

Abstract
Ovarian fibrosarcomas are extremely rare tumors with little genomic information available to date. In the present report we present the tumoral exome and transcriptome and the germinal exome of an ovarian fibrosarcoma from a 9-years old child. We found a paucity of mutations (0.77/Mb) and CNV alterations. Of these, the most relevant were a point mutation in the metal-binding site of the microRNA-processing DICER1 enzyme and a frame-shift alteration in the tumor suppressor gene NF1. We validated a germinal truncating mutation in DICER1, which was consistent with a DICER1 Syndrome diagnosis, providing the first example of an ovarian fibrosarcoma as the presenting neoplasia in this syndrome. Network and enrichment analyses showed that both a mesenchymal signature and a Hedgehog cascade could be driving the progression of this tumor. We were also able to find a global lincRNA deregulation, as the number of lincRNAs transcripts expressed in the tumor was decreased, with a concomitant upregulation of previously described non-coding transcripts associated with cancer, such as MALAT1, MIR181A1HG, CASC1, XIST and FENDRR. DICER1 Syndrome should be considered as a possible diagnosis in children ovarian fibrosarcoma. The role of lncRNAs in neoplasias associated with DICER1 alterations need to be studied in more detail.
Introduction
Ovarian fibrosarcomas are rare and aggressive tumors. These sex cord-stromal neoplasias are derived from the stromal component of the ovary and give rise to large, highly vascular and mitotically-active tumors. Ovarian fibrosarcomas usually present in older adults, with a median age of presentation of 49 years, sometimes associated to co-morbidities such as Maffucci´s syndrome1, naevoid basal cell carcinoma syndrome2, or other congenital syndromes3. Less than 5% of the reported cases are under 10 years old, including a case in an 8-year-old girl with nevoid basal-cell carcinoma syndrome2,4,5. Very little is known about the molecular characteristics of these tumors.
Recently, germline truncating mutations in the microRNA-processing protein DICER1 gene have been reported in patients with pleuropulmonary blastoma (PPB) or the related familiar DICER1 syndrome which includes, besides PPB, cystic nephroma, Sertoli-Leydig cell tumors, medulloepitheliomas and embryonal rhabdomyosarcomas6. In addition, somatic mutations in the RNAse IIIb domain have been also found in a discrete number of tumors, being common only in non-epithelial ovarian cancers, in which the prevalence can be as a high as 60%7. MicroRNAs are small non-coding RNAs that regulate the degradation of messenger RNAs. MicroRNA precursor (pre-miRNAs) transcripts are cleaved by the endoribonuclease Dicer1 into mature miRNA. Dicer´s RNAse IIIb domain cleaves the 5′-arm of the pre-miRNA and loads it to the RNA-induced silencing complex (RISC)8. It has been previously shown that DICER1 acts via haploinsufficiency since the lack of full DICER1 activity contributes to oncogenesis, without the need of a “hit” in the second allele9. Nevertheless, recent reports suggested that DICER1 could be acting as an oncogenic driver. In this model, an initial loss of the germinal DICER1 activity in patients with DICER syndrome would make primitive cells susceptible to oncogenesis by a second mutation in a RNAse IIIb “hotspot”, altering microRNA processing10.
Due to their rarity, there are no deep characterizations of the molecular alterations in these ovarian fibrosarcomas. In the present article, we present the first genomic report of this rare neoplasia from a 9-years old child and provide evidence that this neoplasia needs to be added as a presentation tumor in DICER1 syndrome patients, supporting an oncogenic role for this gene.
Results
Exome sequencing of paired samples shows a low rate of mutations
A 9-year-old girl, without significant pathologic antecedents, received a diagnosis of an ovarian fibrosarcoma supported by morphology (Fig. 1A–C), and a positive immunohistochemistry staining for vimentin (100% positive cells) and negative for inhibin (Fig. 1D,E). In order to get insight into the genomic alterations of the rare pediatric tumor, we sequenced paired exomes of tumor and normal (white blood cells) tissues from the patient. We obtained 34 single nucleotide variants (SNV) and 286 small insertions and deletions (InDels) (Supplementary Data). The low mutation rate (0.77/Mb) found is typical of pediatric tumors, as reported elsewhere11. We then created a high confidence (HC) gene list that included only mutations in coding regions with a high probability of being deleterious, as assessed by the CADD algorithm12 (see Methods). Figure 2A and Table 1 shows that 16 mutations complied with the aforementioned criteria. To validate these mutations, we employed RNASeq from the tumor samples. We were able to corroborate 5 out of the 16 HC mutations. Most of the remaining mutations presented a very low or absent expression in the tumor or, alternatively, low allelic fraction in the initial exome analysis, possible due to clonal heterogeneity. Nevertheless, we were able to validate expressed mutations in two previously reported cancer drivers: DICER1 and NF1. DICER1 gene presented a missense mutation predicted to change a glutamic acid for a glycine in the residue 1813 of the protein (Fig. 2B). Interestingly, mutations in this metal-binding site abolish the RNAse IIIb activity of the enzyme, changing the specificity of its microRNA-processing ability13. Mutations in this site have been found in other tumor types7, although no alterations have been described in fibrosarcomas. We also validated a frame-shift mutation in NF1, a tumor-suppressor gene that is involved in the generation of multiple subtypes of soft-tissue sarcomas14 and has been associated with poor response to chemotherapy and targeted therapy15. This mutation is predicted to produce a functional truncated protein (Fig. 2C). We could not find a second alteration in the remaining allele of this gene. Nevertheless, the expression levels of its main isoform, ENST00000356175.7_NF1-002 were significatively downregulated (Fold change: 0.045, PPEE = 0).
Figure 1.

Ovarian fibrosarcoma pathology. (A) Hematoxylin-Eosin staining at 40x, (B) 100x and (C) 200x. (D) Immunohistochemistry of Vimentin. (E) Immunohistochemistry of Inhibin. Scale bars are shown at the right bottom of each micrograph.
Figure 2.
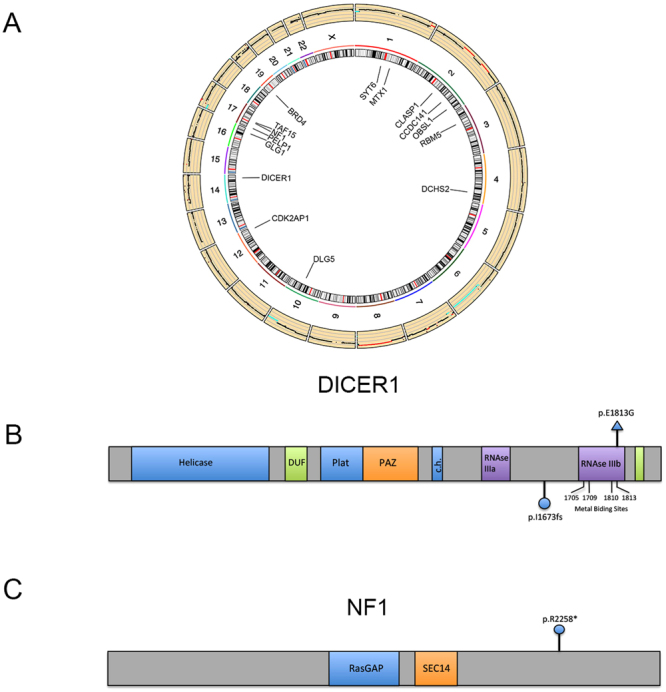
Mutational landscape of the ovarian fibrosarcoma. (A) Circos plot showing, at the outermost ring, the copy-number alterations found in the tumor. Mutations (SNV and InDels) in codifying genes are shown at the center. (B) Diagram of the DICER1 gene, showing its main domains. The mutation found in the tumor is marked at the upper side of the diagram, whereas the mutation found in the germinal DNA is depicted below it. (C) Diagram of the NF1 gene, showing its main domains. The mutation found in the tumor is shown.
Table 1.
Filtered SNV and InDel from the tumor exome.
| Gene | Variation class | Variation type | Effect | AF |
|---|---|---|---|---|
| BRD4 | Missense Mutation | SNV | p.A1243D | 0.333 |
| CCDC141 | Missense Mutation | SNV | p.K992T | 0.123 |
| CDK2AP1 | Missense Mutation | SNV | p.R99K | 0.098 |
| CLASP1 | Missense Mutation | SNV | p.R276L | 0.426 |
| DCHS2 | Missense Mutation | SNV | p.R799H | 0.185 |
| DICER1 | Missense Mutation | SNV | p.E1813G | 0.435 |
| DLG5 | Nonsense Mutation | SNV | p.R530* | 0.394 |
| GLG1 | Splice Site | SNV | p.P147L | 0.106 |
| MTX1 | Frame Shift Del | DEL | p.F157fs | 0.714 |
| NF1 | Nonsense Mutation | SNV | p.R2258* | 0.069 |
| OBSL1 | Missense Mutation | SNV | p.R994C | 0.161 |
| PELP1 | Frame Shift Del | DEL | p.P1025fs | 0.333 |
| RBM5 | Missense Mutation | SNV | p.G729S | 0.457 |
| SCARF1 | Frame Shift Ins | INS | p.G303fs | 0.250 |
| SYT6 | Missense Mutation | SNV | p.S404V | 0.244 |
| TAF15 | Missense Mutation | SNV | p.R165L | 0.420 |
Del: Deletion. SNV: Single Nucleotide Variation. AF: Allelic Frequency.
Copy number variation analysis
We then analyzed the data for the presence of Copy Number Variants (CNV). Figure 3A shows the presence of amplifications in large regions of chromosomes 1, 2, 7, 17 and, more important, large regions of chromosome 8. It has been shown that chromosome 8 trisomy is a marker that distinguishes ovary fibromas from fibrosarcomas16. We also found large deletions affecting the long arm of chromosomes 6 and 10 and the short arm of chromosome 17. To obtain a pathologically relevant CNV gene list we filtered the genes within the variant regions against a curated cancer drivers list17. Table 2 shows the genes affected by CNV in the tumor. Interestingly, we found amplifications of MYC, which have been commonly reported for sarcomas18 and deletions of TP53, another common finding in these tumors19. Interestingly, we found an increase in the expression of c-Myc when compared to normal ovary samples (283 vs 160 FPKM), although it did not reach significance.
Figure 3.

Copy number alterations in the ovarian fibrosarcoma sample. (A) Copy-number profile in the tumor. The normalized copy number is depicted against the length of each chromosome. The mean copy number is marked with a line. Deletions are depicted below and amplifications above the mean copy number line.
Table 2.
Copy number variation in the tumor exome.
| Gene | Effect | Genotype |
|---|---|---|
| ARID1B | Deletion | 0:−1 |
| BRAF | Amplification | 3:AAB |
| CASP8 | Amplification | 4:AAAB |
| EZH2 | Amplification | 3:AAB |
| FGFR2 | Deletion | 0:−1 |
| IDH1 | Amplification | 3:AAB |
| MAP2K4 | Deletion | 0:−1 |
| MET | Amplification | 3:AAB |
| MLL3 | Amplification | 3:AAB |
| MYC | Amplification | 3:AAB |
| NCOR1 | Amplification | 8:AAAAAABB |
| NFE2L2 | Amplification | 3:AAB |
| PRDM1 | Deletion | 0:−1 |
| SF3B1 | Amplification | 4:AAAB |
| SMO | Amplification | 3:AAB |
| TNFAIP3 | Deletion | 0:−1 |
| TP53 | Deletion | 0:−1 |
Genotype: Allele number/Allele genotype. NC: Not calculated.
Mutations in non-coding RNAs
We also found several mutations in non-coding regions, including annotated lincRNAs (Table S2). None of these mutations have been reported in recent series of recurrent lincRNAs alterations in cancer20.
Micro-satellite instability status
In a recent survey of cancer cell lines, it was reported that 4 of 781 of these lines presented a DICER1 truncating mutation. Interestingly, all of them presented also microsatellite instability (MSI)9. For this reason, we analyzed the MSI status in this patient using the MSIseq package21 and found that the tumor indeed presented an MSI-High profile, being negative to a POLE-mutated phenotype. To validate this finding, we performed PCR and capillary electrophoresis of five commonly used MSI markers (BAT25, BAT26, NR21, NR22 and NR24) and found micro-satellite instability in three of the five markers analyzed (Table 3). We could not find mutations in MLH1, MSH2, MSH6 or PMS2 genes, although we cannot exclude a possible epigenetic mechanism of inactivation of MLH1 as the reason for the MSI phenotype.
Table 3.
MSI markers.
| Marker | Germinal size | Tumor size |
|---|---|---|
| BAT25 | 145 bp | 140 bp |
| BAT26 | 140 bp | 132 bp |
| NR21 | 120 bp | 120 bp |
| NR22 | 160 bp | 164 bp |
| NR24 | 150 bp | 146 bp |
The size of each marker is depicted for the germinal and tumor sample in base pairs (bp).
Ovary fibrosarcoma’s transcriptome presents a mesenchymal network signature and an enriched Sonic Hedgehog network
We then compared the transcriptome data with a panel of previously reported normal fibroblast RNASeq experiments. We found 1193 Differentially-Expressed (DE) genes (Posterior Probability of Differential Expression (PPDE) >0.95 and Fold Change > 2) (Supplementary Data). To get more insight into the relevance of these transcriptional events, we performed a model network analysis using the Ingenuity Pathway Analysis tool (IPA, Qiagen Redwood City). Fig. S1 shows the enrichment of several processes on three main categories (Diseases, Molecular and Cellular Functions and Physiological Systems Functions). As expected, we found that the top enrichment in the Diseases category was Cancer, whereas postranslational modifications and muscular system were the most significant finding in molecular and physiological functions, respectively (Fig. 4A). Interestingly, the muscular system enrichment was driven by a group of genes responsible for regulating cascades related to mesenchymal development, as supported by a network upstream regulator analysis showing that a group of mesenchymal master regulators (including MYOCD, TBX5 and HAND2, Table 4) were leading this transcriptional cassette (Fig. 4B).
Figure 4.

Network analysis of expression data. (A) The top enriched function in each category is plotted with the log p-value. The significance threshold is marked. (B) Mesenchymal network derived from the upstream/master regulators found in the expression dataset. (C) Gene Set Enrichment Analysis (GSEA) of the mesenchymal hallmark signature. Left Panel: The enrichment score is compared with the up-regulated (left) and down-regulated (right) genes. The False Discovery Rate is also shown. Right Panel: The top regulated genes found in the analysis. Lighter gray, higher expression. Darker gray, lower expression. (D) The Hedgehog pathway is enriched in the ovarian fibrosarcoma sample. Gray tones represent expressional changes.
Table 4.
Upstream regulators.
| Gene | p-value | Variation type | Effect |
|---|---|---|---|
| MYOCD | 1.23E−06 | SNV | p.A1243D |
| HAND2 | 3.93E−05 | SNV | p.K992T |
| TBX5 | 3.93E−05 | SNV | p.R99K |
| GATA4 | 1.74E−04 | SNV | p.R276L |
| UPF2 | 3.64E−04 | SNV | p.R799H |
| VEGFA | 4.60E−04 | SNV | p.E1813G |
| NEUROG1 | 5.44E−04 | SNV | p.R530* |
| TENM1 | 5.57E−04 | SNV | p.P147L |
| DRAP1 | 6.16E−04 | DEL | p.F157fs |
| NMNAT1 | 8.93E−04 | SNV | p.R2258* |
| MED12 | 1.04E−03 | SNV | p.R994C |
| MYOC | 1.17E−03 | DEL | p.P1025fs |
| estrogen receptor | 1.22E−03 | SNV | p.G729S |
| NOTCH3 | 1.31E−03 | INS | p.G303fs |
| GLI1 | 1.61E−03 | SNV | p.S404V |
| ZNF217 | 1.86E−03 | SNV | p.R165L |
Del: Deletion. SNV: Single Nucleotide Variation.
An additional analysis using the Gene Set Enrichment Analysis (GSEA) algorithm22 also revealed the enrichment of a myogenesis signature (Fig. 4C), further supporting the involvement of a mesenchymal transcription signature in the ovarian fibrosarcoma progression. Among the top regulated networks, we also found a GLI1-driven cascade (Fig. 4D), which could be one of the key pathways involved in this neoplasia, due to the previous report of mutations in members of the Sonic Hedgehog pathway and the involvement of GLI1 as one of the top upstream regulators discovered in our gene set (Table 4). We also analyzed the data for the presence of fusion transcripts, but we were unable to validate the positive hits found.
microRNAs deregulation in ovarian fibrosarcoma
We next sought to assess the microRNAs expression landscape in the fibrosarcoma sample. As expected, we found that a larger number of pre-miRNAs were deregulated, when compared to mature miRNAs (Fig. S1A). The top networks associated with the regulated genes were associated with MYC and the Argonaute proteins AGO1 and AGO3 (Fig. S2B and Supplementary Data).
lncRNAs deregulation in ovarian fibrosarcoma
It has been recently described that DICER1 is able to control hundreds of long non-coding RNAs in a genome-wide fashion23. In this study, gene deletion or mutation of the RNAse III catalytic residues of DICER1 impaired the expression of hundreds of lincRNAs in mouse embryonic stem cells by a c-Myc-dependent mechanism. To explore if this regulation could also be present in cancer, and in particular in the fibrosarcoma sample under study, we compared the ratio of expressed lincRNAs versus mRNAs in our sample with a panel of normal fibroblasts. Consistent with this hypothesis, we found a clear depletion in the number of expressed lincRNAs (Fisher exact test p < 2.2−16) and Fig. 5A. A similar depletion was found when we performed a comparison with panels of normal and tumoral ovary samples (Fisher exact test p < 2.2−16) Fig. S3A. Unexpectedly, lincRNAs expression was higher in the tumoral sample than in the control fibroblasts or normal ovary cell lines, pointing toward a specific regulation in this tumor (Figs 5B,C and S3). This is supported by the presence of several oncogenic lincRNAs in the top expressed and DE non-coding RNA genes (Table 5), including MALAT1, MIR181A1HG, CASC1, XIST and FENDRR. Interestingly, several tumor samples presented also higher lincRNAs expression and, coincidentally, these tumors tended to have higher (although statistically non-significative) DICER1 expression (Fig. S3).
Figure 5.

Long-intergenic non-coding RNAs are deregulated in the fibrosarcoma sample. (A) Plot showing the ratio of lincRNAs versus mRNAs in the fibrosarcoma (FS) and control fibroblasts samples. (B) Boxplots showing the lincRNAs expression, in Fragments Per Kilobase of Exon per Million Fragments Mapped (FPKM), of each analyzed sample. The median, upper and lower quartiles and outliers are depicted for the fibrosarcoma (FS) and control fibroblasts samples. (C) Boxplots showing the mRNAs expression in FPKM, of each analyzed sample. The median, upper and lower quartiles and outliers are depicted for the fibrosarcoma (FS) and control fibroblasts samples.
Table 5.
Differentially expressed lincRNAs.
| lincRNA | PPDE | Fold Change |
|---|---|---|
| LINC01087 | 1.00 | 33072 |
| LINC01266 | 1.00 | 46690 |
| XIST 39 | 1.00 | 4464627 |
| MIR181A1HG 40 | 1.00 | 817 |
| MALAT1 41 | 1.00 | 35 |
| RP11-384P7.7 | 1.00 | 377 |
| RP11-701H24.4 | 1.00 | 93 |
| RMRP 42 | 1.00 | 904540 |
| TSIX 43 | 1.00 | 6021 |
| CASC15 44 | 1.00 | 54 |
| UG0898H09 | 1.00 | 3517 |
| SCARNA2 | 1.00 | 3335 |
| CH507-513H4.5 | 1.00 | 7249 |
| RP11-693J15.5 | 1.00 | 665 |
| LINC01597 | 1.00 | 262 |
| RP11-923I11.6 | 1.00 | 55 |
| CTD-2545M3.8 | 1.00 | 580 |
| FENDRR 45 | 1.00 | 88 |
| RP11-13K12.1 | 1.00 | 15564 |
| LINC00626 46 | 1.00 | 2189 |
| DKFZP434L187 | 1.00 | 863 |
| LINC00184 | 1.00 | 55 |
| RP11-268G12.1 | 1.00 | 925 |
| PWAR6 | 1.00 | 17 |
| RP11-191L9.4 47 | 1.00 | 11673 |
| LINC00645 | 1.00 | 11673 |
| AC083843.1 | 1.00 | 17 |
| RP11-706O15.5 | 1.00 | 46 |
| FIRRE 48 | 1.00 | 232 |
| RP11-138A9.1 | 1.00 | 1625 |
| LINC01579 | 1.00 | 122 |
| MIR99AHG 49 | 1.00 | 34 |
| RP11-244M2.1 | 1.00 | 38 |
| RP1-78B3.1 | 1.00 | 104 |
| AC096669.3 | 1.00 | 9728 |
| RP11-13K12.5 | 1.00 | 9728 |
| CTA-280A3.2 | 1.00 | 9728 |
| RP11-348P10.2 | 1.00 | 13 |
| NBAT1 50 | 0.99 | 72 |
| RP11-161M6.2 51 | 0.99 | 46 |
| RP11-448A19.1 | 0.99 | 10 |
| PKIA-AS1 | 0.99 | 258 |
| CTC-444N24.8 | 0.97 | 6 |
| CTD-2311B13.1 | 0.97 | 7782 |
| RP11-453M23.1 | 0.97 | 7782 |
| FAM95C | 0.97 | 7782 |
| RP11-37B2.1 | 0.96 | 5 |
PPDE: Posterior probability of differential expression. LincRNAs involved in cancer are marked in bold, with the associated reference.
Germinal mutations analysis confirms DICER1 Syndrome
We then analyzed the germinal line for putative pathological variants that could be accounting for the early presentation of this rare tumor. We established a tiered classification, after filtering variants using the criteria stated in Methods. We were able to find two tier 1 mutations (mutations in genes previously reported as dominant-acting in pediatric hereditary tumors) in our data. Of these mutations, the PALB2 alteration has been reported as likely benign in ClinVar (ID 142310) so we excluded it as a pathogenic contributor. Remarkably, the remaining mutation affected DICER1 gene. This mutation, not been reported in public databases, consisted in a frame shift insertion, predicted to produce a functional truncated protein (Fig. 2B). This finding was verified by Sanger sequencing in both tumoral and germinal DNA, as well as in RNA derived from the tumor (Fig. 6A–C and not shown). Although the MuTect algorithm called a dinucleotide insertion, these assays showed a single-base insertion. The presence of a DICER1 functional truncating germinal mutation associated with an additional tumor mutation event in the RNAse IIIb domain of this enzyme is characteristic of the associated neoplasias characteristic of the recently described DICER1 Syndrome (OMIM 601200)9. To validate this finding, we sequenced the altered region in germinal DNA derived from the parents and a sibling. Figure 6D shows that only the proband presented the mutation in DICER, pointing toward a de novo mutation in this gene as the cause for the syndrome, although we cannot exclude germline mosaicism in one of the parents.
Figure 6.

DICER1 mutations. Sequence traces from Sanger sequencing of the (A) Tumoral and (B) Germinal DNA. The arrow shows the insertion point, where a duplicate signal starts. (C) Nucleotide alignment of wild type (WT) and mutated (MT) sequences derived from the exome data, with the sequences obtained from RNA (RNA and RNAa). The inserted adenine is shown in cursive. (D) Sequence traces from the mother (i), father (ii) and brother (iii) of the affected individual.
Discussion
In the present article, we report the genomic characterization of a fibrosarcoma arising from the ovary of a 9 years-old child. It has been previously reported that adult ovary fibrosarcomas present structural alterations, including chromosomal number aberrations such as trisomy of the 12 and 8 chromosomes. Trisomy 8 has been postulated as a marker for distinguishing fibroma from fibrosarcoma16. In the present report we were able to show higher copy number of large regions in chromosome 8, consistent with its proposed role as a fibrosarcoma marker.
MYC is one of the most recurrent alterations in sarcomas18. A recent report has shown that a DICER1-microRNA-Myc circuit is responsible for the steady-state transcription of hundreds of long non-coding RNAs23. Since it has been reported lncRNAs are important in the progression of pediatric sarcomas24, it is tempting to speculate that the amplification of MYC could be required for fibrosarcoma progression in the specific setting of DICER1 germinal inactivation. In this report, the authors found that DICER1 RNAse III domain is responsible for the expression of a large number of these non-coding RNAs, in particular those responsible for maintaining pluripotency. This model closely resembles the progression of DICER1 Syndrome tumors, where an initial truncating mutation is later complemented with a second point mutation in the RNAse IIIb domain, giving rise to a diverse array of tumors, including the new association we report here. In their experiments, the authors found that the effect of DICER1 knockout/mutation depends on the regulation of a specific miRNA (miR-295) and the upregulation of c-Myc. Interestingly, we found that c-Myc was amplificated in the fibrosarcoma sample, when compared to normal fibroblasts, although we could not detect a significative increase in its expression. The initial oncogenic insult could be the loss of one DICER1 allele, which could change the expression of a subset of oncogenic lincRNAs that may drive the oncogenic initiation. The second and more specific loss would require the amplification of c-Myc to further drive cancer progression, in order to have minimal Myc expression levels. In vitro and in vivo models testing this are clearly needed to test this hypothesis.
In our data, we found a paucity of mutations, as reported for other pediatric tumors. The main possible drivers for this neoplasia were DICER1, NF1 and MYC. The first two presented mutations, whereas the latter two were altered by structural changes. As mentioned, we did not found alterations in the previously reported PTCH1 or Sonic Hedgehog pathway, although we cannot discard that epigenetic changes could be participating in the progression of the disease. This is supported by the transcriptome results, in which a significative deregulation of this pathway was found. DICER1 mutations are rare in cancer patients. Most of them are associated with infrequent embryonal or primitive tumors such as sex cord-stromal tumors of the ovary, embryonal rhabdomyosarcomas, Wilms tumors, etc., pointing toward a specific development pathway requirement for its cancer driver´s activity. More important, several of these mutations (although not all), are associated with germinal DICER1 mutations, in which the germinal allele generally presents a truncation mutation. Since no LOH has been found, and the second allele retains a modified activity, it has been proposed that DICER1 acts as an oncogene driver, with an altered microRNA processing activity acting as the oncogenic trigger7. A recent survey of cancer cell lines showed that 4 of 781 of these lines presented a DICER1 truncating mutation. All of them presented also microsatellite instability (MSI)9, so the authors conclude that DICER1 mutations were unlikely to be drivers. Nevertheless, in our patient the somatic mutation was found in a hot spot, consistent with its proposed oncogenic role. We tried to assert the reason for the MSI-H phenotype, but were unable to find mutations in MLH1, MSH2, MSH6 or PMS2 genes. Nevertheless, we cannot exclude a possible epigenetic mechanism of inactivation of MLH1.
Methods
The project received ethical and scientific approval from Instituto Nacional de Medicina Genomica, Hospital Infantil de Mexico and Comision Federal para la Proteccion contra Riesgos Sanitarios (COFEPRIS) committees. After obtaining informed consent from the parents and the corresponding children assent, a sample derived from the surgical specimen was snap-frozen and a portion of it subjected to histopathological assessment. A blood sample was also obtained at the same time. The tumoral sample presented more than 80% neoplasic cellularity.
Clinical Case
A 9-year-old girl, without a significant pathologic clinical history, was admitted at the Hospital Infantil de México Federico Gómez with one month´s history of moderate progressive abdominal pain localized in mesogastrium, vomiting and weight loss. A heterogeneous ovarian mass of 17 × 9,7 cm was discovered by CT scan image. The patient underwent surgical excision of the mass. On laparotomy, surgeons found a right ovarian tumor. The pathology specific presented a typical herringbone pattern, high mitogenic index (4 mitoses in 10 high-magnification fields), extensive hemorrhagic and necrotic areas and nuclear pleomorfism. The final histopathology diagnosis was fusocellular sarcoma compatible with fibrosarcoma. Immunohistochemistry was performed against Vimentin (MA5-11883, Invitrogen, CA, USA; 1:100 dilution) and Inhibin (5692, Bio SB, CA, USA; 1:100 dilution) as reported previously25. The patient had two siblings, her brother was diagnosed with bilateral renal tumors: Wilms´ tumor and a possible metanephric adenoma whereas her sister is healthy at the moment. We were unable to obtain paternity tests.
Exome analysis
Genomic DNA from the patient was extracted using commercially available kits from primary tumor (DNeasy Blood & Tissue Kit, Qiagen, CDMX, Mexico) and from peripheral blood (Puregene Blood Kit, Qiagen, CDMX, Mex) according to manufacturer protocols. The DNA was subjected to exome purification and sequencing at the Broad´s Institute, following previously described protocols26. The mean coverage obtained after alignment was 89% at 20x for the germinal DNA and 88% at 20x for the tumoral DNA. Sequencing was performed using an Illumina HiSeq 2000 with the V3 Sequencing kits and the Illumina 1.3.4 pipeline (Illumina, CA). For variant calling, we used MuTect ver 1.1.427 in High Confidence mode (HC) for SNV and InDelocator for small insertions and deletions. Variants were annotated with Oncotator28. Non-coding variants, with the exception of splice-site mutations were excluded. SNVs and InDels were further filtered by predicted protein functional impact using Combined Annotation-Dependent Depletion algorithm (CADD)12, with a cutoff of 1.5. Genes included in the CNV regions were filtered against a list of previously reported genes affected by CNV17. Germinal variants were called using Broad´s Institute Best Practices approach, where the BAM files were re-calibrated with the HaplotypeCaller using a joint genotyping approach with 60 additional normal exomes. After quality filtering we excluded variants with a Minor Allele Frequency (MAF) of 1% or greater allelic in Amerindian/European/African/Asian populations (1000 genomes, EXAC, dbSNP and local databases). We then established a tiered approach, where variants were assigned to tier 1 group if they were present in genes responsible for autosomal dominant cancer syndrome and tier 2 if have been associated with recessive cancer syndromes. All variants were manually checked with IGV.
Transcriptome analyses
RNA was isolated using TRIzol reagent (Thermo Fisher Scientific, MA, USA). RNA with a RIN of 8 was used to construct a library using Illumina´s TruSeq RNA kit, following the manufacturer´s instructions. The paired-end library was sequenced using a GAIIx equipment (Illumina, CA) in a 72 bp configuration. After quality control and trimming, the reads were aligned with the STAR aligner29 and the resulting SAM was further processed with the PICARD tool30 to recalibrate reads. Finally, we called and filtered variants using the Haplotype Caller from GATK. Fusion transcripts were obtained with the TopHat-fusion pipeline31. To quantify differentially-expressed transcripts, we realigned and processed.
RNASeq data from 10 normal human fibroblasts samples32 (GEO dataset GSE51518 from the NCBI) together with the ovarian fibrosarcoma data and used RSEM33 and the R package EBSeq34 to normalize, quantitate and compare the expression data. microRNA analysis was performed using a miRNA 4.0 array (Affymetrix, Santa Clara, CA). A normal ovary cell line cel file was obtained from the GEO information system (GSE7644935) for comparison. Quality control, background subtracted, quantile normalized and log2- transformed using robust multi-array analysis (RMA) and differential expression analysis of the three cel files were done with Partek v 6.6. Candidate miRNAs were considered to be differentially-regulated if they presented a Fold Change > = 2, p-values < 0.05 and FDR less than 0.05.
Structural variants
Copy-number variants (CNV) were predicted using Control-Freec ver 8.036, calculating variant changes by exon and using a breakpoint threshold of 1.5. Translocations were assessed using Delly37, using standard parameters.
MSI Status
MSI status was infered from raw tumoral SNV and microInDels data using the MSIseq package21, using the author´s 526 tumoral exomes training database. MSI was validated using a panel of 5 markers that include BAT25, BAT26, NR21, NR22 AND NR2438; PCR of 5 markers in germinal and tumor sample DNA was performed as follows: denaturation 95 °C for 10 m, followed to 35 cycles (95 °C for 30 s, 50 °C for 30 s and 72° for 30 s) (Table S1). PCR products size was analyzed with the Agilent 4200 TapeStation System (Agilent Technologies).
Sanger sequencing
DNA derived from leukocytes or tumoral tissue was subjected to PCR with the following conditions: denaturation 95 °C for 10 m, followed by 35 cycles (95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s), using the following primers with added M13 sequences to facilitate sequencing (Table S1). PCR products were gel-purified using QIAquick Gel Extraction kit (Qiagen, CA) and sequenced in a 3730XL DNA analyzer using the Big Dye direct sequencing kit (Applied Biosystems, CA).
Compliance with ethical standards
Ethical approval was obtained from Instituto Nacional de Medicina Genomica Research and Ethical boards, Hospital Infantil del Mexico research and ethical boards, and Federal COFEPRIS (Comision Federal para la Proteccion contra Riesgos Sanitarios). Informed consent was obtained from all participants. All procedures were carried out in accordance to the ethical guidelines of all the mentioned Research and Ethical Boards.
Electronic supplementary material
Acknowledgements
We wish to thank the Broad´s Institute sequencing facility for their effort and help, Dr. Maria Cortes for her support and sample/data coordination, Dr. Todd Golub for his help in providing exome sequencing of our samples, Alfredo Mendoza and Raul Mojica from INMEGEN´s sequencing and microarray facility for their support. We thank Dr. Guillermo Ramon from the Hospital Infantil de Mexico and Dr Joel Roman from LabPat laboratory for their pathologic assessment of the fibrosarcoma sample. This project was supported in part by the Slim Initiative in Genomic Medicine, a project funded by the Carlos Slim Foundation and by CONACyT grant 161619. AT was supported by a postdoctoral fellowship (25660) from CONACyT’s grant FC-2015/449.
Author Contributions
All authors were involved in writing the paper and gave final approval of the submitted and published versions. G.M.-C. and J.M.-Z. conceived the study; J.G., carried out laboratory experiments; J.M.-Z., A.T. and V.M. performed bioinformatics analyses, M.Z., L.J.-V., P.L.-D.-V., K.R.-M. performed clinical assessment and contributed to data collection and management, E.M.-C. performed genetic evaluations, N.B.G. performed pathology analyses, G.M.-C. and J.M.-Z. coordinated the study.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-21663-9.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jorge Melendez-Zajgla, Email: jmelendez@inmegen.gob.mx.
Gabriela E. Mercado-Celis, Email: gemercadocelis@gmail.com
References
- 1.Christman JE, Ballon SC. Ovarian fibrosarcoma associated with Maffucci’s syndrome. Gynecol Oncol. 1990;37:290–291. doi: 10.1016/0090-8258(90)90350-T. [DOI] [PubMed] [Google Scholar]
- 2.Kraemer BB, Silva EG, Sneige N. Fibrosarcoma of ovary. A new component in the nevoid basal-cell carcinoma syndrome. Am J Surg Pathol. 1984;8:231–236. doi: 10.1097/00000478-198403000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Kruger S, Schmidt H, Kupker W, Rath FW, Feller AC. Fibrosarcoma associated with a benign cystic teratoma of the ovary. Gynecol Oncol. 2002;84:150–154. doi: 10.1006/gyno.2001.6408. [DOI] [PubMed] [Google Scholar]
- 4.Nieminen U, Numers CV, Purola E. Primary sarcoma of the ovary. Acta Obstet Gynecol Scand. 1969;48:423–432. doi: 10.3109/00016346909156657. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh AK. Bilateral fibrosarcoma of the ovary following hysterectomy. Am J Obstet Gynecol. 1972;112:1136–1138. doi: 10.1016/0002-9378(72)90196-2. [DOI] [PubMed] [Google Scholar]
- 6.Stewart CJ, Charles A, Foulkes WD. Gynecologic Manifestations of the DICER1 Syndrome. Surg Pathol Clin. 2016;9:227–241. doi: 10.1016/j.path.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Heravi-Moussavi A, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med. 2012;366:234–242. doi: 10.1056/NEJMoa1102903. [DOI] [PubMed] [Google Scholar]
- 8.Foulkes WD, Priest JR, Duchaine TF. DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer. 2014;14:662–672. doi: 10.1038/nrc3802. [DOI] [PubMed] [Google Scholar]
- 9.Slade I, et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet. 2011;48:273–278. doi: 10.1136/jmg.2010.083790. [DOI] [PubMed] [Google Scholar]
- 10.Anglesio MS, et al. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J Pathol. 2013;229:400–409. doi: 10.1002/path.4135. [DOI] [PubMed] [Google Scholar]
- 11.Lawrence MS, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klein S, et al. Expanding the phenotype of mutations in DICER1: mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J Med Genet. 2014;51:294–302. doi: 10.1136/jmedgenet-2013-101943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dodd RD, et al. NF1 deletion generates multiple subtypes of soft-tissue sarcoma that respond to MEK inhibition. Mol Cancer Ther. 2013;12:1906–1917. doi: 10.1158/1535-7163.MCT-13-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15:290–301. doi: 10.1038/nrc3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuji T, Kawauchi S, Utsunomiya T, Nagata Y, Tsuneyoshi M. Fibrosarcoma versus cellular fibroma of the ovary: a comparative study of their proliferative activity and chromosome aberrations using MIB-1 immunostaining, DNA flow cytometry, and fluorescence in situ hybridization. Am J Surg Pathol. 1997;21:52–59. doi: 10.1097/00000478-199701000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith SC, et al. CIC-DUX sarcomas demonstrate frequent MYC amplification and ETS-family transcription factor expression. Mod Pathol. 2015;28:57–68. doi: 10.1038/modpathol.2014.83. [DOI] [PubMed] [Google Scholar]
- 19.Mulligan LM, Matlashewski GJ, Scrable HJ, Cavenee WK. Mechanisms of p53 loss in human sarcomas. Proc Natl Acad Sci USA. 1990;87:5863–5867. doi: 10.1073/pnas.87.15.5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lanzos, A. et al. Discovery of Cancer Driver Long Noncoding RNAs across 1112 Tumour Genomes: New Candidates and Distinguishing Features. bioRxiv, 065805, (2016). [DOI] [PMC free article] [PubMed]
- 21.Huang MN, et al. MSIseq: Software for Assessing Microsatellite Instability from Catalogs of Somatic Mutations. Sci Rep. 2015;5:13321. doi: 10.1038/srep13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng GX, Do BT, Webster DE, Khavari PA, Chang HY. Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat Struct Mol Biol. 2014;21:585–590. doi: 10.1038/nsmb.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marques Howarth M, et al. Long noncoding RNA EWSAT1-mediated gene repression facilitates Ewing sarcoma oncogenesis. J Clin Invest. 2014;124:5275–5290. doi: 10.1172/JCI72124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arellano-Llamas A, et al. High Smac/DIABLO expression is associated with early local recurrence of cervical cancer. BMC Cancer. 2006;6:256. doi: 10.1186/1471-2407-6-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ojesina AI, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cibulskis K, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramos AH, et al. Oncotator: cancer variant annotation tool. Hum Mutat. 2015;36:E2423–2429. doi: 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A, et al. Bioinformatics. 2013. STAR: ultrafast universal RNA-seq aligner; pp. 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Broad Institute. Picard, http://broadinstitute.github.io/picard (2015).
- 31.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung M, et al. Longitudinal epigenetic and gene expression profiles analyzed by three-component analysis reveal down-regulation of genes involved in protein translation in human aging. Nucleic Acids Res. 2015;43:e100. doi: 10.1093/nar/gkv473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leng N, et al. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics. 2013;29:1035–1043. doi: 10.1093/bioinformatics/btt087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanlikilicer P, et al. Ubiquitous Release of Exosomal Tumor Suppressor miR-6126 from Ovarian Cancer Cells. Cancer Res. 2016;76:7194–7207. doi: 10.1158/0008-5472.CAN-16-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boeva V, et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012;28:423–425. doi: 10.1093/bioinformatics/btr670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rausch T, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28:i333–i339. doi: 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Umar A, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weakley SM, Wang H, Yao Q, Chen C. Expression and function of a large non-coding RNA gene XIST in human cancer. World J Surg. 2011;35:1751–1756. doi: 10.1007/s00268-010-0951-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frederic L, et al. Next Generation RNA Sequencing of Acute Promyelocytic Leukemia (APL) Identifies Novel Long Non Coding RNAs Including New Variants of MIR181A1HG That Are Differentially Expressed during Myeloid Differentiation. Blood. 2014;124:1041. [Google Scholar]
- 41.Ren D, et al. Novel insight into MALAT-1 in cancer: Therapeutic targets and clinical applications. Oncol Lett. 2016;11:1621–1630. doi: 10.3892/ol.2016.4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shao Y, et al. LncRNA-RMRP promotes carcinogenesis by acting as a miR-206 sponge and is used as a novel biomarker for gastric cancer. Oncotarget. 2016;7:37812–37824. doi: 10.18632/oncotarget.9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chow JC, Hall LL, Clemson CM, Lawrence JB, Brown CJ. Characterization of expression at the human XIST locus in somatic, embryonal carcinoma, and transgenic cell lines. Genomics. 2003;82:309–322. doi: 10.1016/S0888-7543(03)00170-8. [DOI] [PubMed] [Google Scholar]
- 44.Russell MR, et al. CASC15-S Is a Tumor Suppressor lncRNA at the 6p22 Neuroblastoma Susceptibility Locus. Cancer Res. 2015;75:3155–3166. doi: 10.1158/0008-5472.CAN-14-3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu TP, et al. Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fibronectin1 expression. J Hematol Oncol. 2014;7:63. doi: 10.1186/s13045-014-0063-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu H, et al. Identification and validation of long noncoding RNA biomarkers in human non-small-cell lung carcinomas. J Thorac Oncol. 2015;10:645–654. doi: 10.1097/JTO.0000000000000470. [DOI] [PubMed] [Google Scholar]
- 47.Jing, W., Ye-qing, H., Bin, X. & Ming, C. Effection of lncRNA RP11-191L9.4 on the migration and invasion of prostate cancer cell PC-3. cnki03, (2016).
- 48.Qiu JJ, Yan JB. Long non-coding RNA LINC01296 is a potential prognostic biomarker in patients with colorectal cancer. Tumour Biol. 2015;36:7175–7183. doi: 10.1007/s13277-015-3448-5. [DOI] [PubMed] [Google Scholar]
- 49.Emmrich S, et al. LincRNAs MONC and MIR100HG act as oncogenes in acute megakaryoblastic leukemia. Mol Cancer. 2014;13:171. doi: 10.1186/1476-4598-13-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu P, et al. NBAT1 suppresses breast cancer metastasis by regulating DKK1 via PRC2. Oncotarget. 2015;6:32410–32425. doi: 10.18632/oncotarget.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Grembergen O, et al. Portraying breast cancers with long noncoding RNAs. Sci Adv. 2016;2:e1600220. doi: 10.1126/sciadv.1600220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.