

Abstract
Enzymes that modify the proteome, referred to as post‐translational modifying (PTM) enzymes, are central regulators of cellular signaling. Determining the substrate specificity of PTM enzymes is a critical step in unraveling their biological functions both in normal physiological processes and in disease states. Advances in peptide chemistry over the last century have enabled the rapid generation of peptide libraries for querying substrate recognition by PTM enzymes. In this article, we highlight various peptide‐based approaches for analysis of PTM enzyme substrate specificity. We focus on the application of these technologies to proteases and also discuss specific examples in which they have been used to uncover the substrate specificity of other types of PTM enzymes, such as kinases. In particular, we highlight our multiplex substrate profiling by mass spectrometry (MSP‐MS) assay, which uses a rationally designed, physicochemically diverse library of tetradecapeptides. We show how this method has been applied to PTM enzymes to uncover biological function, and guide substrate and inhibitor design. We also briefly discuss how this technique can be combined with other methods to gain a systems‐level understanding of PTM enzyme regulation and function.
Keywords: post‐translation modifying enzymes, proteases, kinases, substrate specificity, peptide libraries, peptide synthesis, mass spectrometry
Abbreviations
- PTM
post‐translational modification
- MSP‐MS
multiplex substrate profiling by mass spectrometry
- SPPS
solid‐phase peptide synthesis
- HPLC
high‐performance liquid chromatography
- FRET
Förster resonance energy transfer
- PS‐SCL
positional scanning‐synthetic combinatorial library
- HyCoSuL
hybrid combinatorial substrate library
- CoSeSuL
counter selection substrate library
- LC‐MS/MS
liquid chromatography with tandem mass spectrometry
Introduction
The primary mechanism by which the diversity of the proteome is increased is through the post‐translational modification of proteins. PTM enzymes are responsible for over 200 kinds of modifications of protein substrates and can be divided into two distinct mechanistic categories: (1) enzymes that hydrolyze peptide bonds (proteases) and (2) enzymes that covalently modify amino acid side chains. PTM enzymes constitute over 5% of the human genome but most have yet to be fully characterized.1 An important aspect of understanding the functions of these enzymes requires developing in vitro assays in which their specificity and activity can be monitored. Although a variety of assays exist for profiling PTM enzyme specificity, there is particular value in assays in which post‐translational modifications of peptide substrates are quantitatively and directly measured. To facilitate this type of assay format, researchers have taken advantage of synthetic peptide chemistry to develop large and diverse peptide libraries.
Peptide synthesis was pioneered by the work of Emil Fischer and Ernest Fourneau who synthesized the dipeptide glycylglycine in 1901. This work laid the foundation for subsequent advances in peptide synthesis with the Nobel Prize in Chemistry being awarded to Bruce Merrifield in 1984 for the development of solid‐phase peptide synthesis (SPPS).2 Merrifield's strategy involved assembly of a peptide chain in a stepwise manner with one end of the nascent peptide anchored to a solid resin until completion of synthesis. Covalent attachment of the growing peptide chain to a solid support renders it insoluble, which facilitates easier transition between synthetic steps, such as washing away excess reactant and byproduct. SPPS has been further streamlined over the last several decades and Fmoc SPPS is currently the most widely used synthetic strategy.3 Generating synthetic peptides using this technology gained popularity when biologists recognized that synthetic peptides could be used for antibody selection and production.4 Fmoc SPPS is now easily accomplished using highly automated workflows.5, 6
In this review, we first discuss how SPPS has been applied to generate large, highly diverse peptide libraries for the analysis of protease substrate specificity. We provide an overview of the various methods and describe several applications of how these methods have been applied to develop selective protease substrates and inhibitors. We extend this to a discussion of how SPPS has enabled the development of peptide libraries for determining the specificity of other types of PTM enzymes. Proteome‐derived peptide libraries and phage and bacterial display have also been widely applied for analysis of protease substrate specificity; however, these technologies are not the focus of the current review and have been reviewed elsewhere.7, 8, 9
Peptide‐Based Technologies for Analysis of Protease Specificity
Proteases are one of the largest classes of PTM enzymes, with over 550 encoded in the human genome.10 These enzymes are essential for normal cellular functions and are implicated in a variety of diseases, such as cancer, neurodegeneration, and blood clotting disorders. Because of the size and importance of this enzyme class, substantial effort has been put into the development of peptide‐based technologies for determining protease substrate specificity. Proteases generally recognize substrates in an extended linear conformation, making this class of enzymes particularly amenable to analysis with peptide‐based profiling methods.11
Traditionally, identification of protease substrates relied on relatively small collections of synthetic peptides with sequences derived from proteins that were known to be proteolyzed. Peptides would be incubated with a target protease and their cleavage assessed, generally through high‐performance liquid chromatography (HPLC) with mass spectrometry for cleavage site identification.12 Once initial substrates were identified, new substrates with variations at select positions would be synthesized to explore subsite specificity. The development of colorimetric and fluorescent peptide substrates simplified cleavage assessment,13, 14, 15 however, defining protease substrate specificity remained an iterative and tedious process. Over the last two decades, this process has been transformed by the development of large and highly diverse peptide libraries. Table 1 summarizes some of the peptide‐based technologies for analysis of protease specificity. The information determined through these approaches can be used for a number of important applications. For example, selective substrates can be designed that enable the real‐time monitoring of proteolysis in vitro and in vivo. Peptide substrates also can be converted into protease inhibitors through coupling to an electrophilic warhead. Furthermore, as proteolytic enzymes recognize their substrates as linear motifs of extended beta strands, specificity information can be used to prioritize potential endogenous substrates. For example, a number of computational approaches have been developed to predict caspase and granzyme B substrates using specificity data determined through peptide‐based profiling methodologies.16, 17 Synthetic substrate synthesis, inhibitor design, and endogenous substrate identification are all critical steps in improving our understanding of the biological role of a given protease in both cellular function and pathogenesis.
Table 1.
Peptide‐Based Protease Activity Profiling Technologies
| Peptide‐Based Profiling Technology | Advantages | Disadvantages |
|---|---|---|
| On‐bead FRET libraries |
High sequence diversity. Useful for analysis of subsite cooperativity. |
Requires prior knowledge of specificity. On‐bead immobilization can produce artifacts. New libraries need to be synthesized for each protease. |
| Positional scanning‐synthetic combinatorial libraries |
High sequence diversity, especially for newer libraries incorporating unnatural amino acids. Simple and validated in‐solution fluorescent assay. |
Generally limited to nonprime specificity profiling. Does not provide information related to subsite cooperativity. |
| Electrophile‐based libraries |
High sequence diversity. Already contain electrophilic warhead for conversion into protease inhibitor. |
Generally limited to nonprime specificity profiling. Primarily limited to serine, threonine, and cysteine proteases. |
| Peptide microarrays |
Can be used to determine kinetic parameters for substrate hydrolysis. Able to analyze protease activity in complex biological samples. Useful for analysis of subsite cooperativity. |
Limited sequence diversity. Immobilization of peptide substrates can produce artifacts. Limited to nonprime specificity profiling. |
| Mixture‐based oriented peptide libraries |
High sequence diversity. Can be used to profile prime and nonprime specificity. |
New libraries often need to be synthesized for nonprime side profiling. Primarily limited to proteases with strong prime side specificity. Requires enrichment step. |
| Multiplex substrate profiling by mass spectrometry |
Can profile prime and nonprime specificity. Can be used to determine kinetic parameters for hydrolysis of substrates. Useful for analysis of subsite cooperativity. Global activity in complex samples can be analyzed. Rationally designed peptide library. |
Relativity limited sequence diversity. Activity is not monitored in real‐time. |
On‐Bead Fluorescent Peptide Libraries
In the late 1980s, Kahne et al. developed a radioassay to monitor the hydrolysis of bead‐bound peptides.18 Although this assay was used to assess the half‐life of an amide bond in neutral water, which remarkably was determined to be ∼7 years, the authors suggested that this technique could also be used to monitor proteolysis. Meldal et al. were the first to carry that idea forward with the development of an on‐bead, combinatorial peptide library for assessing protease specificity.19 These combinatorial libraries were constructed through split peptide synthesis, leading to a single peptide sequence being present on each bead. The bead‐conjugated peptides were constructed with a C‐terminal fluorophore and N‐terminal quencher, resulting in fluorescence quenching through Förster resonance energy transfer (FRET) prior to release of the fluorophore via proteolytic cleavage.20, 21 Positions C‐terminal and N‐terminal to the site of proteolytic cleavage are commonly referred to as prime (P′) and nonprime (P) positions, respectively.22 Following treatment with a protease of interest, the fluorescent beads were isolated and subjected to Edman degradation where N‐terminal residues are labeled and sequentially released to reconstruct the amino acid sequence. However, because each bead contained a mixture of intact and cleaved peptide, it was not possible to differentiate residues released from the native or neo‐N‐termini generated through proteolysis. This generally meant that the cleavage site needed to be predefined to accurately determine if amino acids detected during Edman degradation were from the intact or cleaved peptide. Once the ratio of intact to cleaved peptide was determined, this was used to calculate percent conversion and estimate catalytic efficiency.19
Positional Scanning Substrate Libraries
One of the more widely applied, fluorescence‐based techniques for analysis of protease specificity involves the generation of positional scanning‐synthetic combinatorial libraries (PS‐SCLs). PS‐SCLs consist of distinct pools of peptides in which an amino acid in one position is fixed within each pool, while the other positions contain a mixture of amino acids. Initial PS‐SCLs were used to determine the P4‐P2 specificity of proteases and incorporated a 7‐amino‐4‐methylcoumarin (AMC) fluorophore at the P1′ position [Fig. 1(A)].23 These libraries were restricted to certain P1 residues, such as aspartic acid and lysine, because the SPPS protocol required attachment of the growing peptide chain to the solid support through the P1 amino acid side chain. The development of a bifunctional 7‐amino‐4‐carbamoylmethylcoumarin (ACC) fluorophore, which can be directly attached to a solid support, enabled the development of PS‐SCLs with diversity at the P1 position (Fig. 1).24, 25 P1 diversity significantly increased the number of proteases amenable to analysis through the PS‐SCL approach. PS‐SCLs have been used to profile the P4‐P1 specificity of diverse proteases, including cysteine cathepsins,26 kallikreins,27 caspases,28 and granzymes.29
Figure 1.
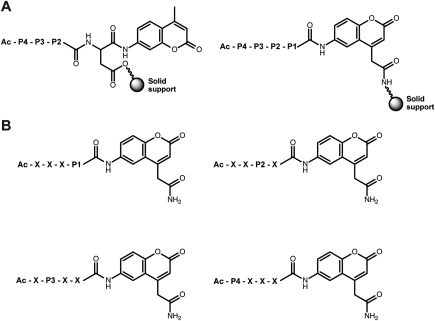
Construction of positional scanning‐synthetic combinatorial libraries for analysis of prime side specificity. (A) During solid‐phase peptide synthesis, 7‐amino‐4‐methylcoumarin (AMC)‐based positional scanning‐synthetic combinatorial libraries are generally conjugated to the solid support via the side chain of the P1 amino acid, while 7‐amino‐4‐carbamoylmethylcoumarin (ACC)‐based libraries are conjugated directly through the fluorophore. (B) The four Pn sublibraries each contain 20 distinct pools of substrates, where one amino acid is fixed at Pn position and the remaining positions (X) contain an equimolar mixture of amino acids.
Drag et al. recently reported the development of ACC positional scanning libraries incorporating up to 110 unnatural amino acids in the P1 to P4 positions.30, 31 These extended libraries, termed hybrid combinatorial substrate libraries (HyCoSuL), were used to identify a neutrophil elastase substrate with the highest reported catalytic efficiency.30 The increased chemical space explored through HyCoSuL has also enabled the development of selective caspase substrates.31 Furthermore, a Counter Selection Substrate Library (CoSeSuL) approach against caspases has been used to develop highly selective legumain probes.32
PS‐SCLs have also been designed to profile prime side specificity through the incorporation of FRET‐based quenching.33, 34, 35, 36, 37, 38 FRET‐based PS‐SCLs contain a fluorophore and quencher separated by several amino acids. Unlike AMC‐ and ACC‐based PS‐SCLs, fluorescence occurs following proteolytic cleavage between any of the amino acids in the peptide substrate. Therefore, mass spectrometry is required to validate the site of cleavage and reconstruct a specificity profile. Recently, Poreba et al. combined FRET‐ and non‐FRET‐based approaches to determine the optimal nonprime and prime side specificity of serine, cysteine, and metalloproteases.39
Electrophile‐Based Libraries
An alternative design for positional scanning libraries incorporates electrophilic “warheads” that have been widely used for activity‐based profiling of enzymes.40, 41 The electrophile is placed at the P1 position and covalently labels the active‐site nucleophile of the target protease. Electrophile‐based libraries contain diversity in the P4–P2 positions and generally use a radiolabel for quantitation of protease labeling.42, 43 These libraries have been successfully used to profile a number of proteases, including cysteine cathepsins, calpains,42 and the proteasome subunits.43 Electrophile‐based libraries are currently limited to cysteine, serine, and threonine proteases because of the requirement of an active site nucleophile and cannot be used to determine nonprime side specificity. However, a unique advantage of these libraries is that they can be readily converted into protease inhibitors, as specificity information is determined in the context of the electrophilic warhead that can be used in an inhibitor. Although beyond the scope of this review, large libraries of electrophile‐containing compounds and fragments have been applied for identifying PTM enzyme inhibitors.44, 45
Microarray Peptide Libraries
Fluorescent peptide libraries are also commonly used in microarray formats to profile protease substrate specificity.46, 47 Peptide microarrays consist of fluorescent substrates that are spatially separated on a microarray surface either by direct covalent attachment or through individual nanodroplets.46, 48, 49 Unlike PS‐SCLs, which use pools of fluorescent substrates, cleavage of individual substrate sequences can be directly assessed with microarrays. This enables the highly multiplexed determination of kinetic parameters for each of the typically hundreds of substrates that are evaluated in a given experiment. Moreover, proteases often exhibit subsite cooperativity due to shared determinants of substrate specificity among binding pockets and the optimal positioning of amino acids within the target sequence. Such subsite cooperativity information is lost when using techniques that rely on pools of substrates, but is readily assessable with microarrays because of the spatial separation of individual substrates. One drawback of the peptide microarrays developed to date is the relatively low sequence diversity as compared to positional scanning methods. With a few notable exceptions,50 peptide microarrays have been most successful when exploring nonprime specificity and other methods are required to query prime side specificity.
Mixture‐Based Oriented Peptide Libraries
Edman degradation of mixture‐based oriented peptide libraries has been used to determine both prime side and nonprime side protease specificity within the same assay format. This profiling strategy developed by Turk et al. uses two separate peptide libraries.51 First, a fully randomized 12‐mer peptide library is synthesized with acetylated N‐termini. This library is partially degraded with a protease of interest, releasing C‐terminal cleavage products with free N‐termini. Edman degradation of the C‐terminal cleavage products is used to determine the frequency of each amino acid at each of the prime side positions. A second 12‐mer peptide library is then synthesized that incorporates a randomized N‐terminal 6‐mer and the most favorable P1′ to P6′ amino acids for the C‐terminal portion. The predefined prime side sequence directs cleavage to the middle of each peptide in the library. All peptides also contain a C‐terminal biotin and free N‐termini. Following protease treatment, C‐terminal cleavage products are removed with immobilized avidin and the remaining N‐terminal products are sequenced by Edman degradation to determine the nonprime side specificity. This approach has been successfully applied to a range of proteases, including matrix metalloproteases,51, 52 anthrax lethal factor,53 and the serine proteases HtrA1/2.54, 55 Although this is a highly versatile technique, one limitation is that new peptide libraries generally need to be synthesized for each investigated protease. Furthermore, these libraries work best for proteases that have strong prime side specificity determinants, as a sequence is required to direct cleavage to the middle of each peptide in the second peptide library.
Multiplex Substrate Profiling by Mass Spectrometry
Mass spectrometry in combination with proteome‐derived substrate libraries has been successfully applied to define protease specificity.56, 57, 58, 59, 60, 61 These “degradomics” methods use liquid chromatography with tandem mass spectrometry (LC‐MS/MS) to identify the site of proteolytic cleavage within proteome‐derived peptides and can define prime side and nonprime side specificity determinants in a single assay. These techniques are quite powerful, but require chemical labeling steps for enrichment and identification of neo‐termini and present a challenge in extracting kinetic parameters.
MSP‐MS was developed to provide simple, yet highly sensitive and quantitative assay for assessing the extended substrate specificity of proteases. This technique currently uses a library of 228 synthetic tetradecapeptides that contain maximal physicochemical diversity within a minimal sequence space.62, 63 This library was designed based on the observation that most proteases require two optimally positioned amino acids for substrate recognition and cleavage. This phenomenon is generally referred to as the “two‐site hypothesis.” For example, the specificity of granzyme B is dominated by a preference for isoleucine at the P4 position and aspartic acid at the P1 position.24, 62 As is evident in the crystal structure of granzyme B, two prominent cavities on the enzyme surface (S1 and S4) accommodate these residues and are the primary determinants of substrate recognition (Fig. 2).64 Though these sites are not the only determinants of enzyme efficiency, they contribute to greater than 70% of the binding energy required for substrate recognition and turnover. Numerous proteases appear to follow the two‐site hypothesis with the sites being juxtaposed, for example, on either side of the scissile bond or separated in space along the substrate by one or two amino acids. Therefore, physicochemical diversity in the MSP‐MS library was generated through incorporation of all neighbor (XY) and near‐neighbor (X*Y, X**Y) amino acid pairings. This simple and chemically defined library enables facile extraction of kinetic parameters for each substrate and is readily amenable to profiling the specificity of purified proteases or complex biological samples without the need for enrichment strategies.
Figure 2.
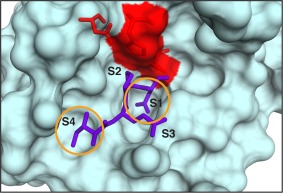
The ecotin peptide (purple) binds to the active site of granzyme B in a linear conformation.64 The catalytic triad is shown in red. Recognition of the peptide, IEPD (written P4‐P1), is dominated by the S4 and S1 pockets (circled in yellow).
For MSP‐MS specificity determination, a recombinant protease or other biological sample of interest is incubated with the peptide library and aliquots are removed at multiple time points (Fig. 3). Cleavage sites within library peptides are then identified through LC‐MS/MS analysis of each time point and specificity is visualized using a sequence logo, which displays protease amino acid preference relative to the site of cleavage. Label‐free quantitation of both parent peptides and their corresponding cleavage products over time can be used to determine kinetic parameters for substrate hydrolysis. This information is critical for the prioritization of optimal sequences for substrate and inhibitor design.
Figure 3.
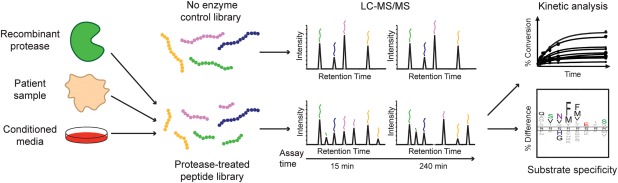
MSP‐MS workflow for protease specificity determination. A recombinant protease, patient sample, conditioned media, or other complex, protease‐containing biological sample is added to the MSP‐MS peptide library. Aliquots are removed at specific time points and peptide cleavage is assessed through LC‐MS/MS analysis. Cleavage‐site identification can be used to construct a sequence logo representation of the global substrate specificity. Cleavage product quantification enables the kinetic analysis of individual substrate cleavage events.
Substrate specificities of a wide range of proteases from all major classes have been interrogated using the MSP‐MS assay. Furthermore, because the termini of the peptides are unmodified, the library is well suited for profiling exopeptidase specificity. In particular, the MSP‐MS assay has been used to profile carboxypeptidases, such as PRCP,62 which are generally not amenable to analysis with most of the previously discussed methods that use peptides modified with reporter groups. Furthermore, the MSP‐MS assay has been used to identify the prime side specificity determinants of aminopeptidases, such as aminopeptidase N.65
The ability of the MSP‐MS library to readily profile mixtures of proteases has transformed our ability to quantitatively characterize proteolytic activity in complex biological systems. The assay was recently used to profile the catalytic subunits of the Plasmodium falciparum proteasome.66 The specificity differences between this and the human proteasome were then used to rationally design selective peptidic inhibitors that attenuated malaria development in vivo. The MSP‐MS assay has been used with protease inhibitors, gene deletions, and immunodepletion in combination with traditional proteomic methods to identify component proteases that are highly active in complex biological samples. This has enabled the “deconvolution” of proteolytic signatures from fungal pathogens,63, 67, 68 parasitic organisms,69, 70 cancer cell lines,62, 71 and patient samples,72, 73 and allowed for the prioritization of proteases based on their functional contribution to the global substrate specificity profile. For example, the MSP‐MS assay was used to analyze the global activity signatures of the opportunistic fungal pathogen Candida albicans in the planktonic and biofilm states.67 Comparison of the activity signatures from each state coupled to inhibitor and proteomic analysis revealed that two specific secreted aspartyl proteases (Saps) are upregulated during biofilm growth and are critical to biofilm formation in vitro and in vivo. MSP‐MS was also recently used to identify two human aspartyl proteases that are selectively upregulated in cystic precursor lesions of pancreatic cancer.73 This strategy enabled the development of a highly accurate diagnostic assay using fluorogenic substrates for differentiating benign and premalignant lesions within a cohort of patient cyst fluid samples.
Kinase Specificity Analysis Using Peptide Library‐Based Methods
The MSP‐MS assay was designed and validated using proteases primarily because they cleave linear peptides and predominately rely on primary amino acid sequence for substrate recognition. Similarly, kinases typically phosphorylate unstructured regions of proteins, and their specificity is strongly dependent upon the amino acid residues surrounding the phosphoacceptor site.74 Available crystal structures of eukaryotic kinases reveal that many kinases, as with proteases, bind their substrates in an extended, linear conformation.75, 76 Computational efforts using these crystal structures have been able to successfully identify endogenous substrates, highlighting the importance of linear peptide sequences in kinase substrate specificity.77
There are numerous peptide‐based approaches for profiling the substrate specificity of kinases.77, 78, 79, 80, 81, 82 These methods primarily employ fluorescence, radioactivity, or colorimetry to detect enzyme activity. For kinase‐directed PS‐SCLs, a series of biotinylated peptides are generated. The central phosphorylatable residue remains fixed and all other positions are varied to query amino acid preferences.77 These peptide libraries are assayed in a multiwell format with the kinase of interest and radiolabeled ATP. Aliquots of the reaction are then transferred to a streptavidin‐coated membrane, and phosphorylation of each peptide substrate is measured via radiography.83 Quantification of the amount of phosphorylation is then used to determine the specificity of the kinase of interest.
The MSP‐MS assay presents a significant improvement upon traditional methods available for profiling kinases. Reporter groups used in other techniques can interfere with kinase‐substrate recognition and radioactivity‐based methods have costly disposal and present health hazards. The label‐free and unbiased design of the MSP‐MS library has made it applicable to the analysis of a wide variety of kinases.84
Kinases without previously known substrate preference have been profiled using MSP‐MS, allowing for the discovery of their key substrate specificity determinants. In addition, the high sensitivity of this assay can allow for profiling of picomolar amounts of kinase.84 This enables the profiling of a kinase from a single immunoprecipitation experiment, which is highly advantageous if the enzyme cannot be readily expressed and purified. The MSP‐MS assay has also been used to obtain kinetic parameters for phosphorylation of individual peptides within the library. This capability has proven useful for analyzing the effect of interacting factors on kinase substrate specificity and catalytic efficiency. For example, MSP‐MS was recently used to interrogate the P‐TEFb–HIV‐1 Tat interaction. P‐TEFb, a human kinase that is integral to Tat's transactivation, phosphorylates RNA Polymerase II and two negative elongation factors to overcome the stalled RNA Pol II complex, which then allows transcription of the integrated viral genome to continue. There is controversy as to which site within RNA Pol II that P‐TEFb phosphorylates, and what effect Tat has on specificity and phosphorylation rate.85, 86, 87 MSP‐MS analysis of recombinant and immunoprecipitated P‐TEFb revealed that P‐TEFb phosphorylates serine 5 within the RNA Pol II C‐terminal domain. Analysis of P‐TEFb with the addition of Tat revealed that Tat selectively increased the catalytic efficiency of P‐TEFb toward peptides that most closely resembled RNA Pol II serine 5.84
MSP‐MS Analysis of Additional PTM Enzymes
There are ∼200 types of PTM enzymes, and many of these enzymes have yet to be characterized.1 In addition to being able to detect proteolytic cleavage and phosphorylation events, the MSP‐MS assay could allow for profiling of a variety of other PTMs [Fig. 4(A)]. The mass spectrometry‐based detection strategy enables high adaptability because most peptide modifications can be readily identified using existing MS data analysis packages. As described, this technology is particularly well suited for the specificity analysis of PTM enzymes that recognize peptide substrates in an extended, linear conformation. For example, crystal structures of the kinase CDK2/cyclin A and the histone acetyltransferase HAT1 bound to peptide substrates revealed that substrates adopt a linear conformation in the enzyme active site [Fig. 4(B,C)].88, 89 Initial MSP‐MS experiments with other types of PTM enzymes have already been carried out. O‐GlcNAc transferase (OGT) was profiled using the MSP‐MS assay, resulting in “HexNAc” modifications at serine and threonine residues on five library peptides (unpublished data). Protein arginine N‐methyltransferase (PRMT1), an arginine‐specific histone methyltransferase, was also assayed using the MSP‐MS library. PRMT1 methylated a single peptide in the library at an arginine residue adjacent to a glycine. This “RG” motif aligns with the known PRMT1 substrate motif, “RGG.”90 These preliminary results underscore that a simplified library of linear peptides can be used to obtain relevant PTM specificity information.
Figure 4.
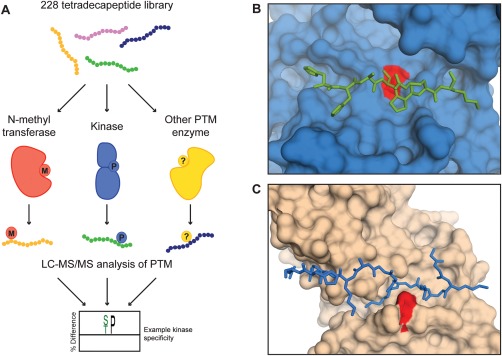
Application of the MSP‐MS library to other PTM enzymes. (A) A general scheme for the specificity analysis of PTM enzymes using the MSP‐MS assay. PTM enzymes are incubated with the peptide library and modification is detected through LC‐MS/MS analysis. (B) The kinase CDK2/cyclin A (shown in blue) recognizes a peptide substrate (depicted as green sticks) in a linear conformation.89 The active site lysine residue is colored red. (C) Similarly, the histone acetyltransferase HAT1 also binds a linear substrate.88
Conclusion
Peptide‐based technologies have significantly expanded our ability to profile the substrate specificity of proteases and other PTM enzymes. The technologies discussed in this review can provide complementary information when applied together, enabling a more complete understanding of the specificity of a particular enzyme. The specificity information discovered can be used for a number of important applications. As mentioned previously, PTM enzyme specificity has facilitated the design of highly selective chemical probes, such as protease inhibitors or fluorescent substrates.30, 66 These types of probes have been particularly useful in enabling the noninvasive detection of a variety of cancers.91, 92 Specificity information can also be used to help better define the biological roles of a given PTM enzyme. For example, PTM enzyme specificity has been used to identify the likely site of post‐translational modification within endogenous substrates.16, 17, 93 This is particularly important when used in combination with degradomic‐based methods or other techniques that identify large potential substrate repertoires. More generally, it is increasingly recognized that PTM enzymes, and particularly proteases, regulate biological systems through large, interconnected enzyme‐inhibitor networks.94, 95, 96 Determining specificity is a critical aspect of identifying likely interaction partners within the context of these networks. This, in turn, helps to define novel enzymatic cascades, allowing for the development of a systems‐level understanding of PTM enzyme biology.
Peptide‐based enzyme profiling technologies have progressed significantly since the advent of SPPS and we expect that they will continue to be critical for improving our understanding of the vast repertoire of cellular functions that PTM enzymes regulate.
Acknowledgments
The authors would like to thank Peter J. Rohweder for assistance and review of the article. Mass spectrometry was performed in collaboration with the UCSF Mass Spectrometry Facility (directed by Alma Burlingame and supported by NIH and the Adelson Medical Research Foundation). The authors declare no conflicts of interest.
Sam L. Ivry and Nicole O. Meyer contributed equally to this work.
Charles Craik is the winner of the 2016 Emil Thomas Kaiser Award.
References
- 1. Walsh CT, Garneau‐Tsodikova S, Gatto GJ (2005) Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed 44:7342–7372. [DOI] [PubMed] [Google Scholar]
- 2. Merrifield RB (1985) Solid phase synthesis (Nobel Lecture). Angew Chem Int Ed 24:799–810. [Google Scholar]
- 3. Behrendt R, White P, Offer J (2016) Advances in Fmoc solid‐phase peptide synthesis. J Pept Sci 22:4–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walter G (1986) Production and use of antibodies against synthetic peptides. J Immunol Methods 88:149–161. [DOI] [PubMed] [Google Scholar]
- 5. Eberle AN, Atherton E, Dryland A, Sheppard RC (1986) Peptide synthesis. Part 9. Solid‐phase synthesis of melanin concentrating hormone using a continuous‐flow polyamide method. J Chem Soc Perkin Trans 1:361. [Google Scholar]
- 6. Mijalis AJ, Thomas DA, Simon MD, Adamo A, Beaumont R, Jensen KF, Pentelute BL (2017) A fully automated flow‐based approach for accelerated peptide synthesis. Nat Chem Biol 13:464–466. [DOI] [PubMed] [Google Scholar]
- 7. López‐Otín C, Overall CM (2002) Protease degradomics: a new challenge for proteomics. Nat Rev Mol Cell Biol 3:509–519. [DOI] [PubMed] [Google Scholar]
- 8. auf dem Keller U, Schilling O (2010) Proteomic techniques and activity‐based probes for the system‐wide study of proteolysis. Biochimie 92:1705–1714. [DOI] [PubMed] [Google Scholar]
- 9. Vizovišek M, Vidmar R, Fonović M, Turk B (2016) Current trends and challenges in proteomic identification of protease substrates. Biochimie 122:77–87. [DOI] [PubMed] [Google Scholar]
- 10. Puente XS, Sánchez LM, Overall CM, López‐Otín C (2003) Human and mouse proteases: a comparative genomic approach. Nat Rev Genet 4:544–558. [DOI] [PubMed] [Google Scholar]
- 11. Diamond SL (2007) Methods for mapping protease specificity. Curr Opin Chem Biol 11:46–51. [DOI] [PubMed] [Google Scholar]
- 12. Stentz FB, Kitabchi AE, Schilling JW, Schronk LR, Seyer JM (1989) Identification of insulin intermediates and sites of cleavage of native insulin by insulin protease from human fibroblasts. J Biol Chem 264:20275–20282. [PubMed] [Google Scholar]
- 13. Zimmerman M, Yurewicz E, Patel G (1976) A new fluorogenic substrate for chymotrypsin. Anal Biochem 70:258–262. [DOI] [PubMed] [Google Scholar]
- 14. Zimmerman M, Ashe B, Yurewicz EC, Patel G (1977) Sensitive assays for trypsin, elastase, and chymotrypsin using new fluorogenic substrates. Anal Biochem 78:47–51. [DOI] [PubMed] [Google Scholar]
- 15. Mares‐Guia M, Shaw E, Cohen W (1967) Studies on the active center of trypsin. J Biol Chem 242:5777–5782. [PubMed] [Google Scholar]
- 16. Backes C, Kuentzer J, Lenhof H‐P, Comtesse N, Meese E (2005) GraBCas: a bioinformatics tool for score‐based prediction of Caspase‐ and Granzyme B‐cleavage sites in protein sequences. Nucleic Acids Res 33:W208–W213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barkan DT, Hostetter DR, Mahrus S, Pieper U, Wells JA, Craik CS, Sali A (2010) Prediction of protease substrates using sequence and structure features. Bioinformatics 26:1714–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kahne D, Still WC (1988) Hydrolysis of a peptide bond in neutral water. J Am Chem Soc 110:7529–7534. [Google Scholar]
- 19. Meldal M, Svendsen I, Breddam K, Auzanneau FI (1994) Portion‐mixing peptide libraries of quenched fluorogenic substrates for complete subsite mapping of endoprotease specificity. Proc Natl Acad Sci USA 91:3314–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosse G, Kueng E, Page MG, Schauer‐Vukasinovic V, Giller T, Lahm HW, Hunziker P, Schlatter D (2000) Rapid identification of substrates for novel proteases using a combinatorial peptide library. J Comb Chem 2:461–466. [DOI] [PubMed] [Google Scholar]
- 21. Ekici OD, Zhu J, Chung IYW, Paetzel M, Dalbey RE, Pei D (2009) Profiling the substrate specificity of viral protease VP4 by a FRET‐based peptide library approach. Biochemistry 48:5753–5759. [DOI] [PubMed] [Google Scholar]
- 22. Schechter I, Berger A (1967) On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27:157–162. [DOI] [PubMed] [Google Scholar]
- 23. Rano TA, Timkey T, Peterson EP, Rotonda J, Nicholson DW, Becker JW, Chapman KT, Thornberry NA (1997) A combinatorial approach for determining protease specificities: application to interleukin‐1beta converting enzyme (ICE). Chem Biol 4:149–155. [DOI] [PubMed] [Google Scholar]
- 24. Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS (2000) Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc Natl Acad Sci USA 97:7754–7759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Backes BJ, Harris JL, Leonetti F, Craik CS, Ellman JA (2000) Synthesis of positional‐scanning libraries of fluorogenic peptide substrates to define the extended substrate specificity of plasmin and thrombin. Nat Biotechnol 18:187–193. [DOI] [PubMed] [Google Scholar]
- 26. Choe Y, Leonetti F, Greenbaum DC, Lecaille F, Bogyo M, Brömme D, Ellman JA, Craik CS (2006) Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J Biol Chem 281:12824–12832. [DOI] [PubMed] [Google Scholar]
- 27. Debela M, Magdolen V, Schechter N, Valachova M, Lottspeich F, Craik CS, Choe Y, Bode W, Goettig P (2006) Specificity profiling of seven human tissue kallikreins reveals individual subsite preferences. J Biol Chem 281:25678–25688. [DOI] [PubMed] [Google Scholar]
- 28. Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia‐Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. J Biol Chem 272:17907–17911. [DOI] [PubMed] [Google Scholar]
- 29. Mahrus S, Craik CS (2005) Selective chemical functional probes of granzymes A and B reveal granzyme B is a major effector of natural killer cell‐mediated lysis of target cells. Chem Biol 12:567–577. [DOI] [PubMed] [Google Scholar]
- 30. Kasperkiewicz P, Poreba M, Snipas SJ, Parker H, Winterbourn CC, Salvesen GS, Drag M (2014) Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc Natl Acad Sci USA 111:2518–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Poreba M, Kasperkiewicz P, Snipas SJ, Fasci D, Salvesen GS, Drag M (2014) Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates. Cell Death Differ 21:1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poreba M, Solberg R, Rut W, Lunde NN, Kasperkiewicz P, Snipas SJ, Mihelic M, Turk D, Turk B, Salvesen GS, Drag M (2016) Counter selection substrate library strategy for developing specific protease substrates and probes. Cell Chem Biol 23:1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cotrin SS, Puzer L, De Souza Judice WA, Juliano L, Carmona AK, Juliano MA (2004) Positional‐scanning combinatorial libraries of fluorescence resonance energy transfer peptides to define substrate specificity of carboxydipeptidases: assays with human cathepsin B. Anal Biochem 335:244–252. [DOI] [PubMed] [Google Scholar]
- 34. Bersanetti PA, Andrade MCC, Casarini DE, Juliano MA, Nchinda AT, Sturrock ED, Juliano L, Carmona AK (2004) Positional‐scanning combinatorial libraries of fluorescence resonance energy transfer peptides for defining substrate specificity of the angiotensin I‐converting enzyme and development of selective C‐domain substrates. Biochemistry 43:15729–15736. [DOI] [PubMed] [Google Scholar]
- 35. Tanskul S, Oda K, Oyama H, Noparatnaraporn N, Tsunemi M, Takada K (2003) Substrate specificity of alkaline serine proteinase isolated from photosynthetic bacterium, Rubrivivax gelatinosus KDDS1. Biochem Biophys Res Commun 309:547–551. [DOI] [PubMed] [Google Scholar]
- 36. Puzer L, Cotrin SS, Alves MFM, Egborge T, Araújo MS, Juliano MA, Juliano L, Brömme D, Carmona AK (2004) Comparative substrate specificity analysis of recombinant human cathepsin V and cathepsin L. Arch Biochem Biophys 430:274–283. [DOI] [PubMed] [Google Scholar]
- 37. Wysocka M, Lesner A, Majkowska G, Legowska A, Guzow K, Rolka K, Wiczk W (2010) The new fluorogenic substrates of neutrophil proteinase 3 optimized in prime site region. Anal Biochem 399:196–201. [DOI] [PubMed] [Google Scholar]
- 38. Wysocka M, Wojtysiak A, Okońska M, Gruba N, Jarząb M, Wenta T, Lipińska B, Grzywa R, Sieńczyk M, Rolka K, Lesner A (2015) Design and synthesis of new substrates of HtrA2 protease. Anal Biochem 475:44–52. [DOI] [PubMed] [Google Scholar]
- 39. Poreba M, Szalek A, Rut W, Kasperkiewicz P, Rutkowska‐Wlodarczyk I, Snipas SJ, Itoh Y, Turk D, Turk B, Overall CM, Kaczmarek L, Salvesen GS, Drag M (2017) Highly sensitive and adaptable fluorescence‐quenched pair discloses the substrate specificity profiles in diverse protease families. Sci Rep 7:43135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanman LE, Bogyo M (2014) Activity‐based profiling of proteases. Annu Rev Biochem 83:249–273. [DOI] [PubMed] [Google Scholar]
- 41. Niphakis MJ, Cravatt BF (2014) Enzyme inhibitor discovery by activity‐based protein profiling. Annu Rev Biochem 83:341–377. [DOI] [PubMed] [Google Scholar]
- 42. Greenbaum DC, Arnold WD, Lu F, Hayrapetian L, Baruch A, Krumrine J, Toba S, Chehade K, Brömme D, Kuntz ID, Bogyo M (2002) Small molecule affinity fingerprinting: a tool for enzyme family subclassification, target identification, and inhibitor design. Chem Biol 9:1085–1094. [DOI] [PubMed] [Google Scholar]
- 43. Nazif T, Bogyo M (2001) Global analysis of proteasomal substrate specificity using positional‐scanning libraries of covalent inhibitors. Proc Natl Acad Sci USA 98:2967–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bachovchin DA, Koblan LW, Wu W, Liu Y, Li Y, Zhao P, Woznica I, Shu Y, Lai JH, Poplawski SE, Kiritsy CP, Healey SE, DiMare M, Sanford DG, Munford RS, Bachovchin WW, Golub TR (2014) A high‐throughput, multiplexed assay for superfamily‐wide profiling of enzyme activity. Nat Chem Biol 10:656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Backus KM, Correia BE, Lum KM, Forli S, Horning BD, González‐Páez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF (2016) Proteome‐wide covalent ligand discovery in native biological systems. Nature 534:570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Salisbury CM, Maly DJ, Ellman JA (2002) Peptide microarrays for the determination of protease substrate specificity. J Am Chem Soc 124:14868–14870. [DOI] [PubMed] [Google Scholar]
- 47. Winssinger N, Damoiseaux R, Tully D, Geierstanger B, Burdick K, Harris JL (2004) PNA‐encoded protease substrate microarrays. Chem Biol 128:189–190. [DOI] [PubMed] [Google Scholar]
- 48. Gosalia DN, Salisbury CM, Ellman JA, Diamond SL (2005) High throughput substrate specificity profiling of serine and cysteine proteases using solution‐phase fluorogenic peptide microarrays. Mol Cell Proteom 4:626–636. [DOI] [PubMed] [Google Scholar]
- 49. Gosalia DN, Salisbury CM, Maly DJ, Ellman JA, Diamond SL (2005) Profiling serine protease substrate specificity with solution phase fluorogenic peptide microarrays. Proteomics 5:1292–1298. [DOI] [PubMed] [Google Scholar]
- 50. Barrios AM, Craik CS (2002) Scanning the prime‐site substrate specificity of proteolytic enzymes: a novel assay based on ligand‐enhanced lanthanide ion fluorescence. Bioorgan Med Chem Lett 12:3619–3623. [DOI] [PubMed] [Google Scholar]
- 51. Turk BE, Huang LL, Piro ET, Cantley LC (2001) Determination of protease cleavage site motifs using mixture‐based oriented peptide libraries. Nat Biotechnol 19:661–667. [DOI] [PubMed] [Google Scholar]
- 52. Park HI, Turk BE, Gerkema FE, Cantley LC, Sang QXA (2002) Peptide substrate specificities and protein cleavage sites of human endometase/matrilysin‐2/matrix metalloproteinase‐26. J Biol Chem 277:35168–35175. [DOI] [PubMed] [Google Scholar]
- 53. Turk BE, Wong TY, Schwarzenbacher R, Jarrell ET, Leppla SH, Collier RJ, Liddington RC, Cantley LC (2004) The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat Struct Mol Biol 11:60–66. [DOI] [PubMed] [Google Scholar]
- 54. Martins LM, Turk BE, Cowling V, Borg A, Jarrell ET, Cantley LC, Downward J (2003) Binding specificity and regulation of the serine protease and PDZ domains of HtrA2/Omi. J Biol Chem 278:49417–49427. [DOI] [PubMed] [Google Scholar]
- 55. Chien J, He X, Shridhar V (2009) Identification of tubulins as substrates of serine protease HtrA1 by mixture‐based oriented peptide library screening. J Cell Biochem 107:253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schilling O, Overall CM (2008) Proteome‐derived, database‐searchable peptide libraries for identifying protease cleavage sites. Nat Biotechnol 26:685–694. [DOI] [PubMed] [Google Scholar]
- 57. Biniossek ML, Niemer M, Maksimchuk K, Mayer B, Fuchs J, Huesgen PF, McCafferty DG, Turk B, Fritz G, Mayer J, Haecker G, Mach L, Schilling O (2016) Identification of protease specificity by combining proteome‐derived peptide libraries and quantitative proteomics. Mol Cell Proteom 15:2515–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Staes A, Van Damme P, Helsens K, Demol H, Vandekerckhove J, Gevaert K (2008) Improved recovery of proteome‐informative, protein N‐terminal peptides by combined fractional diagonal chromatography (COFRADIC). Proteomics 8:1362–1370. [DOI] [PubMed] [Google Scholar]
- 59. Kleifeld O, Doucet A, auf dem Keller U, Prudova A, Schilling O, Kainthan RK, Starr AE, Foster LJ, Kizhakkedathu JN, Overall CM (2010) Isotopic labeling of terminal amines in complex samples identifies protein N‐termini and protease cleavage products. Nat Biotechnol 28:281–288. [DOI] [PubMed] [Google Scholar]
- 60. Vizovišek M, Vidmar R, Van Quickelberghe E, Impens F, Andjelković U, Sobotič B, Stoka V, Gevaert K, Turk B, Fonović M (2015) Fast profiling of protease specificity reveals similar substrate specificities for cathepsins K, L and S. Proteomics 15:12. [DOI] [PubMed] [Google Scholar]
- 61. Mahrus S, Trinidad JC, Barkan DT, Sali A, Burlingame AL, Wells JA (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134:866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. O'Donoghue AJ, Eroy‐reveles AA, Knudsen GM, Ingram J, Zhou M, Statnekov JB, Greninger AL, Hostetter DR, Qu G, Maltby DA, Anderson MO, DeRisi JL, McKerrow JH, Burlingame AL, Craik CS (2012) Global identification of peptidase specificity by multiplex substrate profiling. Nat Methods 9:1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. O'Donoghue AJ, Knudsen GM, Beekman C, Perry JA, Johnson AD, DeRisi JL, Craik CS, Bennett RJ (2015) Destructin‐1 is a collagen‐degrading endopeptidase secreted by Pseudogymnoascus destructans, the causative agent of white‐nose syndrome. Proc Natl Acad Sci USA 112:E3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Waugh SM, Harris JL, Fletterick R, Craik CS (2000) The structure of the pro‐apoptotic protease granzyme B reveals the molecular determinants of its specificity. Nat Struct Biol 7:762–765. [DOI] [PubMed] [Google Scholar]
- 65. Joshi S, Chen L, Winter MB, Lin Y‐L, Yang Y, Shapovalova M, Smith PM, Liu C, Li F, LeBeau AM (2017) The rational design of therapeutic peptides for aminopeptidase N using a substrate‐based approach. Sci Rep 7:1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li H, O'Donoghue AJ, van der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, da Fonseca PCA, Bogyo M (2016) Structure‐ and function‐based design of Plasmodium‐selective proteasome inhibitors. Nature 530:233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Winter MB, Salcedo EC, Lohse MB, Hartooni N, Gulati M, Sanchez H, Takagi J, Hube B, Andes DR, Johnson AD, Craik CS, Nobile CJ (2016) Global identification of biofilm‐specific proteolysis in Candida albicans . MBio 7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Clarke SC, Dumesic PA, Homer CM, O'Donoghue AJ, La Greca F, Pallova L, Majer P, Madhani HD, Craik CS, May RC (2016) Integrated activity and genetic profiling of secreted peptidases in Cryptococcus neoformans reveals an aspartyl peptidase required for low pH survival and virulence. PLoS Pathog 12:e1006051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Goupil LS, Ivry SL, Hsieh I, Suzuki BM, Craik CS, O'Donoghue AJ, McKerrow JH (2016) Cysteine and aspartyl proteases contribute to protein digestion in the gut of freshwater Planaria. PLoS Negl Trop Dis 10:e0004893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dvořák J, Fajtová P, Ulrychová L, Leontovyč A, Rojo‐Arreola L, Suzuki BM, Horn M, Mareš M, Craik CS, Caffrey CR, O'Donoghue AJ (2015) Excretion/secretion products from Schistosoma mansoni adults, eggs and schistosomula have unique peptidase specificity profiles. Biochimie 122:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Donoghue AJO, Ivry SL, Chaudhury C, Hostetter DR, Hanahan D, Craik CS (2016) Procathepsin E is highly abundant but minimally active in pancreatic ductal adenocarcinoma tumors. Biol Chem 397:871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. O'Donoghue AJ, Jin Y, Knudsen GM, Perera NC, Jenne DE, Murphy JE, Craik CS, Hermiston TW (2013) Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PLoS One 8:e75141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ivry SL, Sharib JM, Dominguez DA, Roy N, Hatcher SE, Yip‐Schneider M, Schmidt CM, Brand RE, Park WG, Hebrok M, Kim GE, O'Donoghue AJ, Kirkwood KS, Craik CS (2017) Global protease activity profiling provides differential diagnosis of pancreatic cysts. Clin Cancer Res 23:4865–4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Songyang Z, Lu KP, Kwon YT, Tsai L‐H, Filhol O, Cochet C, Brickey DA, Soderling TR, Bartleson C, Graves DJ, DeMaggio AJ, Hoekstra MF, Blenis J, Hunter T, Cantley LC (1996) A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin‐ dependent kinase II, CDK5, and Erk1. Mol Cell Biol 16:6486–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Brinkworth RI, Breinl RA, Kobe B (2003) Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc Natl Acad Sci USA 100:74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Johnson LN, Lowe ED, Noble MEM, Owen DJ (1998) The structural basis for substrate recognition and control by protein kinases. FEBS Lett 430:1–11. [DOI] [PubMed] [Google Scholar]
- 77. Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE (2004) A rapid method for determining protein kinase phosphorylation specificity. Nat Methods 1:27–29. [DOI] [PubMed] [Google Scholar]
- 78. Wu JJ, Phan H, Lam KS (1998) Comparison of the intrinsic kinase activity and substrate specificity of c‐Abl and Bcr‐Abl. Bioorgan Med Chem Lett 8:2279–2284. [DOI] [PubMed] [Google Scholar]
- 79. Rychlewski L, Kschischo M, Dong L, Schutkowski M, Reimer U (2004) Target specificity analysis of the Abl kinase using peptide microarray data. J Mol Biol 336:307–311. [DOI] [PubMed] [Google Scholar]
- 80. Rodriguez M, Li SSC, Harper JW, Songyang Z (2004) An Oriented Peptide Array Library (OPAL) strategy to study protem‐protein interactions. J Biol Chem 279:8802–8807. [DOI] [PubMed] [Google Scholar]
- 81. Luo K, Zhou P, Lodish HF (1995) The specificity of the transforming growth factor beta receptor kinases determined by a spatially addressable peptide library. Proc Natl Acad Sci USA 92:11761–11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pearson RB, Kemp BE (1991) Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol 200:62–81. [DOI] [PubMed] [Google Scholar]
- 83. Chen C, Turk BE (2010) Analysis of serine‐threonine kinase specificity using arrayed positional scanning peptide libraries. Curr Protoc Mol Biol Chapter 18: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Meyer NO, O'Donoghue AJ, Schulze‐Gahmen U, Ravalin M, Moss SM, Winter MB, Knudsen GM, Craik CS (2017) Multiplex substrate profiling by mass spectrometry for kinases as a method for revealing quantitative substrate motifs. Anal Chem 89:4550–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schüller R, Forné I, Straub T, Schreieck A, Texier Y, Shah N, Decker T‐M, Cramer P, Imhof A, Eick D (2016) Heptad‐specific phosphorylation of RNA polymerase II CTD. Mol Cell 61:305–314. [DOI] [PubMed] [Google Scholar]
- 86. He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T, Zhou Q (2010) HIV‐1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV‐1 transcription. Mol Cell 38:428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Czudnochowski N, Bösken CA, Geyer M (2012) Serine‐7 but not serine‐5 phosphorylation primes RNA polymerase II CTD for P‐TEFb recognition. Nat Commun 3:842. [DOI] [PubMed] [Google Scholar]
- 88. Alterio V, De Simone G, Monti SM, Scozzafava A, Supuran CT (2007) Carbonic anhydrase inhibitors: inhibition of human, bacterial, and archaeal isozymes with benzene‐1,3‐disulfonamides‐solution and crystallographic studies. Bioorganic Med Chem Lett 17:4201–4207. [DOI] [PubMed] [Google Scholar]
- 89. Cook A, Lowe ED, Chrysina ED, Skamnaki VT, Oikonomakos NG, Johnson LN (2002) Structural studies on phospho‐CDK2/cyclin A bound to nitrate, a transition state analogue: implications for the protein kinase mechanism. Biochemistry 41:7301–7311. [DOI] [PubMed] [Google Scholar]
- 90. Wooderchak WL, Zang T, Zhou ZS, Acuña M, Tahara SM, Hevel JM (2008) Substrate profiling of PRMT1 reveals amino acid sequences that extend beyond the “RGG” paradigm. Biochemistry 47:9456–9466. [DOI] [PubMed] [Google Scholar]
- 91. Blum G, von Degenfeld G, Merchant MJ, Blau HM, Bogyo M (2007) Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity‐based probes. Nat Chem Biol 3:668–677. [DOI] [PubMed] [Google Scholar]
- 92. Whitley MJ, Cardona DM, Lazarides AL, Spasojevic I, Ferrer JM, Cahill J, Lee C‐L, Snuderl M, Blazer DG, Hwang ES, Greenup RA, Mosca PJ, Mito JK, Cuneo KC, Larrier NA, O'Reilly EK, Riedel RF, Eward WC, Strasfeld DB, Fukumura D, Jain RK, Lee WD, Griffith LG, Bawendi MG, Kirsch DG, Brigman BE (2016) A mouse‐human phase 1 co‐clinical trial of a protease‐activated fluorescent probe for imaging cancer. Sci Transl Med 8:320ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Song R, Oren D. a, Franco D, Seaman MS, Ho DD (2013) Strategic addition of an N‐linked glycan to a monoclonal antibody improves its HIV‐1‐neutralizing activity. Nat Biotechnol 31:1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Overall CM, Dean RA (2006) Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev 25:69–75. [DOI] [PubMed] [Google Scholar]
- 95. Fortelny N, Cox JH, Kappelhoff R, Starr AE, Lange PF, Pavlidis P, Overall CM (2014) Network analyses reveal pervasive functional regulation between proteases in the human protease web. PLoS Biol 12:e1001869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mason SD, Joyce JA (2011) Proteolytic networks in cancer. Trends Cell Biol 21:228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]