

Abstract
Nanolipoprotein particles (NLPs), composed of membrane scaffold proteins and lipids, have been used to support membrane proteins in a native‐like bilayer environment for biochemical and structural studies. Traditionally, these NLPs have been prepared by the controlled removal of detergent from a detergent‐solubilized protein‐lipid mixture. Recently, an alternative method has been developed using direct cell‐free expression of the membrane scaffold protein in the presence of preformed lipid vesicles, which spontaneously produces NLPs without the need for detergent at any stage. Using SANS/SAXS, we show here that NLPs produced by this cell‐free expression method are structurally indistinguishable from those produced using detergent removal methodologies. This further supports the utility of single step cell‐free methods for the production of lipid binding proteins. In addition, detailed structural information describing these NLPs can be obtained by fitting a capped core‐shell cylinder type model to all SANS/SAXS data simultaneously.
Keywords: NLPs, nanodiscs, nanolipoprotein particles, SANS, SAXS, small‐angle scattering, cell‐free expression
Introduction
Nanolipoprotein particles (NLPs), also known as “Nanodiscs,” are disc‐shaped patches of membrane bilayer surrounded at the perimeter by an Apolipoprotein A‐I (ApoA‐I)‐derived “belt” protein, referred to as a “membrane scaffold protein” (MSP). The MSP acts as a scaffold to stabilize the bilayer and fixes the disc diameter between roughly 10 and 60 nm, depending on the MSP variant selected.1 Membrane proteins are typically packaged into NLPs by combining the detergent‐solubilized MSPs, lipids, and purified membrane protein. Subsequent removal of the detergent then causes the discs to spontaneously form, capturing the membrane protein in the disc bilayer2 (we refer to this procedure as the “traditional” NLP preparation process). These NLP‐packaged membrane proteins then remain in solution and can be studied by traditional solution biochemical methods.
While the traditional disc packaging methods have been successful in many cases, optimizing the process of reconstitution for a new membrane protein target can be cumbersome and labor‐intensive, and it is not easily amenable to high‐throughput methods for the analysis of many different membrane proteins or constructs at once. Therefore, we have recently developed an alternative approach, where the membrane protein and MSP are simultaneously co‐expressed in the presence of lipid vesicles in a cell‐free expression system.3 The cell‐free expression system causes NLPs to form spontaneously, with enclosed membrane proteins, without the need for detergent solubilization or pre‐purification of the membrane protein. Since the starting material is DNA rather than purified proteins, the cell‐free expression method is amenable to high‐throughput screening of multiple protein constructs or functional conditions.4 Cell‐free expression systems also make it intrinsically simple to introduce additives, cofactors, binding partners, or other necessary factors during expression, for example, to aid stability or function for particular membrane proteins.
Although the cell‐free method has certain intrinsic advantages, the NLPs produced in this manner have not been as well‐characterized as those utilizing more traditional assembly methods. These traditional methods, first described by Bayburt et al.,2 include the use of detergents and temperature shifts during the assembly process. Since all the components are the same in both cases, the only question is whether the dramatically different self‐assembly processes lead to any differences in the final structural arrangement. To that end, we performed simultaneous Small‐Angle Neutron Scattering and Small‐Angle X‐ray Scattering (SANS/SAXS) on NLPs assembled from 1,2‐dimyristoyl‐sn‐glycero‐3‐phosphocholine (DMPC) lipids produced via traditional in vitro methods, and by the cell‐free method. We also analyzed NLPs produced using the cell‐free method in the presence of a PEG5k‐CA8 telodendrimer additive, which has been shown to improve solution stability and monodispersity of the NLPs.5, 6 Finally, after verifying the structural equivalence of the three NLP preparations, we fit an appropriate core‐shell disc‐type model to the scattering data to verify the geometry and molecular composition of NLPs. To improve the robustness of the model fitting, we also included SANS data from an NLP sample prepared using tail‐deuterated DMPC.
Results
NLP assembly and purification
Three NLP samples were prepared for comparison studies: (1) by standard in vitro assembly using detergent removal from a mixture of purified components (“ivNLPs”); (2) by cell‐free expression (“cfNLPs”); and (3) by cell‐free expression in the presence of a PEG5k‐CA8 telodendrimer additive (“t‐cfNLPs”) (Fig. 1). After an initial round of immobilized metal affinity chromatography (IMAC) to isolate NLPs from free lipids and other reaction components, sample purity was >95% as determined by SDS‐PAGE (Fig. S1). Samples were then lyophilized until needed for SANS/SAXS.
Figure 1.
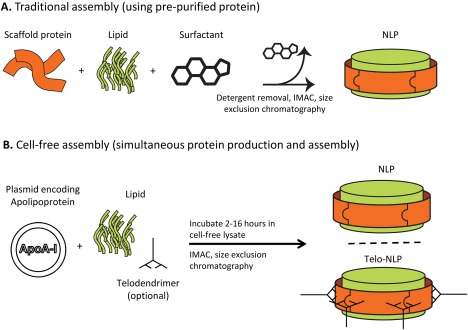
NLP assembly methods. (A) Scaffold protein of interest, lipids, and detergent are mixed, and detergent is removed to allow NLPs to form. (B) Plasmid encoding the scaffold protein of interest is mixed with lipids (and telodendrimers, for telodendrimer‐NLP discs) in a cell‐free reaction chamber. In both (A) and (B), correctly formed NLPs/telodendrimer‐NLPs are purified using SEC.
Upon reconstitution, size exclusion chromatography (SEC) was used to isolate correctly formed NLPs for SANS/SAXS data collection. By comparison with gel filtration standards (Bio‐Rad1), we determined the Stokes radius of the principal species in all samples to be roughly 5 nm (Fig. 2), consistent with the expected size of correctly formed NLPs assembled using ApoA‐I‐derived MSPs. In initial SEC runs performed in 100% D2O, additional minor peaks/tailing was observed in most samples, corresponding to aggregates or to unpackaged MSPs. Thus, the central peak fractions were carefully isolated for scattering analysis. Fractions of 100 µL were collected in 96‐well plates, and 4 peak fractions (0.4 mL) were pooled (see brackets, Fig. 2). For the ivNLP sample that was re‐run on SEC in 42% D2O after 1 week (discussed below), a single peak was present with no evidence of disassembly or re‐aggregation.
Figure 2.
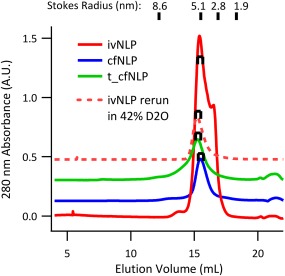
Size exclusion chromatography (SEC) of NLPs. Correctly formed NLPs were isolated by SEC, and fractions from the central portion of the predominant peak (shown with brackets) were collected for SANS/SAXS. Plots of UV absorbance are shown at true scale, but vertically offset for clarity and may include baseline subtraction.
Direct comparison of SANS/SAXS data between NLPs assembled by different methods
Before model fitting, we directly compared normalized SAXS and SANS data sets (in 100% D2O buffer) from different NLP preparations to each other (Fig. 3). Except in the very low‐q part of the SAXS curves (see below), no significant differences were present between the data sets, and we concluded that there were no significant structural differences between ivNLP, cfNLP, and t‐cfNLP samples in solution. Both the low‐q part of the SANS curves (sensitive to overall disc dimensions and composition) as well as the positions and depth of the SAXS minima (more sensitive to MSP and lipid headgroup layer dimensions, placement, and polydispersity) were the same between all three NLP samples.
Figure 3.
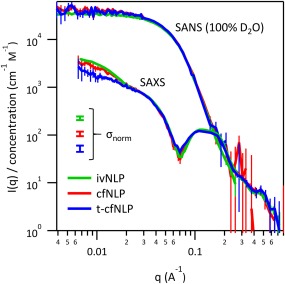
Comparison of SANS/SAXS data from different NLP preparations. Data were placed on an absolute intensity scale and normalized by molar concentration of MSP. Incoherent backgrounds were subtracted from the SANS data. No significant differences were seen between the scattering curves of NLPs prepared by different methods, and we concluded that there were no detectible structural differences between the NLPs. Small differences seen at very low q are discussed in the text. Error bars are plus or minus one standard deviation. Error bars on data points represent the uncertainty due to counting statistics. In addition, the uncertainty due to normalizing by protein concentration (σnorm) is illustrated with separate error bars; these represent a systematic vertical shift of the entire curve within this error.
In the SAXS data, a small additional rise in scattering intensity at very low‐q was observed, and was variable in magnitude between the three samples. This signal was completely absent by SANS, which extends lower in q and thus should have been even more sensitive to such scattering. We therefore interpreted the X‐ray signal as due to either a capillary subtraction error, or to a small amount of aggregation that occurred in the samples as they were being manipulated for SAXS (more likely in this case given the variable nature of the upturn). While scattering techniques minimize the potential for artifacts, protein aggregation can still occur under native‐like solution conditions over time, or due to stresses such as temperature shifts, agitation, or exposure to interfaces. Many protein SAXS beamlines address this using in‐line SEC (“SEC‐SAXS”), which was however not available on our home source. We thus restricted further SAXS data analysis to the Q range higher than the onset of the upturn.
Fitting of structural models to SANS/SAXS data
The relatively regular particle morphology of the empty NLP platform allows for the use of analytical descriptions of the scattering data, in this case a capped core‐shell cylinder model. When such analytical models are available, they allow for the incorporation of prior knowledge and restraints, and can therefore provide much richer and more robust information than ab initio models (e.g., simple bead models). Since the ivNLP, cfNLP, and t‐cfNLP data were found to be equivalent, we used the sample of highest concentration (ivNLP) to perform this analysis. For this sample, the previous SAXS and 100% D2O SANS data sets were complimented by two additional SANS datasets: the first collected after exchanging the same ivNLP sample into 42% D2O buffer, and the other by assembling a separate NLP sample from tail‐deuterated DMPC followed by SEC in 100% D2O buffer. This yielded four complementary data sets (100% D2O SANS, 42% D2O SANS, tail‐deuterated DMPC SANS in 100% D2O, and SAXS) which were then used for a global, constrained model fit (Fig. 4). The molecular composition and layer dimensions were constrained to be equal for all four data sets, with molecular scattering lengths fixed appropriately to the sample conditions (i.e., for neutron vs. X‐ray, and for differentially deuterated components). The very low‐q part of the SAXS curve (as discussed above) was excluded from fitting. The resulting model was a disc with an overall effective diameter of 85.1 ± 0.3 Å and height of 46.2 ± 0.2 Å. Fit values for the headgroup layer height and belt thickness were 10.47 ± 0.09 Å and 7.1 ± 0.2 Å, respectively. NLPs were found to contain 134.5 ± 0.7 DMPC lipid molecules and 2.08 ± 0.02 MSP molecules, with 8.7 ± 0.3 water molecules per lipid contained in the headgroup layer and 259 ± 23 water molecules per MSP in the protein rim layer. Tabulated fit parameters, with an illustration of their uncertainties and pairwise correlations, are shown in Figure 5.
Figure 4.
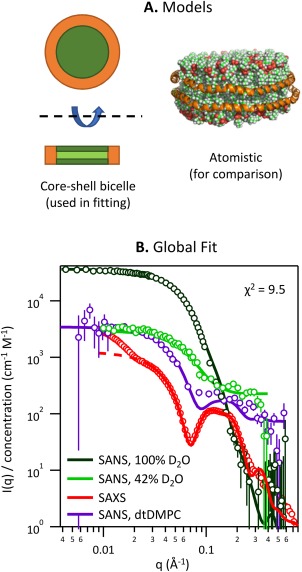
Simultaneous model fitting of SANS/SAXS data. (A) The fit model consisted of a core layer with capping layers above and below, surrounded at the perimeter by a rim. An atomistic rendering of the model is also shown for illustration, but was not used in fitting. (B) The model was simultaneously fit to four complementary scattering data sets. Error bars are plus or minus one standard deviation. For display purposes, the number of data points was reduced by binning. “dtDMPC” refers to tail‐deuterated DMPC.
Figure 5.
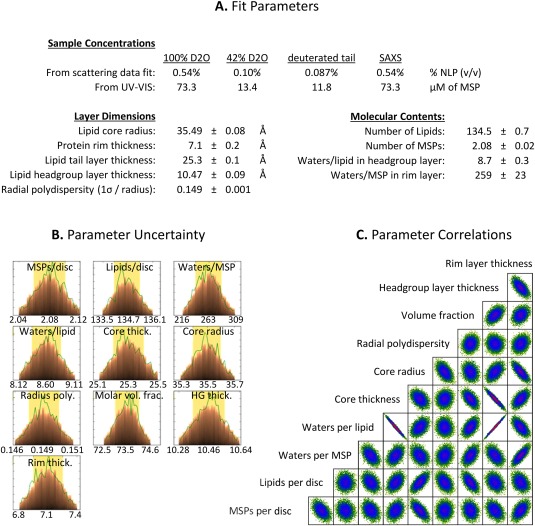
Fit Parameters and Uncertainty Analysis. (A) Structural parameters derived from fitting the core‐shell bicelle model simultaneously to SANS/SAXS curves are shown. Parameter uncertainties in the table are 68% confidence intervals based on counting statistics and do not account for systematic errors. (B) The parameter likelihood distributions obtained during fitting using the DREAM algorithm. The most probable value and 95% confidence intervals are labeled. (C) Parameter correlations from the DREAM algorithm. Axis boundaries are omitted from (C) but are the same as those labeled in (B).
Deposition of scattering data and models
Reduced scattering data sets have been deposited in the Small Angle Scattering Biological Data Bank (SASBDB). Deposited data sets have been solvent‐subtracted (SAXS) or incoherent background‐subtracted (SANS), but not scaled by molar concentration, and are thus reported on an absolute scale in units of cm−1. Accession codes are: SASDC99 (100% D2O SANS), SASDCC9 (42% D2O SANS), and SASDCA9 (SAXS) for the ivNLP sample; SASDCF9 (100% D2O SANS) and SASDCD9 (SAXS) for the cfNLP sample; SASDCG9 (100% D2O SANS) and SASDCE9 (SAXS) for the t‐cfNLP sample; and SASDCB9 for the NLPs assembled using tail‐deuterated DMPC (100% D2O SANS). In addition, the model code has been deposited at the SasView Model Marketplace (http://marketplace.sasview.org) with model name “nanodisc_simple.”
Discussion and Conclusions
In these studies, we have used simultaneous SANS/SAXS to examine NLPs produced using several different methods. Specifically, we compared traditional in vitro assembly methods to co‐translational cell‐free assembly with and without a telodendrimer additive. Different techniques can give different dimensions for NLPs, depending on sample preparation, the sensitivity of the technique to different features of the sample, and the specific quantity being measured by a given technique (Table 1). SANS/SAXS is particularly suitable for samples such as these since it allows them to be probed under relevant solution conditions, without any potentially artifact‐inducing sample preparation treatments such as vitrification, staining, surface binding, and introduction of extrinsic labels. Direct, model‐free comparison of the scattering data (Fig. 3) demonstrated clearly that discs with similar structures were produced as the predominant species by all three methods. Thus, it can be expected that cell‐free NLP preparation methods should have, in principle, the same utility for packaging membrane proteins as the traditional assembly methods relying on detergent removal.
Table 1.
Nanoparticle Dimensions Reported by Different Techniquesa
| DLS (2*Rh, nm) | AFM (D, nm) | EM (D, nm) | SAXS/SANS (D, nm) | |
|---|---|---|---|---|
| ivNLP | 12–14a | 10–201 | 12–201 | 8.5 ± 0.6b |
| cfNLP | 12–23a | 17–2322 | 15–25a | 8.5 ± 0.6b |
| t‐cfNLP | 23–305 | n.d. | 105 | 8.5 ± 0.6b |
Note in particular that DLS reports a hydrodynamic radius (Rh), which is quantitatively different, but related to, the disc diameter (D) reported here from SAXS/SANS data.
Present work, data not shown.
Present work.
Although comparison of the data alone was sufficient to verify that the three NLP samples had similar disc structures, we also fit analytical models to the data in order to extract dimensions and molecular compositions. A capped core‐shell disc model was found to be suitable, and allowed for the determination of the NLP dimensions and composition. These parameters were largely consistent with previously reported values.7, 8 Importantly, the number of belt proteins and lipids derived from model fitting (2.08 and 135, respectively) were similar to expected values of 2 and roughly 150, respectively.9, 10 It has been found that the stoichiometry of lipid incorporation in NLPs can vary across subfractions of an SEC peak,11 so the selective pooling of small portions of an SEC peak can result in slightly different lipid stoichiometry than what may be reported for less stringent fraction pooling.
All parameters determined from the global model fit were physically reasonable in general. Parameters affected by the inclusion of water molecules were less well‐determined than others, due to inherent ambiguities in defining where a bound water layer ends and bulk solvent begins. For instance, the headgroup layer thickness was highly correlated with the number of waters per lipid [Fig. 5(C)], which is reasonable since increasing the thickness of the headgroup layer means that there is room for additional hydration waters to fill the layer. However, this also means that the exact thickness of the headgroup layer and number of waters included are not precisely determined. In addition, the protein rim layer was slightly thinner than might be expected (7.1 Å, vs. at least 11 Å for the diameter of an MSP alpha helix). We attribute this discrepancy to the difference between our simplified model and the detailed structure of the actual system. Specifically, in NLPs, it is thought that the MSP covers only the hydrophobic portion of the lipid bilayer (the “core” layer); whereas in the “core‐shell bicelle” model, the rim is forced to extend over both the core and capping layers. Making the rim thinner partly compensates for this extra length; although in the higher‐resolution (high‐q) parts of the scattering data, discrepancies would be expected, and are indeed seen (Fig. 4, red SAXS curve, at high q values). In addition, this simplified model rim may partly explain the large number of waters present in the rim layer (20% by volume); these waters are compensating for the fact that the part of the rim adjacent to the lipid headgroups should actually contain solvent.
The NLP dimensions measured here by scattering were generally comparable to those previously measured by other methods (Table 1). Interestingly, there were no apparent differences in scattering data between samples with and without incorporated telodendrimer. This suggests that the PEG chains are disordered and highly hydrated, thus lacking sufficient contrast to be detectable in static scattering experiments. However, such an extended polymer would contribute significant hydrodynamic drag, leading to differences in the hydrodynamic radius between t‐cfNLP and cfNLP. This difference is clearly seen via DLS (Table 1). Thus, one can obtain a more complete understanding of the system by using these complementary techniques: SAXS/SANS to measure absolute dimensions, and DLS the hydrodynamic behavior.
It is important to note that all scattering data sets were placed on an absolute scale, and normalized for concentration using independent UV measurements. While an overall scaling parameter was fitted (the value of which can be used to determine the NLP volume fractions in solution), data sets were not allowed to scale independently from each other; relative differences in scattering intensity could be accounted for in the model only by altering the underlying dimensions and molecular contents. Requiring the model to fit multiple independent, absolutely scaled data sets in this way greatly improves the uniqueness and accuracy of the model fit. If NLP volume fractions are estimated independently by other techniques and fixed during analysis, it is possible to perform fits to a single scattering data set; however, fitting the volume fraction itself generally requires two data sets at a minimum (e.g., SAXS and SANS; SANS at 100% and 42% D2O; etc.). Given a fixed volume fraction, fits to single data sets are typically “better” in the sense that they have lower χ2 values; but many fit parameters are highly correlated, which leads to high uncertainties unless some parameters are fixed using a priori assumptions or other measurement techniques. The addition of more data sets to a simultaneous fit leads to higher χ2 values, since systematic errors (for example, in the sample concentrations or absolute scaling) then cannot be fully compensated for by the model. Nevertheless, this reduces overall systematic biases, thus improving the accuracy of the derived fit parameters. The additional constraints from complementary scattering data sets also greatly reduce the correlations between fit parameters. Therefore, we included as many independent data sets as possible in our analysis, even when some had much higher uncertainties than others (due to low sample concentrations or high incoherent backgrounds). Finally, in these simultaneous fits, the use of molecular quantities as fit parameters (rather than directly fitting SLDs) was found to be a useful “bookkeeping” procedure in order to appropriately constrain the relationships between SLDs of related layers (e.g., lipid headgroups and tails) and related sample types (e.g., the same particles in SAXS vs. SANS). Others have used a similar molecular fitting approach for the analysis of NLPs10, 12 and similar systems such as peptide discs13 by small‐angle scattering. Actual SLDs derived from these molecular quantities and layer dimensions are illustrated in Figure S3.
Our data demonstrates that homogeneous NLPs can be produced using cell‐free approaches, and that these NLPs are equivalent in their molecular contents and dimensions to traditional NLP preparations. Disc formation was robust even in the absence of controlling the protein to lipid ratios of the cell‐free reactions. Furthermore, this co‐translational method avoids the need for processes that include the addition and removal of detergents. We found that NLPs could be lyophilized and recovered without apparent deleterious effects, even in high concentrations of imidazole. Finally, we showed that the scattering from NLPs could be accounted for using a simple analytical model, which allows for the measurement of detailed solution structural parameters for NLPs prepared from DMPC lipids.
Having thus verified that the self‐assembly of the empty NLP platform is not affected by this new approach, future studies can examine the effect of membrane protein inclusion. The scattering analysis will also have to evolve, as completely analytical approaches will no longer be possible. However, the detailed quantitative analysis presented here, and by others,10 will provide a strong foundation for building what will likely be a hybrid analysis; that is, combining the analytical disc model with either atomistic or bead modeling to handle the complex membrane protein structural envelope.12
Materials and Methods
Chemicals and buffers
Chemicals were from Sigma unless stated otherwise. IMAC buffer was 50 mM NaH2PO4 and 300 mM NaCl, adjusted to pH 8.0 with NaOH. SEC buffer was prepared by dissolving pouches of pre‐mixed phosphate‐buffered saline (PBS) powder (Sigma P3813) to a final concentration of 1× using either 100% or 42% D2O. Tail‐deuterated d54‐DMPC was purchased from Avanti Polar Lipids (860345P).
Plasmids
The pIVEX2.4d vector encoded a His‐tagged, truncated ApoA‐I protein (D49ApoAI) for nickel affinity purification as previously described.3 Briefly, this plasmid encodes ApoA‐I with 49 amino acids truncated from the amino terminus, relative to the first amino acid of human Proapolipoprotein A‐I. The histidine tag is encoded on the N‐terminus. The protein sequence of the ApoA‐I used in the cell‐free experiment is slightly different from the protein sequence of the ApoA‐I used in the in vitro assembly experiment due to the difference in the cloning procedure. Nevertheless, since these constructs have been used in prior work, we maintained the same constructs for comparability. The extra amino acids are in a non‐structural region that is not expected to affect NLP formation. Full amino acid sequences of constructs are provided in the Supplementary Information.
Preparation of DMPC vesicles with and without telodendrimers
We have previously detailed the use of telodendrimers as a nano‐delivery tool.5 PEG5k‐CA8 was prepared according to published methods.14 Small unilamellar vesicles of DMPC (Avanti Polar Lipids, Alabaster, Alabama) were prepared by probe sonication of a 20 mg/mL aqueous solution of DMPC until optical clarity was achieved; typically, 3 intervals of 30 seconds were sufficient. After sonication, the samples were centrifuged at 14,100g for 1 min to remove metal contamination from the probe tip. For the DMPC/telodendrimer mixtures, a solution of 20 mg/mL DMPC and another of 2 mg/mL telodendrimer were mixed at a volume ratio of 1:1. The same procedures were used, with molar equivalent amounts of DMPC, when preparing NLPs from tail‐deuterated DMPC.
E. coli expression and NLP assembly
The expression clone to produce D49ApoAI with the N‐terminal 6 × His and Thioredoxin tag was used. Production and purification of ApoA‐I has been described in detail elsewhere.15
NLP formation and purification was performed using a modification of a protocol described in detail previously.1 Briefly, DMPC in chloroform was added to an Eppendorf tube. Chloroform was evaporated in a stream of nitrogen with constant rotation to distribute the lipid evenly along the tube wall. The tube was placed under vacuum to completely remove the chloroform. DMPC was re‐suspended in 30 mM cholate and PBS. D49ApoAI was then added to the PBS/DMPC/cholate solution. The mixture was then incubated at 25°C and 1400 rpm for 1 h on a thermomixer (Eppendorf). Biobeads (Bio‐Rad) were then added to the assembly mixture at 0.4 g biobeads per mg protein and incubated at 25°C and 1100 rpm on a thermomixer overnight, resulting in NLP formation through the removal of detergent.
Cell free expression
Large scale reactions (1 mL) were carried out using the E. coli HY Kit (Biotechrabbit GmbH, Hannover, Germany). Reaction components were combined as specified by the manufacturer. 1 µg of D49ApoA1 DNA was added to each 1 mL reaction. A total of 200 µL DMPC or 400 µL DMPC/telodendrimer mixture was then added. The reactions were incubated at 30°C, with shaking at 300 rpm for 14–18 h.
Affinity purification of NLP‐related complexes
IMAC was used to isolate the NLP assemblies of interest (D49ApoA1) from the cell‐free reaction mixtures. 1 mL of a 50% slurry of cOmplete His‐Tag Purification Resin (Roche, Basel, Switzerland) was equilibrated with IMAC buffer supplemented with 10 mM imidazole in a 2‐mL microcentrifuge tube. The total cell free reaction (1 mL) was mixed with the equilibrated resin, and was incubated/nutated at 4°C for 1 h. The solution was added to a 10 mL column and then washed with IMAC buffer supplemented with 20 mM imidazole. Four resin bed volumes (BV) of wash buffer were used for a total of 12 BVs of washing. The His‐tagged proteins of interest were eluted in six 300 µL fractions of IMAC buffer containing 250 mM imidazole (a seventh fraction was eluted with 500 mM imidazole). All elutions were analyzed by SDS‐PAGE and peak elutions were combined.
Size‐exclusion chromatography of NLP‐related complexes
Pooled fractions from IMAC were supplemented with 200 mM trehalose and lyophilized before shipment for SANS/SAXS data collection. Approximately 24 h before data collection, samples were reconstituted with D2O to a volume equal to their pre‐lyophilization volume. Samples were then concentrated to 0.5 mL using 100 kDa spin concentrators (Millipore). SEC was performed using a Superdex 200 10/300 GL column (GE Healthcare) equilibrated in SEC buffer, which had been prepared using 100% D2O. Elution was followed by monitoring solution absorbance at 280 nm. Fractions were stringently selected and pooled from the central portion of each SEC peak to obtain the best possible monodispersity and sample quality for SANS/SAXS. Data collection was performed directly on pooled fractions without concentrating them further.
After performing SANS/SAXS on all NLP samples in 100% D2O, the ivNLP sample was buffer‐exchanged into 42% D2O to obtain SANS data at another contrast, chosen to be near the match point of the MSP. This was done by subjecting the sample to another round of SEC, this time using PBS prepared with 42% D2O. Peak fractions were selected and pooled for SANS as above. Approximately 1 week passed from initial SANS data collection in 100% D2O until buffer‐exchange and collection of the new SANS data in 42% D2O.
Data collection and reduction
SANS data were collected on beamlines NG7 and NGB3016 at the NIST Center for Neutron Research (NCNR), Gaithersburg, MD. Samples were inserted into 1 mm path length cylindrical Helma quartz Suprasil cuvettes and data collected at 5°C below the liquid/gel transition temperature for the specified lipid (i.e., collection at 20°C for non‐deuterated DMPC, and 15°C for tail‐deuterated DMPC), with the exception of the ivNLP sample in 42% D2O, which had to be collected at 25°C due to instrument constraints on the day of collection. SANS data were reduced and placed on an absolute scale in the usual way using software developed by the NCNR.17 SANS data reduced using the NCNR procedures includes an estimate of the Q‐dependent instrument resolution, and this information was used during fitting to smear the calculated SANS intensity curves.
SAXS data were collected in‐house using a hybrid instrument, which is essentially a combination of the SAXSLAB GANESHA 300XL (optics, flight path and detector) with a Rigaku 007HF Cu Kα rotating anode source. Samples were flowed successively into a fixed capillary for SAXS data collection, allowing optimal buffer subtraction and absolute scaling. Data were collected for 3 h per distance at sample‐to‐detector distances of 1305, 705, and 355 mm. During exposure, 25 μL of sample were continuously oscillated in a fixed capillary at 20°C. SAXS data were reduced using a version of the software BioXTAS RAW18 that was supplied and configured by the instrument manufacturer, and were placed on an absolute intensity scale by calibration against the known low‐q scattering of H2O in the same capillary at 20°C.19
SANS/SAXS data were measured nearly simultaneously (within 4–24 h for any given sample) on aliquots taken from a parent sample. Since the different NLP solutions were not adjusted to match concentrations (e.g., by spin‐concentration or dilution) after pooling SEC fractions, the ivNLP, cfNLP, t‐cfNLP, and tail‐deuterated NLP sample concentrations differed from each other during scattering data collection. Therefore, scattering data sets were normalized by the molar concentration of MSP to enable direct comparison and analysis (giving absolute intensity units of cm−1 M−1). The molar concentration of MSP was determined using absorbance at 280 nm with calculated extinction coefficients. For SANS data comparisons, incoherent backgrounds were subtracted before normalization. These scaled and background‐subtracted data were used in all further analysis, and are referred to as “normalized” data hereafter.
Data analysis and model fitting
In order to determine the molecular composition and geometry of the NLPs, we used the software SasView (version 4.0.1)20 to fit a capped core‐shell disc model to the normalized scattering data. This model includes a core, top and bottom faces, and rim (see Fig. 4 diagram and Fig. S2) with adjustable thicknesses and scattering length densities (SLDs), and is referred to in SasView as the “core‐shell bicelle model.” The default version of the model was modified to use the number of molecules of lipids, MSPs, and waters as fit parameters, rather than directly fitting the SLDs of the disc layers. Instead, SLDs were calculated as a function of the layer volume, number of molecules in the layer, and the scattering lengths of each molecule. Waters of hydration of the lipid headgroups and MSPs were included in the molecular contents of the layers, and exchangeable hydrogens in the MSPs were taken into account (see Supplementary Material for exact definitions of the SLDs in terms of molecular contents and layer dimension). This effectively constrains the relationships between SLDs of the different layers to physically meaningful values: for example, ensuring that the number of lipid headgroups equals the number of tails; and in the case of multiple data sets, ensuring that the contrast of different layers is related in a meaningful way to the data collection conditions (i.e., SAXS vs. SANS, different solvent D2O levels).
Solvent SLDs were fixed to their calculated values during fitting. Initial guesses for fit parameters were refined to convergence without polydispersity, at which point polydispersity in the disc radius was introduced into the model. After fixing all other parameters, the polydispersity was optimized as a fit parameter. Finally, the DREAM optimizer21 was used to obtain final fit values and uncertainties, along with information about the pairwise correlations between parameters. Correlated parameters are accounted for by the DREAM algorithm when reporting uncertainties.
Supplementary Material
The file “Supplementary Information.docx” includes complete protein sequences for the ApoA‐I constructs used in these studies, as well as an SDS‐PAGE analysis of NLP purity after IMAC. In addition, it lists equations explicitly defining how the model scattering length densities were calculated from other model parameters. An accurately scaled schematic is also included, illustrating the final fit model geometry and SLDs.
Supporting information
Supporting Information
Acknowledgments
Access to NGB30 was provided by the Center for High Resolution Neutron Scattering, a partnership between the National Institute of Standards and Technology and the National Science Foundation under Agreement No. DMR‐1508249. This work benefited from the use of the SasView application, originally developed under NSF Award DMR‐0520547. SasView also contains code developed with funding from the EU Horizon 2020 programme under the SINE2020 project Grant No 654000. We thank Alexander Grishaev for assistance with SAXS instrumentation and helpful discussions. The Biomolecular Labeling Lab (BL2) at the Institute for Bioscience and Biotechnology Research (IBBR) was utilized for protein purification. Research was also supported by the National Institutes of Health under award numbers R21AI120925, R01CA155642 and R01GM117342. Part of this work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE‐AC52‐07NA27344.
Summary Statement: Embedding membrane proteins in nanolipoprotein particles (NLPs) is a method to maintain them in a soluble, functional form for biochemical studies. A recently developed method to assemble NLPs is via co‐translational cell‐free expression, a technique that is simpler in some cases than traditional assembly methods. In this work, we use small‐angle scattering to demonstrate that cell‐free expression produces NLPs that are structurally equivalent to traditionally produced NLPs.
Footnote
Certain trade names and company products are identified in order to specify adequately the experimental procedure. In no case does such identification imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the products are necessarily the best for the purpose.
References
- 1. Chromy BA, Arroyo E, Blanchette CD, Bench G, Benner H, Cappuccio JA, Coleman MA, Henderson PT, Hinz AK, Kuhn EA, Pesavento JB, Segelke BW, Sulchek TA, Tarasow T, Walsworth VL, Hoeprich PD (2007) Different apolipoproteins impact nanolipoprotein particle formation. J Am Chem Soc 129:14348–14354. [DOI] [PubMed] [Google Scholar]
- 2. Bayburt TH, Carlson JW, Sligar SG (1998) Reconstitution and imaging of a membrane protein in a nanometer‐size phospholipid bilayer. J Struct Biol 123:37–44. [DOI] [PubMed] [Google Scholar]
- 3. Cappuccio JA, Blanchette CD, Sulchek TA, Arroyo ES, Kralj JM, Hinz AK, Kuhn EA, Chromy BA, Segelke BW, Rothschild KJ, Fletcher JE, Katzen F, Peterson TC, Kudlicki WA, Bench G, Hoeprich PD, Coleman MA (2008) Cell‐free co‐expression of functional membrane proteins and apolipoprotein, forming soluble nanolipoprotein particles. Mol Cell Proteomics 7:2246–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Katzen F, Chang G, Kudlicki W (2005) The past, present and future of cell‐free protein synthesis. Trends Biotechnol 23:150–156. [DOI] [PubMed] [Google Scholar]
- 5. He W, Luo J, Bourguet F, Xing L, Yi SK, Gao T, Blanchette C, Henderson PT, Kuhn E, Malfatti M, Murphy WJ, Cheng RH, Lam KS, Coleman MA (2013) Controlling the diameter, monodispersity, and solubility of ApoA1 nanolipoprotein particles using telodendrimer chemistry. Protein Sci 22:1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He W, Felderman M, Evans AC, Geng J, Homan D, Bourguet F, Fischer NO, Li Y, Lam KS, Noy A, Xing L, Cheng RH, Rasley A, Blanchette CD, Kamrud K, Wang N, Gouvis H, Peterson TC, Hubby B, Coleman MA (2017) Cell‐free production of a functional oligomeric form of a Chlamydia major outer‐membrane protein (MOMP) for vaccine development. J Biol Chem 292:15121–15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blanchette CD, Segelke BW, Fischer N, Corzett MH, Kuhn EA, Cappuccio JA, Benner WH, Coleman MA, Chromy BA, Bench G, Hoeprich PD, Sulchek TA (2009) Characterization and purification of polydisperse reconstituted lipoproteins and nanolipoprotein particles. Int J Mol Sci 10:2958–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blanchette CD, Cappuccio JA, Kuhn EA, Segelke BW, Benner WH, Chromy BA, Coleman MA, Bench G, Hoeprich PD, Sulchek TA (2009) Atomic force microscopy differentiates discrete size distributions between membrane protein containing and empty nanolipoprotein particles. Biochim Biophys Acta 1788:724–731. [DOI] [PubMed] [Google Scholar]
- 9. Marty MT, Zhang H, Cui W, Blankenship RE, Gross ML, Sligar SG (2012) Native mass spectrometry characterization of intact nanodisc lipoprotein complexes. Anal Chem 84:8957–8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skar‐Gislinge N, Arleth L (2011) Small‐angle scattering from phospholipid nanodiscs: derivation and refinement of a molecular constrained analytical model form factor. Phys Chem Chem Phys 13:3161–3170. [DOI] [PubMed] [Google Scholar]
- 11. Skar‐Gislinge N (2014) Developing nanodiscs as a tool for low resolution studies of membrane proteins. PhD. Thesis, University of Copenhagen, Denmark.
- 12. Kynde SA, Skar‐Gislinge N, Pedersen MC, Midtgaard SR, Simonsen JB, Schweins R, Mortensen K, Arleth L (2014) Small‐angle scattering gives direct structural information about a membrane protein inside a lipid environment. Acta Cryst 70:371–383. [DOI] [PubMed] [Google Scholar]
- 13. Midtgaard SR, Pedersen MC, Kirkensgaard JJK, Sørensen KK, Mortensen K, Jensen KJ, Arleth L (2014) Self‐assembling peptides form nanodiscs that stabilize membrane proteins. Soft Matter 10:738–752. [DOI] [PubMed] [Google Scholar]
- 14. Luo J, Xiao K, Li Y, Lee JS, Shi L, Tan Y, Xing L, Holland Cheng R, Liu G, Lam KS (2010) Well‐defined, size‐tunable, multifunctional micelles for efficient paclitaxel delivery for cancer treatment. Bioconjug Chem 21:1216–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bayburt TH, Grinkova YV, Sligar SG (2002) Self‐assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett 2:853–856. [Google Scholar]
- 16. Glinka CJ, Barker JG, Hammouda B, Krueger S, Moyer JJ, Orts WJ (1998) The 30 m small‐angle neutron scattering instruments at the National Institute of Standards and Technology. J Appl Cryst 31:430–445. [Google Scholar]
- 17. Kline SR (2006) Reduction and analysis of SANS and USANS data using IGOR Pro. J Appl Cryst 39:895–900. [Google Scholar]
- 18. Nielsen SS, Toft KN, Snakenborg D, Jeppesen MG, Jacobsen JK, Vestergaard B, Kutter JP, Arleth L (2009) BioXTAS RAW, a software program for high‐throughput automated small‐angle X‐ray scattering data reduction and preliminary analysis. J Appl Cryst 42:959–964. [Google Scholar]
- 19. Dreiss CA, Jack KS, Parker AP (2006) On the absolute calibration of bench‐top small‐angle X‐ray scattering instruments: a comparison of different standard methods. J Appl Cryst 39:32–38. [Google Scholar]
- 20. Doucet M, Cho JH, Alina G, King S, Butler P, Kienzle P, Krzywon J, Jackson A, Richter T, Gonzales M, Nielsen T, Ferraz‐Leal R, Markvardsen A, Heenan R, Juhas P, Bakke A (2016) SasView 4.0. doi:10.5281/zenodo.159083.
- 21. Vrugt JA, ter Braak CJF, Diks CGH, Robinson BA, Hyman JM, Higdon D (2009) Accelerating Markov chain Monte Carlo simulation by differential evolution with self‐adaptive randomized subspace sampling. Int J Nonlinear Sci Numer Simul 10:273–290. [Google Scholar]
- 22. Gao T, Blanchette CD, He W, Bourguet F, Ly S, Katzen F, Kudlicki WA, Henderson PT, Laurence TA, Huser T, Coleman MA (2011) Characterizing diffusion dynamics of a membrane protein associated with nanolipoproteins using fluorescence correlation spectroscopy. Protein Sci 20:437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information