

Abstract
The ER resident chaperone molecule GRP78 has been shown to translocate to the cell surface where it associates with Cripto and signals cell growth, playing a still partially understood role in tumorigenesis. Consequently, a better understanding of GRP78 topology and structure at the surface of cancer cells represents an important step in the development of a new class of therapeutics. Here, we used a set of programs for creation of a complex containing GRP78 and Cripto proteins. We elucidated possible interactions of GRP78, Cripto, and their complex with the membrane. Using molecular dynamics simulations, we demonstrated that Cripto binding to GRP78 completely changes the dynamics of its behavior on the membrane, not allowing GRP78 to disconnect from it, thus enabling GRP78 tumorigenic functions.
Keywords: GRP78 protein, Cripto protein, GRP78–Cripto complex, cell membrane, cancer, protein–membrane interactions, molecular dynamics
Introduction
The endoplasmic reticulum (ER) resident chaperone molecule GRP78 is the master regulator of the ER stress response. It regulates the activation of three ER transmembrane‐bound sensors, IRE1α, ATF6, and PERK.1 Under normal circumstances, these three sensors remain inactive through luminal association with GRP78. However, upon stress conditions within the cell, GRP78 dissociates from these three sensors, allowing each of them to activate downstream signaling cascades to normalize protein folding and secretion. GRP78 plays numerous roles in the tumorigenesis of various organs.2 GRP78 can translocate to the surface of cancer cells where it serves as a signaling molecule for cell growth by activating PI3K.3, 4, 5, 6 The nature of surface‐expressed GRP78 is poorly understood, but several reports indicate that it associates with Cripto, a multifunctional cell surface protein encoded by the CFC1B gene with important roles in vertebrate embryogenesis and the progression of human tumors.7, 8 Cripto and GRP78 interact at the cell surfaces of cancer cells, and their interaction is most likely independent of prior association within the ER. Thus, elucidation of the molecular interaction between GRP78 and Cripto is a key to comprehending the initiation of growth signals, and interaction that has until now received very little attention.
Because surface‐located GRP78 is important in tumorigenesis, a better understanding of its topology and structure at the surface of cancer cells represents an important step in the development of a new class of therapeutics, such as small molecules or antibodies. However, any such development requires a detailed enumeration of the residues that interact with its ligand Cripto and of the sites that are potentially accessible after the interaction with Cripto has taken place. Here, we report our initial efforts to define the structural characteristics of the membrane interaction between GRP78 and Cripto.
Results
Homology modeling
We used the crystal structure of GRP78 (PDB ID 3ldl). It consists of two functional domains: a nucleotide‐binding domain (NBD) and a substrate‐binding domain (SBD). The NBD binds and hydrolyzes ATP, and the SBD binds polypeptides.9 It was shown before that the GRP78 construct (Δ19–68 GRP78) binds to Cripto;10 therefore, the GRP78 residues 19–68 are important. The N terminal of the crystal structure of GRP78 resolved residues start from Asp26; therefore, seven essential residues of the SBD is lacking. We created a homology model using the MOE software11 and the full sequence of GRP78.12 The missed part of the N terminal is MKLSLVAAMLLLLSAARAEEEDKKE. Because the only area of interest is the residues 19–68, we replaced only eight amino acids: AEEEDKKE. An image of the homology model is shown in Figure 1.
Figure 1.
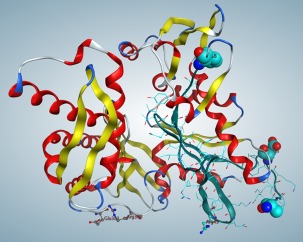
Homology model of GRP78. Spheres show the beginning and end of probable docking area. Eight residues (AEEEDKKE) were added to the N terminal. Cyan ribbon and side chains—the region of experimentally conformed GRP78–Cripto interactions.
Docking of Cripto with GRP78
Using HEX protein docking program,13 we found possible docking configurations between the homology model of GRP78 and Cripto. HEX suggested 128 docking solutions for the complex. The docking score for each complex was calculated as a function of the degrees of freedom in the search, and can be interpreted as a scaled representation of the interaction energy, which was minimized. Previously it has been derived from experiments9 that the region in GRP78 with the greatest probability for docking with Cripto is between the residues Glu19 and Thr68. That is why we selected the GRP78–Cripto complex docking solutions in which the established region of importance of GRP78 (Glu19–Thr68) was within a feasible distance to Cripto to further study. Furthermore, docking solutions with at least two points within the complex at which GRP78 and Cripto were close enough to have notable interactions, such as solution 3, were marked for further review. More details of interactions between the residues of these molecules are presented in Table S1.
Orientation of the GRP78–Cripto complex on the membrane
The next step in our study was to find a possible location of the GRP78–Cripto complex on the membrane. We examined all the selected GRP78–Cripto complex solutions produced by the docking for their possible affinity to the membrane bilayer. To predict membrane interaction, we used the Membrane Optimal Docking Area (MODA)14 and Position of Proteins in Membrane (PPM)15, 16 servers (see Materials and Methods section). Our prediction resulted in numerical values assigned to each surface residue representing the likelihood of that residue being involved in a membrane interaction. MODA scores above 40 were expected to be membrane interaction sites, scores between 20 and 40 are possible interactions, and scores below 20 were generally viewed as unlikely interface residues. Table 1 shows the residues interacting with the membrane in the complex and in the separated individual molecules GRP78 and Cripto. The membrane affinity prediction scores for an isolated GRP78 without the docked Cripto showed relatively weak affinity and low likelihood for interaction with the membrane, while Cripto individually had very high scores, notable for around 60% of the residues, showing its generally strong attraction to the bilayer (Table 1).
Table 1.
Docking Results for Single GRP78 and Cripto with the Membrane Obtained from MODA Programa
| GRP78 | Cripto | ||||
|---|---|---|---|---|---|
| Residue | MODA | Residue | MODA | ||
| Number | Name | Score | Number | Name | Score |
| 45 | PHE | 221.983 | 1 | LYS | 0 |
| 46 | LYS | 2.01714 | 2 | GLU | 0 |
| 47 | ASN | 60.9471 | 3 | HIS | 0 |
| 48 | GLY | 302.422 | 4 | CYS | 0 |
| 49 | ARG | 60.4073 | 5 | GLY | 0 |
| 50 | VAL | 0 | 6 | SER | 84.7529 |
| 51 | GLU | 4.46043 | 7 | ILE | 274.646 |
| 106 | PRO | 8.97215 | 8 | LEU | 156.356 |
| 107 | SER | 0 | 9 | HIS | 416.001 |
| 108 | VAL | 0 | 10 | GLY | 235.593 |
| 109 | GLN | 84.7406 | 11 | THR | 594.654 |
| 110 | GLN | 64.8705 | 12 | TRP | 1549.68 |
| 111 | ASP | 0 | 13 | LEU | 1120.77 |
| 112 | ILE | 57.2149 | 14 | PRO | 322.692 |
| 113 | LYS | 380.697 | 15 | LYS | 162.077 |
| 114 | PHE | 83.8182 | 16 | LYS | 256.907 |
| 160 | TYR | 1.35072 | 17 | CYS | 0 |
| 196 | MET | 19.7399 | 18 | SER | 0 |
| 197 | ARG | 351.685 | 19 | LEU | 393.391 |
| 198 | ILE | 11.1251 | 20 | CYS | 504.358 |
| 213 | LYS | 68.816 | 21 | ARG | 805.983 |
| 240 | GLY | 11.0225 | 22 | CYS | 315.019 |
| 241 | VAL | 3.26102 | 23 | TRP | 544.076 |
| 242 | PHE | 5.22858 | 24 | HIS | 0 |
| 396 | TYR | 56.9854 | 25 | GLY | 27.0712 |
| 397 | GLY | 0 | 26 | GLN | 314.199 |
| 398 | ALA | 0 | 27 | LEU | 60.2914 |
| 399 | ALA | 0 | 28 | HIS | 10.7785 |
| 400 | VAL | 0 | 29 | CYS | 1.7354 |
| 401 | GLN | 36.7045 | 30 | LEU | 4.44926 |
| 402 | ALA | 0 | 31 | PRO | 98.648 |
| 403 | GLY | 431.994 | 32 | GLN | 300.976 |
| 404 | VAL | 212.022 | 33 | THR | 110.815 |
| 405 | LEU | 244.877 | 34 | PHE | 430.754 |
| 406 | SER | 181.862 | 35 | LEU | 137.598 |
| 36 | PRO | 54.3661 | |||
| 37 | GLY | 0 | |||
| 38 | CYS | 0 | |||
| 39 | ASP | 0 | |||
Name—three‐letter residue name, MODA Score—the MODA scoring values related to the natural tendency of each individual residue within the protein with a known 3D structure to interact with the membrane. Residues with scores above 40 are expected to represent a membrane interaction site, while residues with scores 20–40 should undergo further inspection to whether they interact with the membrane.
Analyzing the 128 solutions of the docking residues and orientation of each solution's complex on the membrane, we selected four (3, 90, 98, and 34) in which Cripto's residues interacted with the 19–68 region of GRP78, which was experimentally shown by Kelber and co‐authors.10 The docking residues involved in the interactions between GRP78 and Cripto in the complex and the distances of the closest pairs of heavy atoms are shown in Table S1.
The criteria used to select the complexes included the presence of the experimentally confirmed residues at the binding site (residues 19–68 on GRP78) and the likeliness of the complex to bind to the cell membrane, which was calculated using the PPM server. As it was shown by Maheshwari and Brylinski,17 existing experimental binding‐site information significantly improves computational docking results. Docking results for GRP78 and Cripto, as well as results for the four selected solutions of the complex, are shown in Table 2. After analysis, solution 98 was chosen for further validation (see more details in Table 3). Images of GRP78, Cripto, and the complex (solution 98) binding individually to the membrane are shown in Figure 2(A–D). Residues of the complexes interacting with the membrane are presented in Table S2.
Table 2.
Docking Results for GRP78, Cripto, and Selected Solutions for GRP78–Cripto Complex with Membrane Obtained with the PPM Program
| Protein/Solution | Depth/Hydrophobic Thickness, Å | G transfer kcal/mol | Tilt Angle, ° | Embedded residues |
|---|---|---|---|---|
| Cripto | 3.8 ± 1.9 | −4.1 | 74 ± 14 | 12–14 |
| GRP78 | 0.5 ± 2.8 | −1.9 | 87 ± 13 | 303 |
| Complex 3 | 4.7 ± 0.8 | −5.7 | 87 ± 6 | 12–14 |
| Complex 34 | 1.3 ± 1.7 | −2.6 | 82 ± 20 | 14 |
| Complex 90 | 1.5 ± 1.8 | −3.5 | 74 ± 8 | 1314 |
| Complex 98 | 4.3 ± 1.8 | −2.8 | 78 ± 8 | 14,16 |
Table 3.
Docking Results for Solution 98 of GRP78–Cripto Complex
| Solution | Depth /Hydrophobic thickness, Å | ΔG transfer kcal/mol | Tilt angle, ° | Embedded residues (Cripto) | GRP78 residues close to membrane | Distance to membrane, Å |
|---|---|---|---|---|---|---|
| 98 | 4.3 ± 1.8 | −2.8 | 78 ± 8 | 14,16 | 303 | 6.75 |
| 380 | 2.56 | |||||
| 383 | 2.45 | |||||
| 386 | 4.13 |
Figure 2.
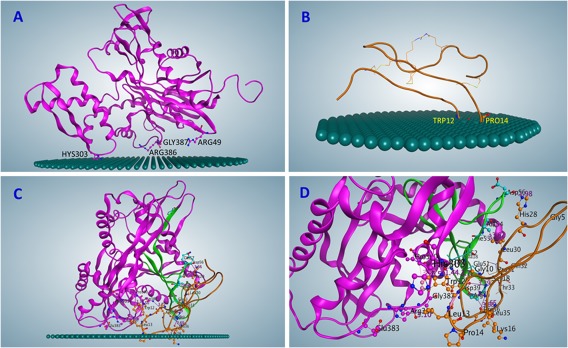
Orientation of proteins on membrane obtained with PPM modeling (displayed on MOE). (A) PPM model of GRP78 on the membrane. (B) PPM model of Cripto on the membrane. (C) PPM model of GRP78–Cripto, Solution 98, on the membrane. (D) GRP78–Cripto interaction area (close‐up, membrane removed).
Dynamic properties of GRP78‐Cripto complex
Molecular dynamic (MD) simulations of the GRP78–Cripto complex near the cell membrane were conducted to study the behavior of the complex and its interaction energy with the lipid bilayer. The GRP78 protein and the GRP78–Cripto complex were both simulated in MD for 120 ns, with the resulting coordinates saved every 10 ps for analysis. Both simulations began with a starting position corresponding to the results from the PPM server. For GRP78 individually, the simulation showed the protein having only weak attractions to the membrane at 25 ns of MD [Table S3 and Fig. 3(A) and 4(A)], and by 50 ns, the protein was completely detached [Fig. 4(B)]. For the GRP78–Cripto complex, the simulation showed the complex adjusting to the membrane. By 50 ns, Cripto residues made notable contact with the membrane [Fig. 4(C)], and adjustment of the complex on the membrane continued through 120 ns [Fig. 3(B–C) and 4(D)] to decrease the distances and increase the interactions between the Cripto residues and the membrane.
Figure 3.
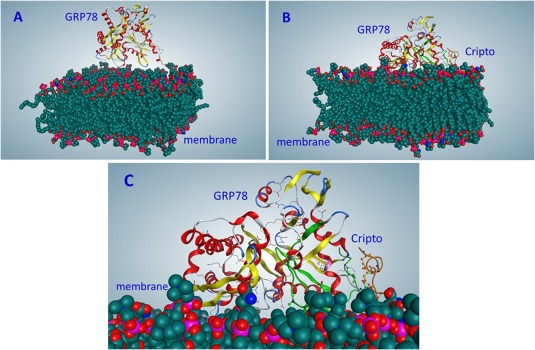
Interactions of proteins with membrane (on results of molecular dynamics). (A) GRP78, 25 ns. (B) GRP78–Cripto complex (Solution 98), 120 ns. (C) Close‐up of Figure 3(B).
Figure 4.
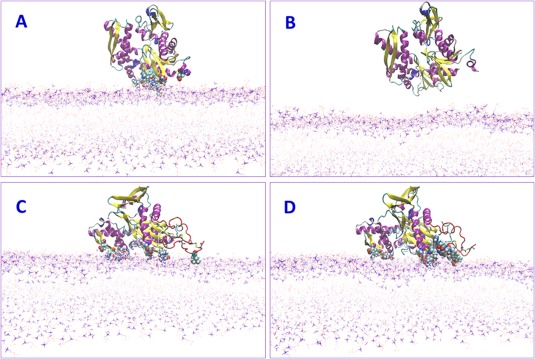
Snapshots of molecular dynamics (MD) of proteins on the membrane. (A) GRP78 at 25 ns. CPK‐residues contacting the membrane. (B) GRP78 at 50 ns already disconnected from the membrane. (C) GRP78–Cripto complex at 50 ns still contacting the membrane. (D) GRP78–Cripto complex at 120 ns MD stabilized on the membrane. CPK—heavy atoms interacting with the membrane.
During the molecular dynamics simulation, the energy of interaction of the complex with the membrane notably increased between 25 and 50 ns, and stabilized by 120 ns (Fig. 5). For the single GRP78, the energy of interaction with the membrane improves until around 35 ns, and then sharply decreases and goes to zero with GRP78 disconnecting with the membrane. The interactions of the GRP78 residues with the membrane at 25 ns are presented in Table S3.
Figure 5.
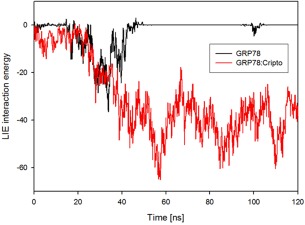
Energies of interaction of GRP78 and GRP78–Cripto complex with the membrane during MD simulation. One can see that GRP78 completely disconnected from membrane, while energy of GRP78–Cripto complex is stabilized.
By the end of the recorded simulation (120 ns) for the GRP78–Cripto complex on the membrane, the original docking position was adjusted. The interactions between the GRP78–Cripto complex and the membrane during the molecular dynamics simulation are shown in Table S4.
Discussion
The GRP78 protein is important in tumorigenesis not only as integral component of the ER stress response in the endoplasmic reticulum, but also as it is translocated to the cell surface where it mediates growth signals.4, 5 Cripto has been previously shown to be a ligand for the surface‐expressed GRP78.6, 7 Here, we explored the possibility that Cripto, when binding to GRP78, creates a GRP78–Cripto complex that attaches to the membrane, not allowing GRP78 to disconnect from it, enabling its tumorigenic functions. We reviewed the set of possible docking configurations of GRP78 with Cripto to find their possible affinity to the membrane. Membrane interaction studies of GRP78 alone and in the complex with the Cripto predicted the weak interaction of GRP78 with the membrane. On the contrary, Cripto by itself was predicted to have very strong interactions with the membrane. For the GRP78–Cripto complex, the highest predicted membrane‐interaction values representing the affinity to the membrane were from the Cripto residues, rather than the GRP78 residues, which had only weak attractions to the membrane. Consequently, membrane‐interaction study showed that the complex would be attached to the membrane as a peripheral protein with most of the embedded residues being from the Cripto region of the complex. These results suggest that it is the Cripto bound to the GRP78 which plays a crucial role in the attachment of the entire complex to the membrane at the initial stage of complex formation. To study the possible stability of this complex on the membrane, we conducted MD simulations of GRP78 alone and in the complex with Cripto. The MD simulation shows that GRP78 alone from its starting position with weak attraction to the membrane soon detaches completely after 50 ns of MD. The MD simulation of the GRP78–Cripto complex, from the same starting position with GRP78 weakly interacting with the membrane, shows that the entire complex immediately adjusts due to the strong attraction of Cripto to the membrane. By 75 ns, Cripto and GRP78 have notable contacts with the membrane, and up to 100–120 ns, the complex continues adjusting as distances between Cripto and the membrane are decreased, and the interactions increase. The improved docking between GRP78 and Cripto within the complex is also noted through specific interactions between the residues, and throughout the simulation, GRP78 and Cripto remain attached to one another.
In general, we demonstrated that the Cripto protein stabilizes the position of GRP78 on the membrane, making it possible for it to fulfill its function on the surface of cancer cells, where it serves as a signaling molecule for cell growth by the activation of PI3K.
Materials and Methods
Approach overview
To study the mechanism of interaction of the GRP78 with the cell membrane and Cripto, we needed to define the significant residues of interaction. The flowchart of approach is shown in Figure 6. On the first step, using the MOE program, we restored the missing GRP78 residues that most likely bind to Cripto10 and created a homology model. On the second step, we simulated the docking of GRP78 to Cripto, using the HEX docking program. Obtained solutions were analyzed to select those that correspond to experimental data.10 The third step consisted of locating where GRP78, Cripto, and the selected GRP78–Cripto complexes bound to the membrane. The fourth step included verifying binding energies with the MODA server and finding the best solution. Then, on the fifth step, molecular dynamics simulations were conducted.
Figure 6.
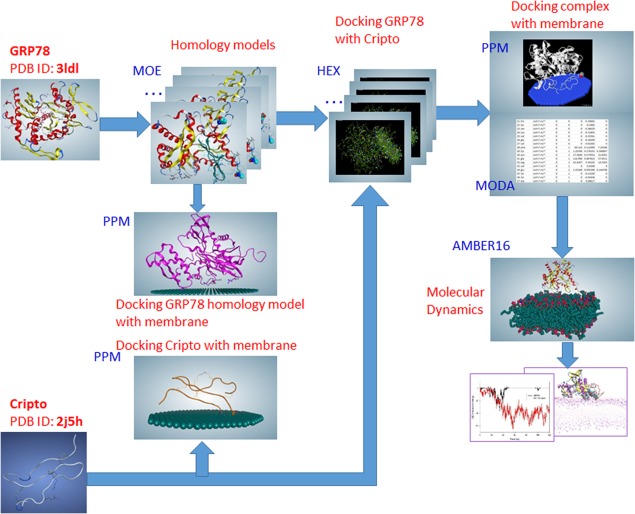
Flowchart of the approach: MOE (homology models); HEX (docking positions); PPM and MODA, (interactions with the membrane); Molecular Dynamics (stability of the complex on the membrane).
Homology modeling
The substrate‐binding domain of the GRP78 crystal structure started from Asp26; therefore, seven essential residues of the N terminal (UniProt # P11021) were absent. To add them, we conducted homology modeling of the GRP78 protein N‐terminal fragment using the Molecular Operating Environment (MOE) software, version 2015.10, CCG, Montreal, Canada).11 The PDB Search module of the program was used to search for a suitable template for homology modeling. The module implemented a two‐stage search strategy: (1) a fast scan to create an initial list of candidates and (2) a HMMER (Hidden Markov Models algorithm) to determine family membership. For a template search, the Domain Motif Search method was used. Found fragments were analyzed for similarity, and sequences with higher scores were selected and aligned. Homology models were built with the Homology Model module, using Amber10:EHT forcefield with a Reaction Field (R‐Field). A homology model with better RMSD and contact energy was chosen.
Docking methods
Docking of GRP78 and Cripto was conducted with the HEX 8.0.0 software.13 HEX is a docking and superposition program using spherical polar Fourier (SPF) correlations to accelerate the calculations, and it has built‐in graphics to view the results. Besides the protein–protein interactions, HEX also calculates protein–ligand docking, assuming the ligand is rigid, and superposing pairs of molecules on the base of their 3D shapes. Docking solutions of GRP78 and Cripto, which were defined as a receptor and a ligand, respectively, inputted as PDB files, were identified based on the calculations of the docking and superimposition program HEX. The interactive graphics display defaulted the two molecules with the molecular skeleton and side chains visible while the hydrogens, H‐bonds, residue ID, animated ribbons, harmonic surfaces, solid surfaces, and solid model remain hidden.
The reference state, or starting position, was established by taking the frequency of interactions from a decoy set of near‐native conformations with solely good docking complementarity. Given the data for the crystal structure of the complex position desired, a separate complex structure PDB file may also be uploaded and used as the reference orientation to evaluate the accuracy of the docking prediction. Calculating the correlation of surface shape and electrostatic charge between GRP78 and Cripto to obtain an overall docking score, all the feasible conformations within a 3D space are identified and modeled, the process of which is accelerated by using the conventional fast Fourier Transform.
The docking process used the parameters such that the steric scan (N = 18) phase will be performed at (1 + 40)/0.8 = 51.25 intermolecular separations, in ± steps of 0.80 Å starting from the distance R: 22.1 Å. The final search (N = 25) phase will be applied to the highest scoring scan orientations in steps of 0.8 Å. The rotational search will use angular increments of 7.5° in each of the two ligand and receptor rotational angles up to 180°, and in steps of 5.5° about the twist angle up to 360°. A 0.6 Å Cartesian grid is used to sample the molecular surface skin coefficients numerically, and the program then proceeds to calculate the docking correlation scores at each incremented position. The best 2000 maximum orientations with the lowest energy are then displayed in the graphical interface.
Study of protein–membrane interactions
The preliminary position of the GRP78–Cripto complex on the membrane was estimated using the Position of Proteins in Membrane (PPM) server, version 2.0.15 The method15 uses the Orientations of Proteins in Membranes (OPM) database16 of known membrane‐interacting proteins and their 3D structural classification, topology, and intracellular localization sorted into superfamilies, families, and membrane classes to calculate the spatial positions of proteins in membranes. Resulting information includes membrane penetration depth and ΔG transfer energy (protein transfer energy from water to the lipid bilayer), calculated as the sum of the area of the protein accounting for van‐der‐Waals and H‐bonding interactions and the electrostatic interactions. The GRP78–Cripto complex from the docking calculations was submitted to the PPM server. Calculations excluded heteroatoms, water, and detergents, and used the outer topology model for the membrane PPM server outputs contained: hydrophobic thickness, tilt angle, transfer energy, a table of embedded residues, and a coordinate file for the protein positioned in the lipid bilayer. The GRP78–Cripto complex is declared by the PPM program as a peripheral protein, reflected in the minimum transfer energy being around −15 kcal/mol.
To validate the rotational and translational position of the GRP78–Cripto complex on the membrane resulted from PPM server, the Membrane Optimal Docking Area (MODA) method14 was used. MODA detects and calculates interactions between the lipid bilayer and the residues of proteins contacting the membrane. MODA considers multiple aspects of a protein and the bilayer, including electrostatic interactions, hydrophobic groups inserted in the bilayer, specific lipid binding, and membrane curvature. When calculating the position and characteristics of the GRP78–Cripto complex on the membrane, it was uploaded as a single PDB file. Based mostly on the complex's overall 3D shape, a surface patch of a 5 Å distance from the protein's atoms is generated to predict the strength of interaction with the membrane. By establishing a sphere of influence of a given distance, the membrane interactions at that point were calculated with consideration of the residues in the vicinity as well as the general complex shape.
Molecular dynamics
Both the GRP78–Cripto complex and the individual GRP78 protein on the membrane were simulated using the AMBER16 software.18 Simulations of GRP78 in a complex with Cripto were based on the docking of Cripto to the crystal structure of GRP78. The GRP78–Cripto complex was placed in close proximity to an equilibrated bilayer consisting of 20% DOPS (1,2‐Dioleoyl‐sn‐glycero‐3‐phosphoserine) and 80% DOPC (1,2‐Dioleoyl‐sn‐glycero‐3‐phosphocholine). The complex was oriented so that the residues known to interact with the membrane were facing the bilayer. The system was solvated in a water box encompassing the protein with extensions in the XY‐dimensions identical to the lipid bilayer. Starting coordinates for simulations of GRP78 in absence of Cripto were prepared by replacing Cripto with water molecules while keeping the rest of the system identical to the GRP78–Cripto complex. Simulations were performed using the GPU‐accelerated PMEMD module19 implemented in the AMBER16 molecular dynamics software package,18 applying the ff14SB (http://pubs.acs.org/doi/abs/10.1021/acs.jctc.5b00255) and lipid1720 forcefields. Periodic boundary conditions with particle mesh Ewald summation21 of electrostatic interactions were applied and van‐der‐Waals interactions were truncated with a 10 Å cutoff. SHAKE was used to constrain bonds involving hydrogen atoms. After 10 000 steps of minimization, the systems were subjected to 5 ps of gradual constant volume heating (NVT) from 0 to 100 K followed by 100 ps gradual constant pressure heating (NPT) applying anisotropic Berendsen coupling22 with reference pressure set to 1 bar. Temperature was regulated with the Langewin thermostat23 gradually over 100 ps from 100 to 303K. Weak restraints maintained by a force constant of 10 kcal mol−1 Å−2 were applied to the proteins and lipids throughout both heating steps. The systems were finally simulated at constant pressure conditions (NPT) without restraints for 120 ns at 303 K and the resulting coordinates were saved every 10 ps for analysis.
Supporting information
Supporting Information
Statement: This research article describes the predicted configuration of complex formed by the ER resident chaperone molecule GRP78 and the Cryptic family protein 1B (Cripto/CFC1B). This novel study demonstrates that the binding of Cripto to GRP78 stabilizes the complex's binding to the cell membrane, paving the way to cancerogenic activity caused by this protein.
References
- 1. Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. [DOI] [PubMed] [Google Scholar]
- 2. Lee AS (2014) Glucose‐regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer 14:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Misra UK, Gonzalez‐Gronow M, Gawdi G, Wang F, Pizzo SV (2004) A novel receptor function for the heat shock protein Grp78: silencing of Grp78 gene expression attenuates alpha2M*‐induced signalling. Cell Signal 16:929–938. [DOI] [PubMed] [Google Scholar]
- 4. Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, Pasqualini R (2004) Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell 6:275–284. [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y, Liu R, Ni M, Gill P, Lee AS (2010) Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem 285:15065–15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Y, Tseng CC, Tsai YL, Fu X, Schiff R, Lee AS (2013) Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS One 8:e80071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC (2008) GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor β signaling and enhance cell growth. Mol Cell Biol 28:666–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Spike BT, Kelber Jonathan A, Booker Evan, Kalathur Madhuri, Rodewald Rose, Lipianskaya Julia, La Justin, He Marielle, Wright Tracy, Klemke Richard, Wahl Geoffrey M, Gray Peter C (2014) CRIPTO/GRP78 signaling maintains fetal and adult mammary stem cells ex vivo. Stem Cell Rep 2:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang J, Nune M, Zong Y, Zhou L, Liu Q (2015) Close and allosteric opening of the polypeptide‐binding site in a Human Hsp70 chaperone BiP. Structure 23:2191–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kelber JA, Panopoulos AD, Shani G, Booker EC, Belmonte JC, Vale WW, Gray PC (2009) Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways. Oncogene 28:2324–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maier JKX, Labute P (2014) Assessment of fully automated antibody homology modeling protocols in molecular operating environment. Proteins 82:1599–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. UniProtKB ‐ P11021 (GRP78_HUMAN), http://www.uniprot.org/uniprot/P11021. Last modified June 22, 2017.
- 13. Macindoe G, Mavridis L, Venkatraman V, Devignes M‐D, Ritchie DW (2010) HexServer: an FFT‐based protein docking server powered by graphics processors. Nucleic Acids Res 38:W445–W449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kufareva I, Lenoir M, Dancea F, Sridhar P, Raush E, Bissig C, Gruenberg J, Abagyan R, Overduin M (2014) Discovery of novel membrane binding structures and functions. Biochem Cell Biol 92:555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40:D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI (2006) OPM: orientations of proteins in membranes database. Bioinformatics 22:623–625. [DOI] [PubMed] [Google Scholar]
- 17. Maheshwari S, Brylinski M (2015) Predicted binding site information improves model ranking in protein docking using experimental and computer generated target structures. BMC Struct Biol 15:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Case DA, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Greene D, Homeyer N, Izadi S, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Liu J, Luchko T, Luo R, Mermelstein D, Merz KM, Monard G, Nguyen H, Omelyan I, Onufriev A, Pan F, Qi R, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello‐Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, York DM, Kollman PA (2017) AMBER 2017, University of California: San Francisco. [Google Scholar]
- 19. Salomon‐Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theory Comput 9:3878–3888. [DOI] [PubMed] [Google Scholar]
- 20. Skjevik ÅA, Dickson CJ, Madej BD, Teigen K, Gould I R, Walker RC. A comprehensive AMBER force field for the simulation of zwitterionic and anionic lipids. Manuscript in preparation.
- 21. Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092. [Google Scholar]
- 22. Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81:3684–3690. [Google Scholar]
- 23. Loncharich RJ, Brooks BR, Pastor RW (1992) Langevin dynamics of peptides: the frictional dependence of isomerization rates of N‐acetylalanyl‐N'‐methylamide. Biopolymers 32:523–535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information