

Abstract
Group A rotaviruses (RVAs) with distinct G and P genotype combinations have been reported globally. We report the genome composition and possible origin of seven G8P[4] and five G2P[4] human RVA strains based on the genetic evolution of all 11 genome segments at the nucleotide level. Twelve RVA ELISA positive stool samples collected in the representative countries of Eastern, Southern and West Africa during the 2007–2012 surveillance seasons were subjected to sequencing using the Ion Torrent PGM and Illumina MiSeq platforms. A reference-based assembly was performed using CLC Bio’s clc_ref_assemble_long program, and full-genome consensus sequences were obtained. With the exception of the neutralising antigen, VP7, all study strains exhibited the DS-1-like genome constellation (P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2) and clustered phylogenetically with reference strains having a DS-1-like genetic backbone. Comparison of the nucleotide and amino acid sequences with selected global cognate genome segments revealed nucleotide and amino acid sequence identities of 81.7–100 % and 90.6–100 %, respectively, with NSP4 gene segment showing the most diversity among the strains. Bayesian analyses of all gene sequences to estimate the time of divergence of the lineage indicated that divergence times ranged from 16 to 44 years, except for the NSP4 gene where the lineage seemed to arise in the more distant past at an estimated 203 years ago. However, the long-term effects of changes found within the NSP4 genome segment should be further explored, and thus we recommend continued whole-genome analyses from larger sample sets to determine the evolutionary mechanisms of the DS-1-like strains collected in Africa.
Keywords: Rotavirus, Whole-genome analyses, G8P[4], G2P[4]
Introduction
Group A rotaviruses (RVAs) are a major cause of acute gastroenteritis in infants and young children globally. They are associated with approximately 453,000 childhood deaths annually, the majority of which occur in African and Asian countries [1]. The genome of RVA is composed of 11 segments of double-stranded RNA that primarily encodes six structural (VP) and six non-structural (NSP) proteins. Viral evolution occurs through both the selection of point mutations and by reassortment of the segmented RVA genome, which is a powerful evolutionary mechanism to increase viral genetic diversity [2].
The two outer viral proteins, VP7 (glycoprotein, G) and VP4 (protease-sensitive, P), are known to elicit neutralising antibody responses and segregate independently since they are encoded by different RNA segments. Based on this, RVA strains have been conventionally classified into G and P types [2]. More recently, RVAs have been classified using whole-genome nucleotide sequences with a genotype descriptor of Gx-P[x]-Ix-Rx-Cx-Mx-Ax-Nx-Tx-Ex-Hx (where x = genotype number) representing genome segments encoding VP7-VP4-VP6-VP1-VP2-VP3-NSP1-NSP2-NSP3-NSP4-NSP5, respectively [3-5]. Currently, at least 27G, 35P, 16I, 9R, 9C, 8M, 18A, 9N, 12T, 14E and 11H genotypes have been recognised from both human and animal RVA strains using the complete genome constellation classification system recommended by the Rotavirus Classification Working Group [5, 6]. Of these, rotaviruses with genotypes G1 through G6, G8 through G12 and G20, as well as P[1] through P[11], P[14], P[19], P[25] and P[28], have been identified in humans [7].
Whole-genome (codon-complete) sequencing has become widely established in the recent RVA research, because it offers a platform for genotyping all 11 genome segments of RVA, and thus provides insight into RVA genetic evolution and reassortment events [7, 8]. Strains with genotype 1 (I1-R1-C1-M1-A1-N1-T1-E1-H1 [Walike]) and 2 (I2-R2-C2-M2-A2-N2-T2-E2-H2 [DS-1-like]) constellations represent the two primary human RVA pathogens that spread rapidly among the human populations. RVA with genotype 3 (I3-R3-C3-M3-A3/A12-N3-T3-E3-H3/H6 [AU-1-like]) genetic backbones are rarely detected in humans [9]. The variation among RVA genome constellations is greater in developing countries than in developed countries [10, 11].
The G8P[10] prototype strain (69M) was first reported in Indonesia in 1985 [12]. In Africa, the first G8 was detected as a mixed G1 and G8 infection in a child from Nigeria presenting with diarrhoea [13]. The G8 genotype has since been reported as the predominant genotype in some African countries such as Malawi, where the predominance of G8 strains was first described in the late 1990s [14], and Kenya, where G8 strains were reported during annual seasonal RVA surveillance [15]. Most human RVA G8 strains contain DS-1-like genetic backbone with occasional reassortment events and account for up to 27 % of the total circulating strains in some African countries, such as Malawi [14, 16]. Some G8 strains have been reported to belong to the Wa-like genotype constellation [9, 17, 18].
Among humans, RVA VP4 genotype P[8] predominates worldwide, followed by P[4]. Human RVA strains with P[4] genotypes have been reported primarily in association with G2 and G8 VP7 genotypes [19]. Most often, RVA strains with a G2P[4] genotype have a DS-1-like genotype constellation. In this study, we analysed the complete coding regions of seven G8P[4] and five G2P[4] strains from seven African countries to determine the evolutionary mechanisms giving rise to genome constellations having G8 and G2 strains sharing the VP4 P[4] genotype.
Materials and methods
Sample collection and RNA extraction
The Medical Research Council/Diarrhoeal Pathogens Research Unit (MRC/DPRU), a World Health Organisation (WHO) Rotavirus Regional Reference Laboratory in South Africa/WHO (RRL-SA), has been conducting annual rotavirus genotyping training workshops since 1998 that involves several African countries. Stool samples used during these workshops are collected from hospitalised children below the age of five years presenting with acute gastroenteritis.
Stool samples were initially screened for RVA with a commercially available rotavirus kit (ProSpecT Rotavirus Microplate Kit, England) and stored at +4 °C in sentinel or national laboratories. Samples were subsequently transferred to WHO RRL-SA as part of the WHO African surveillance rotavirus network and used for rotavirus molecular characterisation and were eventually stored at −20 °C for future use, if and when necessary. For this study, archival samples that had displayed some interesting results after their initial molecular characterisation by polyacrylamide gel electrophoresis and that also represented a diverse geographical distribution were retrieved for full-length genome analysis. The dsRNA genome was extracted from seven G8P[4] and five G2P[4] strains following previously described methods [11, 18].
RNA sequencing
A 1:30 dilution of 0.9 μl of extracted RVA RNA was used in each of 11 one-step RT-PCR reactions (QIAGEN OneStep RT-PCR Kit, QIAGEN, Hilden, Germany) to amplify full-length RVA RNA genome segments using segment-specific primers ordered from Integrated DNA Technologies (Coralville, IA, USA; Supplementary data 1). The thermocycling parameters included 30 min at 45 °C for reverse transcription, followed by 50 cycles of denaturation (10 s, 94 °C), annealing (1 min, 55 °C) and extension (3 min, 68 °C). RTPCR products were verified on 1 % agarose gels, and excess primers and dNTPs were removed by treatment with exonuclease I (New England BioLabs, Ipswich, MA, USA) and shrimp alkaline phosphatase (Affymetrix, Santa Clara, CA, USA) at 37 °C for 60 min, followed by incubation at 72 °C for 15 min. The products were quantitated using a SYBR green dsDNA detection assay (SYBR Green I Nucleic Acid Gel Stain, Thermo Fisher Scientific, Waltham, MA, USA), and all 11 RT-PCR products for each genome were pooled in equimolar amounts. Pooled amplicons were then sequenced using the Illumina and/or Ion Torrent sequencing platforms.
Illumina libraries were prepared using the Nextera DNA Sample Preparation Kit (Illumina, Inc., SanDiego, CA, USA) with half-reaction volumes. Briefly, 25 ng of pooled DNA amplicons was tagmented at 55 °C for 5 min. Tagmented DNA was cleaned with the ZR-96 DNA Clean & Concentrator Kit (Zymo Research Corporation, Irvine, CA, USA) and eluted in 25 μl resuspension buffer. Illumina sequencing adapters and barcodes were added to tagmented DNA via PCR amplification, which used 20 μl tagmented DNA combined with 7.5 μl Nextera PCR Master Mix, 2.5 μl Nextera PCR Primer Cocktail, and 2.5 μl of each index primer (Integrated DNA Technologies, Coralville, IA, USA) for a total volume of 35 μl per reaction. Thermocycling was performed with an initial extension for 3 min at 72 °C, followed by five cycles of PCR as per the Nextera DNA Sample Preparation Kit protocol (i.e. denaturation for 10 s at 98 °C, annealing for 30 s at 63 °C, and extension for 3 min at 72 °C) to create a dual-indexed library for each sample. After PCR amplification, 10 μl of each library was pooled into a 1.5-μl tube and the pool was cleaned two times with Ampure XP Reagent (Beckman Coulter, Inc., Brea, CA, USA) to remove all leftover primers and small DNA fragments. The first cleaning used 1.2 × volume Ampure Reagent, and the second cleaning used 0.6 × volume Ampure Reagent. The cleaned pool was sequenced on the Illumina MiSeq v2 instrument (Illumina, Inc., San Diego, CA, USA) with 300-bp paired-end reads.
Ion Torrent libraries were prepared by shearing pooled RVA amplicons for 12 min, and Ion Torrent compatible barcoded adapters were ligated to the sheared DNA using the Ion Xpress Plus Fragment Library Kit (Thermo Fisher Scientific, Waltham, MA, USA) to create 200-bp libraries. Barcoded libraries were pooled in equal volumes and cleaned with the Ampure XP Reagent (Beckman Coulter, Inc., Brae, CA, USA). Real-time PCR was performed on the pooled, barcoded libraries to assess their quality and to determine the template dilution factor for emulsion PCR. The pool was diluted appropriately and amplified on Ion Sphere Particles (ISPs) using the Ion One Touch instrument (Thermo Fisher Scientific, Waltham, MA, USA). The pool was cleaned and enriched for template-positive ISPs on the Ion One Touch ES instrument (Thermo Fisher Scientific, Waltham, MA, USA) and sequencing was performed on the Ion Torrent PGM platform using an Ion 316 chip (Thermo Fisher Scientific, Waltham, MA, USA).
Additionally, for some samples, pooled RVA RT-PCR products were randomly amplified using a sequence-independent single-primer amplification (SISPA) method [20]. Briefly, 100 ng of amplified pooled amplicons was denatured in the presence of DMSO and a chimeric oligonucleotide containing a known 22-nt barcode sequence followed by a 3′ random hexamer. A Klenow reaction was prepared with the denatured DNA template by adding NE Buffer 2, 3′ –5′ exo- Klenow (New England BioLabs, Ipswich, MA, USA), and dNTPs (Thermo Fisher Scientific, Waltham, MA, USA). The Klenow reaction was incubated at 37 °C for 60 min, followed by incubation at 75 °C for 10 min. The resulting cDNA was amplified with 35 cycles of PCR using Promega Go Taq Hot Start Polymerase (Promega Corporation, Madison, WI, USA), with denaturation for 30 s at 94 °C, annealing for 30 s at 55 °C and extension for 48 s at 68 °C. PCR reactions contained primers corresponding to the known 22-nt barcode sequence from the oligonucleotide utilised in the previous Klenow step. SISPA products were normalised and pooled into a single reaction, which were purified using the QIAquick PCR Purification Kit (QIAGEN, Hilden, Germany). The sample was further gel purified to select for SISPA products of 300–500 bp in size and sequenced on the Illumina MiSeq v2 instrument (Illumina, Inc., San Diego, CA, USA) with paired-end 250-bp reads.
The sequencing reads from the Ion Torrent PGM and Illumina MiSeq v2 instrument were sorted by barcode, trimmed and de novo assembled using CLC Bio’s clc_novo_assemble program (QIAGEN, Hilden, Germany), and the resulting contigs were searched against custom full-length RVA segment nt databases to find the closest reference sequence for each segment (http://www.clcbio.com/products/clc-assemblycell/). All sequence reads were then mapped to the selected reference RVA segments using CLC Bio’s clc_ref_assemble_long program. At lociwhere both Ion Torrent and Illumina sequence data agreed on a variation (as compared to the reference sequence), the reference sequence was updated to reflect the difference. A final mapping of all the next-generation sequences to the updated reference sequences was performed with CLC Bio’s clc_ref_assemble_long program. Sequences were submitted to NCBI’s GenBank and assigned accession numbers (Supplementary data 2).
Whole-genome phylogenetic analysis
Complete open reading frames were obtained for all 12 RVA genome segments from each study strain, as well as for various reference genomes from GenBank (see Supplementary data 2 for reference strain accession numbers by segment). Genotypes for each genome segment were determined using the RotaC v2.0 webserver [21]. Nucleotide sequence alignments for each genome segment were constructed using the MUSCLE algorithm implemented in MEGA 5.1 [22].
Maximum likelihood phylogenies were inferred for each gene segment based on their nucleotide sequences and included the African G8P[4] and G2P[4] strains, as well as the corresponding genome segment sequences from selected rotavirus strains available in GenBank (Fig. 1a–l). The best substitution models were selected based on the corrected Akaike Information Criterion (AICc) value as implemented in MEGA 5.1. Models used in this study were GTR+I+G (NSP1, NSP3 and VP7-G2), GTR+G (NSP4 and NSP5), TN93+G+I (NSP2, VP1, VP2 and VP6), T92+G+I (VP3), HKY+G+I (VP4) and T92+G (VP7-G8). In addition, multiple sequence alignments for genome segments were constructed, and nucleotide and amino acid similarity matrices were prepared (Supplementary data 3A–L). Bayesian MCMC calculations were carried out in BEAST v2.1.1 as described previously [23, 24]. Divergence dates were estimated for lineages containing the African strains of interest, and genotypic lineages were defined as tight phylogenetic clusters, when bootstrap support was > 70. The evolutionary rate for all 11 genome segments was calculated.
Fig. 1.
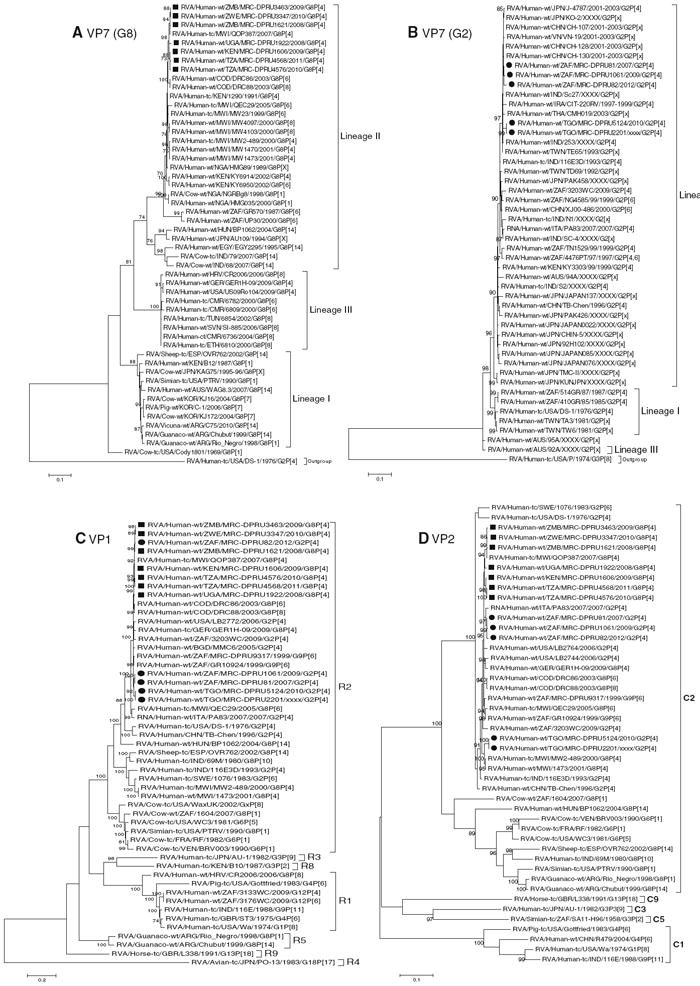


a–l Maximum likelihood phylogenetic trees based on the full-length coding nucleotide sequences of the rotavirus genes: a VP7 (G8), b VP7 (G2), c VP1, d VP2, e VP3, f VP4, g VP6, h NSP1, i NSP2, j NSP3, k NSP4 and l NSP5. Bootstrap values above 70 are provided for 500 replicates. The clades for each viral protein genotype are labelled in each tree. For the VP7 and VP4 trees, representative strains from different lineages were used to determine in which lineage the study strains clustered. The black squares represent the G8 study strains and the black circles indicate the G2 study strain in all trees
Results
Sequence analyses
The nucleotide and deduced amino acid sequences for the eleven gene segments, encoding VP1–VP4, VP6–VP7, and NSP1–NSP5, from the 12 African strains (seven G8P[4] and five G2P[4]) were determined, and the nomenclature and genotype constellations for the study strains are provided in Table 1. Using the RotaC online genotyping tool to assign genotypes to the genome segments encoding VP6-VP1-VP2-VP3-NSP1-NSP2-NSP3-NSP4-NSP5, the seven G8P[4] strains and five G2P[4] strains exhibited the characteristics of DS-1-like genetic backbone constellations (I2-R2-C2-M2-A2-N2-T2-E2-H2).
Table 1.
Genotype constellations for the rotavirus study strains and reference strains from GenBank
| Genome constellations
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S9 (VP7) | S4 (VP4) | S6 (VP6) | S1 (VP1) | S2 (VP2) | S3 (VP3) | S5 (NSP1) | S8 (NSP2) | S7 (NSP3) | S10 (NSP4) | S11 (NSP5) | |
| DS-1-like study strains | |||||||||||
| RVA/Human-wt/KEN/MRC-DPRU1606/2009/G8P[4]a | G8 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/ZMB/MRC-DPRU1621/2008/G8P[4]a | G8 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/ZMB/MRC-DPRU3463/2009/G8P[4]a | G8 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/UGA/MRC-DPRU1922/2008/G8P[4]a | G8 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/ZWE/MRC-DPRU3347/2010/G8P[4]a | G8 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/TZA/MRC-DPRU4568/2011/G8P[4]a | G8 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/TZA/MRC-DPRU4576/2010/G8P[4]a | G8 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/ZAF/MRC-DPRU1061/2009/G2P[4]a | G2 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/ZAF/MRC-DPRU81/2007/G2P[4]a | G2 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/ZAF/MRC-DPRU82/2012/G2P[4]a | G2 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/TGO/MRC-DPRU5124/2010/G2P[4]a | G2 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/TGO/MRC-DPRU2201/2010/G2P[4]a | G2 | P[4] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| Wa-like reference strain | |||||||||||
| RVA/Human-wt/CMR/MRC-DPRU1424/2009/G9P[8]b | G9 | P[8] | I1 | R1 | C1 | M1 | A1 | N1 | T1 | E1 | H1 |
| Reassortant DS-1-like reference strain | |||||||||||
| RVA/Human-wt/ZAF/MRC-DPRU9317/1999/G9P[6]c | G9 | P[6] | I2 | R2 | C2 | M2 | A2 | N1 | T2 | E2 | H2 |
Bold values indicated to visualise genome constellation pattern (Wa-like) and italic values indicated to visualise genome constellation pattern (DS-1-like)
S genome segment, VP viral structural protein, NSP viral non-structural protein, RVA Group A rotaviruses, WT wild-type, KEN Kenya, ZMB Zambia, ZAF South Africa, UGA Uganda, ZWE Zimbabwe, TZA Tanzania, TGO Togo, CMR Cameroon, MRC Medical Research Council, DPRU Diarrhoeal Pathogens Research Unit
All study rotavirus strains contain a DS-1-like genetic backbone (italic)
Reference strain contains a Wa-like genetic backbone (bold) [11]
Reference reassortant strain contains a mixture of DS-1-like and Wa-like (S8) genotypes within its genetic backbone
The sequence diversity data show the maximum diversity in the NSP4 encoding genome segment, which is also reflected in the extremely large divergence time for this gene within the DS-1-like group. The other ten genome segments do not show such high sequence diversity compared with other strains in GenBank, which is reflected in their more recent estimated divergence times (data not shown).
Nucleotide and amino acid sequence analyses of the G8 and G2 VP7 encoding genome segment
Comparison of the coding nucleotide and deduced amino acid sequences of the seven African G8P[4] strains showed that they share nucleotide and amino acid identities in the range of 97.1–100 % among themselves and a fairly moderate relatedness to previously reported G8 strains with various P genotypes from humans and animals (81.5–100 %). Absolute nucleotide and amino acid identities were shared between the G8P[4] strains MRC-DPRU3463 and MRC-DPRU3347. They were also significantly closely related to the Malawian strain RVA/Human-tc/MWI/QOP387/2007/G8P[4], sharing nucleotide and amino acid identities of 99.8 and 100 %, respectively (Supplementary data 3A). Phylogenetic analysis of the G8 rotaviruses based on their VP7 nucleotide sequence showed that the G8 study strains and other published G8 strains form three separate lineages. All the African G8 study strains are in lineage II and sub-cluster with other previously reported African G8 strains from Malawi, the DRC, Kenya and Nigeria, irrespective of their VP4 genotypes (Fig. 1a).
The five African G2P[4] study strains share nucleotide and amino acid identities of 97.1–99.7 % and 98.3–100 %, respectively. Comparative analysis of the G2 nucleotide and amino acid sequences of these African G2 study strains with cognate sequences of older and contemporary G2 strains from GenBank showed moderate to high nucleotide and amino acid identities in the ranges of 91.9–99.7 % and 94.1–100 %, respectively (Supplementary data 3B). Phylogenetic analysis of the G2 study strains, along with several published G2 strains, revealed that all African G2 study strains cluster together in lineage II and are most closely related to G2 strains from China, Iraq, Thailand and India (Fig. 1b).
Nucleotide and amino acid sequence analyses of the VP4 encoding genome segment
A distance matrix analysis indicated that the VP4 encoding genome segment of the seven G8P[4] and five G2P[4] study strains shares nucleotide and amino acid identities with each other in the ranges of 92.8–9.7 % and 95.5–99.9 %, respectively. The VP4 encoding genome segments are highly identical (98.6–99.9 %) among the G8P[4] study strains, but are less identical (92.9–99.6 %) among the G2P[4] study strains. However, nucleotide and amino acid sequence comparisons between the VP4 encoding genome segment of the 12 African study strains and those from GenBank showed a moderate to high sequence identity in the range of 91.9–99.7 % (Supplementary data 3C). Phylogenetic analysis showed that the 12 African study strains cluster into two separate lineages, irrespective of their VP7 genotypes. The study strains MRC-DPRU1606 (G8P[4]), MRC-DPRU1621 (G8P[4]), MRC-DPRU1922 (G8P[4]), MRC-DPRU3347 (G8P[4]), MRC-DPRU3463, MRC-DPRU82 (G2P[4]), MRC-DPRU4568 (G2P[4]) and MRC-DPRU 4576 (G8P[4]) are in lineage II, whereas strains MRC-DPRU2201 (G2P[4]), MRC-DPRU5124 (G2P[4]), MRC-DPRU81 (G2P[4]), and MRC-DPRU1061 (G2P[4]) cluster in lineage III (Fig. 1f).
Nucleotide and amino acid sequence analyses of the VP1, VP2, VP3 and VP6 encoding genome segments
Phylogenetic analyses of the nucleotide sequences coding for VP1, VP2, VP3 and VP6 for the seven G8P[4] and five G2P[4] African study strains demonstrated that each of these encoding genome segments groups in several clusters together with strains isolated worldwide (Fig. 1c–e, g). The African strains show a close genetic relationship with cognate gene sequences of previously described G8P[4], G2P[4], G8P[14], G6P[5], G9P[6], G6P[1] and G8P[10] strains detected in Malawi, the DRC, the USA, Hungary, South Africa, France, Venezuela and India. Nucleotide (amino acid) identity values among the study strains range from 97.1–99.5 % (98.8–99.9 %), 96.5–100 % (99.5–100 %), 97.2–100 %(94.4–99.5 %) and 96.1–100 %(99–100 %) for VP1, VP2, VP3 and VP6, respectively. Complete nucleotide and amino acid identity (100 %) is shared between strains MRC-DPRU4576 and MRC-DPRU4568 in their VP2 and VP6 encoding genome segments. However, when the nucleotide and amino acid identities of the VP1–VP3 and VP6encoding genome sequences for the African study strains were compared with corresponding sequences from strains belonging to previously described VP1–VP3 and VP6 genotypes, all of them were more closely related to strains in the R2, C2, M2 and I2 genotypes, respectively (Supplementary data 3D–G). Further comparison showed that, within each genotype, the African study strains have maximum nucleotide (amino acid) identities of 84.1–97.7 % (95.6–99.7 %) forVP1, 84.2–99.8 %(96.8–99.9 %) for VP2, 97.2–100 % (94.4–99.5 %) for VP3 and 86.7–99.7 % (96.1–100 %) for VP6.
Nucleotide and amino acid sequence analyses of the NSP1, NSP2, NSP3, NSP4 and NSP5 encoding genome segments
Phylogenetic analyses demonstrated that the NSP1–NSP5 coding nucleotide sequences of the seven G8P[4] and five G2P[4] African study strains group into small, separate subclusters of genotypes A2, N2, T2, E2 and H2, respectively, together with other strains from around the world (Fig. 1h–l). The African study strains showed close genetic association with cognate corresponding genome segment sequences of previously described DS-1-like G8P[8], G2P[4], G8P[6] and G9P[6] strains. Nucleotide (amino acid) identity values among these African study strains range from 97.0–99.9 % (96.7–100 %), 96.2–99.9 % (97.5–100 %), 97.2–100 % (97.0–100 %), 85.0–99.8 % (92.4–100 %) and 97.5–100 % (96.8–100 %) for NSP1, NSP2, NSP3, NSP4 and NSP5, respectively (Supplementary data 3H–L). Complete nucleotide and amino acid identities are shared between strains MRC-DPRU4576 and MRC-DPRU4568 in their NSP3 encoding genome segments. Comparison of the nucleotide and amino acid identities between the NSP1–NSP5 encoding genome sequences of the African study strains to corresponding gene sequences for strains belonging to already identified NSP1–NSP5 genotypes, all of them were more closely related to strains in the A2, N2, T2, E2 and H2 genotypes, respectively. Within each of these genotypes, the study strains shared maximum nucleotide (amino acid) identities of 91.6–99.6 % (91–99.4 %), 85.5–99.8 % (91.8–99.7 %), 89.9–99.6 % (93.4–99.7 %), 81.7–99.4 % (90.6–100 %) and 94.6–99.6 % (94.1–100 %), respectively.
Further analysis of the deduced amino acid sequences of the NSP4 encoding genome segment for the African study strains showed that several amino acid substitutions exist in the previously described antigenic sites and enterotoxin domain of NSP4, when aligned with the prototype DS-1 strain and other strains belonging to the NSP4 genotype E2 [25]. Most of the substitutions were found between residues 128 and 161, which include the enterotoxin domain (amino acid residues 114–135) of NSP4, as well as the antigenic sites AS II (residues 136–150) and AS I (residues 151–169) (Supplementary data 4).
Discussion
The current study contributes to the ongoing monitoring and molecular characterisation of RVA strains circulating in Africa, where interspecies RVA transmission between animals and humans is common and where RVA vaccine effectiveness has remained relatively low compared to data from the developed countries. Although the whole-genome RVA classification system has been widely adopted to classify circulating rotaviruses globally [3-5, 9], there are no or only a limited number of G8P[4] and G2P[4] RVA strains that have been completely sequenced from some of the countries included in the present study. Whole-genome data for G8P[4] and G2P[4] strains in other African countries are also limited to a few studies [16, 18, 26, 27]. Therefore, an assessment of the nucleotide and amino acid sequences from the complete genomes of the seven G8P[4] and five G2P[4] rotavirus strains reported here has considered data on DS-1-like genotype constellations in Kenya, Zimbabwe, Tanzania, Togo, Uganda, Zambia and South Africa relative to what is available in other countries of Africa and globally.
The G8 genotype is primarily reported in association with several VP4 genotypes, including P[4], P[6], P[8] and P[14]. In developing countries, the G8 genotype has been shown to circulate and even predominate in some countries, such as Malawi and Kenya, primarily in combination with P[4], P[6] and P[8] [10, 14, 15]. In Kenya, the whole-genome sequence of one G8P[1] rotavirus strain has been previously characterised and was reported to have originated from reassortment events between artiodactyl-like and human rotavirus strains [17, 28]. G8P[4] strains are considered medically important in Kenya in association with diarrhoeal diseases and have previously been detected in Kenya using RT-PCR assays that target the genes encoding the two RVA outer capsid proteins, VP7 and VP4 [15, 29]. In contrast, to the best of our knowledge, no G8P[4] complete genomes have been previously reported from Zimbabwe, Uganda, Zambia or Tanzania, even though the RVA G and P types from these countries are reported annually as part of the ongoing African rotavirus surveillance network [30, 31].
All seven G8P[4] study strains exhibited intra-genotype diversity across the genome and grouped in various lineages that were shared with other global DS-1-like strains. They were also closely related to other human and animal DS-1-like strains and may have evolved over the last couple of decades through genetic reassortment events with other rotavirus strains, thus changing their VP4 genotype while maintaining a conserved DS-1-like genotype constellation for their other RVA genome segments. Likewise, a study done in Malawi reported an almost identical scenario [16]. Comparative analyses of the G8P[4] study strains from Kenya, Tanzania, Uganda, Zambia and Zimbabwe revealed close evolutionary relationships in all 11 genome segments to other human G8 strains containing the P[4], P[6] and P[10] genotypes that were primarily isolated from the DRC, Malawi, the USA and China. With the exception of the two outer capsid proteins, VP7 and VP4, the remaining nine gene segments had the closest evolutionary relationship to human and animal rotaviruses belonging in the DS-1-like genotype. Phylogenetic analyses showed evidence for relatedness in most genome segments between the G8P[4] study strains and the previously analysed G9P[6] reassortant strains RVA/Human-wt/ZAF/MRC-DPRU9317/1999/G9P[6] and RVA/Human-wt/ZAF/GR10924/1999/G9P[6], which both contain a DS-1-like genetic backbone [11, 18]. Comparison of nucleotide sequences from the study strains with selected global cognate genome segments revealed that the nucleotide (amino acid) identities ranged from 81.7–100 % (90.6–100 %), with the greatest diversity among their NSP4 gene segments.
The G2P[4] strains in this study have a highly conserved genome with an overall lesser genetic divergence in all genome segments, when compared with selected reference DS-1-like strains, which has been previously reported for other G2P[4] strains [19]. However, four of the G2P[4] study strains fell into lineage III, while one was part of lineage II which demonstrated some reassortment within the VP4. Previously, whole-genome analyses of only one wild-type G2P[4] strain, RVA/Human-wt/ZAF/3203WC/2009/G2P[4], from South Africa was published [18] and nearly complete genome sequences of two Kenyan G2P[4] strains, AK26 and D205 [32]. Although the G2 viruses are highly conserved, they have been reported to have arisen from a reassortment event occurring between G3P[4] strains co-circulating with G2P[4] strains under a DS-1-like genetic backbone, as reported for a single human rotavirus strain circulating in India [33]. Additional genetic variants of the RVA G2P[4] genotype have also been identified and are reported to circulate in Brazil; these variants most likely arose through the occurrence of point mutations, reassortment events and widespread global gene flow [34].
Bayesian analyses to estimate times of divergence for the lineages containing the G8P[4] and G2P[4] African study strains from other known DS-1-like strains also gave similar results to those of maximum likelihood analyses. For all genes, the divergence times ranged from 16 to 44 years ago, except for the NSP4 gene where the lineage seemed to diverge in the much more distant past, about 203 years ago (data not shown). Such high sequence diversity in the NSP4 encoding genome segment has been previously reported in Cameroon G9 strains [35]. This suggests that the NSP4 genome segment comes from a much older lineage. All African study strains showed numerous amino acid substitutions in the enterotoxin domain (amino acids 114–135) of NSP4. These changes occur at amino acid positions 128, 132, 134 and 136, which are within the region reported to be responsible for altered pathogenesis mediated by the NSP4 protein [36]. Several residue substitutions in the four previously described NSP4 antigenic sites were also observed [25]. Antigenic sites AS IV (amino acids 1–24), AS III (amino acids 112–133), AS II (amino acids 136–150) and AS I (amino acids 151–169) had 1, 4, 6 and 3 amino acid substitutions, respectively. NSP4 is a transmembrane glycoprotein known to be involved in virus assembly and is capable of inducing diarrhoea in infant mice [26]. It is possible that the observed substitutions may affect the conformation or activity of NSP4 and may also alter the ability of host responses to neutralise the enterotoxic function of the NSP4 gene segment [26].
Conclusion
This study provides whole-genome sequence data of G8P[4] and G2P[4] strains from seven different African countries. With the exception of the NSP4 encoding genome segment of these African study strains, which is highly diverse when compared with the DS-1 reference strain and other strains belonging to the NSP4 genotype E2, the remaining ten gene segments were closely related in both their nucleotide and amino acid sequences. Estimated times of divergence for the lineages containing the African study strains from other known DS-1-like strains also gave similar results for the NSP4 and other encoding genome segments. For all the genome segments, divergence times ranged from 16 to 44 years ago, except for the NSP4 encoding genome segment for which the lineage seems to have diverged in the more distant past, about 203 years. The long-term effects of changes found within the NSP4 encoding genome segment should be continuously explored, and we therefore recommend continued whole genome analyses from larger sample sets to better understand the evolutionary mechanisms of the DS-1-like group collected in African countries. This data would also help to inform improved laboratory detection methods. In addition, similar full-genome sequence studies of cognate animal RVA strains are needed to unquestionably determine the specific origin of those genes relative to both human and animal rotavirus strains.
Supplementary Material
Acknowledgments
This project was funded by the South African Medical Research Council, the Poliomyelitis Research Foundation in SouthAfrica, and federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract number HHSN272200900007C. We thank Lorens Maake, Kebareng Rakau, Nonkululeko Magagula and Ina Peenze for technical assistance at the MRC/Diarrhoeal Pathogens Research Unit. We also thank the Virology Group at the JCVI, especially Asmik Akopov, Nadia Fedorova and Susmita Shrivastava for their technical assistance in the laboratory and bioinformatics support. In addition, we thank all of the sample collection teams from the different countries that were involved in this study.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s11262-014-1091-7) contains supplementary material, which is available to authorised users.
Contributor Information
Martin M. Nyaga, South African Medical Research Council/UL Diarrhoeal Pathogens Research Unit (MRC/DPRU), Department of Virology, University of Limpopo (Medunsa Campus) and National Health Laboratory Service, PO Box 173, Medunsa, Pretoria 0204, South Africa
Karla M. Stucker, The J. Craig Venter Institute, Rockville, MD, USA
Mathew D. Esona, South African Medical Research Council/UL Diarrhoeal Pathogens Research Unit (MRC/DPRU), Department of Virology, University of Limpopo (Medunsa Campus) and National Health Laboratory Service, PO Box 173, Medunsa, Pretoria 0204, South Africa Gastroenteritis and Respiratory Viruses Laboratory Branch, Division of Viral Diseases, NCIRD, CDC, Atlanta, GA, USA.
Khuzwayo C. Jere, South African Medical Research Council/UL Diarrhoeal Pathogens Research Unit (MRC/DPRU), Department of Virology, University of Limpopo (Medunsa Campus) and National Health Laboratory Service, PO Box 173, Medunsa, Pretoria 0204, South Africa Institute of Infection and Global Health, Department of Clinical Infection, Microbiology and Immunology, University of Liverpool, Merseyside, UK.
Bakari Mwinyi, Kenyatta National Hospital, Nairobi, Kenya.
Annie Shonhai, Department of Medical Microbiology, Virology Section, University of Zimbabwe, Harare, Zimbabwe.
Enyonam Tsolenyanu, Department of Paediatrics, Sylvanus Olympio Teaching Hospital of Lome, Lome, Togo.
Augustine Mulindwa, Mulago National Referral Hospital, Kampala, Uganda.
Julia N. Chibumbya, Virology Laboratory, University Teaching Hospital, Lusaka, Zambia
Hokororo Adolfine, Bugando Medical Centre, Mwanza, Tanzania.
Rebecca A. Halpin, The J. Craig Venter Institute, Rockville, MD, USA
Sunando Roy, Gastroenteritis and Respiratory Viruses Laboratory Branch, Division of Viral Diseases, NCIRD, CDC, Atlanta, GA, USA.
Timothy B. Stockwell, The J. Craig Venter Institute, Rockville, MD, USA
Chipo Berejena, Department of Medical Microbiology, Virology Section, University of Zimbabwe, Harare, Zimbabwe.
Mapaseka L. Seheri, South African Medical Research Council/UL Diarrhoeal Pathogens Research Unit (MRC/DPRU), Department of Virology, University of Limpopo (Medunsa Campus) and National Health Laboratory Service, PO Box 173, Medunsa, Pretoria 0204, South Africa
Jason M. Mwenda, Regional Office for Africa, World Health Organization, Brazzaville, Republic of Congo
A. Duncan Steele, South African Medical Research Council/UL Diarrhoeal Pathogens Research Unit (MRC/DPRU), Department of Virology, University of Limpopo (Medunsa Campus) and National Health Laboratory Service, PO Box 173, Medunsa, Pretoria 0204, South Africa; Global Health Program, Enteric and Diarrhoeal Diseases Programme, Bill and Melinda Gates Foundation, Seattle, WA, USA.
David E. Wentworth, The J. Craig Venter Institute, Rockville, MD, USA
M. Jeffrey Mphahlele, South African Medical Research Council/UL Diarrhoeal Pathogens Research Unit (MRC/DPRU), Department of Virology, University of Limpopo (Medunsa Campus) and National Health Laboratory Service, PO Box 173, Medunsa, Pretoria 0204, South Africa.
References
- 1.Tate JE, Burton AH, Boschi-Pinto C, Steele AD, Duque J, Parashar UD WHO-coordinated Global Rotavirus Surveillance Network. 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. Lancet Infect Dis. 2012;2:136–141. doi: 10.1016/S1473-3099(11)70253-5. [DOI] [PubMed] [Google Scholar]
- 2.Estes MK, Kapikian AZ. In: Fields Virology. Knipe DM, editor. Lippincott Williams and Wilkins; Philadelphia: 2007. pp. 1917–1957. [Google Scholar]
- 3.Matthijnssens J, Ciarlet M, Heiman E, Arijs I, Delbeke T, McDonald SM, Palombo EA, Iturriza-Gómara M, Maes P, Patton JT, Rahman M, Van Ranst M. Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J Virol. 2008;82:3204–3219. doi: 10.1128/JVI.02257-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthijnssens J, Ciarlet M, Rahman M, Attoui H, Bányai K, Estes MK, Gentsch JR, Iturriza-Gómara M, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Patton JT, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Desselberger U, van Ranst M. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch Virol. 2008;153:1621–1629. doi: 10.1007/s00705-008-0155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Bányai K, Brister JR, Buesa J, Esona MD, Estes MK, Gentsch JR, Iturriza-Gómara M, Johne R, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Parreño V, Rahman M, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Patton JT, Desselberger U, van Ranst M. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG) Arch Virol. 2011;156:1397–1413. doi: 10.1007/s00705-011-1006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jere KC, Esona MD, Ali YH, Peenze I, Roy S, Bowen MD, Saeed IK, Khalafalla AI, Nyaga MM, Mphahlele JM, Steele D, Seheri ML. Novel NSP1 genotype characterised in an African camel G8P[11] rotavirus strain. Infect Genet Evol. 2014;21:58–66. doi: 10.1016/j.meegid.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Matthijnssens J, Bilcke J, Ciarlet M, Martella V, Bányai K, Rahman M, Zeller M, Beutels P, van Damme P, van Ranst M. Rotavirus disease and vaccination: impact on genotype diversity. Future Microbiol. 2009;10:1303–1316. doi: 10.2217/fmb.09.96. [DOI] [PubMed] [Google Scholar]
- 8.Trojnar E, Sachsenröder J, Twardziok S, Reetz J, Otto PH, Johne R. Identification of an avian group A rotavirus containing a novel VP4 gene with a close relationship to those of mammalian rotavirus. Curr Opin Virol. 2013;4:426–433. doi: 10.1099/vir.0.047381-0. [DOI] [PubMed] [Google Scholar]
- 9.Matthijnssens J, van Ranst M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr Opin Virol. 2012;2:426–433. doi: 10.1016/j.coviro.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Bányai K, László B, Duque J, Steele AD, Nelson EA, Gentsch JR, Parashar UD. Systematic review of regional and temporal trends in global rotavirus strain diversity in the pre rotavirus vaccine era: insights for understanding the impact of rotavirus vaccination programs. Vaccine. 2012;30:122–130. doi: 10.1016/j.vaccine.2011.09.111. [DOI] [PubMed] [Google Scholar]
- 11.Nyaga MM, Jere KC, Peenze I, Mlera L, van Dijk AA, Seheri ML, Mphahlele MJ. Sequence analysis of the whole genomes of five African human G9 rotavirus strains. Infect Genet Evol. 2013;16:62–77. doi: 10.1016/j.meegid.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 12.Matsuno S, Hasegawa A, Mukoyama A, Inouye S. A candidate for a new serotype of human rotavirus. J Virol. 1985;2:623–624. doi: 10.1128/jvi.54.2.623-624.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adah MI, Rohwedder A, Olaleye OD, Werchau H. Sequence analysis of VP7 gene of two Nigerian rotavirus strains. Acta Virol. 1996;40:187–193. [PubMed] [Google Scholar]
- 14.Cunliffe NA, Ngwira BM, Dove W, Thindwa BD, Turner AM, Broadhead RL, Molyneux ME, Hart CA. Epidemiology of rotavirus infection in children in Blantyre, Malawi, 1997-2007. J Infect Dis. 2010;202:168–174. doi: 10.1086/653577. [DOI] [PubMed] [Google Scholar]
- 15.Kiulia NM, Kamenwa R, Irimu G, Nyangao JO, Gatheru Z, Nyachieo A, Steele AD, Mwenda JM. The epidemiology of human rotavirus associated with diarrhoea in Kenyan children: a review. J Trop Pediatr. 2008;54:401–405. doi: 10.1093/tropej/fmn052. [DOI] [PubMed] [Google Scholar]
- 16.Nakagomi T, Doan YH, Dove W, Ngwira B, Iturriza-Gómara M, Nakagomi O, Cunliffe NA. G8 rotaviruses with conserved genotype constellations detected in Malawi over 10 years (1997-2007) display frequent gene reassortment among strains co-circulating in humans. J Gen Virol. 2013;94:1273–1295. doi: 10.1099/vir.0.050625-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh S, Kobayashi N. Whole-genomic analysis of rotavirus strains: current status and future prospects. Future Microbiol. 2011;9:1049–1065. doi: 10.2217/fmb.11.90. [DOI] [PubMed] [Google Scholar]
- 18.Jere KC, Mlera L, O’Neill HG, Potgieter AC, Page NA, Seheri ML, van Dijk AA. Whole genome analyses of African G2, G8, G9, and G12 rotavirus strains using sequence-independent amplification and 454® pyrosequencing. J Med Virol. 2011;83:2018–2042. doi: 10.1002/jmv.22207. [DOI] [PubMed] [Google Scholar]
- 19.Bányai K, László B, Duque J, Steele AD, Nelson EA, Gentsch JR, Parashar UD. Systematic review of regional and temporal trends in global rotavirus strain diversity in the pre rotavirus vaccine era: insights for understanding the impact of rotavirus vaccination programs. Vaccine. 2012;30:22–30. doi: 10.1016/j.vaccine.2011.09.111. [DOI] [PubMed] [Google Scholar]
- 20.Djikeng A, Halpin R, Kuzmickas R, Depasse J, Feldblyum J, Sengamalay N, Afonso C, Zhang X, Anderson NG, Ghedin E, Spiro DJ. Viral genome sequencing by random priming methods. BMC Genomics. 2008;9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maes P, Matthijnssens J, Rahman M, van Ranst M. BMC Microbial. 2009;9:238. doi: 10.1186/1471-2180-9-238. 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matthijnssens J, Heylen E, Zeller M, Rahman M, Lemey P, Van Ranst M. Phylodynamic analyses of rotavirus genotypes G9 and G12 underscore their potential for swift global spread. Mol Biol Evol. 2010;27:2431–2436. doi: 10.1093/molbev/msq137. [DOI] [PubMed] [Google Scholar]
- 24.Drummond AJ, Rambaut A. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ball JM, Mitchell DM, Gibbons TF, Parr RD. Rotavirus NSP4: a multifunctional viral enterotoxin. Viral Immunol. 2005;18:27–40. doi: 10.1089/vim.2005.18.27. [DOI] [PubMed] [Google Scholar]
- 26.Esona MD, Geyer A, Page N, Trabelsi A, Fodha I, Aminu M, Agbaya VA, Tsion B, Kerin TK, Armah GE, Steele AD, Glass RI, Gentsch JR. Genomic characterization of human rotavirus G8 strains from the African rotavirus network: relationship to animal rotaviruses. J Med Virol. 2009;81:937–951. doi: 10.1002/jmv.21468. [DOI] [PubMed] [Google Scholar]
- 27.Matthijnssens J, Rahman M, Yang X, Delbeke T, Arijs I, Kabue JP, Muyembe JJ, van Ranst M. G8 rotavirus strains isolated in the Democratic Republic of Congo belong to the DS-1-like genogroup. J Clin Microbiol. 2006;44:1801–1809. doi: 10.1128/JCM.44.5.1801-1809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosh S, Gatheru Z, Nyangao J, Adachi N, Urushibara N, Kobayashi N. Full genomic analysis of a G8P[1] rotavirus strain isolated from an asymptomatic infant in Kenya provides evidence for an artiodactyl-to-human interspecies transmission event. J Med Virol. 2011;83:367–376. doi: 10.1002/jmv.21974. [DOI] [PubMed] [Google Scholar]
- 29.Nyangao J, Page N, Esona M, Peenze I, Gatheru Z, Tukei P, Steele AD. Characterization of human rotavirus strains from children with diarrhea in Nairobi and Kisumu, Kenya, between 2000 and 2002. J Infect Dis. 2010;202:187–192. doi: 10.1086/653564. [DOI] [PubMed] [Google Scholar]
- 30.Mwenda JM, Ntoto KM, Abebe A, Enweronu-Laryea C, Amina I, Mchomvu J, Kisakye A, Mpabalwani EM, Pazvakavambwa I, Armah GE, Seheri LM, Kiulia NM, Page N, Widdowson MA, Steele AD. Burden and epidemiology of rotavirus diarrhoea in selected African countries: preliminary results from the African Rotavirus Surveillance Network. J Infect Dis. 2010;202:5–11. doi: 10.1086/653557. [DOI] [PubMed] [Google Scholar]
- 31.Seheri M, Nemarude L, Peenze I, Netshifhefhe L, Nyaga MM, Ngobeni HG, Maphalala G, Maake LL, Steele AD, Mwenda JM, Mphahlele JM. Update of rotavirus strains circulating in Africa from 2007 through 2011. Pediatr Infect Dis J. 2014;1:76–84. doi: 10.1097/INF.0000000000000053. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh S, Adachi N, Gatheru Z, Nyangao J, Yamamoto D, Ishino M, Urushibara N, Kobayashi N. Whole-genome analysis reveals the complex evolutionary dynamics of Kenyan G2P[4] human rotavirus strains. J Gen Virol. 2011;92:2201–2208. doi: 10.1099/vir.0.033001-0. [DOI] [PubMed] [Google Scholar]
- 33.Tran TN, Nakagomi T, Nakagomi O. Evidence for genetic reassortment between human rotaviruses by full genome sequencing of G3P[4] and G2P[4] Strains co-circulating in India. Trop Med Health. 2013;41:13–20. doi: 10.2149/tmh.2012-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gómez MM, de Mendoncça MC, Volotão Ede M, Tort LF, da Silva MF, Cristina J, Leite JP. Rotavirus A genotype P[4]G2: genetic diversity and reassortment events among strains circulating in Brazil between 2005 and 2009. J Med Virol. 2011;83:1093–1106. doi: 10.1002/jmv.22071. [DOI] [PubMed] [Google Scholar]
- 35.Esona MD, Mijatovic-Rustempasic S, Foytich K, Roy S, Banyai K, Armah GE, Steele AD, Volotão EM, Gomez MM, Silva MF, Gautam R, Quaye O, Tam KI, Forbi JC, Seheri M, Page N, Nyangao J, Ndze VN, Aminu M, Bowen MD, Gentsch JR. Human G9P[8] rotavirus strains circulating in Cameroon, 1999–2000: genetic relationships with other G9 strains and detection of a new G9 subtype. Infect Genet Evol. 2013;18:315–324. doi: 10.1016/j.meegid.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang M, Zeng CQ, Dong Y, Ball JM, Saif LJ, Morris AP, Estes MK. Mutations in rotavirus nonstructural glycoprotein NSP4 are associated with altered virus virulence. J virol. 1998;72:3666–3672. doi: 10.1128/jvi.72.5.3666-3672.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.