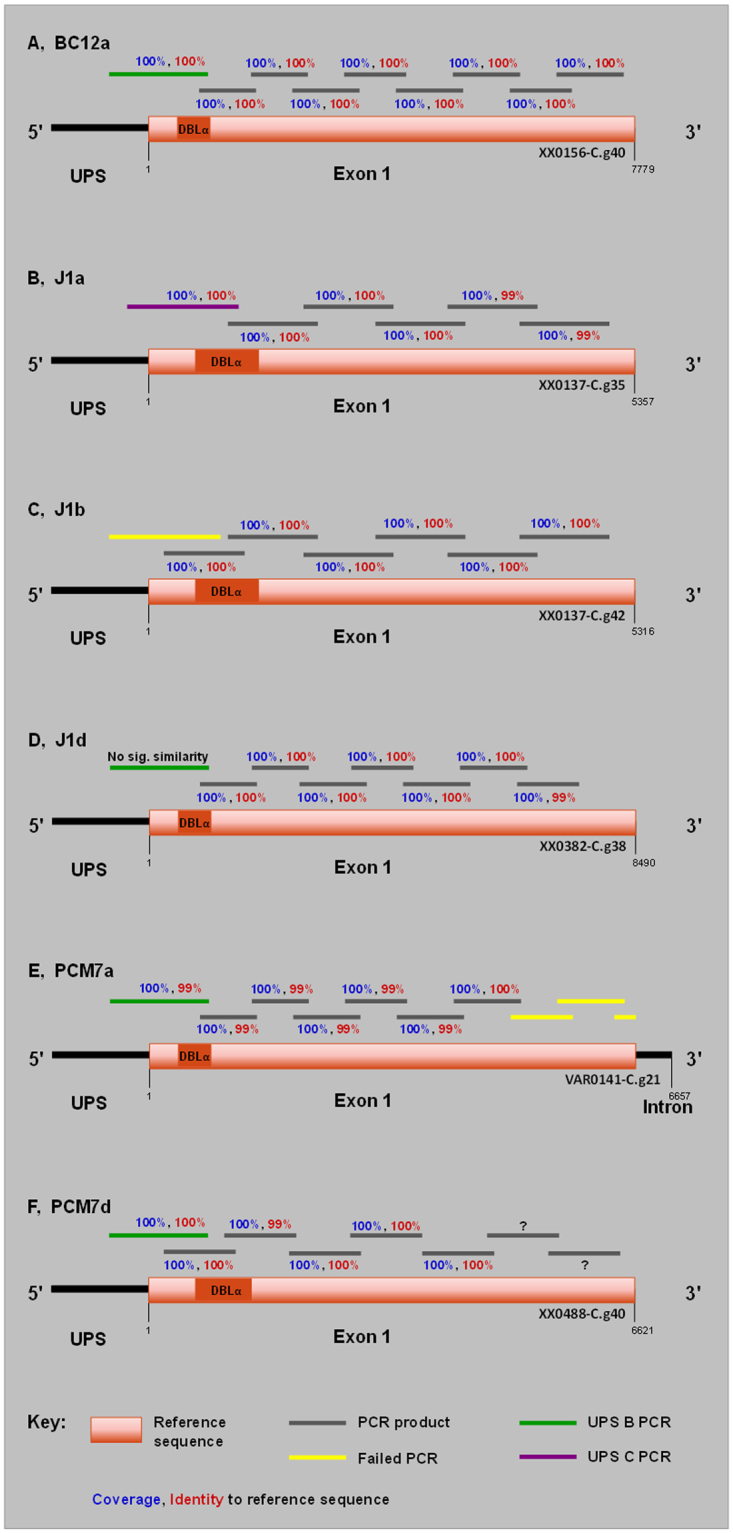
Schematic of var gene reconstruction using Pf3k var gene database matches to newly identified DBLα tags. The highly expressed DBLα tags of BC12 (A), J1 (B–D) and PCM7 (E,F) were BLAST searched against the Pf3k var gene database. Resulting matches were treated as reference sequences (orange) for PCR primer design resulting in ~1 kb overlapping products amplified from genomic DNA, represented by grey bars. PCR products were cloned, sequenced and compared to their respective reference sequences, with percentage coverage (blue) and identity (red) shown. Upstream typing was also performed using UPS A, B and C specific primers6 in combination with a tag-specific primer. Sequence similarity of UPS fragments to that of the coding reference sequence is shown. Failed PCRs are shown in yellow. Question marks in F denote amplified products of highly AT-rich sequence, suggesting priming occurred within an intron. All primer sequences are listed in Table S1.
Definition of PfEMP1 domain structure by sequence analysis
The domain composition of PfEMP1 proteins determines host receptor binding, with different domain types and subtypes known to mediate binding specificity. We analysed the reconstructed var gene sequences to obtain their PfEMP1 domain structure. Domain boundaries and types were defined by the VarDom 1.0 Server and each domain was BLAST searched against the 7 genomes data set4 to identify subtypes. Domain structures are represented schematically in Fig. 3. All are of DBLα type 0 with varying sub-classifications, a type almost exclusively associated with UPS B and C var genes4, concordant with our UPS typing. Similarly, the six genes analysed all contain CIDRα subtypes predicted to bind CD36, a phenotype of UPS B and C type var genes17. BC12a, J1a and J1d contain DBLβ5 which is predicted to mediate binding to ICAM-118,19. The DBLβ domains of PCM7a and PCM7d had BLAST hits to two different domain subtypes – PCM7a: 5/8 and PCM7d: 3/5. Three DBLβ3 domains tested have been shown to bind ICAM-1 but several others do not18–20. Interestingly, J1b does not contain a DBLβ domain, suggesting that it is not involved in ICAM-1 binding. The J1b DBLα tag represented 12% of var gene expression in the J1 isolate (Fig. 1B) and may be the result of a switching event after ICAM-1 selection.
Figure 3.

PfEMP1 domain and DC annotation of var genes reconstructed using the Pf3k database. Domain boundaries were defined by the VarDom 1.0 Server and individual domains BLAST searched against the 7 genomes dataset in Rask et al.4. The top hits were analysed and domain subtypes identified based on consensus sequence and coverage. Where multiple subtypes were identified, both are shown if they are of equal standing (e.g. 0.4/12) or excluded if more than two subtypes displayed similar coverage. Dashed lines outline domain cassettes (DC) with the DC number shown in the far-right column. ND: not defined, no matches by BLAST search.
Sequence to phenotype analysis of DBLβ domains predicted to bind ICAM-1
We analysed the 5 newly identified DBLβ domains of the ICAM-1 binding patient isolates in more detail by comparing them to known DBLβ sequences. Due to the highly recombinogenic nature of var genes, phylogenetic trees do not adequately capture the evolutionary history of DBL domains. We therefore chose to carry out a global sequence similarity approach by constructing a blast distance matrix of all DBLβ domains identified in the Rask et al. dataset4 together with the newly identified sequences obtained here from BC12a, J1a, J1d, PCM7a and PCM7d. The results demonstrate the diverse nature of the DBLβ domains (Fig. 4) with no specific clustering of the ICAM-1 binding domains.
Figure 4.

Blast distance matrix of DBLβ domains. Visualization of all-against-all blast of all DBLβ domains described in the 7 genomes dataset4 and the DBLβ domains identified in this study from ICAM-1 binding patient isolates BC12, J1 and PCM7. The two columns of blue barcodes show the clustering of the newly obtained DBLβ domains and the known ICAM-1 binding domains including the DBLβ domains of the dominantly expressed var genes of BC12, J1 and PCM7.
The majority of DBLβ binding studies have been carried out on the IT4 parasite isolate. Therefore, a second comparison was performed that was restricted to our newly identified DBLβ domains together with those from IT418, five from the 3D7 clone and one from the Dd2 clone that have been tested for ICAM-1 binding (Fig. 5). In this more limited comparison, 12/15 of the ICAM-1 binders cluster very well together at the top of the figure. Of the three that do not, ITvars16 and 41 are next to each other but as far as possible from the main cluster and Dd2var32 appears alone in the middle. 10/15 of the ICAM-1 binding domains are of the UPS B type, three are UPS A and two are UPS C. Overall, it appears that within this more limited group, sequence similarity is an important contributor to ICAM-1 specificity.
Figure 5.

Blast distance matrix of newly identified DBLβ domains and those previously tested for ICAM-1 binding. Visualization of an all-against-all blast of all IT4 DBLβ domains, five DBLβ domains from 3D7 and one from Dd2 tested for ICAM-1 binding and the dominantly expressed DBLβ domains identified in this study. To each domain we associated annotation on the right, ICAM-1 binding, DBLβ types and the UPS types (as from the VarDom 1.0 Server). ICAM-1 binding domains are highlighted within the columns ICAM-1 binder with a *, non-binders with a - and those not tested for ICAM-1 binding is empty18–20,39. UPS type is indicated in boxes at branch ends, along with the DBLβ type.
Confirmation of ICAM-1 binding of BC12a and J1a recombinant DBLβ domains
BC12a and J1a DBLβ domains were expressed in an E. coli expression system and purified to homogeneity (Fig. 6A and B). Correct protein folding was confirmed by circular dichroism spectroscopy showing the presence of a predominantly α-helical protein, as expected for a DBL domain21 (Fig. 6C). Surface plasmon resonance (SPR) was used to test binding of BC12aDBLβ and J1aDBLβ to ICAM-1D1D5 and obtain kinetic parameters of the interaction. We included the previously characterised IT4var13DBLβ 22 for comparison. All SPR experiments were carried out using purified monomeric fractions shown in Fig. 6A and B. Binding kinetics of IT4var13DBLβ to ICAM-1D1D5 display a fast association rate and a slow dissociation rate, with a KD of 3.45 nM (Fig. 6F and Table 2), comparable to published values for this domain22. BC12aDBLβ and J1aDBLβ both bind to ICAM-1D1D5 with mid-nanomolar affinities of 197 nM and 51.7 nM respectively (Table 2). While these affinities are lower than those of IT4var13DBLβ, due to slower on-rates and faster off-rates, this variability in affinity of PfEMP1 domains for a single ligand is indeed often seen in the case of other domains that bind to ICAM-1, EPCR or CD365,22–24.
Figure 6.

DBLβ domains of BC12a and J1a bind ICAM-1D1D5 with nanomolar affinity. Size exclusion chromatographs and purity by SDS-PAGE are shown for BC12DBLβ (A) and J1aDBLβ (B). The majority of BC12DBLβ is present in aggregated form with a small proportion of monomeric protein (A). J1aDBLβ is predominantly monomeric with some aggregated protein evidenced by presence of a large shoulder (B). mAu, miliabsorbance units. The purified monomeric samples used in the SPR studies are shown in the gels to the right of the chromatographs in (A) and (B), with full gels shown in Fig. S1 (BC12) and Fig. S2 (J1a). (C) CD analysis of secondary structure. CD spectra were recorded between 195 and 260 nm at 20 °C. For each protein, four measurements were averaged and corrected for buffer absorption. D-F, ICAM-1D1D5 was coupled to a sensor chip surface (1100 RU) and DBLβ domains were injected at 30 µl/min with an association time of 240 seconds and a dissociation time of 600 seconds. Shown are sensorgrams for the binding to ICAM-1D1D5 of BC12aDBLβ (D), J1aDBLβ (E) and IT4var13DBLβ (F). Data (black lines) are modelled to a 1:1 global interaction model (red lines). Shown are sensorgrams for the sequential binding of IT4var13DBLβ and BC12aDBLβ to ICAM-1D1D5 (G) and IT4var41DBLβ and J1aDBLβ to ICAM-1D1D5 (H), blue lines. The response to the second domain alone is shown in red. Data for Fig. 6 is shown in S2–S4 Data.
Table 2.
Kinetic parameters derived from SPR experiments.
| Interaction | ka (X 104 M−1 s−1) | kd (X 10−4 s−1) | KD (nM) |
|---|---|---|---|
| ICAM-1 D1D5:: BC12aDBLβ | 0.48 | 9.44 | 197 |
| ICAM-1 D1D5:: J1aDBLβ | 2.96 | 15.3 | 51.7 |
| ICAM-1 D1D5:: IT4var13DBLβ | 3.61 | 1.25 | 3.45 |
We also used an SPR competition assay to test whether the DBLβ domains have an overlapping binding surface. This involves sequential injection of DBLβ domains over the ICAM-1D1D5 surface with the expectation that if the domains have separate binding surfaces, an additive response will be observed, whereas a shared binding surface will not increase the response on addition of the second domain. We performed this experiment with the injection of IT4var13DBLβ followed by BC12aDBLβ (Fig. 6G) and, in a separate experiment, with the injection of IT4var41DBLβ followed by J1aDBLβ (Fig. 6H). In each case, the injection of the second domain did not result in additional response units to that of the first domain, suggesting that the DBLβ domains tested here have an overlapping binding site.
Binding of BC12aDBLβ, J1aDBLβ and IT4var13DBLβ to ICAM-1 mutant proteins
In an effort to better understand the differences in binding kinetics between the DBLβ domains, we used SPR to test binding of recombinant BC12aDBLβ, J1aDBLβ and IT4var13DBLβ to four ICAM-1 mutant proteins (ICAM-1S22A, ICAM-1Kilifi, ICAM-1L42A and ICAM-1L44A) that have been shown to have differential effects on binding of different parasite isolates25,26. Each of the four ICAM-1 mutations obliterated binding of BC12aDBLβ with only minimal signal detected (Table 3 and Fig. S3). Binding to all mutants is <8% of the full-length five domain reference ICAM-1 (ICAM-1D1D5) binding, although it should be noted that signals are close to background noise levels and, therefore, may not be accurate and are presented as representative values only. Similarly, J1aDBLβ only minimally interacted with the ICAM-1 mutant proteins (Table 3 and Fig. S4). Binding to ICAM-1Kilifi, ICAM-1L42A and ICAM-1L44A is ≤5% of wild type ICAM-1D1D5 binding. ICAM-1S22A binding is slightly higher at 11% of wild type binding but is still very low.
Table 3.
The effect of ICAM-1 mutations on DBLβ domain binding.
| % ICAM-1D1D5 binding | ||||
|---|---|---|---|---|
| ICAM-1S22A | ICAM-1Kilifi | ICAM-1L42A | ICAM-1L44A | |
| BC12aDBLβ | 2.9 | 7.7 | 6.7 | 6.7 |
| J1aDBLβ | 10.9 | 5.4 | 1.4 | 2.7 |
| IT4var13DBLβ | 94.5 | 8.9 | 3.1 | 10 |
IT4var13DBLβ also had minimal interaction with ICAM-1Kilifi, ICAM-1L42A and ICAM-1L44A which is ≤10% of ICAM-1D1D5 binding (Table 3 and Fig. S5). However, IT4var13DBLβ did bind to ICAM-1S22A with 9.82 nM affinity. Therefore, the S22A mutation decreases IT4var13DBLβ binding affinity for ICAM-1 but only by 5% (Table 3). This is a minor effect compared to BC12aDBLβ and J1aDBLβ for which binding is reduced by ≥90%, suggesting that this residue is not critical for IT4var13DBLβ binding.
Overall, the mutant ICAM-1 binding assays suggest that the DBLβ domains share an overlapping but not identical binding surface in the area of the mutations. The Kilifi, L42A and L44A mutations dramatically reduce the binding ability of all three DBLβ domains. The S22A mutation has a similarly dramatic effect on BC12aDBLβ and J1aDBLβ binding but has minimal effect on IT4var13DBLβ binding, suggesting a specific difference in contact residues in this area.
Flow adhesion assays
The effect of the ICAM-1 mutations on recombinant DBLβ domain binding does not reflect the initial parasite binding study which assessed IE binding in static adhesion assays26. In light of this, we performed flow adhesion assays to test binding of BC12, J1 and IT4var13 parasite isolates to ICAM-1D1D5 and the four ICAM-1 mutant proteins (Fig. S6) (Data for the flow assays is shown in S1 Data). In the majority of cases, the ICAM-1 mutations resulted in a 16–56% reduction in binding of all three parasite lines, not reaching statistical significance (Fig. S6(A) and (B)). Exceptions are the L42A and L44A mutations. ICAM-1L42A resulted in a 78 and 76% reduction of J1 and IT4var13 binding, respectively, and ICAM-1L44A resulted in an 88% reduction of IT4var13 binding. RT-qPCR confirmed the dominantly expressed var gene of BC12 to be BC12a and IT4var13 to be var13 (Fig. S6(C)). The dominant var of J1 is J1a which represents 62% of expressed vars with a number of other var genes making up the remaining 38% (Fig. S6). This includes the previously identified secondary tags J1d and J1b and also includes J1f which is represented at a higher level than previously observed (Fig. 1B). Thus, while there are common outcomes in binding between parasite flow adhesion assays and recombinant DBLβ domain SPR assays, such as the impact of the L42A and L44A mutations, these findings also reveal differences in results from the two formats.
Discussion
Identifying expressed var genes in Plasmodium falciparum infected erythrocytes often starts using DBLα tag primers15 that target conserved homology blocks of the DBLα domain and are thought to amplify the majority of var genes. Indeed, such primers are frequently used in surveys of var gene diversity6,27–29. However, universal amplification by these primers cannot be known for certain due to the high variability of var genes. This, combined with reliance on cloning, which can carry bias towards some sequences, results in uncertainty that all expressed DBLα tags have been identified. However, in our data the most frequently cloned tag did prove to be the dominantly expressed tag in all isolates, indicating that any bias only affects secondary tags, at least in these culture-adapted isolates. The over-representation of some minor tags identified by cloning has been observed previously30 and highlights the importance of the more quantitative RT-qPCR method. Repeating ICAM-1 selection and RNA extraction increases confidence in this method, as was performed on BC12 and PCM7 isolates (3 replicates each). However, this proved difficult with isolate J1 (1 RNA extraction) which tended to produce gametocytes. This was also the case with several other isolates from the initial characterisation study26 from which sufficient RNA could not be extracted due to gametocytogenesis (data not shown).
We performed ICAM-1 selection of these isolates to identify new ICAM-1 binding var genes but this is not necessarily selecting for a clonal population, as multiple var genes can still be expressed within a selected culture. Therefore, expression of secondary tags in J1 and PCM7 can be explained by multiple var genes within an isolate with the ability to bind ICAM-1, as seen with multiple IT4 var genes18. Alternatively, var gene switching may have taken place after selection which occurs in an ordered manner and involves initially switching to numerous genes before a single dominant gene emerges31. We minimised the effect of switching by extracting RNA as quickly as possible after ICAM-1 selection. Whether the secondary var genes identified here also have the ability to bind ICAM-1 is yet to be determined.
The var gene database, which is a result of the Pf3k genome sequencing project, is a new resource with the potential to provide valuable information on an unprecedented scale. However, the copious amount of ‘omics’ data now being generated presents the challenge of interpretation and of identifying practical applications. We wanted to test whether this resource can predict full length var genes from their short ~400 bp DBLα tags. The results presented here from ICAM-1 binding patient isolates were remarkable, with the majority of successfully cloned fragments sharing ≥99% identity with the reference sequence (Fig. 2). The two cases of differential sequence (J1d_1-567 and PCM7a_4798-6762) occurred at a single point in the sequence suggesting a gene recombination event or possibly a sequence assembly error. The former is likely as it has been reported that mitotic recombination frequently occurs in var genes2,32. Interestingly, recombination occurs between domains of the same type resulting in in-frame products and domain structure preservation, which produces viable genes2, concordant with our results. A later search of the database containing 2512 parasite genomes found the presence of PCM7a_4798-6762 sequence and, in addition, the full length (PCM7a_1-6762) sequence (data not shown). This is perhaps unsurprising as the Pf3k project is ongoing and the database is constantly expanding. It must also be kept in mind that these genes are constantly undergoing recombination events, both mitotic2,32 and meiotic in the mosquito vector33. Therefore, although the sequences might be known, the order in which they are combined in any one gene is still unpredictable.
Limitations of the database include the varying number of hits to each DBLα tag, in particular J1a for which there were 153 hits, and the length of contiguous sequence, which varies in the database and may only provide partial sequence. In practice, there are ways to overcome some of the limitations, for example, we were able to sequence the 3′ end of var genes of J1b and PCM7a using a conserved exon 2 primer16. However, we were unable to clone and sequence exon 2 PCR products of BC12a, J1a, J1d or PCM7d and could only make size predictions of all except the PCM7d products. The un-sequenced region of J1a is the longest and is the result of having 153 returned database hits which differed significantly from the original tag sequence in the 3′ end (although several of these returned hits were identical to each other) making specific primer design impossible in this region.
Classification of var genes is based on a combination of upstream sequence, genomic location and direction of transcription3,34. Group A genes have been associated with severe disease35–37 as well as group B in some studies37,38. The majority of ICAM-1 binding var genes identified in the reference strains IT4 and 3D7 are group B and C type18,19 with only two group A ICAM-1 binders identified in 3D7: PF11_052139,40 and PFD1235w20,35. Both UPS A ICAM-1 binding genes have been shown to induce adhesion blocking (rat) antibodies to their recombinant domains20,40, with PFD1235w specific antibodies additionally able to block binding of other DC4 expressing parasites, displaying cross-reactivity20. The dominantly expressed var genes of our ICAM-1 binding patient isolates are UPS B type and one UPS C type, an unsurprising result given that group A var genes are seldom expressed in vitro19,41,42. This is thought to be related to high switching bias towards centrally located and/or short var genes (4 extracellular domains), all of which are UPS B or C, rather than the specific avoidance of UPS A gene expression43. Assuming IT4 and 3D7 strains are representative of all parasites then we expect only 0–2 group A ICAM-1 binding var genes per genome and their identification from these patient isolates would require alternative methods to those employed here. For example, whole genome sequencing would identify the full var repertoires of these isolates and allow analysis of their predicted binding functions. However, sequence predictions alone cannot fully predict ICAM-1 binding.
The recombinogenic nature of var genes precludes their accurate analysis by phylogenetics. We tried to overcome this difficulty in associating sequence data with phenotype by using a similarity method that does not relate to evolutionary history, but clusters by global identity. It is interesting that the limited set of ICAM-1-binding domains do tend to cluster based on sequence identity but represent different DBLβ and UPS types (Fig. 5). However, a comparison of amino-acids from our newly sequenced DBLβ domains with those identified as important in ICAM-1 binding of UPS A, DC4-containing PfEMP144 revealed very little sequence conservation (data not shown), suggesting the binding of UPS B and C var genes to ICAM-1 is mediated by distinct amino-acids. Further evidence of distinct UPS A binding sites is provided by blocking of binding to ICAM-1 by antibody recognising DC4 DBLβ domains in this class of variants but not being blocked by antibody recognising other DBLβ ICAM-1-binding domains (ITvar16 DBLβ5)20. This could point to two distinct origins of ICAM-1 binding, segregating UPS A from UPS B and C var genes44. Such segregation would be maintained by the mechanisms of var gene recombination which occur between domains of the same type and are mainly restricted to within UPS groups due to chromosomal location and orientation of transcription2,3,45,46.
We successfully expressed and purified the newly identified DBLβ domains BC12aDBLβ and J1aDBLβ, from the dominantly expressed var genes of the ICAM-1 selected patient isolates BC12 and J1, respectively, to >90% purity and performed SPR to assess ICAM-1 binding, along with the previously characterised IT4var13DBLβ. BC12aDBLβ, J1aDBLβ and IT4var13DBLβ, that all bind ICAM-1D1D5 with nanomolar affinity. IT4var13DBLβ has the strongest interaction, followed by J1aDBLβ and BC12aDBLβ (KD values: 3.45, 51.7, 197 nM, respectively, Fig. 6 and Table 2). These KD values are similar to those found previously for five IT4 DBLβ domains, tested in comparable SPR assays, which ranged from 2.6 nM for IT4var13DBLβ (comparable to our 3.45 nM value for this domain) to 144 nM for IT4var31DBLβ 22. The UPS A type, DC4 DBLβ domain was also found to bind ICAM-1 with nanomolar affinity (7.9 nM) in an SPR assay where the DBLβ domain was immobilised on the chip and ICAM-1D1D2 passed over44. All three domains studied here are DBLβ5 type and these findings fit with previous observations that, to date, all domains of this type mediate ICAM-1 binding19. The difference in binding kinetics between IT4var13DBLβ and J1aDBLβ is very interesting. They have a similar fast association rate which can be thought of as necessary to overcome the flow rate of the capillaries to mediate IE binding. Indeed, ICAM-1 has been shown to improve efficiency of IE capture under flow conditions47. However, they differ in their dissociation rates. IT4var13DBLβ has a slow dissociation rate which fits with stationary adhesion of the IE. J1aDBLβ, however, has a very fast dissociation rate which could suggest that this domain could mediate rolling adhesion, in which the IE constantly attaches and detaches from the capillary wall in a rolling motion47,48, although care needs to be taken in extrapolating these types of data to the situation in vivo.
The KD value is calculated based on the association and dissociation rates. In this case, the kinetic parameters fit with the response units recorded for each domain, i.e. the highest response was by IT4var13DBLβ, followed by J1aDBLβ then BC12aDBLβ. The lower binding responses recorded for J1aDBLβ and BC12aDBLβ are in agreement with the initial parasite binding study which found both parasite isolates to be low avidity ICAM-1-binders, with BC12 having a lower binding rate than J1 (1362 and 2391 IE/mm2, respectively)26. Interestingly, the IT4var13 parasite line has a lower binding rate (900 IE/mm2) than both BC12 and J1 in static adhesion assays49, a result that is also apparent in our flow adhesion assays (Fig. S6), highlighting differential results between IEs and recombinant protein. There is no clear correlation between KD values of DBLβ domains and the number of bound IEs in static adhesion assays. For example, IT4var16DBLβ has the same KD as J1aDBLβ (51.1 and 51.7 nM, respectively) but parasites expressing IT4var16 have more than twice the binding rate of J1 parasites in static assays (5000 and 2391 IE/mm2, respectively)22,26,49. There could be several explanations for this based on the display of host receptors on plastic and in a membrane, as well as the context of the DBL domain in its corresponding PfEMP1. The binding of an infected erythrocyte to an endothelial surface is far more complex that a simple protein-protein interaction, and many factors, including PfEMP1 architecture, PfEMP1 expression levels and knob-numbers will come into play in addition to the monovalent binding affinities measured in our SPR experiment, making a direct relationship between binding affinity and infected erythrocyte adhesion unlikely.
The DBLβ binding site on ICAM-1 has been identified by mutagenesis studies and a number of ICAM-1 blocking antibodies as the BED side of D125,50,51, plus the structure of the ICAM-1/DC4 (upsA) PfEMP1 complex has been solved52. The four ICAM-1 mutants utilized in this study have been shown to have differential effects on the binding of different parasite isolates25,26. In our SPR assays, the Kilifi (K29M), L42A and L44A mutations dramatically reduce the binding ability of all three recombinant DBLβ domains by ≥90%. The S22A mutation similarly reduces BC12aDBLβ and J1aDBLβ binding by ≥90% but has minimal effect on IT4var13DBLβ binding, suggesting a specific difference in contact residues in this area. When the surface architecture of ICAM-1 is considered (Fig. 7), the S22 residue is near the edge of the area identified as important for DBLβ binding. It is likely that IT4var13DBLβ has a structural difference in the area that contacts here, resulting in the lack of effect of this mutation. This has implications for development of therapeutics such as drugs or antibody therapy, which should focus around the L42 and L44 residues and possibly extend to K29 (mutated in ICAM-1Kilifi) to confer a cross-blocking effect.
Figure 7.
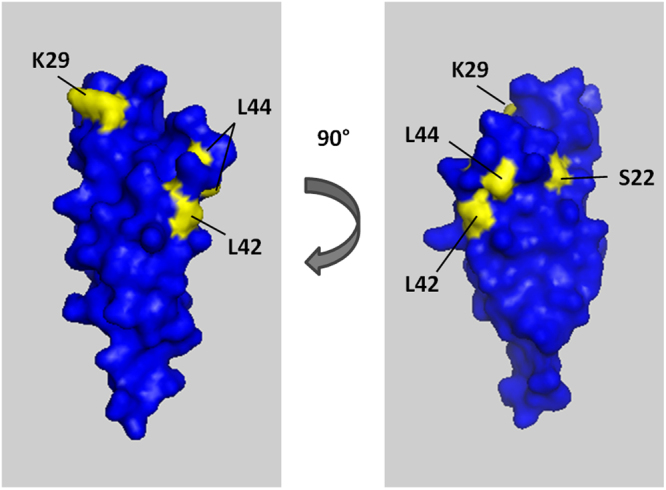
The crystal structure of ICAM-1 D1, showing the surface architecture. Residues of the mutated proteins tested in this study are labelled and coloured yellow. K29 residue is mutated to M in ICAM-1Kilifi. Structure accessed via Protein Data Bank, deposited by61.
In summary, we have shown that it is possible to reconstruct the exon 1 sequence of var genes from short tags by comparison with the large var gene database produced as part of the Pf3K project. This has the potential to open up the field by making full-length PfEMP1 sequences available for analysis in conjunction with clinical and cellular properties of the corresponding patient-derived parasite isolates.
Materials and Methods
Parasite culture
Laboratory-adapted patient isolates BC12, J1 and PCM7, from uncomplicated malaria patients in Thailand53, were collected with consent in previous clinical studies. All patient-derived material, such as white blood cells, have been removed during culture. IT4var13 is a laboratory line that binds to ICAM-1 with high affinity22,54. All parasites were cultured in O+ human erythrocytes at 2% haematocrit, according to standard techniques55 using RPMI-1640 medium (Sigma) supplemented with 37.5 mM HEPES, 7 mM D-glucose, 25 ng/ml gentamicin sulphate, 2 mM L-glutamine and 10% human serum (Haematology Department, Royal Liverpool University Hospital) at pH 7.2, in a gas mixture of 96% nitrogen, 3% carbon dioxide and 1% oxygen.
Parasite selection on ICAM-1
50 µl Protein A Dynabeads (Invitrogen) were washed three times with 500 µl 1% BSA/PBS, using a magnet to retain the beads each time. The beads were resuspended in 1% BSA/PBS and 2.5 µg ICAM-1D1D5 (an Fc fusion protein of ICAM-1 domains 1–525) added to a final volume of 400 µl and incubated at room temperature with 15 rpm rotation for 1 hour. The beads were then washed three times with 500 µl 1% BSA/PBS using a magnet to retain the beads and resuspended in 200 µl 1% BSA/PBS. Parasite cultures were enriched for trophozoite stage using Plasmion (Fresenius Kabi France) by standard techniques56. Enriched parasites were resuspended in 200 µl 1% BSA/PBS and added to the ICAM-1D1D5 labelled Dynabead suspension. The mixture was rotated at 15 rpm for 45 min at room temperature. Two washes were carried out with 500 µl 1% BSA/PBS to remove unbound parasites. The beads were then transferred to a new flask and cultured as described.
RNA extraction and processing
RNA was extracted as described57 with minor modifications. Parasite cultures were pelleted by centrifugation at 600 × g for 5 min. The supernatant was aspirated and the pellet resuspended by tapping before addition of 10 × pellet volume of pre-warmed TRIzol® Reagent (Ambion) and incubated at 37 °C for at least 5 min. TRIzol® lysed parasites were split into 1 ml aliquots and either frozen at −80 °C or RNA extraction continued immediately. To each 1 ml aliquot, 0.2 ml chloroform was added and the tube shaken vigorously for 15 seconds, left to stand for 2–3 min and centrifuged at 1,200 × g for 30 min at 4 °C. The upper aqueous layer was removed to a new 1.5 ml tube whereupon 0.5 ml isopropanol was added and mixed by inverting the tube several times before incubation at 4 °C for at least 2 hours. The tube was mixed again by inverting several times and centrifuged at 16,200 × g for 30 min at 4 °C. The supernatant was carefully removed and the pellet washed in 0.5 ml 75% ethanol in DEPC-H2O (diethyl pyrocarbonate treated H2O, Sigma). After centrifugation at 16,200 × g for 5 min, the supernatant was removed and the pellet left to air dry at room temperature. 50 µl DEPC-H2O was added to the pellet before incubation at 65 °C for 10 min and then placed on ice.
1.2 µg RNA was treated with DNase I (Amplification Grade, Sigma-Aldrich) according to manufacturer’s instructions. cDNA synthesis was performed with Tetro cDNA Synthesis Kit (Bioline) following the manufacturer’s instructions. The reaction comprised 8 µl DNase treated RNA (~480 ng), 1 µl Oligo (dT)18 primer, dNTP at 0.5 mM final concentration, 4 µl 5 × RT Buffer, 10 U RiboSafe RNase Inhibitor, 200 U Tetro Reverse Transcriptase and DEPC-H2O up to 20 µl. The reaction was incubated at 45 °C for 30 min, followed by 85 °C for 5 min to stop the reaction and chilled on ice.
Genomic DNA extraction
Genomic DNA (gDNA) was extracted from parasite cultures by saponin lysis of the RBC and the QIAamp® DNA Blood Mini Kit (Qiagen). Parasite cultures were centrifuged at 600 × g for 5 min and the resulting pellet was resuspended in 5 ml 0.15% saponin and incubated for 5 min on ice, mixing each minute. Parasites were centrifuged at 600 × g for 5 min and washed with PBS 2–3 times. Following the final centrifugation, the parasite pellet was resuspended in 200 µl PBS and transferred to a 1.5 ml tube containing 20 µl Qiagen protease. The QIAamp® DNA Blood Mini Kit (Qiagen) spin protocol was followed with a final elution of gDNA in 100 µl Buffer AE (10 mM Tris- HCl, 0.5 mM EDTA, pH 9).
Primer design and polymerase chain reaction (PCR)
All novel primer sequences were designed with the aid of OligoCalc58. Primer sequences from previously published work are reproduced with the appropriate citation. All primer sequences are listed in Supplementary Table S1 along with annealing temperature.
All PCR reactions were carried out with the proofreading TaKaRa LA Taq® DNA polymerase (Clontech, TaKaRa Bio Inc) according to the manufacturer’s instructions. Each reaction comprised 10 × LA PCR Buffer II, magnesium chloride (MgCl2) at 2.5 mM final concentration, dNTP at 1 mM final concentration, primers at 0.3 mM final concentration each, 2.5 units of TaKaRa LA Taq®, 2 µl template and sterile water up to 50 µl final volume. The DBLα tag was amplified from parasite cDNA using the primers DBLαAF′ and DBLαBR15. Reaction conditions were an initial denaturing step of 95 °C for 3 min, followed by 30 cycles of 95 °C for 30 sec, 47 °C for 30 sec, 65 °C for 30 sec, and a final extension of 65 °C for 3 min. 5′ UPS PCR was carried out using primers unique to the three main UPS types A, B and C6 in combination with a reverse DBLα specific primer designed for RT-qPCR (see Table S1). Reaction conditions were as follows: 95 °C for 3 min, followed by 30 cycles of 95 °C for 30 sec, 52 °C for 30 sec, 65 °C for 30 sec, and a final extension of 65 °C for 3 min. Downstream PCR was carried out using a primer designed to the more conserved exon 216 in combination with an isolate specific forward primer (see Table S1). Reaction conditions: 96 °C for 1 minute, followed by 40 cycles of 98 °C for 6 seconds, 48 °C for 15 seconds, 67 °C for 5 minutes, and a final extension of 68 °C for 10 minutes. PCR of overlapping var gene fragments was carried out using primers listed in Table S1 with annealing temperatures as indication and reaction conditions of 95 °C for 3 min, followed by 30 cycles of 95 °C for 30 sec, 52–55 °C for 30 sec, 65 °C for 1 min, and a final extension of 65 °C for 3 min.
Cloning, plasmid preparation and sequencing
PCR products were cloned into pCRTM4-TOPO® vector (Invitrogen) at 10:1 insert to vector molar ratio according to manufacturer’s instructions. Plasmids were transformed into One Shot® TOP10 E. coli competent cells (Invitrogen), grown overnight and the plasmids isolated using standard methods. Plasmid sequencing was carried out by either the Core Genomic Facility, University of Sheffield or Source Bioscience Sequencing, Rochdale UK. Sequences were visualised with Chromas Lite version 2.1 (Technelysium Pty Ltd, latest version available at http://technelysium.com.au/) and alignments were carried out using ClustalX 2.159 on default settings.
Reverse transcriptase quantitative PCR (RT-qPCR)
RT-qPCR of cDNA was carried out using Brilliant III Ultra-Fast SYBR® Green QPCR Master Mix (Agilent Technologies). Standard curves were generated for each new primer set by performing 10-fold serial dilutions of cDNA to produce five different concentrations of starting material. No-RT and DEPC-H2O reactions were carried out as negative controls. Each reaction comprised 10 µl 2 × SYBR Green QPCR master mix, forward and reverse primers each at 0.5 µM final concentration (see Table S1 for primer sequences), 2 µl template (cDNA, no-RT or DEPC-H2O) and DEPC-H2O to 20 µl final volume. Adenylosuccinate lyase (ASL) was used as an internal control gene60. Reactions were run on an MxPro-Mx3005P machine under the following conditions: 95 °C for 3 min, 40 cycles of 95 °C for 10 seconds, 60 °C for 10 seconds, and a final cycle of 95 °C for 1 min, 55 °C for 30 seconds, 95 °C for 30 seconds.
Primer efficiency was calculated from the gradient of the standard curve using the equation E = 101/−m, where E is efficiency and m is the gradient of the curve. RT-qPCR was repeated on cDNA at a single concentration with the same reagents and conditions (as above) using all primer pairs designed to particular isolates on the same plate. Ct values were normalised against the internal control gene using the equation 2−ΔCt, where ΔCt is the mean Ct value of the gene of interest minus the mean Ct value of the internal control gene. 2−ΔCt values were transformed into percentage of the total for each parasite isolate.
Pf3k var gene database search, primer design and sequencing
Pf3k is a project set up as part of MalariaGEN which aims to sequence 3,000 Plasmodium falciparum genomes and is led by the University of Oxford, the Wellcome Trust Sanger Institute and the Broad Institute (https://www.malariagen.net/projects/parasite/pf3k). As a result, the var genes from over 2,500 clinical isolates are assessable (http://www.sanger.ac.uk/cgi-bin/blast/submitblast/p_falciparum-pf3k). The short DBLα tags generated in this study were BLAST searched against the database containing 1468 parasite genomes with parameters of at least 99% identity and 95% overlap. The returned sequence hits were used as reference genes to design primers (see Table S1) with overlapping products of ~1 kb along the length of the gene. The primers were tested on parasite gDNA (see above for PCR conditions) and the products cloned into pCRTM4-TOPO® vector (Invitrogen) and sequenced as described. Overlapping sequences were assembled to reconstruct each var gene.
Sequence analysis
Reconstructed var gene sequences were translated to amino acid sequence and entered into the VarDom 1.0 Server4 (available at http://www.cbs.dtu.dk/services/VarDom/) in FASTA format to define domain boundaries. The corresponding nucleic acid sequences were then separated into domains and BLAST searched against the entire 7 genomes dataset4. The top six hits were analysed and domain subtypes were identified. Subtypes were included if a consensus between the six hits was reached or if the top hits had appropriate coverage. Where multiple subtypes were identified, they were included if there were two of equal standing (separated by “/”) or excluded if there were more than two matches with similar coverage.
Similarity matrixes were generated as follows. First domains were compared all-against-all with BLASTp, E value 1-e6. The identity of the first hit was normalised by the mean of the two compared domains, resulting in a matrix of identity between all domains, ranging from 0–100. This matrix was visualized in R using the heatmap.2 function from the gplots library. To cluster the domains, the “distance method” from the heatmap.2 function was used.
All previously described DBLβ sequences were downloaded from the VarDom 1.0 Server4.
Recombinant protein expression and purification
DBLβ domains of BC12a and J1a were PCR amplified using primers shown in Table S1, subcloned into a modified pET15b vector and expressed as an N-terminal, hexahistidine tagged protein in Escherichia coli SHuffle 3030 cells (New England Biolabs) at 25 °C for 16 hours. Cells were pelleted and lysed, and the DBLβ domains purified by affinity chromatography using nickel-nitrilotriacetic acid agarose (Ni-NTA, Qiagen) under native conditions. DBLβ domains were further purified by gel filtration on a HiLoad 16/600 Superdex 75 prep grade column (GE healthcare).
IT4var13DBLβ, IT4var41DBLβ22, ICAM-1D1D544, ICAM-1Kilifi, ICAM-1S22A, ICAM-1L42A, ICAM-1L44A25 were expressed and purified as described previously.
CD spectroscopy
Far-UV CD spectroscopy experiments were carried out in a J-815 Spectropolarimeter (Jasco) equipped with a computer-controlled Peltier temperature control unit. Measurements were carried out in 100 mM Na-phosphate buffer, 200 mM NaF, pH 7.2 at a protein concentration of 0.3 mg/ml using a 1 mM path cell. All measurements were taken at 20 °C between 195 and 260 nm wavelengths. Four spectra were recorded for each sample, averaged and corrected for buffer absorption.
Surface plasmon resonance (SPR)
SPR was carried out on a Biacore T200 machine (GE Healthcare). Protein A was immobilised onto a CM5 chip (GE Healthcare) by amine coupling. All experiments were carried out in buffer containing 10 mM HEPES, pH 7.2, 250 mM NaCl and 0.05% Tween- 20, filter sterilised and degassed. ICAM-1 proteins fused to the Fc region of human IgG1 (ICAM-1D1D544, ICAM-1Kilifi, ICAM-1S22A, ICAM-1L42A, ICAM-1L44A25) were captured onto the Protein A surface. Coupling of the different ICAM-1 proteins to the chip was kept comparable at 1108 ± 108 response units (RU). DBLβ domains were flowed over the chip as a twofold dilution series (1000 nM–7.81 nM) at a flow rate of 30 µl/min with an association time of 240 seconds and a dissociation time of 600 seconds. The chip was regenerated after each concentration with the injection of 10 mM Glycine- HCl, pH 1.7 for 120 seconds at a flow rate of 10 µl/min which breaks the Protein A- Fc interaction. The signal from a flow channel without ICAM-1 was subtracted from all measurements. Single stranded DNA (ssDNA, Salmon Sperm DNA sodium salt, AppliChem) was added to BC12aDBLβ to 1 mg/ml final concentration prior to injection to minimise background binding observed during test runs. Injection of ssDNA only had no effect on the signal (data not shown). Sensorgrams were fitted to a global 1:1 interaction model allowing calculation of kinetic values ka, kd and KD using BIAevaluation software 2.0.3 (GE Healthcare).
SPR competition assays were carried out with ICAM-1D1D5 under the same conditions outlined above except that only one concentration of DBLβ was used (adjusted to give the same molar ratio of each protein). The first domain was injected at a flow rate of 30 µl/min with an association time of 240 seconds. The second domain was then injected over the same chip at a flow rate of 30 µl/min with an association time of 240 seconds and a dissociation time of 600 seconds.
Flow adhesion assay
Flow adhesion assays were carried out with the VenaFlux semi-automated microfluidic system and VenaFlux software (Cellix). The Vena8 Fluoro + TM protein biochips (Cellix) used contain 8 channels which allow several proteins to be tested in immediate succession. Individual biochip channels were coated with 4 µl of 50 µg/ml of either ICAM-1D1D5, ICAM-1S22A, ICAM-1Kilifi, ICAM-1L42A or ICAM-1L44A protein and incubated at 37 °C for 1 hour in a humidified petri dish. The protein was then aspirated and the channel blocked with 1% BSA/PBS for either 1 hour at 37 °C or overnight at 4 °C. Channels were warmed to 37 °C before use. Infected erythrocytes (IE) were prepared in binding buffer (RMPI 1640 powder (Invitrogen) dissolved in H2O, pH 7.2) at 3% parasitaemia and 2% haematocrit. The assay followed the Cellix protocol. The protein coated biochip was attached to a microscope stage within a plastic chamber whose temperature was 37 °C. The IE were drawn through the channel at a flow rate of 0.04 Pa for 8 min before washing with binding buffer. Bound parasites were counted in 7–10 microscope fields and the numbers adjusted to IE/mm2.
All methods were carried out in accordance with relevant guidelines and regulations. No patient material was used in this study. Parasite isolates used in this work were laboratory adapted and their derivation, including ethical clearance, is covered in the references cited. Human red blood cells for parasite culture were obtained commercially from the UK Blood Transfusion Service (NHSBT) and used under licence from the UK Human Tissue Authority.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files) or are publicly available at www.malariagen.net/data/pf3k-5.
Electronic supplementary material
Acknowledgements
The authors would like to thank Dr Matt Berriman, Dr Yang Wu and Dr Aymen Madkhali for their support. This publication uses data generated by the Pf3K Project (2016): pilot data release 5. www.malariagen.net/data/pf3k-5. This work was supported by grants from The Wellcome Trust to CIN (104792/Z/14/Z) and AGC (095507/Z/11/Z).
Author Contributions
E.C. and A.G.C. wrote the main manuscript text. E.C. carried out all of the technical work, with support from F.L. (Figs 6 & 7 and Tables 2 & 3) and T.S. (all other Figs). T.D.O. and C.I.N. carried out the BLAST searches and analyses for var gene matches and compiled the var gene database. E.C., M.H., T.O., C.N. and A.G.C. analysed the data. E.C. and T.O. prepared all the figures. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-21591-8.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med. 2013;19:156–167. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claessens A, et al. Generation of Antigenic Diversity in Plasmodium falciparum by Structured Rearrangement of Var Genes During Mitosis. PLoS Genet. 2014;10:e1004812. doi: 10.1371/journal.pgen.1004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardner MJ, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rask, T. S., Hansen, D. A., Theander, T. G., Gorm Pedersen, A. & Lavstsen, T. Plasmodium falciparum erythrocyte membrane protein 1 diversity in seven genomes–divide and conquer. PLoS Comput Biol6, 10.1371/journal.pcbi.1000933 (2010). [DOI] [PMC free article] [PubMed]
- 5.Petter M, Duffy MF. Antigenic Variation in Plasmodium falciparum. Results Probl Cell Differ. 2015;57:47–90. doi: 10.1007/978-3-319-20819-0_3. [DOI] [PubMed] [Google Scholar]
- 6.Mugasa J, Qi W, Rusch S, Rottmann M, Beck HP. Genetic diversity of expressed Plasmodium falciparum var genes from Tanzanian children with severe malaria. Malar J. 2012;11:230. doi: 10.1186/1475-2875-11-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warimwe GM, et al. Prognostic indicators of life-threatening malaria are associated with distinct parasite variant antigen profiles. Sci Transl Med. 2012;4:129ra145. doi: 10.1126/scitranslmed.3003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hviid L, Jensen AT. PfEMP1 - A Parasite Protein Family of Key Importance in Plasmodium falciparum Malaria Immunity and Pathogenesis. Adv Parasitol. 2015;88:51–84. doi: 10.1016/bs.apar.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Bernabeu, M. & Smith, J. D. EPCR and Malaria Severity: The Center of a Perfect Storm. Trends Parasitol, 10.1016/j.pt.2016.11.004 (2016). [DOI] [PMC free article] [PubMed]
- 10.Rowe, J. A., Claessens, A., Corrigan, R. A. & Arman, M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert Rev Mol Med11, e16, 10.1017/S1462399409001082 (2009). [DOI] [PMC free article] [PubMed]
- 11.Sharma L, Shukla G. Placental Malaria: A New Insight into thePathophysiology. Front Med (Lausanne) 2017;4:117. doi: 10.3389/fmed.2017.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turner L, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013;498:502–505. doi: 10.1038/nature12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith JD. The role of PfEMP1 adhesion domain classification in Plasmodium falciparum pathogenesis research. Mol Biochem Parasitol. 2014;195:82–87. doi: 10.1016/j.molbiopara.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jespersen JS, et al. Plasmodium falciparum var genes expressed in children with severe malaria encode CIDRalpha1 domains. EMBO molecular medicine. 2016;8:839–850. doi: 10.15252/emmm.201606188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bull PC, et al. Plasmodium falciparum variant surface antigen expression patterns during malaria. PLoS Pathog. 2005;1:e26. doi: 10.1371/journal.ppat.0010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavstsen T, et al. Plasmodium falciparum erythrocyte membrane protein 1 domain cassettes 8 and 13 are associated with severe malaria in children. Proc Natl Acad Sci USA. 2012;109:E1791–1800. doi: 10.1073/pnas.1120455109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herricks T, Avril M, Janes J, Smith JD, Rathod PK. Clonal variants of Plasmodium falciparum exhibit a narrow range of rolling velocities to host receptor CD36 under dynamic flow conditions. Eukaryot Cell. 2013;12:1490–1498. doi: 10.1128/EC.00148-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howell, D. P. et al. Mapping a common interaction site used by Plasmodium falciparum Duffy binding-like domains to bind diverse host receptors. Mol Microbiol67, 78–87, 10.1111/j.1365-2958.2007.06019.x (2008). [DOI] [PubMed]
- 19.Janes JH, et al. Investigating the host binding signature on the Plasmodium falciparum PfEMP1 protein family. PLoS Pathog. 2011;7:e1002032. doi: 10.1371/journal.ppat.1002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bengtsson A, et al. A novel domain cassette identifies Plasmodium falciparum PfEMP1 proteins binding ICAM-1 and is a target of cross-reactive, adhesion-inhibitory antibodies. J Immunol. 2013;190:240–249. doi: 10.4049/jimmunol.1202578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higgins MK, Carrington M. Sequence variation and structural conservation allows development of novel function and immune evasion in parasite surface protein families. Protein Sci. 2014;23:354–365. doi: 10.1002/pro.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown A, et al. Molecular architecture of a complex between an adhesion protein from the malaria parasite and intracellular adhesion molecule 1. J Biol Chem. 2013;288:5992–6003. doi: 10.1074/jbc.M112.416347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lau CK, et al. Structural conservation despite huge sequence diversity allows EPCR binding by the PfEMP1 family implicated in severe childhood malaria. Cell Host Microbe. 2015;17:118–129. doi: 10.1016/j.chom.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh FL, et al. The structural basis for CD36 binding by the malaria parasite. Nat Commun. 2016;7:12837. doi: 10.1038/ncomms12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tse MT, Chakrabarti K, Gray C, Chitnis CE, Craig A. Divergent binding sites on intercellular adhesion molecule-1 (ICAM-1) for variant Plasmodium falciparum isolates. Mol Microbiol. 2004;51:1039–1049. doi: 10.1046/j.1365-2958.2003.03895.x. [DOI] [PubMed] [Google Scholar]
- 26.Madkhali AM, et al. An analysis of the binding characteristics of a panel of recently selected ICAM-1 binding Plasmodium falciparum patient isolates. PLoS One. 2014;9:e111518. doi: 10.1371/journal.pone.0111518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sulistyaningsih E, Fitri LE, Loscher T, Berens-Riha N. Diversity of the var gene family of Indonesian Plasmodium falciparum isolates. Malar J. 2013;12:80. doi: 10.1186/1475-2875-12-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen DS, et al. A molecular epidemiological study of var gene diversity to characterize the reservoir of Plasmodium falciparum in humans in Africa. PLoS One. 2011;6:e16629. doi: 10.1371/journal.pone.0016629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Albrecht, L. et al. The South American Plasmodium falciparum var gene repertoire is limited, highly shared and possibly lacks several antigenic types. Gene453, 37–44, S0378-1119(10)00015-6 (2010). [DOI] [PubMed]
- 30.Gatton, M. L. et al. Detection sensitivity and quantitation of Plasmodium falciparum var gene transcripts by real-time RT-PCR in comparison with conventional RT-PCR. Am J Trop Med Hyg75, 212–218, 10.4269/ajtmh.2006.75.212 (2006). [PMC free article] [PubMed]
- 31.Recker M, et al. Antigenic variation in Plasmodium falciparum malaria involves a highly structured switching pattern. PLoS Pathog. 2011;7:e1001306. doi: 10.1371/journal.ppat.1001306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bopp SE, et al. Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 2013;9:e1003293. doi: 10.1371/journal.pgen.1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ranford-Cartwright LC, Mwangi JM. Analysis of malaria parasite phenotypes using experimental genetic crosses of Plasmodium falciparum. Int J Parasitol. 2012;42:529–534. doi: 10.1016/j.ijpara.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voss, T. S. et al. Genomic distribution and functional characterisation of two distinct and conserved Plasmodium falciparum var gene 5′ flanking sequences. Mol Biochem Parasitol107, 103–115, 10.1016/S0166-6851(00)00176-6 (2000). [DOI] [PubMed]
- 35.Jensen AT, et al. Plasmodium falciparum associated with severe childhood malaria preferentially expresses PfEMP1 encoded by group A var genes. J Exp Med. 2004;199:1179–1190. doi: 10.1084/jem.20040274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kyriacou, H. M. et al. Differential var gene transcription in Plasmodium falciparum isolates from patients with cerebral malaria compared to hyperparasitaemia. Mol Biochem Parasitol150, 211–218, 10.1016/j.molbiopara.2006.08.005 (2006). [DOI] [PMC free article] [PubMed]
- 37.Rottmann, M. et al. Differential expression of var gene groups is associated with morbidity caused by Plasmodium falciparum infection in Tanzanian children. Infect Immun74, 3904–3911, 10.1128/IAI.02073-05 (2006). [DOI] [PMC free article] [PubMed]
- 38.Kaestli, M. et al. Virulence of malaria is associated with differential expression of Plasmodium falciparum var gene subgroups in a case-control study. J Infect Dis193, 1567–1574, 10.1086/503776 (2006). [DOI] [PMC free article] [PubMed]
- 39.Oleinikov AV, et al. High throughput functional assays of the variant antigen PfEMP1 reveal a single domain in the 3D7 Plasmodium falciparum genome that binds ICAM1 with high affinity and is targeted by naturally acquired neutralizing antibodies. PLoS Pathog. 2009;5:e1000386. doi: 10.1371/journal.ppat.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gullingsrud J, Saveria T, Amos E, Duffy PE, Oleinikov AV. Structure-function-immunogenicity studies of PfEMP1 domain DBL2betaPF11_0521, a malaria parasite ligand for ICAM-1. PLoS One. 2013;8:e61323. doi: 10.1371/journal.pone.0061323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peters, J. M., Fowler, E. V., Krause, D. R., Cheng, Q. & Gatton, M. L. Differential changes in Plasmodium falciparum var transcription during adaptation to culture. J Infect Dis195, 748–755, 10.1086/511436 (2007). [DOI] [PMC free article] [PubMed]
- 42.Ye R, et al. Transcription of the var genes from a freshly-obtained field isolate of Plasmodium falciparum shows more variable switching patterns than long laboratory-adapted isolates. Malar J. 2015;14:66. doi: 10.1186/s12936-015-0565-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noble R, et al. The antigenic switching network of Plasmodium falciparum and its implications for the immuno-epidemiology of malaria. eLife. 2013;2:e01074. doi: 10.7554/eLife.01074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lennartz F, et al. Mapping the Binding Site of a Cross-Reactive Plasmodium falciparum PfEMP1 Monoclonal Antibody Inhibitory of ICAM-1 Binding. J Immunol. 2015;195:3273–3283. doi: 10.4049/jimmunol.1501404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kraemer, S. M. & Smith, J. D. Evidence for the importance of genetic structuring to the structural and functional specialization of the Plasmodium falciparum var gene family. Mol Microbiol50, 1527–1538, 10.1046/j.1365-2958.2003.03814.x (2003). [DOI] [PubMed]
- 46.Kraemer, S. M. et al. Patterns of gene recombination shape var gene repertoires in Plasmodium falciparum: comparisons of geographically diverse isolates. BMC Genomics8, 45, 10.1186/1471-2164-8-45 (2007). [DOI] [PMC free article] [PubMed]
- 47.Gray, C., McCormick, C., Turner, G. & Craig, A. ICAM-1 can play a major role in mediating P. falciparum adhesion to endothelium under flow. Mol Biochem Parasitol 128, 187–193, 10.1016/S0166-6851(03)00075-6 (2003). [DOI] [PubMed]
- 48.Cooke BM, et al. Rolling and stationary cytoadhesion of red blood cells parasitized by Plasmodium falciparum: separate roles for ICAM-1, CD36 and thrombospondin. Br J Haematol. 1994;87:162–170. doi: 10.1111/j.1365-2141.1994.tb04887.x. [DOI] [PubMed] [Google Scholar]
- 49.Madkhali, A. M. Adhesion analysis of different PfEMP1 variants to CD36, ICAM-1 and primary endothelial cells. PhD, University of Liverpool (2015).
- 50.Berendt, A. R. et al. The binding site on ICAM-1 for Plasmodium falciparum-infected erythrocytes overlaps, but is distinct from, the LFA-1-binding site. Cell68, 71–81, 10.1016/0092-8674(92)90207-S (1992). [DOI] [PubMed]
- 51.Ockenhouse, C. F., Betageri, R., Springer, T. A. & Staunton, D. E. Plasmodium falciparum-infected erythrocytes bind ICAM-1 at a site distinct from LFA-1, Mac-1, and human rhinovirus. Cell68, 63–69, 10.1016/0092-8674(92)90206-R (1992). [DOI] [PubMed]
- 52.Lennartz F, et al. Structure-Guided Identification of a Family of Dual Receptor-Binding PfEMP1 that Is Associated with Cerebral Malaria. Cell Host Microbe. 2017;21:403–414. doi: 10.1016/j.chom.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poyomtip T, et al. Polymorphisms of the pfmdr1 but not the pfnhe-1 gene is associated with in vitro quinine sensitivity in Thai isolates of Plasmodium falciparum. Malar J. 2012;11:7. doi: 10.1186/1475-2875-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Avril M, et al. A restricted subset of var genes mediates adherence of Plasmodium falciparum-infected erythrocytes to brain endothelial cells. Proc Natl Acad Sci USA. 2012;109:E1782–1790. doi: 10.1073/pnas.1120534109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- 56.Lelievre, J., Berry, A. & Benoit-Vical, F. An alternative method for Plasmodium culture synchronization. Exp Parasitol109, 195–197, 10.1016/j.exppara.2004.11.012 (2005). [DOI] [PubMed]
- 57.Kyes, S., Pinches, R. & Newbold, C. A simple RNA analysis method shows var and rif multigene family expression patterns in Plasmodium falciparum. Mol Biochem Parasitol 105, 311–315, 10.1016/S0166-6851(99)00193-0 (2000). [DOI] [PubMed]
- 58.Kibbe WA. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007;35:W43–46. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 60.Viebig, N. K. et al. A single member of the Plasmodium falciparum var multigene family determines cytoadhesion to the placental receptor chondroitin sulphate A. EMBO Rep6, 775–781, 10.1038/sj.embor.7400466 (2005). [DOI] [PMC free article] [PubMed]
- 61.Casasnovas JM, Bickford JK, Springer TA. The domain structure of ICAM-1 and the kinetics of binding to rhinovirus. J Virol. 1998;72:6244–6246. doi: 10.1128/jvi.72.7.6244-6246.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its Supplementary Information files) or are publicly available at www.malariagen.net/data/pf3k-5.