

Abstract
Fibrin clot formation is a proteolytic cascade of events with thrombin and plasmin identified as the main proteases cleaving fibrinogen precursor, and the fibrin polymer, respectively. Other proteases may be involved directly in fibrin(ogen) cleavage, clot formation, and resolution, or in the degradation of fibrin‐based scaffolds emerging as useful tools for tissue engineered constructs. Here, cysteine cathepsins are investigated for their putative ability to hydrolyze fibrinogen, since they are potent proteases, first identified in lysosomal protein degradation and known to participate in extracellular proteolysis. To further explore this, we used two independent computational technqiues, molecular docking and bioinformatics sequence analysis (PACMANS), to predict potential binding interactions and sites of hydrolysis between cathepsins K, L, and S and fibrinogen. By comparing the results from these two objective, computational methods, it was determined that cathepsins K, L, and S do bind and cleave fibrinogen α, β, and γ chains at similar and unique sites. These differences were visualized experimentally by the unique cleaved fibrinogen banding patterns after incubation with each of the cathepsins, separately. In conclusion, human cysteine cathepsins K, L, and S are a new class of proteases that should be considered during fibrin(ogen) degradation studies both for disease processes where coagulation is a concern, and also in the implementation and design of bioengineered systems.
Keywords: cathepsins, fibrinogen, fibrinolysis, hydrolysis, molecular docking, bioengineering, cysteine protease, extracellular matrix
Abbreviations
- CatK
cathepsin K
- CatL
cathepsin L
- CatS
cathepsin S
- ECM
extracellular matrix
- Fbgn
fibrinogen
Introduction
Fibrin networks form the basis of a blood clot in response to injury to the endothelial lining of blood vessels to prevent blood loss and maintain homeostasis. Fibrinogen, the fibrin precursor, is a 340 kDa protein that circulates in the blood at high concentrations1, 2, 3, 4 in preparation for an injury event. It is a hexameric protein, composed of two subunits, each with three polypeptide chains (α, β, and γ), held together by disulfide bonds, with a flexible region (E‐domain) and with large globular regions (D‐domain) on the ends.5 It is within the E‐domain that thrombin, a serine protease, binds, and cleaves N‐terminal fibrinopeptides A and B, producing disulfide linkages. In the presence of Factor XIIIa, fibrinogen monomers are converted into an insoluble fibrin network, which at the site of an injury, traps red blood cells, and platelets to prevent blood loss.2, 6 Plasmin is the endogenous enzyme that degrades fibrin during resolution of blood clots.7
While thrombin and plasmin are known proteases involved in the coagulation cascade to cleave fibrinogen and degrade fibrin, we hypothesize that there are other proteases with similar capabilities that have been overlooked, yet could be playing significant roles in fibrin(ogen) polymerization or resolution. One such candidate are cysteine cathepsins, a family of proteases first identified in lysosomes, responsible for protein degradation and amino acid turnover, that are now appreciated to be released from cells and contribute to extracellular protein degradation, matrix proteins, cytokines, and themselves.8, 9, 10 Cathepsins K, L, and S share 60% sequence identity and are elevated in a number of diseases such as sickle cell disease, diabetes, and arthritis11, 12, 13, 14, 15 where hypercoagulation caused by increased fibrinogen proteolysis is a complication.16, 17 Alternatively, dysregulated fibrin degradation can be problematic as well, with clots resolved prior to sufficient tissue repair, which motivated the development of antifibrinolytics to inhibit proteolytic activity of plasmin and plasminogen activators as therapies.18 Significance not only exists for clinical situations, but also for tissue engineering applications as fibrin‐based scaffolds continue to emerge for a wide variety of constructs such as muscle strips of biological machines,19 microphysiological systems,7, 20 and perfusable microvascular networks,21 which can be comprised of cell types that may not naturally produce plasmin, but have still seen degradation of the matrix scaffold.
To investigate the putative binding and cleavage of fibrin(ogen), we used two independent computational methods: (1) molecular docking interactions combined with (2) bioinformatics active site analysis for substrate specificity we call PACMANS22 to identify sites on fibrinogen chains α, β, and γ susceptible to binding and proteolysis by cysteine cathepsins K, L, and S as a first step in generating corroborating molecular support of cathepsin mediated fibrin(ogen)olysis.
Results
Molecular docking predicts primary cathepsin binding sites and PACMANS ranks likelihood of cleavage by cathepsins
ClusPro molecular docking was used to calculate and visualize putative cathepsin‐fibrinogen interactions based on energetically favorable binding confirmations. Our goal was to determine specific residues on fibrinogen with high probability of binding of cathepsin K, L, and S active sites. The active sites of papain family cysteine cathepsins are highly conserved with a cysteine, histidine, and asparagine to form the catalytic triad. For cathepsin K, its catalytic triad is formed of residues C25, H162, and N182,23 for cathepsin L, residues C25, H163, and N187,24 and for cathepsin S, residues C25, H164, and N184.24 ClusPro generated models with similar conformations closely clustered together to produce a single model representative of the group. The structures provided by ClusPro were all energetically favorable and unique (see Materials and Methods), allowing for a thorough and plausible discovery of protein‐substrate interactions.
PACMANS (Protease‐Ase Cleavages from MEROPS ANalyzed Specificities)22 developed by us (and available online: platt.gatech.edu/PACMANS) is an algorithm for users to input a URL link to the MEROPS specificity matrix of a protease (here cathepsin K, L, and S) and the amino acid sequence of a substrate (here fibrinogen α, β, and γ chains), and returns a ranked list scoring likely sites of hydrolysis on the substrate being analyzed. To investigate the potential for cathepsin‐mediated hydrolysis of fibrinogen (α, β, and γ) chains, the top 5% of scored PACMANS sequences were further investigated and compared against the molecular docking results. The full top 5% of scored sequences of individual fibrinogen chains are shown for cathepsin K (Supporting Information Tables S1–S3), cathepsin L (Supporting Information Tables S4–S6), and cathepsin S (Supporting Information Tables S7–S9). Locations that were independently identified by these two methods, molecular docking and specificity preference from PACMANS, were further analyzed to determine mechanistic interactions.
With correlation between putative cleavage sites and molecular docking binding sites, the next step focused on what possible molecular interactions that would make these sites favorable for catalysis. The top scoring sequences from each fibrinogen chain was further analyzed to determine possible interactions between amino acid residues of cathepsin K, L, and S and the cleavage site of each fibrinogen chain. For this study, correlating binding regions on structured, helical portions of the subunits was preferable to binding in the globular regions because cathepsins would have easier access for fibrinogenolysis at these locations in solution.
Cathepsin K cleavage likely in α and γ chains of fibrinogen
Simulated docking showed the active site of cathepsin K directly interacting with the helical subunits in the models, binding in regions that directly correlate to the predicted cleavage sites from PACMANS (Fig. 1). For cathepsin K [Fig. 1(B)], there were five top ranked PACMANS sequences in the α chain with corresponding molecular docking models and four top ranked PACMANS sequences in the β and γ chain. From PACMANS, the greater the score, the more probable the cleavage event and the normalized scoring allowed for comparison between different cathepsins on the same fibrinogen chain. Based on the culled PACMANS scoring information, the α chain in the helical region at residues #111–118 EILR/GDFS was the most likely location for cleavage by cathepsin K to occur (with a score of 0.91). The next top scoring sequences was at residues #90–97 IQLT/YNPD in the γ chain (score of 0.90), #202–209 KQLE/QVIA in the α chain (score of 0.87), and #179–186 HQLY/IDET in the β chain (score of 0.85). Also, interestingly, the top scoring cleavage sequence in the γ chain by cathepsin K, #90–97 IQLT/YNPD (score of 0.90), was five residues shifted from a known plasmin cleavage site on fibrin (QLIK/AIQL).25
Figure 1.
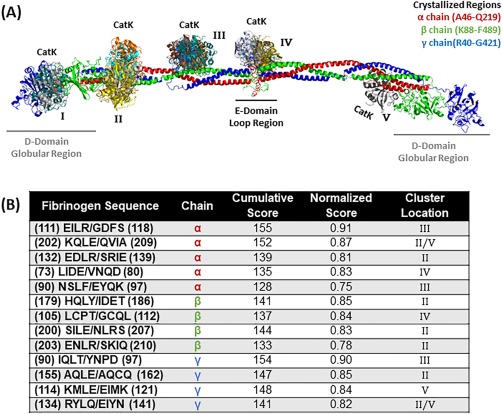
Top scored locations for cathepsin K binding and cleavage of fibrinogen. (A) Molecular docking of cathepsin K on fibrinogen monomer with the α chain is in red, the β chain is in green, and the γ chain is in blue. Cathepsin K bind in various formations and each color indicates a different docking model (note there is some overlap with the model found in cluster location B and E). Structures used were PDB ID 3GHG (fibrinogen) and 4N79 (cathepsin K). (B) Predicted fibrinogen cleavage sites by cathepsin K from PACMANS and molecular docking models.
We focused on highest scoring PACMANS region that correlated with a docking site. For the α chain of fibrinogen [Fig. 1(A)], there was only cathepsin K binding in the helical region. Within the sequence EILR/GDFS, hydrolysis was predicted between R95 and G96 [Fig. 1(B)], in an amphipathic cleavage site [Fig. 2(A)]. There appeared to be a possible electrostatic interaction between the sidechains of E111 of catK and R114 in the cleavage site, [Fig. 2(A)] and this interaction could lock these residues in an extended conformation, exposing the main chain of arginine and glycine for cleavage by the catalytic triad.
Figure 2.
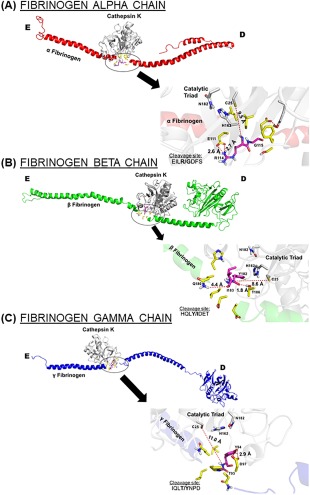
Proposed binding interfaces of cathepsin K on each fibrinogen chain. Amino acid sequences of predicted cleavage sites for cathepsin K on fibrinogen (A) α chain, (B) β chain, and (C) γ chain were visualized in the docking models. The close up is rotate 180° and the bolded site indicates where cleavage was predicted to occur.
Similar interactions were also seen in the β and γ chains [Fig. 2(B,C)]. In the β chain, there was a possible electrostatic interaction between T186 and the backbone of Y182 of the fibrinogen cleavage site HQLY/IDET. Cleavage for this site was predicted between Y182 and I183 and the cleaved isoleucine would be able to form a hydrogen bond with Q180. While the current distance was 4.4 Å, the binding of fibrinogen β chain could promote a conformational change placing these residues within a distance suitable for molecular bonding.
In the highest scoring binding site for the γ chain IQLT/YNPD, Y94 was in position to bind with D97 within a hydrogen bonding distance of 2.9 Å, anchoring this fibrinogen cleavage site [Fig. 2(C)]. The ability for both intra molecular and intermolecular bonding events to stabilize predicted cleavage sites can serve as a molecular basis for successful cathepsin K fibrinogenolysis.
Cathepsin L has highest scored cleavage location in the α chain and greatest number of scored sequences in the β chain
Cathepsin L docking sites had multiple clustering around region IV in the figure, with a cleavage of the α, β, and γ chains of fibrinogen. Region V contained identified putative cathepsin L cleavage sites for all three chains as well. Docking regions I and III, as predicted by ClusPro, did not have corresponding hits in PACMANS scores [Fig. 3(A)], reducing their probability for being the site of cleavage. For cathepsin L [Fig. 3(B)], the highest scoring sequence was found in the α chain at #111–118 EILR/GDFS (score of 0.82), and the top scoring site for cathepsin K cleavage as well [Fig. 2(A)], followed by #185–192 ETVN/SNIP in the β chain (score of 0.80) and SILT/HDSS (score of 0.80) in the γ chain. IQLT/YNPD (score of 0.75) in the fibrinogen γ chain was another likely cleavage site for cathepsin L and was a predicted cathepsin K cleavage site also.
Figure 3.
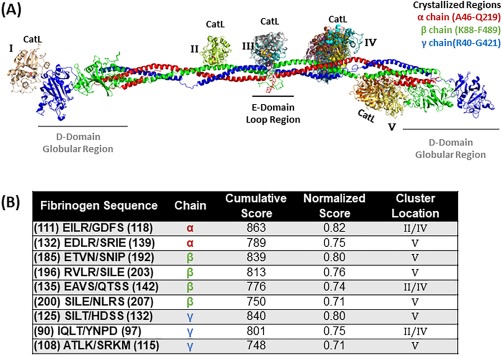
Top scored locations for cathepsin L binding and cleavage of fibrinogen. (A) Molecular docking of cathepsin L on fibrinogen monomer with the α chain is in red, the β chain is in green, and the γ chain is in blue. Cathepsin L binds in various formations and each color indicates a different docking model (note there is some overlap with the model found in cluster location B and D). Structures used were PDB ID 3GHG (fibrinogen) and 5F02 (cathepsin L). (B) Predicted fibrinogen cleavage sites by cathepsin L from PACMANS and molecular docking models.
When cathepsin L binds to the α chain at sequence EILR/GDFS [Fig. 4(A)], the flexible R114 can be stabilized by two cathepsin aspartates, D137 and D160, both within molecular bonding distance to the arginine amino groups, 2.8 and 2.9 Å respectively. An electrostatic interaction could ablate the flexibility of arginine, exposing the scissile bond for successful fibrinogen cleavage. This was a similar stabilizing mechanism when cathepsin L was bound to the β chain [Fig. 4(B)], with E185 in proximity to N188 and S189 in the cleavage site of sequence ETVN/SNIP. For the γ chain [Fig. 4(C)], the cleavage sites do not involve amino acids with flexible side chains, and therefore does not appear to be an amino acid in an advantageous position available for bond stabilization. However, T128 in the cleavage sequence SILT/HDSS is 5.2 Å from S125 and could provide a possible hydrogen bond in a natural environment.
Figure 4.
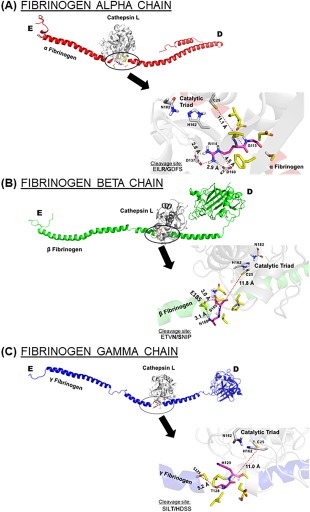
Proposed binding interfaces of cathepsin L on each fibrinogen chain. Amino acid sequences of predicted cleavage sites for cathepsin L on fibrinogen (A) α chain, (B) β chain, and (C) γ chain were visualized in the docking models. The close up is rotate 180° and the bolded site indicates where cleavage was predicted to occur.
Cathepsin S has highly scored locations of cleavage in the γ and β chains in the helical region
For cathepsin S, clusters II, IV, and V contained the most probable sites of binding [Fig. 5(A)] with the most top ranked sequences from PACMANS and molecular docking models in the β chain of fibrinogen, and the highest scoring sequence occurred in the γ chain at AQLE/AQCQ (with a score of 0.86), same as for cathepsins K and L. The next highly scored sequence occurred in the β chain at EAVS/QTSS (score of 0.84) and in the α chain at EILR/GDFS (both with scores of 0.81). In the β chain, the sequence ENLR/SKIQ was also predicted as a cleavage site for both cathepsin S (0.78) and cathepsin K (0.78).
Figure 5.
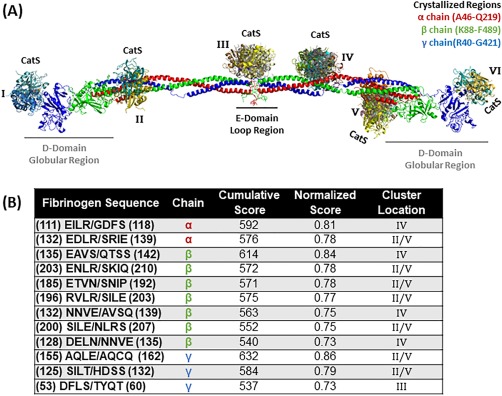
Top scored locations for cathepsin S binding and cleavage of fibrinogen. (A) Molecular docking of cathepsin S on fibrinogen monomer with the α chain is in red, the β chain is in green, and the γ chain is in blue. Cathepsin S binds in various formations and each color indicates a different docking model (note there is some overlap with the model found in cluster location B and E). Structures used were PDB ID 3GHG (fibrinogen) and 4P6E (cathepsin S). (B) Predicted fibrinogen cleavage sites by cathepsin S from PACMANS and molecular docking models.
The intramolecular interactions stabilizing cathepsin S when bound to fibrinogen were similar to those of cathepsins K and L. When cathepsin S binds to the α chain at sequence EILR/GDFS [Fig. 6(A)], the same binding sequence of cathepsin K and cathepsin L with the highest PACMANS score, R114 is stabilized by a hydrogen bond with E111, identical to cathepsin K. For the β sequence EAVS/QTSS [Fig. 6(B)] and γ sequence AQLE/AQCQ [Fig. 6(C)], the scissile bond does not involve flexible side chains, however with the current rigid conformation Q139 can form a hydrogen bond with S143 in the β sequence. In the γ chain, E158 can form a possible hydrogen bond with Q162 in the cleavage sequence [Fig. 6(C)]. The availability of charged polar amino acids for intramolecular stabilization of flexible scissile bonds, and not necessarily for cleavage sites with smaller side chain residues, could provide validity to the idea that cleavage by cathepsins can occur with intra‐molecular bonding as a mechanism to facilitate successful cleavage of flexible scissile bonds.
Figure 6.

Proposed binding interfaces of cathepsin S on each fibrinogen chain. Amino acid sequences of predicted cleavage sites for cathepsin S on fibrinogen (A) α chain, (B) β chain, and (C) γ chain were visualized in the docking models. The close up is rotate 180° and the bolded site indicates where cleavage was predicted to occur.
Cathepsins K, L, and S cleave all three fibrinogen chains
To experimentally confirm cathepsins K, L, and S fibrinogen cleavage, equal amounts of fibrinogen were incubated with either cathepsin K, L, or S for 24 hours at 37°C [Fig. 7(A)] followed by reducing SDS‐PAGE. α, β, and γ bands are visible in the control lane and remaining fibrinogen amounts were quantified by densitometry and normalized to controls (no enzyme). There was a significant decrease in the fibrinogen chains after co‐incubation with each cathepsin (P < 0.005), but the banding pattern of degradation was different for each enzyme suggesting different bands being generated by the proteolysis. Of the fibrinogen chains, the α chain was subject to the greatest reduction in band intensity, indicating the highest proteolysis by the three cathepsins (CatK α: 0.15 ± 0.03‐fold, CatL α: 0.09 ± 0.01‐fold, CatS α: 0.06 ± 0.02‐fold) [Fig. 7(B)]. Cathepsin S showed the greatest reduction in all three fibrinogen chains (α: 0.06 ± 0.02‐fold, β: 0.09 ± 0.03‐fold, γ: 0.20 ± 0.11‐fold) compared to cathepsins K and L. The γ chain was the most intact fibrinogen chain after being subject to cathepsin hydrolysis (CatK γ: 0.25 ± 0.05‐fold, CatL γ: 0.16 ± 0.02‐fold, CatS γ: 0.20 ± 0.11‐fold).
Figure 7.
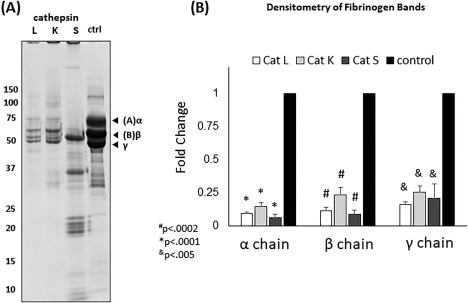
Experimental validation of cleavage of fibrinogen by cathepsins K, L, and S. Fibrinogen was co‐incubated with cathepsins K, L, and S for 24 hours at 37°C. Samples were resolved in a reduced 10% SDS‐PAGE (A). Cathepsins K and L cleave fibrinogen in a similar manner to thrombin. However, cathepsin S degrades most of the Aα and Bβ polypeptide chains of fibrinogen releasing lower molecular weight fragments. (B) Densitometry was used to quantify the bands and revealed that all groups (fibrin, Cat L, Cat K, and Cat S) were statistically significantly less compared to fibrinogen (pH 6). #P < 0.0002, *P < 0.0001, &P < 0.005, n = 3.
Discussion
We computationally assessed where fibrinogen cleavage by cathepsin K, L, and S could be occurring by combining molecular docking and putative bioinformatics cleavage site predictions as two unbiased approaches to objectively determine putative sites based first on electrostatics, and secondly on defined amino acids preferred at the protease active site cleft. The binding interactions of each cathepsin on each fibrinogen chain were visualized as were predicted putative cleavage sites based on the known cathepsin specificities culled from the MEROPS26 database using the PACMANS algorithm. Comparing the proposed binding interactions with predicted cleavage sites corroborates the experimental data of cathepsin‐mediated fibrinogen degradation, identifies putative sites of cleavage, and provides rationale for why the cleavage banding patterns for cathepsins K, L, and S were different from each other.
Much like fibrin(ogen)olytic enzymes thrombin and plasmin, cathepsins K, L, and S showed a preference for binding to the E‐domain and the helical region [Figs. 1(A), 2(A), and 3(A)], the same region where hydrolytic events causing fibrin formation (thrombin) and fibrin degradation (plasmin) occur. The binding conformation was such that the active site of the cathepsin was facing the fibrinogen subunit, indicating that the active site had a high affinity for this region. We were limited by the crystal structure of fibrinogen, which does not include some portions of the fibrinogen subunit due to either resolution or flexibility, however the binding sites proposed by the molecular docking method visualized by the crystal structure were in high agreement with top ranked predicted cleavage sites from PACMANS method, corroborating each other. The preference for the E‐domain, indicated by high PACMANS scores and docking models, provides specific amino acid locations for cathepsin binding and fibrinogen cleavage. The loops that compose the E‐domain indicate a highly flexible region prone to cleavage, and cathepsins may bind here and initiate cleavage. The β chain of fibrinogen, which forms much of the globular region of the D‐domain, has a binding site directly adjacent to this portion of the protein, and was also proximal to the region where crosslinking occurs for fibrin polymerization27, 28 and where there is a known cleavage site for plasmin.
The binding regions on the α, β, and γ chains of the fibrinogen subunit showed a high correlation with the predicted cleavage sites on fibrinogen by PACMANS. Taken together, these results also highlight the redundancy of cathepsins K, L, and S for two α chain amino acid sequences, EILR/GDFS (catK score 0.91, catL score 0.82, catS score 0.81) and EDLR/SRIE (catK score 0.81, catL score 0.75, catS score 0.78), and one β chain sequence, SILE/NLRS (catK score 0.83, catL score 0.71, catS score 0.75), that were highly ranked cleavage sites by all three cathepsins with corroborating support by the molecular docking models as possible interactions. This redundancy for fibrinogen hydrolysis sites mirrors the redundancy found among cathepsins K, L, and S for traditional extracellular matrix substrates,24 and perhaps reflects the conserved nature of their active sites. Cathepsins K and L were predicted to hydrolyze the fibrinogen γ chain at the same cleavage sequence, IQLT/YNPD, a few amino acids away from a known plasmin cleavage site on fibrinogen.
The number of PACMANS predicted cleavage sites with supporting molecular docking models at the same amino acid sequences, indicating corroborating support, were quantified as an indicator of the potential for that specific cathepsin to cleave fibrinogen at multiple sites to generate a greater number of fragment products. The higher the PACMANS score value, the higher the likelihood that cleavage occurs at the scissile bond of the substrate, in this case fibrinogen. The maximum possible score for an “ideal” sequence would be 8.0, which assumes a sequence where the protease will only cleave a specific sequence (i.e., for the scissile bond to be cleaved, only one possible sequence amino acids can be present). For cysteine cathepsins, the maximum score we have seen has been around 2.0 for type I collagen.22 Cysteine cathepsins have redundancy in the sequences/substrates they can cleave, which also reduces the score at each subsite. For example, cathepsin K preferentially accommodates hydrophobic amino acids at the S2 site;29 Leu, Ile, and Val would all have higher frequencies in the P2 position specificity matrix from MEROPS compared to hydrophilic residues, however since all three residues are likely found in the P2 position when cleavage occurs, the maximum score for the S2 site is closer to 0.33 rather than 1, after normalization to compare multiple proteases on one substrate.
There were thirteen predicted cleavage sites for cathepsin K supported by molecular docking models, while cathepsin S had twelve sequences, and cathepsin L had nine sequences. Cathepsin K also had a much greater potential for cleaving multiple locations in the α chain, with five cleavage sequences corroborated molecular docking models compared to the two cleavage sequences for cathepsins L and S in that chain. Likewise, cathepsin S had the greatest potential for cleaving the β chain, with seven cleavage sequences supported by molecular docking models compared to the four cleavage sequences for cathepsins K and L. Cathepsins S and L have a number of overlapping docking models in the β chain EAVS/QTSS (cathepsin S score 0.84, cathepsin L score 0.74), ETVN/SNIP (cathepsin L score 0.80, cathepsin S score 0.78), and RVLR/SILE (cathepsin S score 0.77, cathepsin L score 0.76) and in the γ chain at SILT/HDSS (cathepsin L score 0.80, cathepsin S score 0.79). Also, it is interesting to note that although there was far more congruence in the number of β chain cleavage sequences identified from the molecular docking analysis (four), only one of those sequences was found to be a top ranked PACMANS sequence for hydrolysis. This suggests that there may be binding for cathepsins K, L, and S at that location (docking), but not high priority for hydrolysis (PACMANS). In the α chain, however, there were only two overlapping docking locations, but both were highly ranked cleavage locations for all three cathepsins as well.
The docking models and predicted cleavage sites provide computational support for where cathepsins K, L, and S can bind and cleave fibrinogen. Closer examination illustrated mechanisms of intramolecular stabilization between the cathepsin binding to specific cleavage sites identified by PACMANS (Figs. 2, 4, and 6). Whether the scissile bond involved a flexible or rigid side chain, the presence of nearby polar residues capable of molecular bonding portrayed fibrinogen as an easily accessible cleavage target for cathepsins K, L, and S. While these models used rigid structures, the polarity of these residues supports the hypothesis that these residues and cleavage sites would be readily accessible for cleavage in an aqueous or cellular environment, and indeed, cleavage of fibrinogen by all three cathepsins was demonstrated in Figure 7. Specific confirmation of the binding and cleavage site by mass spectrometry or other analysis of cleavage fragments is still to be done, however, this report experimentally confirms cathepsins K, L, and S degradation of fibrinogen, and provides molecular docking evidence of the stabilization of the intermediates and final products generated during the proteolytic chemical reaction.
The novelty of this study is that it is among the first to directly investigate the involvement of human cysteine cathepsins K, L, and S. This work can be placed with other reports, first from 1994, which demonstrated fibrinogenolytic activity of cathepsin D, an aspartic protease, a different class of enzyme than the cysteine proteases studied here though similar in nomenclature.30 A later report identified a parasite cysteine protease related to cathepsin L that interacts with fibrinogen.31 These are complemented by a more recent re‐examination of papain, a non‐mammalian protease from which the cysteine cathepsins share conserved structure, used as a biochemical reagent for potent hydrolysis of substrates, and found to have fibrinogen clotting ability.32
Defining cysteine cathepsin‐mediated fibrin(ogen)olysis has many implications. As fibrin‐based tissue engineering constructs are used more frequently, and in various cellular environments, it is important for researchers to understand how these synthetic environments can be remodeled, especially by cysteine cathepsins that are produced by a range of cell types. We have recently demonstrated upregulation of cysteine cathepsins during the functional time of a self‐locomoting biological machine constructed of C2C12 cells in a fibrin based scaffold.19 This also opens the door to exploration of cysteine cathepsins in clot formation and clot resolution in a number of disease where cathepsins are known to be upregulated, and particularly in the cardiovascular system, where cathepsins are regulated by shear stress in endothelial cells,33, 34 and could have direct access to fibrinogen circulating in the blood.
Materials and Methods
Molecular docking simulations
Molecular docking was accomplished using the web server ClusPro 2.0.10, 35, 36, 37 The atomic co‐ordinates of human fibrinogen (PDB ID: 3GHG), cathepsins K (PDB ID: 4N79), cathepsin L (PDB ID: 5F02), and cathepsin S (PDB ID: 4P6E) were downloaded from the Protein Data Bank.38, 39, 40, 41 Briefly, the crystal structure of cathepsins were stripped of all water molecules and ligands, then uploaded to the server as a ligand. Human fibrinogen, the much larger molecule, was uploaded as a receptor. For the study of individual fibrin chains, the role of ligand and receptor remained constant, with specific chains selected during the upload of the fibrinogen model. Using ClusPro allows for selection of binding simulations that favor binding through electrostatic interactions, hydrophobic interactions and Van Der Waals interactions. The simulations for this work were performed with a balanced approached, meaning an unbiased binding method acknowledging all bonding interactions when developing docking models. The structures were rotated and translated in three dimensions, simulating 109 possible binding interactions. The docking simulations followed a balanced approach, producing 103 energetically favorable conformations, all representative of a possible binding motif. With these conformations, models in which one atom of the ligand was within 1 nm of one receptor atom were selected for clustering. Following selection, the distance between the α‐carbons of the corresponding residues of the selected ligands were calculated. If identical atoms of the ligand in two different conformations had a carbon‐carbon distance within 0.9 nm from each other, then they were clustered into groups. The idea is that in this range, similar conformations would get clustered, and the cluster with the most conformations was considered the most favorable.10, 35, 36, 37 The models were visualized using the PyMol molecular viewer (PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, 2002). We focused on models in which the active site of the ligand was facing the receptor, and discarded possible allosteric alternate binding modes as well as overlapping structures.41, 42 From the crystal structure of fibrinogen, only a portion of the overall structure was captured and further investigated: α chain region included was A46 through Q219, β chain region included was K88 through F489, and γ chain region included was R40 through G421.
Putative cleavage site determination with PACMANS
PACMANS (Protease‐Ase Cleavages from MEROPS ANalyzed Specificities)22 developed by us (and available online: platt.gatech.edu/PACMANS) is an algorithm for users to input a URL link to the MEROPS specificity matrix of a protease (here cathepsin K, L, and S) and the amino acid sequence of a substrate (here fibrinogen α, β, and γ chains), and returns a ranked list scoring likely sites of hydrolysis on the substrate being analyzed. Using a sliding window approach, the program scores the likelihood of hydrolysis occurring between the P1/P1’ scissile bond when a given 8 amino acid segment of the substrate was in the S4 through S4’ subsites of the protease's active site using the protease's specificity matrix characterized by MEROPS. MEROPS specificity matrix contains the frequency that each amino acid has been reported in each subsite of the protease's active site, so the higher the score, the higher the chance that cleavage will occur between the P1/P1’ scissile bond.22, 26 These scores were then normalized based on the number of substrates used to develop the specificity matrix to make the scores more comparable across different proteases of interest.22 The top 5% of scores are then further analyzed for accessibility by the protease and compared with molecular docking results.
Fibrinogenolysis experiment
Human fibrinogen (FIB 3 Plasminogen, von Willebrand Factor and Fibronectin Depleted; Enzyme Research Laboratories) was co‐incubated with recombinant human cathepsins K, L, or S (Enzo) in assay buffer (0.1M sodium phosphate buffer, pH 6.0, 1 mM EDTA) with 2 mM dithiothreitol (DTT) for 24 hours at 37°C. The samples were centrifuged at 7000 x g and the supernatant was saved. The pellet was prepped in 50 mM Tris‐HCl/100 mM NaCl with 25% betamercaptoethanol (OmniPur) and sonicated. Samples were prepared with reduced loading dye (5X—0.05% bromophenol blue, 10% SDS, 1.5 M Tris, 50% glycerol, 25% betamercaptoethanol) and run on 10% SDS‐PAGE gels and stained with Coomassie stain (10% acetic acid, 25% isopropanol, 4.5% Coomassie blue), followed by destaining (10% isopropanol and 10% acetic acid) and imaged with an ImageQuant LAS 4000 (GE Healthcare), and densitometry performed with ImageJ (NIH) to quantify fibrin(ogen) polypeptide chains.
Supporting Information
Tables of the predicted sequencs for each of the proteases and fibrinogen subunits.
Conflict of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author Contributions
MCF, DMW, RDA, and MOP designed the research. MCF, DMW, and SAD performed research and analyzed the data. MCF, DMW, SAD, RDA, and MOP wrote the manuscript.
Supporting information
Supporting Information
Significance: Cathepsins K, L, and S are additional proteases capable of cleaving fibrinogen, as confirmed by two computational methods and one experimental method. Cathepsin‐mediated fibrin(ogen) degradation has significance for cardiovascular diseases, where cathepsins are elevated and may be complicating clot formation or resolution. Also, with the broader use of fibrin‐based scaffolds for tissue‐engineered constructs, cathepsins may be an unexpected culprit in untimely remodeling and degradation.
References
- 1. Mosesson MW (2005) Fibrinogen and fibrin structure and functions. J Thromb Haemost 3:1894–1904. [DOI] [PubMed] [Google Scholar]
- 2. Herrick S, Blanc‐Brude O, Gray A, Laurent G (1999) Fibrinogen. Int J Biochem Cell Biol 31:741–746. [DOI] [PubMed] [Google Scholar]
- 3. Stubbs MT, Oschkinat H, Mayr I, Huber R, Angliker H, Stone SR, Bode W (1992) The interaction of thrombin with fibrinogen. A structural basis for its specificity. Eur J Biochem 206:187–195. [DOI] [PubMed] [Google Scholar]
- 4. Kamath S, Lip GY (2003) Fibrinogen: biochemistry, epidemiology and determinants. QJM 96:711–729. [DOI] [PubMed] [Google Scholar]
- 5. Doolittle RF, Spraggon G, Everse SJ (1998) Three‐dimensional structural studies on fragments of fibrinogen and fibrin. Curr Opin Struct Biol 8:792–798. [DOI] [PubMed] [Google Scholar]
- 6. Weisel JW, Litvinov RI (2017) Fibrin formation, structure and properties. Subcell Biochem 82:405–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whisler JA, Chen MB, Kamm RD (2014) Control of perfusable microvascular network morphology using a multiculture microfluidic system. Tissue Eng Part C, Methods 20:543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berdowska I (2004) Cysteine proteases as disease markers. Clin Chim Acta 342:41–69. [DOI] [PubMed] [Google Scholar]
- 9. Brix K, Dunkhorst A, Mayer K, Jordans S (2008) Cysteine cathepsins: cellular roadmap to different functions. Biochimie 90:194–207. [DOI] [PubMed] [Google Scholar]
- 10. Chapman HA, Riese RJ, Shi GP (1997) Emerging roles for cysteine proteases in human biology. Annu Rev Physiol 59:63–88. [DOI] [PubMed] [Google Scholar]
- 11. Keegan PM, Surapaneni S, Platt MO (2012) Sickle cell disease activates peripheral blood mononuclear cells to induce cathepsins k and v activity in endothelial cells. Anemia 2012:201781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hua Y, Zhang Y, Dolence J, Shi GP, Ren J, Nair S (2013) Cathepsin K knockout mitigates high‐fat diet‐induced cardiac hypertrophy and contractile dysfunction. Diabetes 62:498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu J, Ma L, Yang J, Ren A, Sun Z, Yan G, Sun J, Fu H, Xu W, Hu C, Shi GP (2006) Increased serum cathepsin S in patients with atherosclerosis and diabetes. Atherosclerosis 186:411–419. [DOI] [PubMed] [Google Scholar]
- 14. Yang M, Zhang Y, Pan J, Sun J, Liu J, Libby P, Sukhova GK, Doria A, Katunuma N, Peroni OD, Guerre‐Millo M, Kahn BB, Clement K, Shi GP (2007) Cathepsin L activity controls adipogenesis and glucose tolerance. Nat Cell Biol 9:970–977. [DOI] [PubMed] [Google Scholar]
- 15. Hou WS, Li W, Keyszer G, Weber E, Levy R, Klein MJ, Gravallese EM, Goldring SR, Bromme D (2002) Comparison of cathepsins K and S expression within the rheumatoid and osteoarthritic synovium. Arthritis Rheum 46:663–674. [DOI] [PubMed] [Google Scholar]
- 16. Averett RD, Norton DG, Fan NK, Platt MO (2017) Computational imaging analysis of fibrin matrices with the inclusion of erythrocytes from homozygous SS blood reveals agglomerated and amorphous structures. J Thromb 43:43–51. [DOI] [PubMed] [Google Scholar]
- 17. Norton DG, Fan NK, Goudie MJ, Handa H, Platt MO, Averett RD (2017) Computational imaging analysis of glycated fibrin gels reveals aggregated and anisotropic structures. J Biomed Mater Res A 105:2191–2198. [DOI] [PubMed] [Google Scholar]
- 18. Longstaff C, Kolev K (2015) Basic mechanisms and regulation of fibrinolysis. J Thromb Haemost 13(Suppl 1):S98–105. [DOI] [PubMed] [Google Scholar]
- 19. Cvetkovic C, Ferrall‐Fairbanks MC, Ko E, Grant L, Kong H, Platt MO, Bashir R (2017) Investigating the life expectancy and proteolytic degradation of engineered skeletal muscle biological machines. Sci Rep 7:3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moya ML, Hsu YH, Lee AP, Hughes CC, George SC (2013) In vitro perfused human capillary networks. Tissue Eng Part C Methods 19:730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahmed TA, Dare EV, Hincke M (2008) Fibrin: a versatile scaffold for tissue engineering applications. Tissue Eng Part B Rev 14:199–215. [DOI] [PubMed] [Google Scholar]
- 22. Ferrall‐Fairbanks MC, Barry ZT, Affer M, Shuler MA, Moomaw EW, Platt MO (2017) PACMANS: a bioinformatically informed algorithm to predict, design, and disrupt protease‐on‐protease hydrolysis. Protein Sci 26:880–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Turk B, Turk D, Turk V (2000) Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta 1477:98–111. [DOI] [PubMed] [Google Scholar]
- 24. Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D (2012) Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim Biophys Acta 1824:68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kirschbaum NE, Budzynski AZ (1990) A unique proteolytic fragment of human fibrinogen containing the A alpha COOH‐terminal domain of the native molecule. J Biol Chem 265:13669–13676. [PubMed] [Google Scholar]
- 26. Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40:D343–3D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spraggon G, Everse SJ, Doolittle RF (1997) Crystal structures of fragment D from human fibrinogen and its crosslinked counterpart from fibrin. Nature 389:455–462. [DOI] [PubMed] [Google Scholar]
- 28. Pandya BV, Gabriel JL, O'Brien J, Budzynski AZ (1991) Polymerization site in the beta chain of fibrin: mapping of the B beta 1–55 sequence. Biochemistry 30:162–168. [DOI] [PubMed] [Google Scholar]
- 29. Alves MF, Puzer L, Cotrin SS, Juliano MA, Juliano L, Bromme D, Carmona AK (2003) S3 to S3' subsite specificity of recombinant human cathepsin K and development of selective internally quenched fluorescent substrates. Biochem J 373:981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Simon DI, Ezratty AM, Loscalzo J (1994) The fibrin(ogen)olytic properties of cathepsin D. Biochemistry 33:6555–6563. [DOI] [PubMed] [Google Scholar]
- 31. Costa TF, dos Reis FC, Lima AP (2012) Substrate inhibition and allosteric regulation by heparan sulfate of Trypanosoma brucei cathepsin L. Biochim Biophys Acta 1824:493–501. [DOI] [PubMed] [Google Scholar]
- 32. Doolittle RF (2014) Clotting of mammalian fibrinogens by papain: a re‐examination. Biochemistry 53:6687–6694. [DOI] [PubMed] [Google Scholar]
- 33. Platt MO, Ankeny R, Jo HJ (2006) Laminar shear stress inhibits cathepsin L activity in endothelial cells. Arterioscl Throm Vas 26:E89–E89. [DOI] [PubMed] [Google Scholar]
- 34. Platt MO, Ankeny RF, Shi GP, Weiss D, Vega JD, Taylor WR, Jo H (2007) Expression of cathepsin K is regulated by shear stress in cultured endothelial cells and is increased in endothelium in human atherosclerosis. Am J Physiol Heart Circ Physiol 292:H1479–H1486. [DOI] [PubMed] [Google Scholar]
- 35. Garnero P, Borel O, Byrjalsen I, Ferreras M, Drake FH, McQueney MS, Foged NT, Delmas PD, Delaisse JM (1998) The collagenolytic activity of cathepsin K is unique among mammalian proteinases. J Biol Chem 273:32347–32352. [DOI] [PubMed] [Google Scholar]
- 36. Aguda AH, Panwar P, Du X, Nguyen NT, Brayer GD, Bromme D (2014) Structural basis of collagen fiber degradation by cathepsin K. Proc Natl Acad Sci USA 111:17474–17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bromme D, Li Z, Barnes M, Mehler E (1999) Human cathepsin V functional expression, tissue distribution, electrostatic surface potential, enzymatic characterization, and chromosomal localization. Biochemistry 38:2377–2385. [DOI] [PubMed] [Google Scholar]
- 38. Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: a fully automated algorithm for protein‐protein docking. Nucleic Acids Res 32:W96–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kozakov D, Brenke R, Comeau SR, Vajda S (2006) PIPER: an FFT‐based protein docking program with pairwise potentials. Proteins 65:392–406. [DOI] [PubMed] [Google Scholar]
- 40. Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, Vajda S (2013) How good is automated protein docking? Proteins 81:2159–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S (2017) The ClusPro web server for protein‐protein docking. Nat Protoc 12:255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Comeau SR, Gatchell DW, Vajda S, Camacho CJ (2004) ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20:45–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information