

Abstract
Group A rotaviruses are the major cause of severe gastroenteritis in the young of mammals and birds. This report describes characterization of an unusual G20P[28] rotavirus strain detected in a 24 month old child from Suriname. Genomic sequence analyses revealed that the genotype constellation of the Suriname strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] was G20-P[28]-I13-R13-C13-M12-A23-N13-T15-E20-H15. Genes VP1, VP2, VP3, NSP1, NSP2, NSP3, NSP4 and NSP5 were recently assigned novel genotypes by the Rotavirus Classification Working Group (RCWG). Three of the 11 gene segments (VP7, VP4, VP6) were similar to cognate gene sequences of bat-like human rotavirus strain Ecu534 from Ecuador and the VP7, NSP3 and NSP5 gene segments of strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] were found to be closely related to gene sequences of bat rotavirus strain 3081/BRA detected in Brazil. Although distantly related, the VP1 gene of the study strain and bat strain BatLi09 detected in Cameroon in 2014 are monophyletic. The NSP1 gene was found to be most closely related to human strain QUI-35-F5 from Brazil. These findings suggest that strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] represents a zoonotic infection from a bat host.
Keywords: Rotavirus, G20P[28], Novel genotypes, Suriname
1. Introduction
Group A rotavirus (RVA) is one of the major causes of severe diarrhea and morbidity in young children and animals worldwide. The RVA genome comprises 11 segments of double-stranded RNA (dsRNA) that are enclosed in a triple-layered capsid. Each segment encodes one viral structural protein (VP1, VP2, VP3, VP4, VP6, VP7) and one nonstructural protein (NSP1, NSP2, NSP3, or NSP4), except for segment 11, which encodes NSP5 and, in some strains, NSP6 (Estes and Greenberg, 2013). The Rotavirus Classification Working Group (RCWG) has devised a complete genotype classification system, defining the genotype constellation of RVAs as follows: Gx-P[x]-Ix-Rx-Cx-Mx-Ax-Nx-Tx-Ex-Hx, representing VP7-VP4-VP6-VP1-VP2-VP3-NSP1-NSP2-NSP3-NSP4-NSP5 (Matthijnssens et al., 2011). To date, 35 G, 50 P, 26 I, 21 R, 19C, 19 M, 30 A, 20 N, 21 T, 26 E and 21H genotypes have been identified in human and non-human hosts and classified based on differences in the nucleotide sequence identities of each encoding gene segment, respectively (Abe et al., 2011; Esona et al., 2010; Estes and Greenberg, 2013; Guo et al., 2012; Jere et al., 2014; Matthijnssens et al., 2011; Papp et al., 2012; Trojnar et al., 2013; Yinda et al., 2016), http://rega.kuleuven.be/cev/viralmetagenomics/virus-classification). This classification system has accelerated the comparison of RVA genotypes and increased our understanding of the genetic diversity of RVA.
Interspecies transmission and genetic reassortment between human and animal RVA have been described frequently, and pigs and cattle are considered the major reservoirs for the genetic and antigenic diversity of human RVA strains (Martella et al., 2010; Matthijnssens et al., 2008a). In recent years complete genome based phylogenetic analyses have been used to define the genotype constellations of RVA strains and, based on this, most human RVA strains show high similarity in all segments to either the Wa-like genogroup 1 constellation (Gx-P[x]-I1-R1-C1-M1-A1-N1-T1-E1-H1) or the DS-1-like genogroup 2 constellation (Gx-P[x]-I2-R2-C2-M2-A2-N2-T2-E2-H2) (Matthijnssens et al., 2008a). In addition, a small group of human RVA strains belong to the AU-1-like genogroup 3 constellation (Gx-P[x]-I3-R3-C3-M3-A3-N3-T3-E3-H3) (Matthijnssens et al., 2008b).
Bats are increasingly being recognized an important reservoir for RVA. To date, a few RVA strains, each with distinct genetic diversity have been detected and reported in bats. The nucleotide sequences of these bat RVAs are typically distant from those of human and other mammalian RVAs and have been suggested to be bat specific (Esona et al., 2010; He et al., 2013; Xia et al., 2014; Yinda et al., 2016). However, the VP4 and NSP4 gene segments of bat RVA strain RVA/Bat-wt/KEN/KE4852/2007/G25P[6] detected in Kenya (Esona et al., 2010), gene segments VP7, VP4, VP3, NSP2 and NSP3 of bat RVA strain RVA/Bat-wt/ZMB/LUS12–14/2012/G3P[3] reported in Zambia (Sasaki et al., 2016) and VP6 gene segment of bat RVA strains 2980/BatRVA and 322/BatRVA detected in Kenya (Waruhiu et al., 2017) are highly similar to cognate gene sequences of human RVAs circulating in the same community. This suggests that single or multiple gene reassortment events have occurred between human and bat RVAs. Here we report the molecular characterization of a human G20P[28] RVA strain from Suriname with genes belonging to eight novel genotypes that is possibly the product of reassortment between bat and human RVA strains.
2. Materials and methods
In 2005, Pan American Health Organization (PAHO) initiated regional RVA surveillance in Latin America and the Caribbean. The network was initially established with 7 countries and by the end of 2007, four more countries, including Suriname, were added for a total of 11 countries (de Oliveira et al., 2009). The primary focus of this network was to obtain useful comprehensive RVA disease-burden data for policy makers to help implement the RVA vaccination program. The U.S. Centers of Disease Control and Prevention (CDC) serves as a regional reference laboratory for the network and performs genetic characterization of detected RVA strains.
2.1. Patient and sample collection
In 2013, a 24 months-old girl presented to a physician in Lands Hospital, Suriname with diarrhea (3 episodes per day), vomiting (3 episodes per day) and moderate dehydration. The patient had no record of either RotaTeq® or Rotarix® vaccination and had no record of exposure to animals. A stool sample was collected and forwarded to the CDC for genotyping along with 77 other surveillance samples from Suriname that year.
2.2. RVA dsRNA extraction, purification, cDNA synthesis and amplification; sequencing and genotype assignment
RVA dsRNA for full genome sequencing was extracted from the fecal sample following a previously described method (Potgieter et al., 2009). The sequencing templates and library were prepared by using a sequence-independent whole-genome reverse transcription PCR amplification method (Jere et al., 2011; Potgieter et al., 2009). Next generation sequencing was carried out on a MiSeq sequencer using the Illumina MiSeq reagent kit v.2 with 500 cycles and the standard 250 bp paired-end reads method. Contigs were assembled from the obtained sequence using the de novo assembly command and guided assembly with default parameters in CLC Genomics Workbench 7.0.4 software (http://www.clcbio.com/products/clc-genomics-workbench/). The genotypes were determined according to the guidelines of the RCWG (Matthijnssens et al., 2008b) using the online genotyping tool RotaC (http://rotac.regatools.be) (Maes et al., 2009) and BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Gene segments that were not assigned any genotype by the above-mentioned approach were submitted to the RCWG for genotype assignment.
2.3. Phylogenetic and genetic analyses
For each gene, multiple alignments were made by using the MUSCLE algorithm implemented in MEGA6 software (Tamura et al., 2013), http://www.megasoftware.net/). Once aligned, the DNA Model Test program implemented in MEGA version 6 was used to identify the optimal evolutionary models that best fit the sequence datasets. Using Corrected Akaike Information Criterion (AICc), the following models were found to best fit the sequence data for the indicated genes: GTR + G + I (VP1, VP2, VP3, VP4, VP6, NSP1, NSP2, and NSP3), GTR + G (NSP4), HKY + G (NSP5), and HKY + G + I (VP7). With these models, maximum-likelihood trees were constructed using MEGA 6 with 500 bootstrap replicates to estimate branch support. Nucleotide and amino acid distance matrices were prepared using the p-distance algorithm of MEGA 6 software (Tamura et al., 2013).
3. Result and discussion
RVA genomic classification studies have revealed the emergence of novel or unusual RVAs carrying several novel gene segments or genotype constellations (Abe et al., 2011; Esona et al., 2010; Jere et al., 2014; Matthijnssens et al., 2011; Papp et al., 2012; Trojnar et al., 2013; Yinda et al., 2016). In this study, we identified and characterized a RVA strain with an unusual genotype constellation and carrying eight recently assigned novel gene segments.
A combination of de novo assembly and subsequent mapping to reference strains was used to obtain the full-length genome of strain RVA/Human-wt/SUR/2014735512/2013/G20P[28]. The complete open reading frames (ORFs) for all 11 gene segments of this strain (Table 1) were deposited in GenBank under accession numbers KX257405-KX257415 for VP7, VP6, VP4, VP3, VP2, VP1, NSP5, NSP4, NSP3, NSP2, and NSP1, respectively.
Table 1.
Size of the Open Reading Frames (ORFs) and deduced amino acid sequences of Suriname strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] compared to genogroup 1 (Wa), genogroup 2 (DS-1) and genogroup 3 (AU-1) strains. Stop codon of each gene segment have been excluded.
| Strain | Genome segments
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| VP1 | VP2 | VP3 | VP4 | VP6 | VP7 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 | |
| ORFs (bp) | |||||||||||
| RVA/Human-wt/SUR/2014735512/2013/G20P[28] | 3264 | 2688 | 2508 | 2328 | 1191 | 978 | 1482 | 951 | 930 | 537 | 615 |
| RVA/Human-tc/USA/Wa/1974/G1P[8] | 3264 | 2670 | 2505 | 2325 | 1191 | 978 | 1456 | 951 | 930 | 525 | 591 |
| RVA/Human-tc/USA/DS-1/1976/G2P[4] | 3264 | 2637 | 2505 | 2325 | 1191 | 978 | 1456 | 951 | 939 | 525 | 600 |
| RVA/Human-tc/JPN/AU-1/1982/G3P[9] | 3264 | 2661 | 2505 | 2325 | 1191 | 978 | 1473 | 951 | 939 | 525 | 594 |
| Proteins (aa) | |||||||||||
| RVA/Human-wt/SUR/2014735512/2013/G20P[28] | 1088 | 896 | 836 | 776 | 397 | 326 | 494 | 317 | 310 | 179 | 205 |
| RVA/Human-tc/USA/Wa/1974/G1P[8] | 1088 | 890 | 835 | 775 | 397 | 326 | 486 | 317 | 310 | 175 | 197 |
| RVA/Human-tc/USA/DS-1/1976/G2P[4] | 1088 | 879 | 835 | 775 | 397 | 326 | 486 | 317 | 313 | 175 | 200 |
| RVA/Human-tc/JPN/AU-1/1982/G3P[9] | 1088 | 887 | 835 | 775 | 397 | 326 | 491 | 317 | 313 | 175 | 198 |
When first detected, the RVA/Human-wt/SUR/2014735512/2013/G20P[28] VP1, VP2, VP3, NSP1, NSP2, NSP3, NSP4 and NSP5 genes, however, were not closely related to the cognate genes of any known RVA strain (Fig. 1). These eight genes shared < 79% nucleotide identity with cognate gene sequences of other RVA strains. They showed the highest nucleotide identities of 75.9%, 79.3%, 71.2%, 75.6%, 71.7%, 73.2%, 67.9% and 75.5% with the VP1, VP2, VP3, NSP1, NSP2, NSP3, NSP4 and NSP5 genes of RVA strains ME848 (KR632623), SA11-H96 (JQ688674), CK20001 (KC443589), QUI-135-F5 (KF185099), BatLi09 (KX268761), RCH272 (KF690133) and DC4455 (KT695003), and SA11-H96 (JQ688683) respectively, which are below the cut-off values defined by RCWG for genotyping (Matthijnssens et al., 2008b). The sequences were submitted to the RCWG, which officially assigned new genotypes to the VP1 (R13), VP2 (C13), VP3 (M12), NSP1 (A23), NSP2 (N13), NSP3 (T15), NSP4 (E20) and NSP5 (H15) genes. Hence, the full genotype constellation of RVA/Human-wt/SUR/2014735512/2013/G20P[28] was designated as G20-P[28]-I13-R13-C13-M12-A23-N13-T15-E20-H15. Since the assignment of new genotypes to the eight genes of the Suriname strain, the NSP3 and NSP5 sequences of a bat RVA strain 3081/BRA from Brazil have been submitted and assigned the same genotypes, T15 and H15, respectively.
Fig. 1.
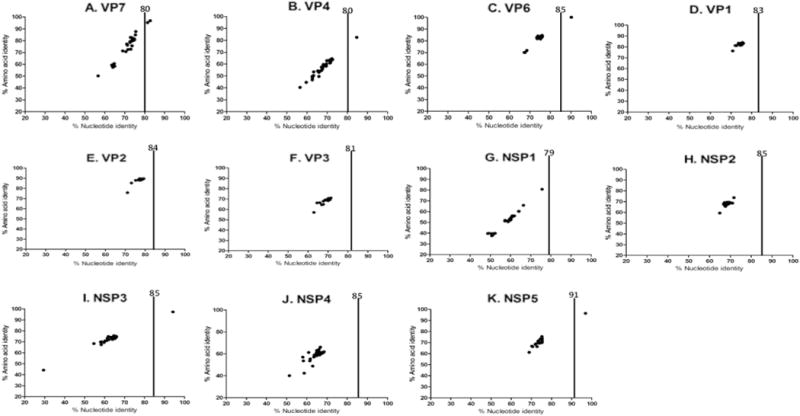
Percentage nucleotide and deduced amino acid homologies of A) viral protein 7 (VP7), B) VP4, C) VP6, D) VP1, E) VP2, F) VP3, G) non-structural protein 1 (NSP1), H) NSP2, I) NSP3, J) NSP4 and K) NSP5 gene segments of strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] compared with cognate genes deposited in GenBank. Vertical lines indicate nucleotide percentage identity cutoff values defining genotypes for 11 rotavirus gene segments. Black dots indicate coordinates for each pairwise comparison when percentage nucleotide identity is plotted against percentage amino acid identity.
Genotype assessment using RotaC revealed that viral genes VP4, VP6, VP7, NSP3 and NSP5 of strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] were classified into previously established human and/or bat RVA genotypes. Analyses of nucleotide and deduced amino acid sequences of gene segments VP4 and VP6 indicated a very close relationship with human rotavirus strain Ecu534 detected in Ecuador in 2006 (Solberg et al., 2009), sharing a nucleotide (amino acid) similarities of 84.6% (97.5%), and 90.2% (100%), respectively (Fig. 1). BLAST searches indicated that the VP7 gene segment of the study strain was also closely related to the Ecuadorian human RVA strain Ecu534 and bat RVA strain 3081/BRA detected in Brazil in 2013 (Asano et al., 2016) and showed nucleotide (amino acid) identities of 81.8% (96.4%) and 81.3% (95.3%), respectively (Fig. 1). The VP7 gene segment of the study strain shared a close genetic relationship to the both human strain Ecu534 and bat strain 3081/BRA. This suggests that the Ecuador strain Ecu534 is not a true human strain and may have originated from a bat.
Also, analyses of nucleotide (amino acid) sequences of NSP3 and NSP5 gene segments indicated that the RVA/Human-wt/SUR/2014735512/2013/G20P[28] NSP3 and NSP5 genes shared 94.2% (97.3%) and 96.8% (96.4%), respectively, to NSP3 and NSP5 genes of bat RVA strain 3081/BRA. Unfortunately, only the VP4, VP7, VP6 genes of the Ecuadorian RVA strain Ecu534 and VP4, VP7, NSP3 and NSP5 genes of bat RVA strain 3081/BRA have been sequenced and deposited in GenBank. However, the VP4 sequence of the bat strain 3081/BRA was < 50% of the VP4 gene and was distantly related (nucleotide < 52.2% and amino acid < 64.4%) to the study strain and strain Ecu534; hence it was omitted in the VP4 gene phylogenetic analysis. Furthermore, eight genes (VP1-VP3, NSP1-NSP5) of strain Ecu534 and seven genes (VP1-VP3, VP6, NSP1, NSP2 and NSP4) of strain 3081/BRA have not been determined and thus are unavailable for analysis.
Phylogenetic analyses of all eleven gene segments revealed that genes VP4, VP6 and VP7 were related to RVA strain Ecu534 detected in Ecuador in 2006 (Fig. 2A–C). Also, the VP7, NSP3 and NSP5 gene segments of strains RVA/Human-wt/SUR/2014735512/2013/G20P [28] clustered with the VP7, NSP3 and NSP5 genes of bat RVA strain 3081/BRA detected in Brazil in 2013. In the phylogeny estimated from gene segment VP1 of strain, RVA/Human-wt/SUR/2014735512/2013/G20P[28] occupied a well-supported group (bootstrap = 95%) with genotype R15 bat RVA strain BatLi09 from Cameroon (Fig. 2D) whereas the NSP1 gene clustered with genotype A23 human RVA strain QUI-35-F5 (bootstrap = 100%), (Fig. 2G). Phylogenetic analysis of the NSP3 gene sequence of strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] resulted in grouping of this strain with genotype T15 bat RVA strain 3081/BRA with very strong bootstrap support (100%; Fig. 2I). The phylogenetic estimate obtained by using NSP5 gene nucleotide data places the study strains in a well-supported group (bootstrap = 100%) with NSP5 gene sequence of bat RVA strain 3081/BRA.
Fig. 2.

Phylograms indicating genetic relationships of complete nucleotide sequences of A) VP7, B) VP4, C) VP6, D) VP1, E) VP2, F) VP3, G) NSP1, H) NSP2, I) NSP3, J) NSP4 and K) NSP5 of strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] (indicated with a black square box) with representatives of known human and animal rotavirus genotypes. Bootstrap values > 70% are indicated at each branch node. Scale bars indicate the number of nucleotide substitutions per site.
The VP2, VP3, NSP2, and NSP4 genes occupied positions at or near the base of their respective trees along with RVA strains from animals (bat, horse, or mouse; Fig. 2E, F, H, J and K).
In the last few years, bats have been recognized as an important natural reservoirs for RNA and DNA virus families and over 130 of these viruses have been found in bats with many being highly pathogenic for both humans and animal livestock (Calisher et al., 2006; Luis et al., 2013; Plowright et al., 2015; Tong et al., 2009). Several improvements in viral detection and characterization methodologies such as high-throughput next generation sequencing have led to a successful discovery and characterization of several novel viruses in bats, including enteric pathogens such as RVA (Luis et al., 2013; Waruhiu et al., 2017). However, since the first detection and characterization of a human-bat reassortant RVA from a Straw-colored Fruit bat in 2010 (Esona et al., 2010), a few of other bat RVA strains, detected and characterized from different bat species and different geographic locations have been reported (Asano et al., 2016; He et al., 2013; Sasaki et al., 2016; Waruhiu et al., 2017; Xia et al., 2013; Yinda et al., 2016). Among the reported bat RVA strains, only bat RVA strain 3081/BRA from Brazil was closely related in the VP7, NSP3 and NSP5 gene segments with the study strain from Suriname. Although interspecies transmission and reassortment events have been described (Esona et al., 2010; Grant et al., 2011; Matthijnssens et al., 2006), the roles of bat RVA in the evolution of human RVA strains and bats as reservoirs for human RVA disease are poorly understood. However, the genetically close relationships of the VP7, VP4, VP6, NSP3 and NSP5 gene segments of the study strain to bat strain 3081/BRA and/or bat-like human strain Ecu534, suggest that RVA/Human-wt/SUR/2014735512/2013/G20P[28] is of bat origin. A few investigators have reported the detection of human RVAs genes in bat RVA strains (Esona et al., 2010; Sasaki et al., 2016; Waruhiu et al., 2017), but this report represents only the second time bat RVAs gene segments have been found in a human RVA strain (Solberg et al., 2009).
In this study we have characterized an unusual human RVA from Suriname with genes belonging to eight recently assigned genotypes and five genes showing relationships to bat and/or bat-like human RVA strains. This Suriname strain represents the third report of the G20 RVA strain detected in animals or humans. The finding that the VP7, VP4, VP6, NSP3 and NSP5 genes of the strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] are nearly identical to the bat strain 3081/BRA and bat-like human strain Ecu534 suggests that these genes, and possibly the strain itself, are of bat origin. Although diarrhea in humans caused by RVA is common in Suriname (de Oliveira et al., 2009), not much is known about the RVA genotypes circulating in animals in this country, particularly bats. To determine whether strain RVA/Human-wt/SUR/2014735512/2013/G20P[28] is emerging in Suriname or was just an incidental detection will require continued RVA strain surveillance.
Acknowledgments
We wish to thank the staff of the Viral Gastroenteritis Branch at the Centers for Disease Control and Prevention and those at Lands Hospital, Suriname for invaluable assistance.
Funding
This work was supported by the Centers for Disease Control and Prevention, Rotavirus Surveillance funds.
Footnotes
Disclaimers
The opinions expressed by authors contributing to this journal do not necessarily reflect the opinions of the Centers for Disease Control and Prevention or the institutions with which the authors are affiliated. Use of trade names is for identification only and does not imply endorsement by any of the groups named above.
Disclosure of potential conflicts of interest
The authors declare that they have no conflict of interest.
References
- Abe M, Ito N, Masatani T, Nakagawa K, Yamaoka S, Kanamaru Y, Suzuki H, Shibano K, Arashi Y, Sugiyama M. Whole genome characterization of new bovine rotavirus G21P[29] and G24P[33] strains provides evidence for interspecies transmission. J Gen Virol. 2011;92:952–960. doi: 10.1099/vir.0.028175-0. [DOI] [PubMed] [Google Scholar]
- Asano KM, Gregori F, Hora AS, Scheffer KC, Fahl WO, Iamamoto K, Mori E, Silva FD, Taniwaki SA, Brandao PE. Group A rotavirus in Brazilian bats: description of novel T15 and H15 genotypes. Arch Virol. 2016;161:3225–3230. doi: 10.1007/s00705-016-3010-9. [DOI] [PubMed] [Google Scholar]
- Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esona MD, Mijatovic-Rustempasic S, Conrardy C, Tong S, Kuzmin IV, Agwanda B, Breiman RF, Banyai K, Niezgoda M, Rupprecht CE, Gentsch JR, Bowen MD. Reassortant group A rotavirus from straw-colored fruit bat (Eidolon helvum) Emerg Infect Dis. 2010;16:1844–1852. doi: 10.3201/eid1612.101089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes MK, Greenberg HB. Rotaviruses. In: Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B, editors. Fields Virology. 6th. Wolters Kluwer/Lippincott, Williams and Wilkins; Philadelphia, PA: 2013. pp. 1347–1401. [Google Scholar]
- Grant L, Esona M, Gentsch J, Watt J, Reid R, Weatherholtz R, Santosham M, Parashar U, O’Brien K. Detection of G3P[3] and G3P[9] rotavirus strains in American Indian children with evidence of gene reassortment between human and animal rotaviruses. J Med Virol. 2011;83:1288–1299. doi: 10.1002/jmv.22076. [DOI] [PubMed] [Google Scholar]
- Guo D, Liu J, Lu Y, Sun Y, Yuan D, Jiang Q, Lin H, Li C, Si C, Qu L. Full genomic analysis of rabbit rotavirus G3P[14] strain N5 in China: identification of a novel VP6 genotype. Infect Genet Evol. 2012;12:1567–1576. doi: 10.1016/j.meegid.2012.06.010. [DOI] [PubMed] [Google Scholar]
- He B, Yang F, Yang W, Zhang Y, Feng Y, Zhou J, Xie J, Feng Y, Bao X, Guo H, Li Y, Xia L, Li N, Matthijnssens J, Zhang H, Tu C. Characterization of a novel G3P[3] rotavirus isolated from a lesser horseshoe bat: a distant relative of feline/canine rotaviruses. J Virol. 2013;87:12357–12366. doi: 10.1128/JVI.02013-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jere KC, Mlera L, Page NA, van Dijk AA, O’Neill HG. Whole genome analysis of multiple rotavirus strains from a single stool specimen using sequence-independent amplification and 454(R) pyrosequencing reveals evidence of inter-genotype genome segment recombination. Infect Genet Evol. 2011;11:2072–2082. doi: 10.1016/j.meegid.2011.09.023. [DOI] [PubMed] [Google Scholar]
- Jere KC, Esona MD, Ali YH, Peenze I, Roy S, Bowen MD, Saeed IK, Khalafalla AI, Nyaga MM, Mphahlele J, Steele D, Seheri ML. Novel NSP1 genotype characterised in an African camel G8P[11] rotavirus strain. Infect Genet Evol. 2014;21:58–66. doi: 10.1016/j.meegid.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Luis AD, Hayman DT, O’Shea TJ, Cryan PM, Gilbert AT, Pulliam JR, Mills JN, Timonin ME, Willis CK, Cunningham AA, Fooks AR, Rupprecht CE, Wood JL, Webb CT. A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc Biol Sci. 2013;280:20122753. doi: 10.1098/rspb.2012.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes P, Matthijnssens J, Rahman M, Van Ranst M. RotaC: a web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiol. 2009;9:238. doi: 10.1186/1471-2180-9-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella V, Banyai K, Matthijnssens J, Buonavoglia C, Ciarlet M. Zoonotic aspects of rotaviruses. Vet Microbiol. 2010;140:246–255. doi: 10.1016/j.vetmic.2009.08.028. [DOI] [PubMed] [Google Scholar]
- Matthijnssens J, Rahman M, Martella V, Xuelei Y, De Vos S, De Leener K, Ciarlet M, Buonavoglia C, Van Ranst M. Full genomic analysis of human rotavirus strain B4106 and lapine rotavirus strain 30/96 provides evidence for interspecies transmission. J Virol. 2006;80:3801–3810. doi: 10.1128/JVI.80.8.3801-3810.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijnssens J, Ciarlet M, Heiman E, Arijs I, Delbeke T, McDonald SM, Palombo EA, Iturriza-Gomara M, Maes P, Patton JT, Rahman M, Van Ranst M. Full genome-based classification of rotaviruses reveals a common origin between human Wa-like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J Virol. 2008a;82:3204–3219. doi: 10.1128/JVI.02257-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijnssens J, Ciarlet M, Rahman M, Attoui H, Banyai K, Estes MK, Gentsch JR, Iturriza-Gomara M, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Patton JT, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Desselberger U, Van Ranst M. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch Virol. 2008b;153:1621–1629. doi: 10.1007/s00705-008-0155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Banyai K, Brister JR, Buesa J, Esona MD, Estes MK, Gentsch JR, Iturriza-Gomara M, Johne R, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Parreno V, Rahman M, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Patton JT, Desselberger U, Van Ranst M. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG) Arch Virol. 2011;156:1397–1413. doi: 10.1007/s00705-011-1006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira LH, Danovaro-Holliday MC, Andrus JK, de Fillipis AM, Gentsch J, Matus CR, Widdowson MA, Rotavirus Surveillance N. Sentinel hospital surveillance for rotavirus in latin american and Caribbean countries. J Infect Dis. 2009;200(Suppl. 1):S131–139. doi: 10.1086/605060. [DOI] [PubMed] [Google Scholar]
- Papp H, Al-Mutairi LZ, Chehadeh W, Farkas SL, Lengyel G, Jakab F, Martella V, Szucs G, Banyai K. Novel NSP4 genotype in a camel G10P[15] rotavirus strain. Acta Microbiol Immunol Hung. 2012;59:411–421. doi: 10.1556/AMicr.59.2012.3.11. [DOI] [PubMed] [Google Scholar]
- Plowright RK, Eby P, Hudson PJ, Smith IL, Westcott D, Bryden WL, Middleton D, Reid PA, McFarlane RA, Martin G, Tabor GM, Skerratt LF, Anderson DL, Crameri G, Quammen D, Jordan D, Freeman P, Wang LF, Epstein JH, Marsh GA, Kung NY, McCallum H. Ecological dynamics of emerging bat virus spillover. Proc Biol Sci. 2015;282:20142124. doi: 10.1098/rspb.2014.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potgieter AC, Page NA, Liebenberg J, Wright IM, Landt O, van Dijk AA. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J Gen Virol. 2009;90:1423–1432. doi: 10.1099/vir.0.009381-0. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Orba Y, Sasaki S, Gonzalez G, Ishii A, Hang’ombe BM, Mweene AS, Ito K, Sawa H. Multi-reassortant G3P[3] group A rotavirus in a horseshoe bat in Zambia. J Gen Virol. 2016;97:2488–2493. doi: 10.1099/jgv.0.000591. [DOI] [PubMed] [Google Scholar]
- Solberg OD, Hasing ME, Trueba G, Eisenberg JN. Characterization of novel VP7, VP4, and VP6 genotypes of a previously untypeable group A rotavirus. Virology. 2009;385:58–67. doi: 10.1016/j.virol.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, Niezgoda M, Haynes L, Agwanda B, Breiman RF, Anderson LJ, Rupprecht CE. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg Infect Dis. 2009;15:482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojnar E, Sachsenroder J, Twardziok S, Reetz J, Otto PH, Johne R. Identification of an avian group A rotavirus containing a novel VP4 gene with a close relationship to those of mammalian rotaviruses. J Gen Virol. 2013;94:136–142. doi: 10.1099/vir.0.047381-0. [DOI] [PubMed] [Google Scholar]
- Waruhiu C, Ommeh S, Obanda V, Agwanda B, Gakuya F, Ge XY, Yang XL, Wu LJ, Zohaib A, Hu B, Shi ZL. Molecular detection of viruses in Kenyan bats and discovery of novel astroviruses, caliciviruses and rotaviruses. Virol Sin. 2017;32:101–114. doi: 10.1007/s12250-016-3930-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia LL, He B, Hu TS, Zhang WD, Wang YY, Xu L, Li N, Qiu W, Yu J, Fan QS, Zhang FQ, Tu CC. Isolation and characterization of rotavirus from bat. Bing Du Xue Bao. 2013;29:632–637. [PubMed] [Google Scholar]
- Xia L, Fan Q, He B, Xu L, Zhang F, Hu T, Wang Y, Li N, Qiu W, Zheng Y, Matthijnssens J, Tu C. The complete genome sequence of a G3P[10] Chinese bat rotavirus suggests multiple bat rotavirus inter-host species transmission events. Infect Genet Evol. 2014;28:1–4. doi: 10.1016/j.meegid.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Yinda CK, Zeller M, Conceicao-Neto N, Maes P, Deboutte W, Beller L, Heylen E, Ghogomu SM, Van Ranst M, Matthijnssens J. Novel highly divergent reassortant bat rotaviruses in Cameroon, without evidence of zoonosis. Sci Rep. 2016;6:34209. doi: 10.1038/srep34209. [DOI] [PMC free article] [PubMed] [Google Scholar]