

Abstract
The dual‐specificity activity of the homeodomain interacting protein kinase 2 (HIPK2) is regulated by cis‐auto‐phosphorylation of tyrosine 361 (Y361) on the activation loop. Inhibition of this process or substitution of Y361 with nonphosphorylatable amino acid residues result in aberrant HIPK2 forms that show altered functionalities, pathological‐like cellular relocalization, and accumulation into cytoplasmic aggresomes. Here, we report an in vitro characterization of wild type HIPK2 kinase domain and of two mutants, one at the regulating Y361 (Y361F, mimicking a form of HIPK2 lacking Y361 phosphorylation) and another at the catalytic lysine 228 (K228A, inactivating the enzyme). Gel filtration and thermal denaturation analyzes along with equilibrium binding experiments and kinase assays performed in the presence or absence of ATP‐competitors were performed. The effects induced by mutations on overall stability, oligomerization and activity support the existence of different conformations of the kinase domain linked to Y361 phosphorylation. In addition, our in vitro data are consistent with both the cross‐talk between the catalytic site and the activation loop of HIPK2 and the aberrant activities and accumulation previously reported for the Y361 nonphosphorylated HIPK2 in mammalian cells.
Keywords: protein kinase, homeodomain interacting protein kinase 2 (HIPK2), posttranslational modification, activation‐loop phosphorylation
Abbreviations
- λ‐PPase
Lambda protein phosphatase
- DYRK
Dual‐specificity tyrosine phosphorylation–regulated kinase
- GST‐HIPK2K228
K228 mutated in A and fused to the GST
- GST
Glutathione S‐transferase
- HIPK2
Homeodomain interacting protein kinase 2
- HIPK2WT or WT
HIPK2 wild type
- HIPK2Y361 or Y361F
Y361 mutated in phenylalanine (F); HIPK2K228 or K228A, K228 mutated in alanine (A)
- Itu
5‐Iodotubercidin; KDom, kinase domain
- K228
lysine 228
- PurvA
Purvalanol A
- S/T or Ser/Thr
serine/threonine
- SB
SB203580
- Stauro
Staurosporine
- TBB
4,5,6,7‐tetrabromobenzimidazole
- TBID
1H‐imidazol‐2‐yl‐4,5,6,7‐tetrabromoisoindoline‐1,3‐dione
- Y or Tyr
tyrosine
- PY361
phosphorylated Y361
- Y361
tyrosine 361
Introduction
The homeodomain interacting protein kinase 2 (HIPK2) is a multidomain, tyrosine(Y)‐regulated serine/threonine (S/T) kinase mainly found in the nucleus. It belongs to the HIPK family, classified as a subfamily of the CMGC kinases.1, 2 By controlling gene expression or protein stability through phosphorylation of specific transcription factors or regulators, HIPK2 contributes to a vast array of biological processes that range from embryonic differentiation and development to cell response to DNA‐damaging agents.3, 4, 5 Dysfunctions of HIPK2 result in the development of diseases and abnormalities linked to increased cell proliferation, as in cancer or fibrosis.6, 7, 8 The pleiotropic function of HIPK2 requires a tight control of its activity, achieved through the complex architecture of its functional domains and a “code” of posttranslational modifications that include ubiquitylation, sumoylation, acetylation, and phosphorylation.9, 10, 11 Among this code, particular attention has been devoted to understand the regulatory effects of phosphorylation in the activation loop, located in the kinase core and conserved among members of the HIPK family.9, 10, 12, 13, 14 Single or multiple phosphorylation events at the activation loop occurring via trans‐ or cis‐auto‐phosphorylation induce conformational changes and regulate the activity of many kinases.15, 16 More specifically, in the dual‐specificity members of the CMGC group of kinases, a cis‐auto‐phosphorylation on specific Y at the activation loop occurs on translational intermediates that form during ribosomal biosynthesis and turns kinase specificity toward S/T residues on substrates.17, 18, 19, 20 The dual‐specificity tyrosine phosphorylation‐regulated kinase 1A (DYRK1A) is the prototype of these dual‐specificity Y phosphorylation‐regulated kinases20, 21, 22 and HIPK2 has been recently shown to be activated through a similar mechanism. Actually, HIPK2 undergoes cis‐auto‐phosphorylation at Y361 (Y354) in the activation loop on a translational intermediate that shows different and transient properties with respect to the mature kinase, i.e., recognition and phosphorylation of a specific Y rather than canonical S/T substrates.9, 10 Similarly to the homologous DYRK1A, this event of phosphorylation in HIPK2 does not determine the inactive‐to‐active transition, but the switch of the mature kinase specificity toward S/T residues of target proteins and HIPK2 itself. Notably, HIPK2 auto‐phosphorylates on S/T residues both inside and outside its activation loop and this activity is also maintained by the isolated kinase domain.9, 10
The relevance of Y361‐cis‐auto‐phosphorylation has been confirmed by the fact that its absence promotes strong Y‐off‐target cis‐auto‐phosphorylation that coupled to a different distribution of the surface charge results in altered activity toward HIPK2 partners. Moreover, pharmacological inhibition of Y361 phosphorylation or the usage of Y361F mutant have been shown to promote a strong delocalization of HIPK2 into the cytoplasm, leading to accumulation into aggresomes.9, 10 Of relevance, this behavior has been extended to each member of the HIPK family23 while a comparable cytoplasmic accumulation has been described for HIPK2 in pathological conditions, such as in some human cancers.10, 24
While a detailed description of the cellular effects of the absence of Y361 phosphorylation has been provided and conformational changes in the regulatory tracts of the kinase domain linked to Y361 phosphorylation have been hypothesized to control HIPK2 dual activity,9, 10 an in vitro characterization is still lacking. Here, we perform an in vitro evaluation of the stability, oligomerization, and activity of the wild type kinase domain (KDom) of HIPK2 (HIPK2WT) in comparison with two phospho‐defective mutants, HIPK2Y361F and HIPK2K228A. The Y361F mutant mimics HIPK2 in its Y361‐unphosphorylated, not mature intermediate, i.e., an aberrant/not‐fully‐active HIPK2, while the HIPK2K228A mutant is mainly inactive and unphosphorylated, given the substitution of the catalytic lysine residue.25, 26 The auto‐phosphorylation pattern of these kinase domains produced in Escherichia coli, as well as of the relative full‐length proteins, has been previously mapped by mass spectrometry.9, 10 As reported, only HIPK2WT is phosphorylated at Y361 (Y354) while Y361F mutation abolishes the possibility for that position to be phosphorylated, and other, though aberrant, Y sites become phosphorylated. Lastly, K228A mutation inactivates the enzyme leading to the absence of phosphorylation. Our in vitro characterization of these HIPK2 KDoms—wild type and phospho‐defective mutants—confirms the cross‐talk between the catalytic site and the activation loop of HIPK2, highlights the existence of different conformations possibly linked to Y361 phosphorylation, and offers a possible explanation for the aberrant functions and the aggresome formation by the Y361 nonphosphorylated HIPK2 observed in mammalian cells.
Results and Discussion
Y361‐phosphorylation affects HIPK2 oligomerization
Recombinant HIPK2 KDoms (HIPK2WT, HIPK2Y361F, and HIPK2K228A) were purified by GST‐ affinity chromatography and, after proteolytic removal of the tag, analyzed by SDS‐PAGE to evaluate protein purity (Supporting Information Fig. S1). The phosphorylation status was assessed by Western blotting (WB) with anti‐phosphoSer/Thr and anti‐phosphoTyr antibodies, before and after Lambda phosphatase (λ‐PPase) treatment [Fig. 1(A)].
Figure 1.
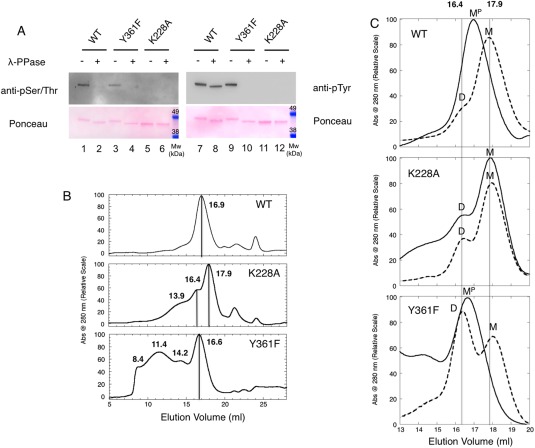
Effects of phosphorylations on HIPK2 KDom oligomerization. (A) Overall phosphorylation and dephosphorylation of HIPK2WT, HIPK2Y361F, and HIPK2K228A. Samples not treated and treated with λ‐PPase were analyzed by immunoassay using both anti‐pSer/Thr and anti‐pTyr antibodies (upper panels). Ponceau staining is also shown as loading control (lower panels). The band shift to a faster mobility of dephosphorylated samples is indicative of the removal of the surface phosphates. (B) Chromatographic profiles. SEC of murine HIPK2WT (WT), HIPK2K228A (K228A), HIPK2Y361F (Y361F). Elution volume is reported at the top of the corresponding peak, straight lines were added to highlight the most abundant eluted species. The minor peaks present in all chromatograms at longer retention times are consistent with the presence of a minute amount of contaminants as shown in Figure S1 that reports an SDS‐PAGE of the more than 90% pure HIPK2 KDom samples. (C) λ‐Phosphatase Assay. SEC of KDom murine HIPK2WT (WT), HIPK2K228A (K228A), and HIPK2Y361F (Y361F). The overlay of the relevant region of the chromatographic profiles of the samples treated (dashed lines) and untreated (solid lines) with λ‐PPase is reported. Once treated, HIPK2WT and HIPK2Y361F elute in two peaks at 16.4 mL and 17.9 mL (straight lines are reported to guide the eyes). M = monomer, MP = monomer with phosphorylations, D = dimer
Size exclusion chromatography (SEC) was first performed on HIPK2WT and HIPK2K228A to assess the overall effect of phosphorylation on the oligomeric state of the kinase domain. Analysis of chromatograms [Fig. 1(B), upper and middle panels] showed that phosphorylated HIPK2WT [Fig. 1(A), lanes 1 and 7] elutes predominantly as monomer (M) (Mw∼ 45.6 kDa) in a peak centred at 16.9 mL, a volume typical of a 40 kDa protein [see also Supporting Information Fig. S2(A)], and confirmed by ultra‐centrifugation (UC) analysis (Supporting Information Table S1). Conversely, the nonphosphorylated HIPK2K228A [Fig. 1(A), lanes 5 and 11] mainly elutes at 17.9 mL with a smaller peak at 16.4 mL, that UC respectively assigned to monomeric and dimeric (D) forms (Supporting Information Table S1). The elution profile also shows an enlarged band centred at a volume of 13.9 mL that suggests the presence of multimeric species. The shape of the elution profiles and the difference between HIPK2WT and HIPK2K228A were observed reproducibly in more than five independent purifications.
To further investigate the effect of phosphorylation on the different elution profiles, HIPK2WT and HIPK2K228A were treated for 30 minutes at 310 K with λ‐PPase or just incubated for 30 minutes at 310 K before SEC. As shown in Figure 1(A), removal of phosphates from HIPK2WT was complete for S/T residues (compare lanes 1 and 2) and, differently to what previously reported on isolated KDom, it was only partial for Y residues (compare lanes 7 and 8), most likely Y361, which is known to be resilient to dephosphorylation in mature full‐length protein.9, 10, 20 The conditions herein explored, that do not include detergents, most likely preserve the fold typical of the mature full‐length protein in the isolated KDom of HIPK2, resulting in a partially buried, not accessible, phosphorylated Y361 residue.
Of note, the electrophoretic mobility of the λ‐PPase treated HIPK2WT became similar to that of the nonphosphorylated HIPK2K228A (compare lanes 1 and 7 with 2 and 8 and 5 and 11, both immunoblot and Ponceau panels) suggesting a negligible contribution of the residual phosphates. Subsequently, the samples were injected into the SEC column and the relative chromatograms recorded before (solid lines) and after (dashed lines) λ‐PPase treatment are compared in Figure 1(C).
As expected, only minor differences were observed on the nonphosphorylated HIPK2K228A before and after λ‐PPase treatment [Fig. 1(C), middle panel]; these differences were presumably linked to protein precipitation, more relevant for higher oligomeric species [Supporting Information Fig. S2(B)]. In contrast, the removal of phosphate groups from HIPK2WT yielded a chromatographic profile [Fig. 1(C), upper panel] that resembles the one from HIPK2K228A, showing a shift of the peak at 17.9 mL and the appearance of a minor shoulder at 16.4 mL. These results indicate that the difference in the elution rate from 16.9 to 17.9 mL of the monomeric phosphorylated (MP) and nonphosphorylated (M) species of HIPK2WT are linked to phosphorylation‐dependent variations of the interaction with the column matrix. In addition, the minor shoulder at 16.4 mL suggests that a small population of dimers appears in the HIPK2WT after its dephosphorylation, though to a lesser amount than observed for HIPK2K228A, suggesting that phosphorylation at Y361, still present after λ‐PPase treatment in HIPK2WT, might prevent dimer formation.
To evaluate the contribution of Y361 phosphorylation on these results, SEC analyzes were performed on the HIPK2Y361F KDom before and after λ‐PPase treatment. Consistent with our previous mass‐spectrometry data,10 WB analyzes showed that HIPK2Y361F is phosphorylated on both S/T and Y residues [Fig. 1(A), compare lines 3 with 4 and 9 with 10]. Treatment with λ‐PPase completely dephosphorylated both types of sites supporting that Y361 is the site resilient to dephosphorylation [Fig. 1(A), compare lines 8 and 10]. SEC [Fig. 1(B), lower panel] shows that untreated HIPK2Y361F elutes with a main peak at 16.6 mL, that UC assigned to a monomer (Supporting Information Table S1). In addition, we reliably observed a peak in the void volume of the column (∼ 8.4 mL) and a few peaks at volumes ascribable to different oligomeric species [Fig. 1(B); Supporting Information Fig. S2(A)], indicating a tendency of the HIPK2Y361F to form high molecular weight oligomers. The elution profile of the λ‐PPase treated HIPK2Y361F [Fig. 1(C), lower panel] constantly showed loss of higher oligomeric species [Supporting Information Fig. S2(B)] and the appearance of a major 17.9 mL (M) and a minor 16.4 mL (D) peaks, corresponding to the nonphosphorylated monomers and dimers observed in the HIPK2K228A and in the λ‐PPase treated HIPK2WT [Fig. 1(C), middle and upper panels]. These findings support a role in HIPK2 KDom in dimer formation of (a) overall phosphorylation, (b) phosphorylation at Y361, and (c) the Y to phenylalanine substitution at position 361.
Concerning the higher oligomeric species (<16.4 mL) present in HIPK2K228A and to a significantly larger extend in HIPK2Y361F, we found that they are endowed with low temperature stability, as they disappear upon incubation at 310 K also in the control experiments without λ‐PPase [Supporting Information Fig. S2(B)]. This result precludes the assessment of the effect of dephosphorylation in the higher oligomeric formation. However, their prevalence in the HIPK2Y361F might be compatible with the previous observation in cellular systems that full‐length HIPK2Y361F mutant and HIPK2WT with nonphosphorylated Y361, obtained by cell treatment with Purvalanol A (PurvA) during protein translation of transfected HIPK2‐expressing plasmid, accumulate into cytoplasmic aggresomes.10
Thermodynamic stability
To assess whether mutations at Y361 and K228 and their overall phosphorylation affect the stability of HIPK2, circular dichroism (CD) thermal denaturation experiments were performed on HIPK2WT and mutant KDoms, following the CD signal at λ = 222 nm. The change in heat capacity, Δc p, was estimated indirectly by following the dependence of the enthalpy change, ΔH Tm, and the melting temperature, T m, at different pH values (Supporting Information Fig. S3), according to Privalov and Fersht.27, 28 The value of Δc p is related to the amount of hydrophobic area exposed to solvent upon unfolding and is constant for a given protein. For HIPK2WT KDom we estimated Δc p = 6099.00 ± 0.02 cal mol−1K−1, compatible with what expected for a globular protein of about 400 folded residues.29 This value was then used as fixed parameter to determine the T m of unfolding for the three HIPK2 KDom variants studied.29
The denaturation curves obtained for HIPK2WT and mutant KDoms closely approach a two‐state transition (Fig. 2). Though several conditions were explored, thermal‐induced denaturation of HIPK2 KDom always resulted in irreversible formation of protein aggregates at high temperature (> 350 K). However, measuring the unfolding parameters for HIPK2WT at different rates of heating yielded similar readouts, suggesting that the kinetics of aggregation does not affect the results.30
Figure 2.
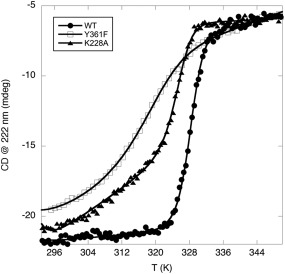
Thermal stability. Thermal denaturation profiles of HIPK2WT (WT, full dots), HIPK2Y361F (Y361F, empty squares), and HIPK2K228A (K228A, full triangles)
Thus, we compared the T m of the three KDoms. As shown in Figure 2, HIPK2WT shows a T m equal to 328.2 ± 0.1 K, HIPK2K228A a T m equal to 325.2 ± 0.1 K, while the HIPK2Y361F a T m equal to 317.0 ± 0.4 K. Therefore, both mutations decrease the HIPK2 KDom stability, but the semi‐conservative mutation at the regulatory Y361, that also affects the overall phosphorylation pattern, introduces a more substantial destabilization of the KDom than the inactivating, nonphosphorylated K228A mutation.
ATP‐competitor binding: Evidence for the cross‐talk between the catalytic pocket and the activation loop
Structural plasticity of the activation loop influences the activity of protein kinases and regulates their function.15, 16 The arrangement that activation loop adopts upon phosphorylation at specific residues allows the interface between N‐ and C‐ lobes of the kinase domain to assume its active conformation, due to a correct organization of critical residues, including the conserved catalytic lysine (e.g., K228 in HIPK2). Consequently, failure in phosphorylation at the activation loop results in inactivation/deregulation of the enzyme due to an incorrect loop conformation.16
To highlight structural differences in the HIPK2 KDom linked to Y361‐phosphorylation, we compared static fluorescence and circular dichroism spectra of HIPK2WT, HIPK2K228A, and HIPK2Y361F KDoms, but negligible differences were detected with this analysis (Supporting Information Fig. S4). Therefore, in a further effort to probe different conformational states of the KDom associated with Y361‐phosphorylation, we measured the binding of the three HIPK2 KDom variants with chemically different ATP‐competitors, i.e., 4,5,6,7‐tetrabromobenzimidazole (TBB), 1H‐imidazol‐2‐yl‐4,5,6,7‐tetrabromoisoindoline‐1,3‐dione (TBID), Purvalanol A (PurvA), SB203580 (SB), staurosporine (Stauro) and 5‐iodotubercidin (Itu) [Fig. 3(A)], that directly interact with the catalytic pocket of the enzyme in the site that is normally occupied by a molecule of ATP.22 Structurally different compounds can in principle affect to a different extent binding affinity and activity of Y361‐phosphorylated and unphosphorylated HIPK2 if the binding sites of the two forms are structurally different.20, 22 Equilibrium binding experiments were performed by measuring the quenching of the intrinsic protein fluorescence upon ligand binding at the active site. The observed saturation curves display a simple hyperbolic transition that accounts for a single binding event [Fig. 3(B); Supporting Information Fig. S5]. The extrapolated dissociation constants K D estimated for HIPK2WT KDom are consistent with previously reported IC50 (Supporting Information Fig. S6 and references31, 32, 33), and show that Stauro and TBB have the highest affinity for HIPK2WT, while Itu and SB yield the lowest ones (Table 1).
Figure 3.
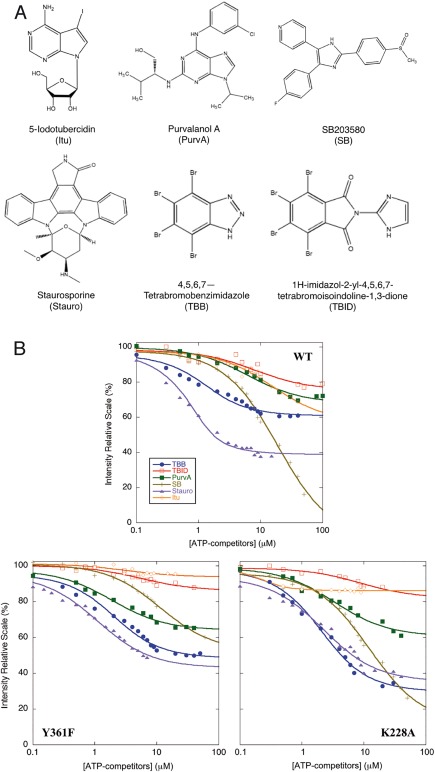
Binding of ATP‐competitors to HIPK2WT and mutants. (A) Chemical structures of ATP‐competitors. The structures of Itu, PurvA, SB, Stauro, TBB and TBID are reported in alphabetic order. (B) Equilibrium binding experiments. The change in the intrinsic fluorescence of the protein that occurs upon ATP‐competitor binding to HIPK2WT (WT), HIPK2Y361F (Y361F), and HIPK2K228A (K228A) is shown. Protein fluorescence intensities at 324, 330, 340, and 342 nm depending on the compound, as detailed in Material and Methods, are reported as function of ATP‐competitor concentration on a logarithmic scale. 100% corresponds to the fluorescence signal of the unbound protein. Determined K D are reported in Table I
Table 1.
Binding Parameters of HIPK2WT, HIPK2Y361F, and HIPK2K228A
| HIPK2WT | HIPK2Y361F | HIPK2K228A | ||||
|---|---|---|---|---|---|---|
| K D (μM) | ΔGbinding (kcal mol−1) | K D (μM) | ΔGbinding (kcal mol−1) | K D (μM) | ΔGbinding (kcal mol−1) | |
| TBB | 0.8 ± 0.2 | −7.86 ± 0.14 | 1.5 ± 0.3 | −7.51 ± 0.11 | 1.8 ± 0.2 | −7.41 ± 0.06 |
| TBID | 8.0 ± 2.8 | −6.57 ± 0.20 | 7.6 ± 2.0 | −6.60 ± 0.15 | 8.7 ± 5.0 | −6.53 ± 0.32 |
| PurvA | 6.2 ± 1.0 | −6.71 ± 0.09 | 1.9 ± 0.3 | −7.38 ± 0.09 | 3.4 ± 0.7 | −7.05 ± 0.12 |
| SB | 18.7 ± 1.7 | −6.10 ± 0.05 | 11.4 ± 0.9 | −6.37 ± 0.04 | 12.0 ± 1.2 | −6.35 ± 0.01 |
| Stauro | 0.50 ± 0.03 | −8.12 ± 0.03 | 1.1 ± 0.2 | −7.68 ± 0.10 | 2.0 ± 0.4 | −7.35 ± 0.11 |
| Itu | 15.6 ± 5.0 | −6.20 ± 0.18 | 3.1 ± 2.6 | −7.10 ± 0.47 | 0.3 ± 0.1 | −8.39 ± 0.18 |
Next, K D values were determined also for Y361F and K228A, reported in Table 1. With respect to the wild type, we observed differences of K D for the binding of PurvA and Itu to HIPK2Y361F and for Itu binding to HIPK2K228A highlighting an effect of these mutations on the structural arrangement of the HIPK2 active site.
It is worth noting that whereas mutation at K228A in the catalytic pocket affects a residue directly involved in ATP binding [Fig. 4(A), site A], mutation at Y361 introduces a perturbation on the activation loop [Fig. 4(A), site B] that is spatially far from the binding site. Indeed, using the DYRK1A KDom in complex with its inhibitor INDY as structural template, we built a model of HIPK2 that allow us to estimate the distance between the catalytic pocket and the activation loop to be above 20 Å [Fig. 4(A)].35 Despite this distance, if sites A and B are structurally coupled, rearrangements of the activation loop (site B) due to phosphorylation at Y361 should affect the binding at the catalytic pocket (site A). Thus, to assess whether the two sites are coupled, we calculated the change in free energy (ΔΔGbinding) for ATP‐competitor binding after mutation at Y361 and K228 (Table 2), using the K D constants determined with the equilibrium binding experiments (Table 1). If the two sites are energetically linked, a perturbation in the binding of ATP‐competitors induced by mutation in site A (i.e., K228A in the catalytic pocket) should correlate with binding perturbations triggered by mutation in site B (i.e., Y361F at the activation loop) [Fig. 4(A)]. As shown in Figure 4(B), a cross‐correlation plot of the ΔΔGbinding values obtained with HIPK2Y361F and with HIPK2K228A (Table 2) reveals a significant correlation between the two sets of binding energies (returning P value of 0.017). This confirms that the two sites are energetically connected and provides a physico‐chemical evidence of a linkage between the Y361 phosphorylation at the activation loop and the conformation of the active site.
Figure 4.
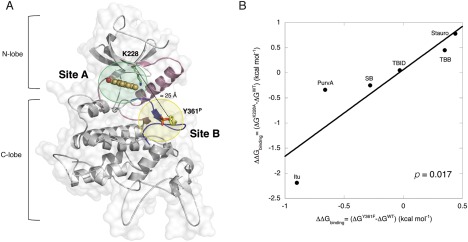
The cross‐talk between the catalytic pocket and the activation loop. (A) Illustrative structural model of the HIPK2WT kinase domain. The structure of the homologous kinase DYRK1A (overall homology 31%) in complex with INDY (orange, ball‐and‐stick mode, PDB ID 3ANQ34) was used as template to model the structure of HIPK2 using the Swiss‐Model server.42 The protein scaffold is represented by light gray ribbons. The activation loop is colored in dark‐blue and is highlighted by a light‐yellow circle (Site B) with phosphorylated Y361 (Y361P) shown in yellow sticks with the phosphate group in red. Helix Cα, the catalytic loop and β3 strand are given in light pink ribbons. K228 is represented in green sticks. The active site of the protein, where ATP‐competitors bind, is highlighted by a light‐green circle (Site A). A dashed line indicates the distance between the Cα carbons of K228 and Y361P. The image was generated using PyMOL (http://pymol.sourceforge.net). (B) Cross‐correlation plot between the free energy of binding parameters for Y361F and K228A mutants of HIPK2. ATP‐competitor binding K D determined at 293 K were used to calculate the change in the free energies upon binding ΔΔGbinding= (ΔGmutant ‐ ΔGwild type) induced by mutation on Y361F and K228A, considering ΔGbinding= RT ln (K D/c 0), with c 0 =1 mol/L. ΔΔGbinding values obtained from K228A mutation are plotted versus the ones obtained in mutant Y361F. The line is the best fit to a linear equation, returning R= 0.89 and R 2 = 0.79; the P value, determined by PRISM 6.0 program, indicates the probability that the two variables are not correlated (P > 0.05 means no correlation)
Table 2.
Changes in ATP‐Competitor Binding Free Energy (ΔΔGbinding) for Single Mutant at Positions Y361 (ΔGY361F ‐ ΔGWT) and K228 (ΔGK228A‐ΔGWT)
|
ΔΔGbinding = (ΔGY361F‐ΔGWT) (kcal mol−1) |
ΔΔGbinding = (ΔGK228A‐ΔGWT) (kcal mol−1) |
|
|---|---|---|
| TBB | 0.35 ± 0.18 | 0.45 ± 0.15 |
| TBID | −0.03 ± 0.25 | 0.05 ± 0.38 |
| PurvA | −0.66 ± 0.13 | −0.34 ± 0.15 |
| SB | −0.28 ± 0.07 | −0.25 ± 0.05 |
| Stauro | 0.44 ± 0.11 | 0.78 ± 0.12 |
| Itu | −0.90 ± 0.50 | −2.19 ± 0.25 |
Kinase activity in the presence of ATP‐competitors highlights the HIPK2 KDom structural plasticity
Lastly, we evaluated the effects of the different ATP‐competitors on the kinase activity of HIPK2WT and its Y361F mutant that, in contrast to the kinase‐defective K228 mutant, was shown to maintain residual and aberrant kinase activity.9, 10 In vitro kinase assays were performed in the presence of [γ‐32 P]‐ATP using myelin binding protein (MBP) and the inactive HIPK2K228A KDom fused to the GST (GST‐HIPK2K228A) as substrates. Even though the HIPK2Y361F KDom showed ∼2‐fold lower kinase activity than HIPK2WT (kcat in Supporting Information Fig. S7), as originally reported for the full‐length proteins,9, 10 we were still able to detect differential effects induced by the addiction of ATP‐competitors to the kinase reactions (Fig. 5, compare ATP‐competitor treated vs. untreated samples).
Figure 5.
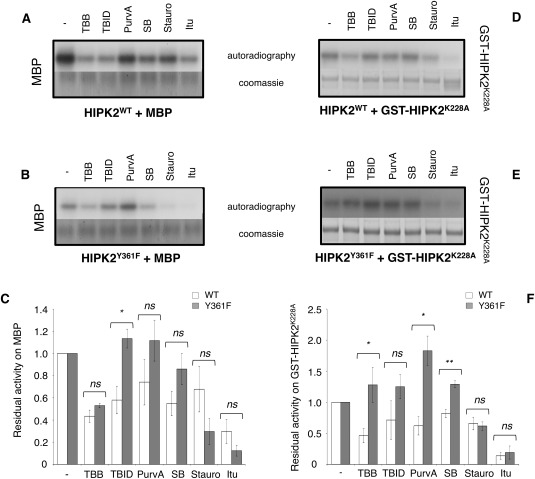
Effect of ATP‐competitors on kinase activity on substrates. Representative autoradiography for HIPK2WT (upper panels, A and D) and HIPK2Y361F (middle panels, B and E) and each substrate (MBP and GST‐HIPK2K228A) are shown. The relative Coomassie blue staining is reported. For MBP the same amount of substrate and the same length of exposure time are shown, for GST‐ HIPK2K228A the substrate was loaded in different amount while the exposure time is the same. Histograms at the bottom (panels C and F) report the residual kinase activity, expressed as a ratio with activity in the absence of ATP‐competitors (‐), of HIPK2WT (WT, white bars) and HIPK2Y361F (Y361F, gray bars) as mean of at least three independent experiments (mean ± Standard Error). ** P < 0.01; * P < 0.05; ns P > 0.05 by Student t test
To highlight different behaviors of HIPK2WT and HIPK2Y361F in response to ATP‐competitors, semi‐quantitative measurement of the residual activity of the KDoms in the presence of the competitors was calculated by normalizing to one the densitometry values of both HIPK2WT and HIPK2Y361F untreated samples [histograms in Fig. 5(C,F)]. Comparison of the residual activities measured from three independent experiments shows that Stauro and Itu similarly inhibited both KDoms, and thus, were not informative for our question. In contrast, TBID, PurvA, and SB affected the activity of HIPK2WT but not of HIPK2Y361F though not always with statistically significant differences [Fig. 5(C,F)], supporting the existence of different conformations between the two KDoms. Surprisingly, TBB similarly inhibited the activity of both KDoms when MBP was the substrate [Fig. 5(C)], while inhibited only HIPK2WT when GST‐HIPK2K228A was used as substrate [Fig. 5(F)], suggesting that ATP‐competitors might differentially affect the kinase activity also depending on the substrate. Therefore, additional kinase assays were performed using p53 as alternative HIPK2 substrate [Fig. 6(A)]. A comparison of the residual kinase activities of HIPK2WT and HIPK2Y361F on the three substrates, MBP, GST‐HIPK2K228A, and p53 [Fig. 6(B)] confirmed that the two KDoms are differently inhibited by different ATP‐competitors also depending on the substrate. Indeed, TBB inhibits HIPK2WT when the substrates are MBP and GST‐HIPK2K228A, whereas it decreases phosphorylation by HIPK2Y361F enzyme of MBP. Finally, TBID induces a pronounced decrease of phosphorylation by HIPK2Y361F when p53 is the substrate.
Figure 6.
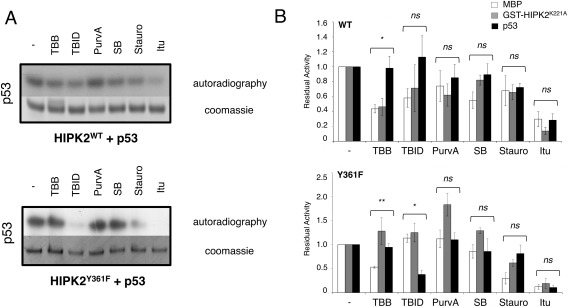
Inhibition activity is substrate dependent. (A) Effect of ATP‐competitors on kinase activity on p53. Representative autoradiography for HIPK2WT (upper panel) and HIPK2Y361F (lower panel) acting on p53 are shown. The relative Coomassie blue staining is also reported. (B) ATP‐competitors affect HIPK2 KDom activity depending on the substrate. Histograms report the residual kinase activity, expressed as a ratio with activity in the absence of ATP‐competitors (‐), of HIPK2WT (top) and HIPK2Y361F (bottom) as mean of at least three independent experiments (mean ± Standard Error). White bars represent kinase residual activity using MBP as nonspecific substrate, gray and black represent respectively the data obtained using GST‐HIPK2K228A and p53 as specific substrates of HIPK2. ** P < 0.01; * P < 0.05; ns P > 0.05 by One‐Way ANOVA (GRAPHPAD PRISM 6.0)
These observations suggest that the conformation of the activation loop, that directly interacts with the kinase targets,16 can change depending on the nature of the substrate thus affecting the structure of the HIPK2 catalytic pocket as a consequence of the cross‐talk between the two sites. Structural changes of the catalytic residues might alter the interaction with the ATP‐competitive compounds, thus modulating their inhibitory activity. Furthermore, in analogy with DYRKs,20, 22 the different sensitivity of HIPK2WT and HIPK2Y361F to structurally diverse ATP‐competitive compounds supports the existence of different conformations of the HIPK2 catalytic site. Based on the semi‐conservative nature of the Y361F mutation, these different conformations can be reasonably ascribed to the phosphorylation state of Y361.
Finally, it is of interest to observe that when GST‐HIPK2K228A is the substrate some compounds such as TBID, PurvA and SB, rather than inhibiting seem to work as activators of the Y361F variant [Fig. 5(D–F)]. Differently from the other substrates, GST‐HIPK2K228A can bind the ATP‐competitors. Therefore, we hypothesize that binding of these compounds to GST‐HIPK2K228A might induce a conformational change in the area surrounding the ATP binding site that could increase solvent exposure of some phosphorylatable residues (i.e., Tyr), making them more accessible and available for phosphorylation by HIPK2Y361F. In other words, the presence of the ATP competitor bound to the GST‐HIPK2K228A active site makes some residues a better substrate for HIPK2Y361F. This phenomenon does not occur with wild type HIPK2 and it represents another evidence that HIPK2WT and HIPK2Y361F are different in structure.
Conclusions
HIPK2 is an interesting molecular target for selective activation or inactivation, depending on the pathology and on the cellular context.4, 7, 36, 37 Nevertheless, the complexity of the system jeopardizes the understanding of how specific modifications contribute to its regulation. Recently, the role of residues located in the kinase domain and in the activation loop that are phosphorylated by the protein itself or by other kinases in response to specific stress signals has been investigated.12, 13, 14, 38 Among those, the Y361 cis‐auto‐phosphorylation that regulates the HIPK2 dual activity has been described as a common mechanism for all members of the HIPK family and a detailed description of the cellular effects of its failing has been provided.9, 10, 23
In this work, we studied the effect of Y361 phosphorylation utilizing the wild type KDom of HIPK2 and its Y361F and K228A mutants. Our data indicate that phosphorylation at Y361 affects the structure of KDom in the regulatory and catalytic tracts. This is consistent with a scenario where activation loop and catalytic pocket are in communication and explore in concert different conformations, possibly also depending on the target protein. Indeed, kinase assays suggest that ATP‐competitive compounds can exert inhibition in a substrate‐specific manner. In addition, we report that the stability and the oligomerization state of the KDom are influenced by the extent of protein phosphorylation and by the amino acid at position 361. Indeed, mutation at Y361 decreases the HIPK2 KDom stability and increases its tendency to form high molecular weight oligomers, resulting in a less active form of the enzyme.
Taken together our data suggest that the absence of phosphorylation at Y361 changes the HIPK2 structure, perturbs its stability, decreases its activity and affects its capability to interact with partners and with itself, increasing its tendency to form aggregates. Therefore, with the caveat that our in vitro observations are limited to the isolated kinase domain, these results offer a biochemical explanation for the abnormal behavior observed in cellular systems with the Y361‐unphosphorylated full‐length form, such as altered activities and specificity on substrates, and accumulation in cytoplasmic aggresomes.9, 10
Materials and Methods
Protein cloning and site‐directed mutagenesis: Expression and purification
The plasmid pGEX‐6P‐1, containing the nucleotide sequence coding for murine HIPK2 wild type (Isoform 1, Reference Sequence: NP_034563.2 (14)) kinase domain (KDom, 165–564) was utilized (HIPK2WT) and expressed in frame with a Glutathione S‐transferase (GST) tag at its N‐terminus, and a PreScission cleavage after the tag.10 Site‐directed mutagenesis was performed by using the QuickChange Lightning kit (Agilent Technologies). Y361 was mutated into phenylanaline (HIPK2Y361F) and K228 was mutated into alanine (HIPK2K228A). Recombinant HIPK2WT KDom and mutants were overexpressed in E. coli and purified as detailed in Supplementary Material. Quality and quantity of purified proteins were evaluated by SDS‐PAGE (Supporting Information Fig. S1) and UV/visible spectra using the theoretical ɛ 280 nm= 48820 M−1 cm−1. Plasmid containing human p53 full‐length quadruple mutant M133L/V203A/N239Y/N268D (p53), N‐terminus fused to a six His‐tag, a lipoyl domain and TEV cleavage site, was expressed in E. coli BL21 (DE3) strain and purified as described previously,39 without removing the tag (Mw 60 kDa). Purified Myelin Basic Protein (MBP, Mw 20 kDa) from mouse was obtained by Sigma Aldrich (St. Louis, Missouri).
λ‐phosphatase assay
Twenty micrograms of HIPK2WT KDom and mutants were incubated at 310 K for 1 hour in the absence and in the presence of 1 μL (400 units) of Lambda protein phosphatase (λ‐PPase, New England BioLabs). Final products were loaded into a BioFox 17/1200 SEC column (Knauer‐Berlin, Ge) coupled with UV‐vis absorbance detector (Smartline 2520, Knauer‐ Berlin, Ge), and eluted in isocratic mode in 50 mM Tris⋅HCl, 300 mM NaCl, 5 mM Tris(2‐carboxyethyl)phosphine (TCEP), 5% Glycerol, pH 8.0. Elution profile was followed at λ = 280 nm at room temperature (RT). Samples treated and not treated with λ‐PPase were analyzed by Western Blot using both anti‐pTyr (Clone 4G10, Millipore) and anti‐pSer/Thr (Cell Signaling Technology) antibodies. Ponceau staining was used as loading control. Differently to previously reported methods for λ‐PPase treatment,9, 10 no detergent was added to the reaction buffer.
CD‐ thermal denaturation
CD thermal denaturation experiments on HIPK2WT KDom and mutants (4 μM) were monitored at 222 nm using a Jasco spectropolarimeter, with heating from 293 to 363 K at a rate of 1 K min−1 in 50 mM potassium phosphate, 150 mM NaCl, 0.4 M Na2SO4, pH 8. At temperature over 350 K, a second transition was observed due to protein aggregation and precipitation. However, measuring the unfolding parameters for HIPK2WT KDom at different rates of heating returned the same apparent unfolding parameters, thus showing that kinetics of aggregation does not affect the results.30 Thermal denaturation was analyzed by fitting the transition region data from 293 to 350 K to a two‐state model.27 Heat capacity change upon thermal denaturation of the wild type, Δc p, has been estimated as described in Supporting Information Figure S3.28
Equilibrium binding experiments
Equilibrium binding experiments of HIPK2WT, HIPK2Y361F, and HIPK2K228A KDom to a set of ATP‐competitors, i.e., 4,5,6,7‐tetrabromobenzimidazole (TBB), 1H‐imidazol‐2‐yl‐4,5,6,7‐tetrabromoisoindoline‐1,3‐dione (TBID), Purvalanol A (PurvA), SB203580 (SB), 5‐Iodotubercidin (Itu), and Staurosporine (Stauro), were performed by measuring quenching of the intrinsic fluorescence of the protein that occurs upon binding. Emission spectra were recorded between 300 and 400 nm with an excitation wavelength of 280 nm using a Fluoromax spectrofluorimeter (Jobin Yvon, Edison, NJ). With the exception of TBID that was a kind gift of Prof. C. Kunick, Braunschweig (Germany), all compounds were purchased by Sigma Aldrich. ATP‐competitor stock solutions were prepared at concentrations ranging from 10 to 40 mM in dimethyl sulfoxide (DMSO). Equilibrium binding experiments were performed at 295 K in 50 mM Hepes and 200 mM NaCl, pH 7.5, using a constant concentration of protein (typically ranging between 0.3 and 1.5 μM, see also Supporting Information Fig. S2), and varying ligand concentration. The final concentration of DMSO was kept below 1%. Since inhibitor absorbance at 280 nm is not negligible, we applied a mathematical model for correction of the inner filter effect to the measured fluorescence intensities:
| (1) |
where i c is the corrected and i m the observed fluorescent intensities as a function of ATP‐competitor concentration (I), and A(λ ex), and A(λ em) are respectively their absorptions at the wavelengths of excitation and emission.40 Each titration curve was fitted to a hyperbolic function and the calculated dissociation constant (K D) was estimated using Kaleidagraph software, by plotting corrected intensities at 324 nm for PurvA, 330 nm for Itu, 340 nm for SB, and 342 nm for TBB, TBID, and Stauro, depending on the compound optical properties, as a function of ATP‐competitor concentration. Since the K D for HIPK2WT binding to TBB and Stauro, and for HIPK2K228A binding to Itu was not markedly higher than the protein concentration used, data in these cases were fitted to a quadratic equation. Correlation analysis of the changes in binding free energy for single mutations at Y361 and K228 with respect to HIPK2WT (ΔΔGbinding) was performed using PRISM 6.0 program (GraphPad, La Jolla).
Kinase assays
Murine HIPK2 KDom wild type and Y361F mutant (1 μg was used to allow a clear visualization and quantification by Coomassie stain as loading control) were preincubated for 60 minutes in the presence of 1 μg of substrate (MBP, p53, GST‐HIPK2K228A) at RT in kinase buffer (20 mM Hepes pH 7.4, 50 mM NaCl, 10 mM MgCl2, 10 mM CaCl2) and 1–1.5% DMSO, either in the absence and in the presence of saturating concentration of ATP‐competitors (>10‐fold of K D, [TBB] = 50 μM, [TBID] = 125 μM, [PurvA] = 100 μM, [SB] = 150 μM, [Stauro] = 30 μM, [Itu] = 180 μM). The GST‐fused form of HIPK2K228A (GST‐HIPK2K228A) was utilized allowing us to distinguish it from the wild type on a SDS‐PAGE due to its larger size. [γ‐32P]‐ATP (≈ 180 kBq) was added (final volume 25 μL), incubating the mixture for 30 minutes at 303 K. The reaction was stopped by 10 minute incubation at 363 K with Laemmli buffer.41 Reaction products were resolved by SDS–PAGE and 32P‐labelled proteins were detected by autoradiography. The detected signals were quantified by densitometric analysis and normalized to the amount of sample loaded into the gel, as determined by Coomassie stain. The relative signal of the samples without ATP‐competitors was used as reference. The reported residual kinase activity of HIPK2WT and HIPK2Y361F is the mean of at least three independent experiments.
Kinase activity of HIPK2WT and HIPK2Y361F was measured on MBP following the incorporation of radiolabeled phosphate from [γ‐ 32P]‐ATP into the substrate.42 See Supporting Information Figure S7 for experimental details and data analysis.
Supporting Information
Supporting Information includes a detailed description of HIPK2WT, HIPK2Y361F, and HIPK2K228A KDom expression and purification protocols and purity assessment by SDS‐PAGE. The comparison of the static fluorescence and CD spectra of the three forms is shown and experimentally detailed. The calibration curve of the SEC column and additional chromatograms as well as the observed sedimentation velocities of the protein samples are reported. Supporting Information also includes raw data for equilibrium binding measurements of ATP‐competitors at 293 K and for TBID also at 310 K, and the experimental details and data analysis for determining the Δc p. Kinetic analysis on the kinase activity of HIPK2WT and HIPK2Y361F on MBP together with experimental details and data analysis are also reported. Supplementary_Material.pdf
Authors’ Contribution
AS and FS performed molecular biology and AS, GP, CC, AG protein expression, purification and quality sample assessment. CZ and DV performed ultracentrifugation analysis. AS, LM and LCM performed gel filtration, western blot, inhibitor binding, thermal denaturation experiments and enzymatic assays. AS, LM, CR, CS, SS, BV and LCM designed the experiments and analyzed data. AS, LM, SS, BV and LCM wrote the manuscript.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting information
Supporting Information
Acknowledgments
We thank Dr Andreas C. Joerger and Prof Conrad Kunick who kindly provided the plasmid containing p53 quadruple mutant and TBID, respectively, and Prof Mario Barteri and Prof Stefano Gianni for their technical assistance during the fluorescence experiments and data analysis.
CNR, Progetto Premiale 2012: Avanzati sistemi biosensoristici per la diagnosi e il follow up della malattia celiaca; FIRC, Triennal Fellowships; AIRC, IG #14592; Sapienza Università di Roma, Avvio alla Ricerca 2016, B52F16001790005, Awards Grant C26H154L5A and H2CU.
Broad audience statement: One of the mechanisms that regulate the activity of HIPK2 is the cis auto‐phosphorylation of a specific tyrosine, Y361, on the activation loop. Here, we show that phosphorylation at Y361 affects the oligomerization state, the stability, and the activity of the kinase domain of HIPK2 and induces structural rearrangements at the activation loop that are transmitted to the catalytic pocket.
Notes: For the sake of clarity, the current NCBI RefSeq for mouse HIPK2 (NP_034563.2) is adopted in this article for HIPK2 amino acid numbering. In brackets the amino acid number used in the relative references is also reported, when the previous HIPK2 NP_001129537.1 was adopted.
Contributor Information
Beatrice Vallone, Email: beatrice.vallone@uniroma1.it.
Linda Celeste Montemiglio, Email: lindac.montemiglio@uniroma1.it.
References
- 1. Kim YH, Choi CY, Lee SJ, Conti MA, Kim Y (1998) Homeodomain‐interacting protein kinases, a novel family of co‐repressors for homeodomain transcription factors. J Biol Chem 273:25875–25879. [DOI] [PubMed] [Google Scholar]
- 2. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298:1912–1934. [DOI] [PubMed] [Google Scholar]
- 3. Rinaldo C, Prodosmo A, Siepi F, Soddu S (2007) HIPK2: a multitalented partner for transcription factors in DNA damage response and development. Biochem Cell Biol 85:411–418. [DOI] [PubMed] [Google Scholar]
- 4. Blaquiere JA, Verheyen EM (2017) Homeodomain‐interacting protein kinases: diverse and complex roles in development and disease. Curr Top Dev Biol 123:73–103. [DOI] [PubMed] [Google Scholar]
- 5. Kuwano Y, Nishida K, Akaike Y, Kurokawa K, Nishikawa T, Masuda K, Rokutan K (2016) Homeodomain‐interacting protein kinase‐2: a critical regulator of the DNA damage response and the epigenome. Int J Mol Sci 17:pii: E1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fan Y, Wang N, Chuang P, He JC (2014) Role of HIPK2 in kidney fibrosis. Kidney Int Suppl 4:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Feng Y, Zhou L, Sun X, Li Q (2017) Homeodomain‐interacting protein kinase 2 (HIPK2): a promising target for anti‐cancer therapies. Oncotarget 8:20452–20461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matt S, Hofmann TG (2016) The DNA damage‐induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci 73:2829–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saul VV, de la Vega L, Milanovic M, Kruger M, Braun T, Fritz‐Wolf K, Becker K, Schmitz ML (2013) HIPK2 kinase actvity depends on cis‐autophosphorylation of its activation loop. J Mol Cell Biol 5:27–38. [DOI] [PubMed] [Google Scholar]
- 10. Siepi F, Gatti V, Camerini S, Crescenzi M, Soddu S (2013) HIPK2 catalytic activity and subcellular localization are regulated by activation‐loop Y354 autophosphorylation. Biochim Biophys Acta 1833:1443–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saul VV, Schmitz ML (2013) Posttranslational modifications regulate HIPK2, a driver of proliferative diseases. J Mol Med 91:1051–1058. [DOI] [PubMed] [Google Scholar]
- 12. Lee S, Shang Y, Redmond SA, Urisman A, Tang AA, Li KH, Burlingame AL, Pak RA, Jovičić A, Gitler AD, Wang J, Gray NS, Seeley WW, Siddique T, Bigio EH, Lee VM, Trojanowski JQ, Chan JR, Huang EJ (2016) Activation of HIPK2 promotes ER stress‐mediated neurodegeneration in amyotrophic lateral sclerosis. Neuron 91:41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Polonio‐Vallon T, Kirkpatrick J, Krijgsveld J, Hofmann TG (2014) Src kinase modulates the apoptotic p53 pathway by altering HIPK2 localization. Cell Cycle 13:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Verdina A, Di Rocco G, Virdia I, Monteonofrio L, Gatti V, Policicchio E, Bruselles A, Tartaglia M, Soddu S (2017) HIPK2‐T566 autophosphorylation diversely contributes to UV‐ and doxorubicin‐induced HIPK2 activation. Oncotarget 8:16744–16754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson LN, Noble ME, Owen DJ (1996) Active and inactive protein kinases: structural basis for regulation. Cell 85:149–158. [DOI] [PubMed] [Google Scholar]
- 16. Nolen B, Taylor S, Ghosh G (2004) Regulation of protein kinases: controlling activity through activation segment conformation. Mol Cell 15:661–675. [DOI] [PubMed] [Google Scholar]
- 17. Becker W, Sippl W (2011) Activation, regulation, and inhibition of DYRK1A. FEBS J 278:246–256. [DOI] [PubMed] [Google Scholar]
- 18. Lindberg RA, Quinn AM, Hunter T (1992) Dual‐specificity protein kinases: will any hydroxyl do?. Trends Biochem Sci 17:1114–1119. [DOI] [PubMed] [Google Scholar]
- 19. Lochhead PA (2009) Protein kinase activation loop autophosphorylation in cis: overcoming a Catch‐22 situation. Sci Signal 2:pe4. [DOI] [PubMed] [Google Scholar]
- 20. Lochhead PA, Sibbet G, Morrice N, Cleghon V (2005) Activation‐loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 121:925–936. [DOI] [PubMed] [Google Scholar]
- 21. Adayev T, Chen‐Hwang MC, Murakami N, Lee E, Bolton DC, Hwang YW (2007) Dual‐specificity tyrosine phosphorylation‐regulated kinase 1A does not require tyrosine phosphorylation for activity in vitro. Biochemistry 46:7614–7624. [DOI] [PubMed] [Google Scholar]
- 22. Walte A, Rüben K, Birner‐Gruenberger R, Preisinger C, Bamberg‐Lemper S, Hilz N, Bracher F, Becker W (2013) Mechanism of dual specificity kinase activity of DYRK1A. FEBS J 280:4495–4511. [DOI] [PubMed] [Google Scholar]
- 23. van der Laden J, Soppa U, Becker W (2015) Effect of tyrosine autophosphorylation on catalytic activity and subcellular localisation of homeodomain‐interacting protein kinases (HIPK). Cell Commun Signal 13:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wee HJ, Voon DC, Bae SC, Ito Y (2008) PEBP2‐beta/CBF‐beta‐dependent phosphorylation of RUNX1 and p300 by HIPK2: implications for leukemogenesis. Blood 112:3777–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, Piaggio G, Fanciulli M, Appella E, Soddu S (2002) Homeodomain‐interacting protein kinase‐2 phosphorylates p53 at Ser46 and mediates apoptosis. Nat Cell Biol 4:11–19. [DOI] [PubMed] [Google Scholar]
- 26. Hofmann TG, Möller A, Sirma H, Zentgraf H, Taya Y, Dröge W, Will H, Schmitz ML (2002) Regulation of p53 activity by its interaction with homeodomain‐interacting protein kinase‐2. Nat Cell Biol 4:1–10. [DOI] [PubMed] [Google Scholar]
- 27. Fersht AR (1999) Structure and mechanism in protein science. New York: Freeman, pp. 508–539. [Google Scholar]
- 28. Privalov PL, Khechinashvili NN (1974) A thermodynamic approach to the problem of stabilization of globular protein structure: a calorimetric study. J Mol Biol 86:665–684. [DOI] [PubMed] [Google Scholar]
- 29. Myers JK, Pace CN, Scholtz JM (1995) Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci 4:2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Greenfield NJ (2006) Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat Protoc 1:2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bain J, Plater L, Elliot M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P (2007) The selectivity of protein kinase inhibitors: a further update. Biochem J 408:297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cozza G, Zanin S, Determann R, Ruzzene M, Kunick C, Pinna LA (2014) Synthesis and properties of a selective inhibitor of homeodomain‐interacting protein kinase 2 (HIPK2). Plos One 9:e89176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pagano MA, Bain J, Kazimierczuk Z, Sarno S, Ruzzene M, Di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F, Pinna LA (2008) The selectivity of inhibitors of protein kinase CK2: an update. Biochem J 415:353–365. [DOI] [PubMed] [Google Scholar]
- 34. Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics 22:195–201. [DOI] [PubMed] [Google Scholar]
- 35. Ogawa Y, Nonaka Y, Goto T, Ohnishi E, Hiramatsu T, Kii I, Yoshida M, Ikura T, Onogi H, Shibuya H, Hosoya T, To N, Hagiwara M (2010) Development of a novel selective inhibitor of the Down syndrome‐related kinase Dyrk1A. Nat Commun 1:86. [DOI] [PubMed] [Google Scholar]
- 36. Nardinocchi L, Puca R, Givol D, D'Orazi G (2010) HIPK2‐a therapeutical target to be (re)activated for tumor suppression: role in p53 activation and HIF‐1α inhibition. Cell Cycle 9:1270–1274. [DOI] [PubMed] [Google Scholar]
- 37. Nugent MM, Lee K, He JC (2015) HIPK2 is a new drug target for anti‐fibrosis therapy in kidney disease. Front Physiol 6:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shang Y, Doan CN, Arnold TD, Lee S, Tang AA, Reichardt LF, Huang EJ (2013) Transcriptional corepressors HIPK1 and HIPK2 control angiogenesis via TGF‐β‐TAK1‐dependent mechanism. Plos Biol 11:e1001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tidow H, Melero R, Mylonas E, Mylonas E, Freund SM, Grossmann JG, Carazo JM, Svergun DI, Valle M, Fersht AR (2007) Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc Natl Acad Sci USA 104:12324–12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luciani X, Mounier S, Redon R, Bois A (2009) A simple correction method of inner filter effects affecting FEEM and its application to the PARAFAC decomposition. Chemometr Intell Lab Syst 96:227–238. [Google Scholar]
- 41. Lazzari C, Prodosmo A, Siepi F, Rinaldo C, Galli F, Gentileschi M, Bartolazzi A, Costanzo A, Sacchi A, Guerrini L, Soddu S (2011) HIPK2 phosphorylates ΔNp63α and promotes its degradation in response to DNA damage. Oncogene 30:4802–4813. [DOI] [PubMed] [Google Scholar]
- 42. Hastie CJ, McLauchlan HJ, Cohen P (2006) Assay of protein kinases using radiolabeled ATP: a protocol. Nat Protoc 1:968–971. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information