

Abstract
Purpose of review
The renin–angiotensin system (RAS) plays a critical role in the pathogenesis of hypertension. Homeostatic actions of the RAS, such as increasing blood pressure (BP) and vasoconstriction, are mediated via type 1 (AT1) receptors for angiotensin II. All components of the RAS are present in the renal proximal tubule, which reabsorbs the bulk of the glomerular filtrate, making this segment of the nephron a location of great interest for solute handling under RAS influence. This review highlights recent studies that illustrate the key role of renal proximal tubule AT1 receptors in BP regulation.
Recent findings
A variety of investigative approaches have demonstrated that angiotensin II signaling via AT1a receptors, specifically in the renal proximal tubule, is a major regulator of BP and sodium homeostasis. Reduction of proximal tubule AT1a receptors led to lower BPs, whereas overexpression generally caused increased BPs.
Summary
AT1a receptors in the proximal tubule are critical to the regulation of BP by the kidney and the RAS. The pattern of BP modulation is associated with alterations in sodium transporters. As a key site for sodium homeostasis, the renal proximal tubule could hence be a potential target in the treatment of hypertension.
Keywords: angiotensin type 1 receptor, hypertension, proximal tubule, renin–angiotensin system
INTRODUCTION
Hypertension affects more than 1 billion people worldwide. In the United States, one-third of adults suffer from hypertension, and only half of these patients have their blood pressure (BP) under control. Ideal targets for BP control are complex [1], yet poorly controlled hypertension increases risk for cardiovascular disease, characterizing it as a condition with a high disease burden and huge healthcare costs [2–4].
A key pharmacologic target in the treatment of hypertension is blockade of the renin–angiotensin system (RAS) with use of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers (ARBs). Both drugs improve morbidity and mortality associated with several cardiovascular diseases [5–8] by effectively reducing effects of angiotensin II (AngII) [9,10]. As a major effector molecule of the RAS, angiotensin II acts via the type 1 (AT1) receptor; activation of AT1R in kidney is an integral part of the BP regulation [11]. Under normal circumstances, the goal of the RAS is to maintain effective circulating volume and tissue perfusion but in pathological conditions, RAS activation can be deleterious to cardiovascular function and BP regulation. Recent studies have characterized the essential and varied functions of AT1 receptors in specific cell types. This review focuses on recent studies of BP regulation by AT1 receptors in the renal proximal tubule and underscores their role in this region as key regulators of BP and sodium homeostasis (Fig. 1).
FIGURE 1.
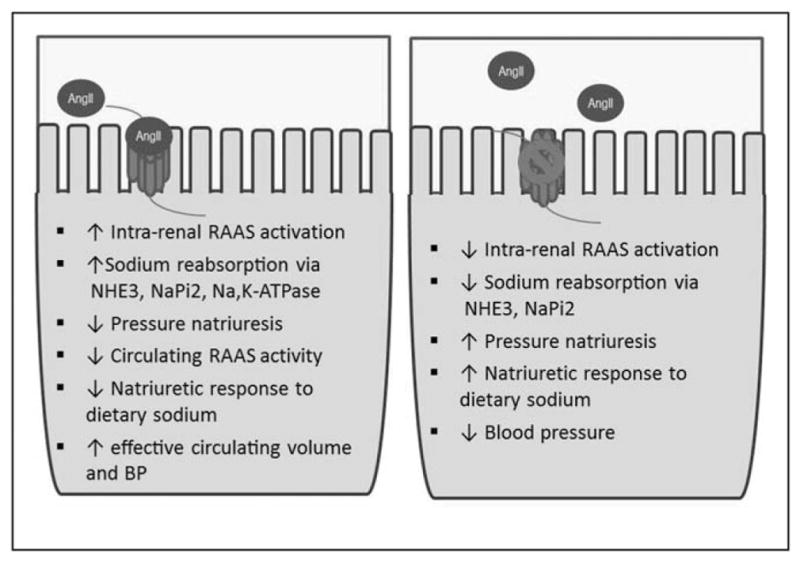
Impact of AT1a receptors in the proximal tubule. Summary of actions of AT1a receptors in BP regulation by the proximal tubule. Left: The presence of AT1a receptors leads to a set of responses culminating in increased BP. Right: The absence or blockade of AT1a receptors results in the opposite phenotype. AngII, angiotensin II; BP, blood pressure; NaPi2, sodium phosphate cotransporter; NHE3, sodium-proton antiporter 3.
BLOOD PRESSURE REGULATION BY THE KIDNEY AND THE RENIN–ANGIOTENSIN SYSTEM
Decades ago, Guyton et al. [12] concluded that regulation of sodium excretion by the kidney was a key determinant of intraarterial pressure and suggested that when BP increases, activation of the pressure natriuresis mechanism facilitates excretion of sodium, which functions as a compensatory system to reduce effective circulating volume and thus lowers BP to normal levels [13]. They described the renal BP regulation as a pressure system of infinite gain, thus, defective handling of sodium by the kidney and consequent dysregulation of body fluid volumes provide a common pathway in pathogenesis of hypertension [13]. Furthermore, Guyton et al. [13] originally suggested a role for RAS in short-term BP regulation, yet the exact role of the RAS in arterial BP regulation was still evolving. Hall et al. [14] showed that infusion of AngII into the renal artery increased BP and that activation of the RAS shifted the pressure-natriuresis curve to the right, that is, where a higher systemic pressure is required to balance sodium output to input. Administering RAS inhibitors restored this relationship between systemic pressure and urinary sodium excretion back to the starting point [14–16]. The dependence of BP upon intact urinary sodium excretion and the pronounced effect of AngII provided insight into a role of the RAS in BP regulation by the kidney.
The kidney contains all components of the RAS, making it an important organ for BP regulation and sodium homeostasis. Renin is produced by the jux-taglomerular apparatus [17] and distal nephron [18,19], angiotensin-converting enzyme (ACE) and ACE2 are highly expressed in the proximal tubule [20] and angiotensin receptors (type 1 and type 2) are localized throughout the kidney [21,22]. Angiotensinogen (Agt) is largely produced in the liver; however, it can be synthesized in the proximal tubule and secreted into the tubular lumen [19,23,24]. Matsusaka et al. [25] recently described the relationship between hepatically derived and renally derived Agt. Their data provide evidence that production is coordinated, and that liver-derived Agt is an important source of renal AngII generation. Furthermore, involvement of intrarenal Agt in mediating hypertension and modulating sodium balance was suggested [26]. Studies of the angiotensin receptors in BP regulation are described below. Our understanding of actions of the RAS has expanded with growing evidence for an intrarenal RAS which can cause hypertension independent of the systemic, circulating RAS [27]. As all RAS components are present in the kidney, it serves to reason that they perform vital functions within this excretory organ. Recent studies, largely in rodents, have shown that there is a renal feed-forward mechanism in AngII-dependent hypertension that stimulates intrarenal AngII generation, with suppression of the systemic RAS [28]. This feed-forward mechanism is caused by AT1 receptor mediated AngII uptake and stimulation of additional renal tubular Agt. Increased renin formation by principal cells of the collecting ducts then convert Agt to AngI, creating even more AngII which exacerbates the sodium retention and hypertension [28]. A fundamental role for intrarenal ACE in the intrarenal RAS was shown in a recent study in which mice lacking ACE in the kidney were unable to produce AngII in the kidney resulting in a blunted hypertensive response to chronic AngII infusion [29]. Taken together, these studies demonstrate the considerable contribution of the intrarenal RAS to sodium handling and BP control.
BLOOD PRESSURE REGULATION BY THE ANGIOTENSIN 1A RECEPTOR IN THE KIDNEY
Throughout the body, AT1 receptors mediate several effects contributing to BP and fluid homeostasis [30,31]. This is best exemplified by the phenotype of mice lacking the AT1a receptor, the dominant AT1 receptor isoform in rodents (AT1b is the other isoform). AT1a receptor-deficient mice demonstrate hypotension and profound sodium sensitivity [32], mimicking the pharmacologic effects of ARB use. In the vasculature, smooth muscle cell AT1a receptors cause vasoconstriction [30]. The direct actions of AT1a receptors contribute to pathogenesis of AngII-induced hypertension by reducing renal blood flow, thereby enhancing sodium retention [33]. When the AT1 receptors are activated in the adrenal cortex, aldosterone is released and drives increased sodium reabsorption in the distal nephron [34]. In the central nervous system, AngII acts on both subtypes of the type 1 receptor, according to their relative cellular distribution, and AT1a receptors exert a predominant effect on cardiovascular function and BP [35,36]. Like in the central nervous system, the relative contribution of AT1b receptors to overall BP regulation is minimal compared with the AT1a receptor. The other major category of the AngII receptors, AT2 receptors, is generally thought to have the opposite effects of AT1 receptors, inducing vasodilatation and natriuresis [37,38] and are described in a recent review by Steckelings et al. [39].
To demonstrate the central role of the kidney AT1 receptor pools in BP regulation, Crowley et al. [11] utilized a kidney cross-transplantation strategy in AT1a knock out and wild-type littermates. This experimental paradigm led to four experimental groups, each with only a single functioning transplanted kidney: kidney knock out, systemic knock out, total knock out, and wild type. Mice lacking AT1a receptors only in the kidney (kidney knock out) presented with lower BP (~20 mmHg), without affecting aldosterone levels. Mice with systemically absent AT1a receptors (systemic knock out, renal AT1a intact) also had similar BP reduction, suggesting AngII actions in extrarenal tissues are also important in determining BP set point. ‘Wild-type’ and ‘ knock out’ surgical controls in this experiment had normal and reduced BPs, respectively.
In follow-up studies, with the same cross-transplant strategy, this same group [40] showed that when AT1a receptors are absent from kidneys during AngII-mediated hypertension, the extrarenal AT1a receptors cannot induce the same level of hypertension and cardiac hypertrophy. Importantly, during AngII infusion, sodium excretion was higher in mice lacking renal AT1a receptors or having no AT1a receptors at all (kidney knock out and total knock out groups) compared with mice with normal expression of AT1a receptors in the kidney (wild type and systemic knock out). These studies support the notion that AT1a receptors in the kidney are primary determinants of hypertension in AngII-dependent hypertension by promoting sodium retention and counteracting pressure natriuresis.
The kidney cross-transplant experiments suggest that AngII induces hypertension mainly through activating renal AT1a receptors. However, there are a vast array of renal cell types and the specific cell lineage responsible (renal epithelia or vasculature) was unknown. We hypothesized that AngII signaling in each segment and/or cell type may exert distinct effects, based on the distinct physiological functions, hormonal control, and expression of RAS components. The proximal tubule was viewed as having high relevance, because two-third of the filtrate is reabsorbed here, it contains all the components of the RAS under local control [41–43], and it is a key locus of the pressure-natriuresis response [44]. Years ago it was shown that AngII in the proximal tubule stimulated fluid and sodium reabsorption by activating AT1 receptors [45,46]. Schuster et al. [45] showed that AngII directly stimulated volume reabsorption in microperfused rabbit proximal convoluted tubules. Beyond effects in the proximal tubule, AT1a receptors in the distal nephron might play a different role. In a recent study by our group, cell-specific deletion of the AT1a receptor from intercalated cells in the collecting duct had no effect on baseline BPs, but exhibited an exaggerated hypertensive response to AngII infusion, indicating quite an opposite effect of these AT1a receptors from those in the proximal tubule [47]. Additionally, knocking out AT1a receptors from principal cells in the collecting duct, the locus of aldosterone stimulation of sodium reabsorption, had no effect on baseline BPs, however, it did attenuate the hypertensive response to AngII [48]. These studies highlight the increasing complexity of the actions of AngII along the nephron.
BLOOD PRESSURE REGULATION BY THE ANGIOTENSIN 1A RECEPTOR IN THE PROXIMAL TUBULE
Multiple investigators have sought to clarify the physiological significance of the proximal tubule RAS in sodium and fluid homeostasis, and together this body of work strongly supports a critical role for proximal tubule AT1a receptors in a complex renal and systemic RAS. Some of the key studies will be reviewed here.
Thomson et al. [49] examined the complex relationship between dietary sodium intake and proximal tubular fluid reabsorption in rats using micropucture. They discovered that high-sodium diet suppressed systemic AngII, as expected, but unexpectedly increased tubular AngII; consistent with this finding, the effect of the ARB losartan on proximal tubule fluid reabsorption was unaffected by a high-salt diet. One conclusion from these studies is that the proximal tubule plays an important role in stabilizing end proximal flow rate rather than effecting natriuresis during high-salt diet. The role of the distal nephron was not directly addressed but is examined in subsequent studies using new mouse models (below).
In 2004, Le et al. [50] reported that in mice with transgenic expression of AT1a receptors under control of the proximal tubule specific γ-glutamyl trans-peptidae promoter (Table 1). Transgenic AT1a receptor expression was applied to mice of a wild-type background with intact receptors and to mice with targeted disruption of one or both of the AT1 receptor genes (Agtr1a and Agtr1b) [51,57]. The transgene did not restore BP or abolish sodium sensitivity in Agtr1a−/− Agtr1b−/− mice but did attenuate the elevated renin expression, a characteristic of AT receptor-deficient mice, and significantly reduced cortical cyst formation in Agtr1a−/− Agtr1b−/− mice. In summary, this model did not demonstrate an effect of proximal tubule-specific AT1a receptors on BP homeostasis, but did demonstrate that partial restoration of proximal tubule AT1a receptors stabilizes renin expression and structural integrity of the renal cortex.
Table 1.
Summary of phenotypes: mouse models with targeted alterations in proximal tubule AT1a receptors
| Model | PT AT1aR deletion (PTKO)a Gurley et al. 2011 [54] |
PT AT1aR overexpression (Tg) or deletion (KO)b Li et al. 2011 [56] |
AT1aR Tg expression in WT and AT1R-deficient micec Le et al. 2004 [50] |
AT1aR transfer ± ECFPAngII in AT1R deficient miced Li et al. 2013 [58▪▪] |
|---|---|---|---|---|
| Baseline SBP | Radiotelemetry: PTKO <controls |
Tailcuff: Tg >NT Radiotelemetry: Tg >NT >KO |
Tailcuff: WT Tg+=WT Tg− KO Tg+=KO Tg− |
Tailcuff: AngII >AT1a receptor alone >AT1a KO |
| AngII hypertension | PTKO <controls (AngII 1000 ng/kg/min × 13d) | Tg >NT (AngII 800 ng/kg/min × 10d) | Not examined | Not examined |
| Sodium handling | Radiotelemetry SBP: PTKO =controls Ur Na excretion during AngII: PTKO >controls | Ur Na excretion: TG >NT =KO |
Tailcuff SBP: KO Tg− =KO Tg+ |
Not examined |
| Sodium transporters | Protein levels Baseline: PTKO had normal NHE3, NaPi2, NKCC, ENaC, NKA AngII infusion: PTKO had lower NHE3, NaPi2a but similar NKCC2. |
mRNA levels Baseline: Tg had normal NHE3, NKCC2, NCC, NaPi2, ENaC, NKA KO had ↓NKCC2 and ↓NCC AngII infusion: NE |
Not examined | Protein levels Baseline: AT1aR-ECFP/AngII mice had higher NHE3 levels |
| Other | Male mice | Female and male mice | Male mice ↓Cortical cyst formation in Tg+ | Male mice |
Phosphoenolpyruvate carboxykinase-Cre recombinase transgene crossed with a line with conditional Agtr1a allele.
PT-specific, androgen-dependent, promotor contruct (kidney androgen regulated protein 2).
γ-glutamyl transpeptidase-driven transgene in WT mice and in mice with disruption of one or both of the murine AT1 receptor genes (Agtr 1a, Agtr 1b)
AT1a receptor transfer with or without ECFP/AngII in AT1aR deficient mice
AngII, angiotensin II; ECFP/AngII, intracellular AngII fusion protein; KO, knock out; NaPi2, sodium phosphate cotransporter; NCC, sodium chloride cotransporter; NE, not examined; NHE3, sodium-proton antiporter 3; NKA, sodium potassium ATPase; NKCC2, Na-K-2Cl cotransporter; NT, nontransgenic controls; PT, proximal tubule; PTKO, proximal tubule knock out (AT1a); Tg, transgenic; WT, wild type.
Gurley et al. [54] developed a mouse model specifically lacking AT1a receptors in the renal proximal tubule with cre–loxp technology (Table 1). Mice bearing a conditional Agtr1a allele [55,52] were generated by the Coffman Laboratory (Duke University), inbred onto the 129/SvEv genetic background, and were crossed with 129/SvEv mice bearing a phosphoenolpyruvate carboxykinase-Cre recombinase transgene (Haase Laboratory, Vanderbilt University, Nashville, Tennessee, USA). Mice lacking AT1a receptors in the renal proximal tubule knock out (PTKO) had significantly lower baseline BPs than controls (~10 mmHg). BPs in both wild-type and knock out groups responded similarly during high and low-sodium diets, indicating that proximal tubule AT1a receptors do not regulate sodium sensitivity. However, at baseline, proximal tubule fluid resorption was significantly lower in PTKO mice compared with controls and protection against hypertension was evident during chronic infusion of AngII. Accompanying this reduced hypertensive response was increased natriuresis in the PTKOs along with reduced expression of key sodium transporter proteins: AngII caused a 30% reduction in the sodium hydrogen exchanger (NHE3) in the wild-type mice and more than 50% reduction in the PTKO group (P <0.05). In addition to NHE3, the sodium phosphate cotransporter 2 (NaPi2) protein levels, unaffected in the wild-type mice, decreased by 36% in the PTKOs (P =0.005). These transporter reductions during AngII are viewed as responses to the elevated BP associated with AngII hypertension and are amplified when there is reduction in signaling through AT1R. Vasoconstrictive responses to acute infusion of AngII were nearly identical in both wild-type and PTKO groups indicating preserved vascular responses in PTKOs. Eliminating the proximal tubule AT1aR prevented the AngII-mediated opposition of the proximal tubule pressure natriuresis response, thus, lower levels of BP are able to bring the mouse into sodium balance. These in-vivo findings highlight the critical role of proximal tubule AT1a receptors and suggest that effectively targeting epithelial functions and sodium reabsorption of the proximal tubule of the kidney could be a useful and promising therapeutic strategy in hypertension.
Li et al. [56] utilized a different molecular approach to examine the effect of AngII on BP in the proximal tubule. Mice with either overexpression or deletion of constitutively active AT1a receptors were generated [53]. This innovative model allowed examination of both gain and loss of function of the AT1R. Androgen administration to female transgenic mice induced the transgene, causing, as expected, overexpression of AT1a and raised BPs compared with the response in nontransgenic mice; Cre recombinase-mediated deletion of proximal tubule AT1a receptors reduced BPs to a level similar to that reported by Gurley et al. [54]. A normal response to AngII was evident in the over-expression model and not examined in the deletion model. There were no apparent changes in expression of NHE3, NaPi2, or the epithelial sodium transporter in this proximal tubule-depletion model.
Work from Li and Zhuo sought to determine the role of proximal tubule intracellular AngII on BP [58▪▪]. Wild-type AT1a receptors were expressed in AT1a deficient mice with or without an intracellular AngII fusion protein (AngII–ECFP). Transfer of only the receptors provoked higher SBP (12 mmHg) indicating a role for systemic AngII, whereas transfer of both receptors and AngII–ECFP protein led to higher proximal tubule AngII levels, even higher BP, and lower sodium excretion. This pattern suggests that, in proximal tubule, both AT1a receptors and intracellular AngII impact BP regulation. The higher BP and signaling responses were prevented by losartan.
Regulation of the AT1 receptor and potential mechanisms
Additional mechanisms by which AT1a receptors are regulated include a role for the AT1 receptor-associated proteins, angiotensin II receptor associated protein (ATRAP) and ARAP1, that determine the density of AT1 receptors by interacting with the carboxylterminus. ATRAP selectively suppresses AngII-mediated pathological activation of the AT1 receptor by inhibiting intracellular AngII signaling [59]. Overexpression of ATRAP can attenuate the effects of chronic AngII on the severity of hypertension by both an increase in natriuresis and suppression of the epithelial sodium transporter [60]. ARAP1 has the opposite effect as it increases AT1 receptor expression by recycling it to the cell membrane, a response we predict would raise BP as over-expression (of AT1R) causes salt-sensitivity and AngII-dependent hypertension [27]. These studies highlight how regulation of the receptor abundance and trafficking could control function.
Several studies, including those highlighted in this review, also examined sodium transport as a downstream function of AngII signaling in the renal proximal tubule. Reduced sodium retention during AT1A receptor blockade or deletion provides potential mechanisms for BP lowering. Additionally, studies by Riquier-Brison et al. [61] illustrate how natriuretic stimuli (angiotensin-converting enzyme inhibitors inhibition of AngII, hypertension, high salt diet,) decrease proximal tubule sodium reabsorption. Modulation of abundance and cellular location of the NHE3 and NaPi2 in the renal proximal tubule are highly likely targets [61,62], as well as distribution of NHE3 and NaPi2 along the apical microvilli of the proximal tubule [61,63]. Finally, we should also consider additional effects of the fine control of sodium handling along the distal nephron and cortical collecting duct as additional agents for integrated sodium homeostasis along the nephron [44,49], for example, mediated by proximal production of AngII induced by proximal tubule AT1a receptors.
CONCLUSION
Several experimental models have demonstrated a nonredundant role for AT1a receptors in the epithelia of the renal proximal tubule to impact and regulate BP and fluid homeostasis. The regulation of systemic BP by the kidney is accomplished by modulation of sodium transporter proteins and sodium and fluid reabsorption. Targeting the proximal tubule may enhance natriuresis to balance elevated distal nephron sodium reabsorption and, thus reduce the BP at which sodium homeostasis can be maintained.
KEY POINTS.
AngII acts via the type 1 angiotensin receptor to execute many of the actions of the RAS.
The renal PT contains all of the components of the RAS and is a key site for the regulation of BP through modulation of tubular fluid reabsorption and regulated expression of sodium transporter proteins via proximal tubule AT1a receptor-stimulated production of AngII.
Reduced AngII signaling in the proximal tubule is associated with lower BP, protection from hypertension, and reduced sodium reabsorption by the nephron.
Targeting the epithelial functions of the proximal tubule could be a useful therapeutic strategy in hypertension.
Acknowledgments
Financial support and sponsorship
The work was supported by the funding from the NIH to S.B.G., A.A.M. (NIH 1R01 DK09838201A1, DK098382, R01 DK083785), and the Duke O’Brien Center for Kidney Research (NIH P30DK096493).
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
- 1.Group SR, Wright JT, Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373:2103–2116. doi: 10.1056/NEJMoa1511939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.NCD Risk Factor Collaboration (NCD-RisC) Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population-based measurement studies with 19. 1 million participants. Lancet. 2017;389:37–55. doi: 10.1016/S0140-6736(16)31919-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merai R, Siegel C, Rakotz M, et al. CDC grand rounds: a public health approach to detect and control hypertension. MMWR Morb Mortal Wkly Rep. 2016;65:1261–1264. doi: 10.15585/mmwr.mm6545a3. [DOI] [PubMed] [Google Scholar]
- 4.Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–2260. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker WL, Coleman CI, Kluger J, et al. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors or angiotensin II-receptor blockers for ischemic heart disease. Ann Intern Med. 2009;151:861–871. doi: 10.7326/0003-4819-151-12-200912150-00162. [DOI] [PubMed] [Google Scholar]
- 6.Pfeffer MA, Swedberg K, Granger CB, et al. CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-overall programme. Lancet. 2003;362:759–766. doi: 10.1016/s0140-6736(03)14282-1. [DOI] [PubMed] [Google Scholar]
- 7.Lewis EJ, Hunsicker LG, Clarke WR, et al. Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 8.Brenner BM, Cooper ME, de Zeeuw D, et al. RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 9.Matchar DB, McCrory DC, Orlando LA, et al. Systematic review: comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers for treating essential hypertension. Ann Intern Med. 2008;148:16–29. doi: 10.7326/0003-4819-148-1-200801010-00189. [DOI] [PubMed] [Google Scholar]
- 10.Husain AG. Drugs, enzymes and receptors of the renin-angiotensin system: celebrating a century of discovery. Sydney: Harwood Academic; 2000. [Google Scholar]
- 11.Crowley SD, Gurley SB, Oliverio MI, et al. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest. 2005;115:1092–1099. doi: 10.1172/JCI200523378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guyton AC, Coleman TG, Cowley AV, Jr, et al. Arterial pressure regulation. Overriding dominance of the kidneys in long-term regulation and in hypertension. Am J Med. 1972;52:584–594. doi: 10.1016/0002-9343(72)90050-2. [DOI] [PubMed] [Google Scholar]
- 13.Guyton AC. Blood pressure control: special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 14.Hall JE, Brands MW, Henegar JR. Angiotensin II and long-term arterial pressure regulation: the overriding dominance of the kidney. J Am Soc Nephrol. 1999;10(Suppl 12):S258–S265. [PubMed] [Google Scholar]
- 15.Hall JE. Control of sodium excretion by angiotensin II: intrarenal mechanisms and blood pressure regulation. Am J Physiol. 1986;250:R960–R972. doi: 10.1152/ajpregu.1986.250.6.R960. [DOI] [PubMed] [Google Scholar]
- 16.Crowley SD, Gurley SB, Coffman TM. AT(1) receptors and control of blood pressure: the kidney and more. Trends Cardiovasc Med. 2007;17:30–34. doi: 10.1016/j.tcm.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Schweda F, Klar J, Narumiya S, et al. Stimulation of renin release by prostaglandin E2 is mediated by EP2 and EP4 receptors in mouse kidneys. Am J Physiol Renal Physiol. 2004;287:F427–F433. doi: 10.1152/ajprenal.00072.2004. [DOI] [PubMed] [Google Scholar]
- 18.Prieto-Carrasquero MC, Harrison-Bernard LM, Kobori H, et al. Enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Hypertension. 2004;44:223–229. doi: 10.1161/01.HYP.0000135678.20725.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohrwasser A, Morgan T, Dillon HF, et al. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension. 1999;34:1265–1274. doi: 10.1161/01.hyp.34.6.1265. [DOI] [PubMed] [Google Scholar]
- 20.Pohl M, Kaminski H, Castrop H, et al. Intrarenal renin angiotensin system revisited: role of megalin-dependent endocytosis along the proximal nephron. J Biol Chem. 2010;285:41935–41946. doi: 10.1074/jbc.M110.150284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouby N, Hus-Citharel A, Marchetti J, et al. Expression of type 1 angiotensin II receptor subtypes and angiotensin II-induced calcium mobilization along the rat nephron. J Am Soc Nephrol. 1997;8:1658–1667. doi: 10.1681/ASN.V8111658. [DOI] [PubMed] [Google Scholar]
- 22.Harrison-Bernard LM, Navar LG, Ho MM, et al. Immunohistochemical localization of ANGII AT1 receptor in adult rat kidney using a monoclonal antibody. Am J Physiol. 1997;273(1 Pt 2):F170–F177. doi: 10.1152/ajprenal.1997.273.1.F170. [DOI] [PubMed] [Google Scholar]
- 23.Kobori H, Harrison-Bernard LM, Navar LG. Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int. 2002;61:579–585. doi: 10.1046/j.1523-1755.2002.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niimura F, Okubo S, Fogo A, Ichikawa I. Temporal and spatial expression pattern of the angiotensinogen gene in mice and rats. Am J Physiol. 1997;272(1 Pt 2):R142–R147. doi: 10.1152/ajpregu.1997.272.1.R142. [DOI] [PubMed] [Google Scholar]
- 25.Matsusaka T, Niimura F, Pastan I, et al. Podocyte injury enhances filtration of liver-derived angiotensinogen and renal angiotensin II generation. Kidney Int. 2014;85:1068–1077. doi: 10.1038/ki.2013.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ying J, Stuart D, Hillas E, et al. Overexpression of mouse angiotensinogen in renal proximal tubule causes salt-sensitive hypertension in mice. Am J Hypertens. 2012;25:684–689. doi: 10.1038/ajh.2012.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen D, Coffman TM. AT1 angiotensin receptors-vascular and renal epithelial pathways for blood pressure regulation. Curr Opin Pharmacol. 2015;21:122–126. doi: 10.1016/j.coph.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Navar LG, Prieto MC, Satou R, Kobori H. Intrarenal angiotensin II and its contribution to the genesis of chronic hypertension. Curr Opin Pharmacol. 2011;11:180–186. doi: 10.1016/j.coph.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, et al. The absence of intrarenal ACE protects against hypertension. J Clin Invest. 2013;123:2011–2023. doi: 10.1172/JCI65460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito M, Oliverio MI, Mannon PJ, et al. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc Natl Acad Sci U S A. 1995;92:3521–3525. doi: 10.1073/pnas.92.8.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sparks MA, Crowley SD, Gurley SB, et al. Classical renin-angiotensin system in kidney physiology. Compr Physiol. 2014;4:1201–1228. doi: 10.1002/cphy.c130040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oliverio MI, Best CF, Smithies O, Coffman TM. Regulation of sodium balance and blood pressure by the AT(1A) receptor for angiotensin II. Hypertension. 2000;35:550–554. doi: 10.1161/01.hyp.35.2.550. [DOI] [PubMed] [Google Scholar]
- 33.Sparks MA, Stegbauer J, Chen D, et al. Vascular type 1A angiotensin ii receptors control BP by regulating renal blood flow and urinary sodium excretion. J Am Soc Nephrol. 2015;26:2953–2962. doi: 10.1681/ASN.2014080816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aguilera G. Role of angiotensin II receptor subtypes on the regulation of aldosterone secretion in the adrenal glomerulosa zone in the rat. Mol Cell Endocrinol. 1992;90:53–60. doi: 10.1016/0303-7207(92)90101-b. [DOI] [PubMed] [Google Scholar]
- 35.Morris MJ, Wilson WL, Starbuck EM, Fitts DA. Forebrain circumventricular organs mediate salt appetite induced by intravenous angiotensin II in rats. Brain Res. 2002;949:42–50. doi: 10.1016/s0006-8993(02)02963-3. [DOI] [PubMed] [Google Scholar]
- 36.Davisson RL, Oliverio MI, Coffman TM, Sigmund CD. Divergent functions of angiotensin II receptor isoforms in the brain. J Clin Invest. 2000;106:103–106. doi: 10.1172/JCI10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carey RM. Blood pressure and the renal actions of AT2 receptors. Curr Hypertens Rep. 2017;19:21. doi: 10.1007/s11906-017-0720-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Padia SH, Carey RM. AT2 receptors: beneficial counter-regulatory role in cardiovascular and renal function. Pflugers Arch. 2013;465:99–110. doi: 10.1007/s00424-012-1146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steckelings UM, Paulis L, Namsolleck P, Unger T. AT2 receptor agonists: hypertension and beyond. Curr Opin Nephrol Hypertens. 2012;21:142–146. doi: 10.1097/MNH.0b013e328350261b. [DOI] [PubMed] [Google Scholar]
- 40.Crowley SD, Gurley SB, Herrera MJ, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990. doi: 10.1073/pnas.0605545103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 42.Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension. 2002;39:316–322. doi: 10.1161/hy0202.103821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weinstein AM. Sodium and chloride transport: proximal nephron. In: Alpern R, Hebert SC, editors. Seldin and Giebisch’s the kidney physiology and pathophysiology. 4. Burlington, MA: Academic Press; 2008. pp. 793–847. [Google Scholar]
- 44.McDonough AA, Nguyen MT. Maintaining balance under pressure: integrated regulation of renal transporters during hypertension. Hypertension. 2015;66:450–455. doi: 10.1161/HYPERTENSIONAHA.115.04593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuster VL, Kokko JP, Jacobson HR. Angiotensin II directly stimulates sodium transport in rabbit proximal convoluted tubules. J Clin Invest. 1984;73:507–515. doi: 10.1172/JCI111237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cogan MG. Angiotensin II: a powerful controller of sodium transport in the early proximal tubule. Hypertension. 1990;15:451–458. doi: 10.1161/01.hyp.15.5.451. [DOI] [PubMed] [Google Scholar]
- 47.Stegbauer J, Chen D, Herrera M, et al. Resistance to hypertension mediated by intercalated cells of the collecting duct. JCI Insight. 2017;2:e92720. doi: 10.1172/jci.insight.92720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen D, Stegbauer J, Sparks MA, et al. Impact of angiotensin type 1A receptors in principal cells of the collecting duct on blood pressure and hypertension. Hypertension. 2016;67:1291–1297. doi: 10.1161/HYPERTENSIONAHA.115.06987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomson SC, Deng A, Wead L, et al. An unexpected role for angiotensin II in the link between dietary salt and proximal reabsorption. J Clin Invest. 2006;116:1110–1116. doi: 10.1172/JCI26092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le TH, Oliverio MI, Kim HS, et al. A gammaGT-AT1A receptor transgene protects renal cortical structure in AT1 receptor-deficient mice. Physiol Genomics. 2004;18:290–298. doi: 10.1152/physiolgenomics.00120.2003. [DOI] [PubMed] [Google Scholar]
- 51.Reid LH, Shesely EG, Kim HS, Smithies O. Cotransformation and gene targeting in mouse embryonic stem cells. Mol Cell Biol. 1991;11:2769–2777. doi: 10.1128/mcb.11.5.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Higgins DF, Kimura K, Bernhardt WM, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Zhou X, Davis DR, et al. An androgen-inducible proximal tubule-specific Cre recombinase transgenic model. Am J Physiol Renal Physiol. 2008;294:F1481–F1486. doi: 10.1152/ajprenal.00064.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gurley SB, Riquier-Brison AD, Schnermann J, et al. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell metabolism. 2011;13:469–475. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006;66:2576–2583. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li H, Weatherford ET, Davis DR, et al. Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1067–R1077. doi: 10.1152/ajpregu.00124.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bronson SK, Plaehn EG, Kluckman KD, et al. Single-copy transgenic mice with chosen-site integration. Proc Natl Acad Sci U S A. 1996;93:9067–9072. doi: 10.1073/pnas.93.17.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58▪▪.Li XC, Zhuo JL. Proximal tubule-dominant transfer of AT(1a) receptors induces blood pressure responses to intracellular angiotensin II in AT(1a) receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol. 2013;304:R588–R598. doi: 10.1152/ajpregu.00338.2012. Combined expression of AT1a receptor and intracellular ligand used to demonstrate that both impact BP regulation in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tanaka Y, Tamura K, Koide Y, et al. The novel angiotensin II type 1 receptor (AT1R)-associated protein ATRAP downregulates AT1R and ameliorates cardiomyocyte hypertrophy. FEBS Lett. 2005;579:1579–1586. doi: 10.1016/j.febslet.2005.01.068. [DOI] [PubMed] [Google Scholar]
- 60.Wakui H, Tamura K, Masuda S, et al. Enhanced angiotensin receptor-associated protein in renal tubule suppresses angiotensin-dependent hypertension. Hypertension. 2013;61:1203–1210. doi: 10.1161/HYPERTENSIONAHA.111.00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Riquier-Brison AD, Leong PK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol. 2010;298:F177–F186. doi: 10.1152/ajprenal.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li XC, Hopfer U, Zhuo JL. AT1 receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through a microtubule-dependent endocytic pathway. Am J Physiol Renal Physiol. 2009;297:F1342–F1352. doi: 10.1152/ajprenal.90734.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McDonough AA. Mechanisms of proximal tubule sodium transport regulation that link extracellular fluid volume and blood pressure. Am J Physiol Regul Integr Comp Physiol. 2010;298:R851–R861. doi: 10.1152/ajpregu.00002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]