

Abstract
Chronic obstructive pulmonary disease (COPD) and cystic fibrosis (CF) share molecular mechanisms that cause the pathological symptoms they have in common. Here, we review evidence suggesting that hyperactivity of the EGFR/ADAM17 axis plays a role in the development of chronic lung disease in both CF and COPD. The ubiquitous transmembrane protease A disintegrin and metalloprotease 17 (ADAM17) forms a functional unit with the EGF receptor (EGFR), in a feedback loop interaction labeled the ADAM17/EGFR axis. In airway epithelial cells, ADAM17 sheds multiple soluble signaling proteins by proteolysis, including EGFR ligands such as amphiregulin (AREG), and proinflammatory mediators such as the interleukin 6 coreceptor (IL-6R). This activity can be enhanced by injury, toxins, and receptor-mediated external triggers. In addition to intracellular kinases, the extracellular glutathione-dependent redox potential controls ADAM17 shedding. Thus, the epithelial ADAM17/EGFR axis serves as a receptor of incoming luminal stress signals, relaying these to neighboring and underlying cells, which plays an important role in the resolution of lung injury and inflammation. We review evidence that congenital CFTR deficiency in CF and reduced CFTR activity in chronic COPD may cause enhanced ADAM17/EGFR signaling through a defect in glutathione secretion. In future studies, these complex interactions and the options for pharmaceutical interventions will be further investigated.
1. Introduction
Airway epithelium, apart from providing a structural barrier against microbes and inhaled particles, also plays an active role in the first line of inflammatory responses [1], and thus emerged as a therapeutic target in chronic lung disease [2]. Airway epithelial cells respond dynamically to bacterial and viral infections [3–5] or inhaled noxious particles by transducing inflammatory signals [6, 7]. They produce cytokines, growth factors, and other inflammatory mediators to orchestrate epithelial repair and adaptation and recruit a range of inflammatory cells including neutrophils and macrophages [1, 8, 9]. Importantly, airway epithelial cells also crosstalk with the underlying connective tissue [10, 11].
This process normally leads to full resolution of inflammation and tissue damage. However, in chronic lung disease, exaggerated trans-signaling can lead to permanent changes of tissue structure and loss of lung function, chronic inflammation, and mucous metaplasia.
Despite their different etiology, cystic fibrosis (CF), a congenital abnormality caused by mutations in a chloride transporter, and chronic obstructive pulmonary disease (COPD), caused by chronic exposure to airborne irritants, share many common features in their pathology detailed in the paragraphs below (Figure 1) [12–14]. Both diseases are characterized by progressive and essentially irreversible lung damage, chronic bronchitis, bacterial colonization, reduced mucociliary clearance, and mucus plugging.
Figure 1.

Common features of CF and COPD. CF and COPD lung disease share many common clinical manifestations despite the differences with respect to etiology. Mutations in the CFTR gene determine CF lung disease. Recently, CS has been shown to affect CFTR channel activity and this is recognized as a factor that may contribute to a CF-like phenotype in COPD. Thus, COPD has been recently described as “acquired CF.”
This leads to the question whether these diseases, while quite different in many aspects, share common molecular mechanisms and therapeutic targets (Figure 1) [15–17]. In this review, we focus on a signaling pathway called the EGFR/ADAM17 axis and its potential role in chronic lung disease, in particular CF and COPD. Similarly, but outside the scope of this review, other forms of chronic inflammatory lung disease, including asthma, can be added to this list.
A disintegrin and metalloprotease 17 (ADAM17), a ubiquitous sheddase formerly known as TACE (Figure 2), that releases a broad spectrum of soluble biologically active ligands from airway epithelial and myeloid cells is recognized as an important transducer of airway epithelial cis- and trans-signaling [18]. As detailed below, ADAM17 interacts with the EGF receptor (EGFR), mainly through the shedding of EGFR ligands, and conversely, EGFR is required for ADAM17 activity, in a feedback signaling cascade labelled the EGFR/ADAM17 axis. This system is highly sensitive to various extracellular triggers, including cigarette smoke and bacterial toxins, resulting in the enhanced shedding of a large repertoire of growth factors, cytokines, and cytokine receptors that are substrates of ADAM17. This in turn leads to paracrine and autocrine receptor activation, which plays an important role in the resolution of airway inflammation and tissue damage.
Figure 2.
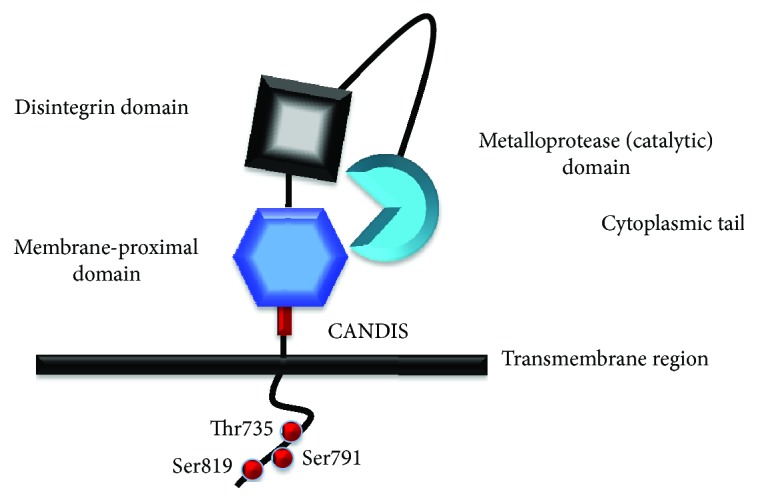
ADAM17 domain structure. ADAM17 is an atypical member of the ADAM family. It has additional disulfide bonds in the metalloprotease domain, and it lacks two calcium binding sites in the disintegrin domain. The membrane proximal domain (MPD), replacing the cysteine-rich and EGF-like domains, with a novel alpha/beta fold has a shorter cysteine-rich segment. The MPD has cysteine residues determining the ADAM17 conformation (open/closed) and ADAM17 protease activity (active/inactive switch). The MPD is in close proximity to the active site likely due to a C-shaped conformation of the extracellular part of mature ADAM17 [99]. ADAM17 lacks an EGF-like domain, so the MPD is followed by the juxtamembrane region “conserved ADAM17 dynamic interaction sequence” (CANDIS) involved in substrate recognition [162]. The trans-membrane region ends with a cytoplasmic tail with phosphorylation sites, which are likely important for ADAM17 activity and trafficking [97–101].
In this review, we focus on the role of the epithelial EGFR/ADAM17 signaling pathway that transmits signals from luminal receptors towards underlying tissue (paracrine) and epithelial cells (autocrine) (Figure 3), affecting inflammation and remodeling. However, EGFR and ADAM17 are ubiquitously expressed, and also the expression of ADAM17 substrates is not limited to epithelial cells. Therefore, the complete signaling cascade also includes fibroblasts, smooth muscle cells, and myeloid cells.
Figure 3.

ADAM17 dependent paracrine and autocrine signaling. A disintegrin and metalloproteinase 17 (ADAM17), also known as a tumor necrosis factor-α converting enzyme (TACE), is involved in the immune defense mechanisms mediated by epithelial cells. ADAM17 releases extracellular domains of transmembrane proteins to produce soluble bioactive signaling proteins taking part in autocrine (activation of receptors within the same epithelial cell layer) and paracrine signaling (activation of cellular receptors on underlying neighboring cells, also termed transactivation). Among the ADAM17 substrates are (1) adhesion proteins (L-selectin, ICAM), (2) transmembrane mucins (MUC-1), (3) membrane-bound cytokines (TNF-α), (4) growth factors (AREG) and other ligands of EGFR (TGF-α, EREG, HB-EGF, and epigen), and (5) cytokine receptors (IL-6R, TNF-R). Ectodomain shedding provides the mechanism for autocrine and paracrine signaling. IL-6R shed from epithelial cells transactivates gp130 on the underlying myofibroblasts, whereas AREG shed from epithelial cells activates EGFR on epithelial cells or on the underlying fibroblasts. The ADAM17 inhibitor TIMP-3 inhibits the ADAM17 proteolytic activity.
The relative contribution of the different cell populations in the EGFR/ADAM17 signaling pathway in humans is still under investigation. Current knowledge is largely based on studies with conditional mutant murine models, in which ADAM17 can be selectively ablated in specific cell types. Knocking out ADAM17 in mesenchymal cells did not produce a detectable developmental phenotype, whereas epithelial ablation resulted in severe abnormalities [19]. Horiuchi et al. showed that the epithelial EGFR/ADAM17 axis plays a major role in tissue regeneration during colonic inflammation and that loss of myeloid expression does not have a major effect. However, myeloid ablation of ADAM17 does reduce the lethality of endotoxin shock induced by intraperitoneal injection [20]. While these studies give important insight in the basic architecture and activity of this system, due caution is required when applied to human tissues. In addition to obvious differences in architecture and function of human and mouse lungs, substrate specificity and expression patterns of members of the ADAM sheddase families and ligand interactions with receptors will differ.
We propose, based on evidence in the literature discussed below, that the molecular mechanism underlying the development of CF and COPD lung disease may involve abnormal activation of the epithelial ADAM17/EGFR axis. To put this in perspective, we first review CF and COPD pathology and the unmet need in clinical intervention. Next, we discuss the role of the EGFR/ADAM17 axis in lung pathology, the molecular mechanisms involved, and potential therapeutic strategies.
2. Cystic Fibrosis: A Congenital Lung Disease with an Early Onset
CF is an autosomal recessive lung disease caused by more than 2000 different mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene [21] with approximately 80,000 patients worldwide. The most common CF mutation is the deletion of a phenylalanine at amino acid position 508 in the NBD1 domain of CFTR protein (F508del) [22]. The CFTR gene encodes a chloride channel mainly expressed in the apical membrane of secretory epithelial cells [23]. CFTR has a key role in maintaining ion and water homeostasis in secretory epithelia [23]. Mutations in CFTR cause a multiorgan CF disease affecting the lungs, intestine, pancreas, liver, sinuses, and reproductive organs [24]. However, under present treatment, the main morbidity and mortality among CF patients is due to lung malfunction; consequently, most available and experimental treatments aim to prevent the progression of CF lung disease [25]. CFTR is also involved in transport of other anions, for instance bicarbonate [26] and glutathione [27, 28]. Bicarbonate is crucial to normal expression of mucins that in CF remain aggregated and poorly solubilized [29]. Glutathione, as a natural antioxidant, reduces oxidative cellular stress [30].
2.1. CF Lung Disease: An Early Disorder of Distal Airways
Advances in imaging and monitoring of the respiratory system in infant patients have revealed that severe CF lung pathology starts early in childhood and progresses irreversibly over time [31–33]. This early onset of lung abnormalities includes bronchiectasis, diagnosed thickening and dilation of the bronchial walls, air trapping, and atelectasis (partial collapse of the lung) [34, 35]. These symptoms occur simultaneously with reduced mucociliary clearance and mucus plugging [36]. Quantitative and standardized tracking of early lung disease progression in infants with CT scans is pursued to advance the comparative analysis and provide the evaluation of the treatment [37, 38].
In a recent micro CT and histological study of end-stage CF lungs, the dilatation and obstruction of distal airways and a severe reduction in the number of functional terminal bronchioles were clearly documented for the first time. This confirms that obstruction and remodeling of peripheral airways is a prominent feature in CF lung disease and therefore are prime targets of experimental therapy [14]. CF mouse models [39] and large CF animal models, like pig [40] and ferret [41], are helpful in the investigation of the mechanism involved in this early onset of CF lung disease and intervention therapy, but all have considerable limitations due to species variation. In addition to state-of-the-art clinical and biomarker studies, there is an urgent need for the development of organotypic and personalized cell culture models in which the complex molecular and cellular interactions involved in CF lung pathology can be studied.
2.2. Mucociliary Transport
In healthy subjects, mucociliary clearance requires CFTR-dependent balanced fluid and proper mucus secretion from surface cells and subepithelial glands. However, high mucus viscosity in CF is caused by reduced CFTR-dependent bicarbonate secretion, required for proper expansion of secreted mucus molecules [42, 43]. Furthermore, CF patients have intrinsically impaired ciliary beat frequency (CBF), which is not only dependent on CFTR-mediated bicarbonate transport but also regulated by soluble adenyl cyclase (sAC) [44]. Together, this impairs effective clearance of bacteria and inhaled particles in CF lung [45, 46]. Importantly, reduced mucociliary clearance is also a feature of COPD, which may be in part related to reduced CFTR activity in smokers' lungs [47].
2.3. Bacterial Infection and Inflammation
Airway epithelial cell cultures from CF patients have reduced air-surface liquid (ASL) height, presumably due to defective CFTR-dependent fluid secretion [48]. Additionally, as a consequence of reduced bicarbonate secretion, CFTR deficiency abnormally acidifies ASL [49] which impairs bacterial killing [50, 51], inhibits the activity of ASL antimicrobials [52], and increases ASL viscosity of newborn CF piglets [45].
Impaired bacterial killing and mucociliary clearance in CF lungs together facilitate colonization with opportunistic pathogens generally harmless in normal individuals [53]. Despite activation of inflammatory responses mediated by the innate and cellular immune system, eradication of bacterial infection is impaired in CF lungs. Instead, bacterial infections induce a massive and chronic recruitment of neutrophils, which ultimately contributes to irreversible airway remodeling, observed as air trapping, bronchiolar obstruction, bronchiectasis, and loss of function [14].
2.4. Sterile Lung Inflammation in CF
While chronic infection is undoubtedly a major issue in CF pathology, it remains contested whether inflammation in CF only results from bacterial infection or is an intrinsic property of CFTR-deficient mucosa. Lung disease and inflammation is already observed in CF infants before bacterial colonization, suggesting that inflammation may precede infection in CF lung. Several reports argue that bacterial infections are indispensable to inflammatory responses, in CF humans [54] or CF pig [55]. However, unchallenged CF mutant mice show inflammation [56]. Also, in the CF ferret model, inflammation and tissue remodeling do not require previous bacterial infections [57]. Such sterile inflammation may be caused by functional abnormalities in CF myeloid cells, in particular macrophages [58, 59], dendritic cells [56], and neutrophils [60] or may be primarily related to abnormal cytokine signaling by CFTR-deficient airway epithelial cells. Several studies suggest that CFTR malfunction leads to overexpression of growth factors and proinflammatory cytokines as a cell-autonomous defect [61–63]
Due to the early onset of CF lung disease and its irreversible nature, it is clear that CF patients require early intervention therapy [32]. Excessive lung inflammation and tissue remodeling observed in CF may be an inherent property of CFTR-deficient lung mucosa. Therefore, it is important to establish the mechanisms involved in CFTR-related inflammatory responses and whether alleviation of inflammatory responses is beneficial in the management of CF lung disease [64].
3. Strategies of CF Therapy
CF lung disease is still the main cause of morbidity and mortality in CF despite intensive treatment [14]. Indirect pharmacological management focuses on anti-inflammatory agents [65, 66], antibiotics [67], and mucolytic agents [68]. Direct pharmacological management of CF disease intends to restore the functional expression of mutated CFTR at the plasma membrane by correcting its folding and gating defect [69]. In 2015, the FDA approved ORKAMBI® for homozygous F508del CFTR patients, comprising fifty percent of the CF population, the combination of CFTR corrector lumacaftor (VX-809) and potentiator ivacaftor (VX-770) in one pill. This therapy improves lung function in patients homozygous for the F508del mutation, although modestly and not in all patients [25, 70]. Therefore, further investigations are in progress to find more effective compounds [71, 72]. So far, the effects of correctors and potentiators on infections and the release of inflammatory mediators have not been broadly investigated in clinical studies. Rowe et al. showed that ivacaftor reduces P. aeruginosa isolated from CF patients carrying a CFTR gating mutation after 6 months treatment. However, the free neutrophil elastase and other inflammation markers like IL-1β, IL-6, and CXCL8 (IL-8) in sputum samples remained unchanged [73]. Therefore, anti-inflammatory, antibacterial, and other additional therapies are still important targets of investigation [74].
In summary, recent breakthroughs in the development of small-molecule compounds targeting the mutant CFTR protein have raised hope to find a cure for CF. However, the presently available CFTR correctors and potentiators are not sufficiently effective in a majority of CF patients, and the search for new compounds and additional therapies is still highly relevant. Though CF is considered monogenetic, the downstream responses to CFTR deficiency and responses to therapeutic intervention are highly variable in the population. The perfect corrector and potentiator combination tailored to the individual patient, supported by additional anti-inflammatory medication (personalized medicine), would likely be the best solution.
4. COPD Is Acquired CF?
Chronic obstructive pulmonary disease (COPD) is the 5th ranking cause of death worldwide. Usually, COPD is characterized by chronic bronchitis and emphysema [75]. Similar to CF, bronchiectasis [76] and peripheral airway thickening are also observed, similar to early and advanced CF lung disease [77]. Some individuals develop lung disease dominated by emphysema, while others exhibit chronic bronchitis. This heterogeneous phenotype likely reflects the contribution of multiple pathogenic mechanisms and the genetic heterogeneity of the population. Once COPD starts to develop, it tends to worsen over time, and so far its progress cannot be controlled effectively in most patients. The most prominent etiological factor leading to COPD is cigarette smoke, and also exposure to fumes, chemicals, and dust [78]. Although COPD and CF differ in primary cause, the spectra of the pathological events overlap considerably (Figure 1). Chronic bacterial infections with frequent exacerbations are observed, including colonization with the opportunistic pathogen Pseudomonas aeruginosa, which are also a hallmark of CF lung disease. However, the strains adapted to COPD lungs appear to differ from CF, suggesting that although similar mechanisms are involved, the luminal milieu in COPD differs from that in CF [79]. Both diseases are characterized by excessive mucus production and insufficient clearance, leading to lower airway obstruction and chronic neutrophilic infiltration. In CF and COPD, airway surface liquid (ASL) dehydration and viscous mucus secretion impair mucociliary clearance, causing chronic inflammation and facilitating recurrent infections [80]. A wide variety of proinflammatory mediators in COPD (like CXCL8, IL-6, and CCL18) overlap with CF-related mediators [74, 81]. There are also parallels on the cellular level which include goblet cell metaplasia, hyperplasia of myoblasts, and extensive extracellular matrix production [17].
The broad spectrum of common features and events observed in CF and COPD encouraged researchers to seek common factors for these diseases. Cigarette smoke decreases CFTR mRNA expression and reduces CFTR protein level through accelerated degradation, leading to impaired mucociliary clearance and depleted ASL in vitro and in vivo [82–84]. Mice treated with cigarette smoke show reduced CFTR activity that can be corrected by the cAMP agonist and phosphodiesterase inhibitor roflumilast, restoring CFTR activity [85]. Furthermore, macrophage phagocytosis and CFTR activity are impaired by cigarette smoke [86]. Similarly, CFTR-deficient macrophages are abnormal [59]. These are short-term and transient effects in normal cells, which do not explain the progressive inflammatory lung disease of COPD patients that stopped smoking. However, several reports show that the abnormal characteristics of COPD epithelial cells, including reduced CFTR expression in situ, persist in culture [87], probably due to a combination of genetic factors and epigenetic imprinting. Furthermore, COPD patients, smokers, and former smokers show signs of persistent reduced CFTR function in upper and lower airways, which may contribute to chronic lung disease [80]. Consequently, several authors described COPD as an “acquired CF” through e reduction of CFTR activity, suggesting that these diseases with different etiology have common therapeutics options [16, 17, 80, 85].
5. Strategies of Therapy in COPD
Based on the previous arguments, CFTR potentiators and correctors, such as those developed to enhance the activity of mutant CFTR, could be useful to treat COPD. However, small molecules designed to target mutant CFTR trafficking and gating may actually reduce activity of normal CFTR upon chronic treatment [88]. Nevertheless, further screening and testing of novel compounds in a personalized setting may lead to improvement of CFTR function and reduction of lung disease in COPD patients.
Chronic inflammation is recognized as the major pathophysiological mechanism of COPD progression, with molecular targets overlapping those of CF (e.g., IL-6, CXCL8, and CCL18) [74]. Thus, chronic airway inflammation remains an important therapeutic target in COPD management. It often persists after cessation of smoking, suggesting that apart from the role of CFTR also epigenetic changes in the resolution of inflammation likely play a role [89, 90]. Presently, several compounds targeting inflammatory responses in COPD are under investigation [91, 92], though none of these have been shown to be beneficial in COPD patients as yet.
Therefore, as in CF, COPD is a complex multifactorial disease, with large variation in the patient population due to largely undefined genetic and environmental factors. A personalized approach using multiple treatments is likely required, but a robust trial strategy is elusive. The advance of personalized 3D culture models, combining induced stem cell (iPSC) and gene editing [93] with microfluidics (“lung-on-a-chip”) technology [94] may allow us to proceed towards better treatment.
6. Epithelial EGFR/ADAM17 Axis: A Potential Therapeutic Target in CF and COPD Lung Disease
The design of anti-inflammatory agents aims mainly to decrease the neutrophil influx into the lung and concomitant inflammatory responses [8]. The importance of airway epithelial cells in inflammatory responses has been recognized for decades, but the signaling network is still under intense investigation [1, 95]. Airway epithelium serves as the first barrier and acts as defense against daily inhaled air pollutants and microbes by mucociliary clearance and secretion of a range of cytokines, cytokine receptors, growth factors, growth factor receptors, and antimicrobial peptides [1, 96]. Airway epithelial cells not only release inflammatory mediators to ASL but also signal to the underlying tissues (myocytes or fibroblasts). One of the mechanisms controlling paracrine and autocrine proinflammatory and profibrotic signaling in airway epithelium is the EGFR/ADAM17 axis.
A disintegrin and metalloproteinase 17 (ADAM17), also known as a tumor necrosis factor-α converting enzyme (TACE), is a transmembrane protein with proteolytic activity. It releases extracellular domains of its substrates, generally transmembrane proteins, to produce soluble bioactive signaling proteins, in a process called shedding. Mature ADAM17 consists of several functional domains (Figure 2), an extracellular metalloprotease domain (catalytic), a disintegrin domain, a membrane proximal domain (MPD) rich in cysteine residues, and a “conserved ADAM-seventeen dynamic interaction sequence” (CANDIS). Unlike other ADAM family members, it lacks an EGF-like domain [97–101]. These extracellular domains are connected to a transmembrane region and cytoplasmic tail with phosphorylation sites that likely are involved in ADAM17 activation or trafficking (Figure 2).
Ectodomain shedding mediated by the metalloprotease domain of ADAM17 provides a mechanism for initiation or inhibition of autocrine/paracrine signaling. So far, 76 proteins have been identified as substrates of ADAM17 [18]. They encompass membrane-bound cytokines (TNF-α), cytokine receptors (IL-6R, TNF-R), growth factors, in particular ligands of EGFR (TGF-α, AREG, EREG, HB-EGF, and epigen), adhesion proteins (L-selectin, ICAM-1), and transmembrane mucins (MUC-1). The shed soluble forms of these proteins are bioactive transducers of cell signaling via activation of cellular receptors on underlying neighboring cells (transactivation/paracrine activation), but they also are involved in activation of the shedding cells and neighboring cells (autocrine activation) (Figure 3).
Because most of the epidermal growth factor receptor (EGFR) ligands are cleaved by ADAM17, this sheddase has emerged as an important transducer of the airway epithelial autocrine and paracrine EGFR signaling (Figure 3). EGFR and ADAM17 are both involved in the broad spectrum of events that is characteristic of both CF and COPD lung disease, like excessive mucus expression [102–104], cytokine secretion [105], airway epithelial cell wound healing [106, 107], abnormal airway proliferation [108], maintenance of barrier integrity, and progressive lung tissue scarring [109]. They both are activated upon bacterial or viral infection and during inflammation [3, 5, 110]. While this is an effective and necessary response, it is suggested that exaggerated airway epithelial signaling in the chronic state may enhance inflammation and may lead to permanent damage to the lung structure.
EGFR functions as a sensor of airway epithelial integrity [111]. When cells have intact tight junctions, EGFR is not activated. But disruption of the epithelial cell integrity, either by mechanical injury or cytokine treatment (TNF-α/IFN-γ), leads to EGFR phosphorylation and concomitant inhibition of protein phosphatase 2A activity [112]. Cigarette smoke exposure of differentiated HBEC also leads to damage of the lung tissue observed as destruction of epithelial cell integrity, loss of E-cadherin/β-catenin complex, and disappearance of cilia [113]. This coincides with phosphorylation and perinuclear trafficking of EGFR [113] suggesting the importance of EGFR in maintenance of epithelial cell barrier integrity. The response of ADAM17 to loss of pulmonary epithelial cell integrity has been also shown by neuregulin-1 (NRG-1) shedding and concomitant activation of human epidermal growth factor receptor-2 (HER2) [114]. This raises the question whether and how EGFR and ADAM17 cooperate in sensing responses to airway injury.
6.1. EGFR and ADAM17 Are Important in Lung Development
Inactivation of ADAM17 by deletion of the zinc binding domain through homologous recombination in mice led to severely hypoplastic lungs at birth, reduced branching morphogenesis and alveolar development, impaired epithelial cell proliferation, and differentiation and delay in vasculogenesis [115]. Due to the early mortality of ADAM-KO mice, several alternative ADAM17 mouse models have been developed, with reduced activity (hypomorphic) or conditional mutants [116]. Conditional knockout ADAM17 mice can serve as a model to investigate the role of ADAM17 in organogenesis, in specific cell types, and in inflammatory responses and tissue remodeling [20, 117]. Induced dysfunction of ADAM17 (ADAM17flox/flox SPC-rtTA TetO-Cre) in developing lung epithelial cells reduced saccular formation, cell proliferation, and lung epithelial cell differentiation, but the mice were born without severe respiratory distress [19]. Knocking out the gene in mesenchymal cells using a Dermo1-Cre transgene did not produce a detectable phenotype [19]. This suggests that epithelial, but not mesenchymal, ADAM17 plays a prominent role in lung development.
Other studies show that conditional knockout mice (ADAM17flox/flox with R26Cre-ER) treated with human neutrophil elastase have attenuated goblet cell metaplasia in comparison to the wild-type mice [118], suggesting that ADAM17 is involved in injury-induced airway metaplasia. Similar to ADAM17-KO mice, EGFR-KO mice survive for up to 8 days after birth and suffer from impaired epithelial development in several organs, including the lungs, underlining the need of a functional EGFR/ADAM17 axis for proper functioning and development of lung epithelium [119].
6.2. ADAM17 and EGFR Crosstalk
ADAM17 works in association with the tyrosine kinase receptor EGFR firstly by shedding most of its ligands, including AREG, HB-EGF, TNF-α, EPGN (epigen), and EREG (epiregulin) [18], resulting in activation of EGFR. In humans, only two EGFR ligands, EGF and betacellulin, are shed by ADAM10 [120, 121], a close relative of ADAM17 [18]. Crosstalk of ADAM17 and EGFR in inflammatory signaling transduction is further defined by the establishment of a positive ADAM17/EGFR feedback loop likely involving activation of ADAM17 via the EGFR/MAPK pathway [110, 122] (Figure 4). The exact molecular mechanism of this feedback signaling has not been firmly established. Moreover, ADAM17 regulates transcription of EGFR mRNA by cleavage of Notch1 and thus increases EGFR expression in a non-small lung carcinoma cell line [123], providing another positive feedback mechanism of the EGFR/ADAM17 axis (Figure 4).
Figure 4.
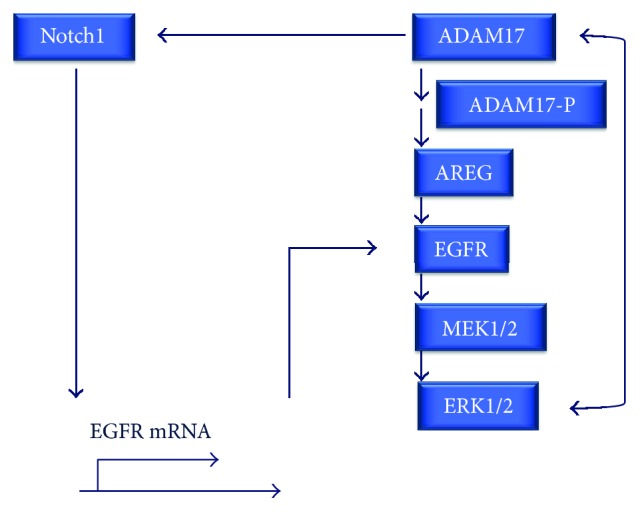
The ADAM17/EGFR axis. The EGFR/ADAM17 positive feedback loop likely involves activation of ADAM17 via the EGFR/MAPK pathway and direct interaction of ERK1/2 with ADAM17. Additionally, by cleavage of Notch1 from a non-small lung carcinoma cell line ADAM17 regulates transcription of EGFR mRNA and increases EGFR expression on the cell surface, providing another mechanism contributing to positive feedback regulation.
In differentiated primary bronchial epithelial cells (HBEC-ALI), inhibitors of EGFR and of ADAM17 completely abolished ADAM17 substrate shedding as well as EGFR-dependent CXCL8 mRNA induction [87], illustrating the strong interrelationship between the key elements of the EGFR/ADAM17 axis (Figure 4).
6.3. The Role of ADAM17 and EGFR in Balanced Regulation of Lung Inflammation and Regeneration
ADAM17 is expressed in human bronchial epithelial cells in the large and small bronchi, in lung smooth muscle cells, lung muscular vessels, alveolar macrophages, perivascular leukocytes, and lung endothelial cells. It has been long recognized as an important regulator of lung tissue homeostasis [116, 124, 125], pro- and anti-inflammatory responses [18], and tissue regeneration [122, 126, 127].
The anti- and proinflammatory properties of the EGFR/ADAM17 signaling pathway are context and cell type dependent [18]. For instance, in airway epithelial cells, ADAM17 together with EGFR induces mRNA expression and protein release of CXCL8, a neutrophil chemotactic factor that promotes inflammation [7, 87, 105]. However, by shedding TNF receptor type 2 (TNFR2) [128], which antagonizes TNF-α, ADAM17 exhibits also anti-inflammatory properties [129]. Thus, it is likely that the type of stimulus and substrate selection determine the effect of the EGFR and ADAM17 activity [130–132].
EGFR/ADAM17 signaling in tissue regeneration encompasses wound healing [122], proliferation [126, 127], differentiation [19, 133], and cell migration [134]. Due to the involvement of EGFR/ADAM17 paracrine and autocrine signaling in several lung disorders [109, 116], modulation of ADAM17 and EGFR activation in both COPD and CF is important, to keep the balance between anti-inflammatory processes and promotion of inflammation, and also between regeneration and excessive tissue remodeling. Indeed, we recently found that cigarette smoke induced shedding of the ADAM17 substrate amphiregulin (AREG), and IL-6R was enhanced in differentiated bronchial cells in culture obtained from COPD patients compared to non-COPD, suggesting that epigenetic factors controlling the activity of the ADAM17/EGFR axis are affected in COPD [87].
6.4. Release of Cytokines, Growth Factors, and Mucins Depends on ADAM17/EGFR Signaling
External stress factors like oxidative stress, viral and bacterial toxins, and CS exposure activate the ADAM17/EGFR signaling pathway. Pathogens inhaled into the airways, or exposure to other stimuli, activate Toll-like receptors (TLR) [135–137] and G-coupled receptors (GPCR) [138] that crosstalk with downstream ADAM17/EGFR signaling. As a result of cigarette smoke extract exposure, secretion of downstream proinflammatory cytokines including CXCL8 [7, 139], growth factors (TGFα, AREG, and HB-EGF) [87, 105], cytokine receptor IL-6R [87], mucins (MUC5AC), and phosphorylated MUC1 is induced in an ADAM17/EGFR-dependent manner [103, 113, 140, 141]. This cascade of events involves mitogen-activated protein kinases (MAPK), MAPK1 (ERK1), MAPK2 (ERK2), and p38-MAPK [3, 142]. Upon activation of the ERK or p38 MAPK pathway, ADAM17 dissociates from an endogenous extracellular tissue inhibitor of metalloproteinase-3 (TIMP-3) [143-145], accumulates on the cell surface [146, 147], and induces release of TGF-α [146]. Acrolein, an active component of cigarette smoke, which induces MUC5AC mRNA in an ADAM17 and EGFR dependent manner, decreases TIMP-3 transcript levels, also suggesting a role of TIMP-3 in ADAM17 activation.
6.5. CFTR Deficiency Affects the EGFR/ADAM17 Axis
Several lines of evidence suggest that CFTR deficiency affects the activity of the EGFR/ADAM17 axis. F508del CFTR mutant mice respond differently from normal to airway injury by naphthalene [148]. A week after injury, we observed a significantly enhanced mRNA expression of amphiregulin (AREG) compared to normal [149]. AREG mRNA expression is dependent both on ADAM17 and EGFR activity in human bronchial epithelial cells [87], suggesting a link between CFTR deficiency and EGFR/ADAM17 activity during resolution of airway injury. Furthermore, inhibition of CFTR with a small molecule (inh-172) in NCI-H292 cells reportedly activates CXCL8 production in an EGFR/ADAM17-dependent way [61]. However, the relationship between ADAM17/EGFR signaling and CFTR deficiency is still poorly understood and may involve a variety of mechanisms [150].
Exaggerated responses mediated by the EGFR/ADAM17/MAPK pathway have also been reported in CFTR-deficient cells compared to CFTR expressing counterparts. The CFTR-deficient cell line IB3 produces more CXCL8 than an isogenic CFTR-expressing cell line. The CFTR-deficient cell line CuFi-1 produces more CXCL8 in response to heat-inactivated P. aeruginosa than a non-CFTR-deficient cell line (NuLi-1), and this involves EGFR phosphorylation and ERK1/2 activation [151]. Most of these studies use undifferentiated, submerged immortalized cell lines [152], compare genetically diverse cell lines (like CuFi and NuLi or IB3 and C38 cells) [61, 151, 153, 154], or use a CFTR inhibitor [61] with reported off-target effects [155]. Further studies in primary human bronchial epithelial cells in air-liquid interface culture, and models that allow comparison of genetically identical populations in parallel, including CFTR-deficient and corrected induced pluripotent stem cells (iPSC) and inducible CFTR-expressing CFBE cells, are currently performed to further support the notion that CFTR is involved in the activity of the EGFR/ADAM17 axis and may contribute to abnormal resolution of injury and inflammation in CF lung disease.
6.6. Redox Potential: A Possible Link between CFTR Deficiency and ADAM17/EGFR Signaling?
The intra- and extracellular redox potential changes in response to physiological processes and in pathophysiological conditions [156]. Reactive oxygen species (ROS), produced during cellular stress [74], are an important regulator of redox state and they are also involved in activation of the EGFR/ADAM17 signaling pathway, affecting TGF-α and AREG release and mucin expression [103, 104, 135]. Some studies point towards the role of NADPH oxidases (NOX), in particular dual oxidase 1 (DUOX1) [104, 135, 157] or dual oxidase 2 (DUOX2) [136], which produce ROS at an extracellular or possibly intravesicular domain. ATP-mediated DUOX1 activation involves a TGF-α/ADAM17/EGFR/ERK signaling pathway [157]. Recent studies indicate a role of DUOX1 in allergen-dependent SRC/EGFR activation in airway cells [158]. However, the mechanism by which ROS affect EGFR/ADAM17 signaling in intact airway cells is likely highly complex and still remains not well understood.
ROS do not only affect receptors, phosphatases, and kinases in the ADAM17/EGFR pathway, but ADAM17 [159] and EGFR [160] themselves are redox-sensitive proteins. EGFR has intracellular cysteine residues in the active site that are targets of ROS and determine EGFR kinase activity, likely through association of EGFR with NADPH oxidase, NOX2 [160]. ADAM17 activity is regulated by thiol-disulfide isomerization in the extracellular MPD domain mediated by protein disulfide isomerase (PDI), an oxidoreductase sensitive to redox changes [161]. PDI, by direct interaction with the membrane proximal domain (MPD) [159], changes the disulfide bridge pattern and thus the conformation of the extracellular protease domain from open active to closed inactive state leading to the inhibition of ADAM17 activity (Figure 5) [101, 162]. Redox-dependent conformational changes likely make ADAM17 sensitive to the extracellular redox potential [159].
Figure 5.

ADAM17 is a redox-sensitive protein. The ADAM17 membrane proximal domain (MPD), which is in close proximity to the active site, is sensitive to extracellular (or intravesicular) redox changes. This redox-sensitive ADAM17 activity is regulated by thiol-disulfide isomerization mediated by protein disulphide isomerase (PDI), an oxidoreductase sensitive to redox changes. PDI changes the disulfide bridge pattern and thus the conformation of the extracellular protease domain from an open active to a closed inactive state, by direct interaction with the membrane proximal domain (MPD), leading to the inhibition of ADAM17 proteolytic activity. Redox sensitive conformational changes likely make ADAM17 sensitive to the extracellular redox potential, which is dependent on glutathione (GSH/GSSH) and ROS.
Since CFTR deficiency is thought to increase ROS levels [163], which would activate ADAM17 and EGFR [122] and inactivate protein phosphatases [164], we propose that redox signaling is a plausible link between EGFR/ADAM17 activity and CFTR deficiency. CFTR deficiency may change the extracellular redox potential to more oxidized in airway, because CFTR is involved in epithelial glutathione transport at the apical membrane [27, 165], which serves as a natural antioxidant [30]. This would in turn enhance the activity of ADAM17 through the mechanism illustrated in Figure 5. Polymorphisms in the glutathione pathway are modifiers of CF lung pathology, emphasizing the importance of this pathway [166]. Further studies are required to establish the relationship between glutathione transport, extracellular redox potential, and the activity of the EGFR/ADAM17 axis in CF airways. This can be achieved by application of fluorescent protein redox probes that can be expressed in different cellular compartments [156] in advanced airway culture models.
7. Role of Amphiregulin in EGFR/ADAM17-Dependent Lung Disease
One of the prominent ADAM17-dependent EGFR agonists produced ubiquitously by the human lung is amphiregulin (AREG) [167] by human airway epithelial cells [87, 105], smooth muscle cells [168], and fibroblasts [111]. AREG is also expressed by infiltrating and resident lung myeloid cells, including activated macrophages [169], eosinophils [170], dendritic cells [171], neutrophils [172], and mast cells [173]. Human AREG is synthetized as an N-glycosylated transmembrane precursor (50 kDa)[174] with a basolateral sorting motif [175] and shed mainly by ADAM17 [110]. Proteolytic cleavage of the AREG extracellular domain releases several AREG-soluble forms, predominantly an α-N-glycosylated 43 kDa form [174], which is one of the EGFR ligands, with lower affinity to EGFR than EGF and TGF-α.
7.1. AREG Is Induced in Lung Disease
AREG is involved in inflammation and repair responses through autocrine and paracrine activation of EGFR, and generally induced in lung disease [176]. In CF sputum samples, elevated levels of AREG have been shown in airway blood neutrophils [172]. In lung biopsies from asthma patients, more AREG is expressed than in healthy controls [167]. Other studies showed that in sputum of asthma patients AREG is upregulated only during an acute attack [177], suggesting its role in quick cellular responses to the triggers. Increase of AREG in sputum samples from children with asthma negatively correlates with lung function [178] and positively correlates with the number of eosinophils [179, 180].
Epithelial secretion of AREG in situ has not been investigated in CF and COPD in comparison to controls. However, Zuo et al. reported enhanced AREG expression in smoking-induced airway lesions [181], and we observed a stronger AREG shedding response to cigarette smoke in cultured airway cells from COPD patients compared to normal [87] suggesting a possible involvement in the progression of lung disease. Current studies are aimed at elucidating the role of CFTR deficiency in AREG shedding.
7.2. Regulation of AREG Transcription and Shedding
In vitro, AREG mRNA expression and protein release are induced upon exposure to different stress factors like histamine [167], diesel exhaust particles [6], cigarette smoke extract exposure [182], cigarette smoke [87], and rhinoviruses [3]. Also, AREG protein secretion is dependent on the EGFR/MAPK pathway in an airway epithelial cell line treated with particulate matter [183]. In differentiated primary airway cells, CS induction of AREG mRNA levels is abolished by ADAM17 and EGFR inhibitors, consistent with a prominent role of the EGFR/ADAM17 axis in AREG signaling and mRNA synthesis [87].
7.3. AREG Affects Mucus and Cytokine Secretion in Asthmatic Patients
In asthmatic patients, AREG produced by mast cells enhances mucus production [184]. AREG-dependent MUC5AC mRNA level induction has been shown upon exposure to particulate matter in NCI-H292 cells [185] and differentiated primary airway cells NHBE-ALI [177]. AREG-dependent secretion of MUC5AC, TGF-β1, and CXCL8 was also observed in epithelial cell line culture supernatants [179]. All these findings suggest an important role of AREG in mucus secretion and cytokine release in airway epithelial cells.
7.4. AREG in Paracrine Signaling and Tissue Remodeling
In addition to its role in epithelial proliferation and differentiation (autocrine), AREG is also involved in fibrotic and inflammatory responses (Figure 3). Conditioned media from AREG-stimulated airway epithelial cells induced expression of CXCL8, VEGF, COX-2, and AREG in human airway smooth muscle cells, providing proof for paracrine crosstalk between epithelial cells and connective tissue in an AREG-dependent manner [168](Figure 6). Inhibition of AREG or EGFR in TGF-β1-stimulated lung fibroblasts diminished AREG-dependent fibroblast proliferation, expression of α-smooth muscle actin and collagen [186], strongly suggesting a role of EGFR/ADAM17/AREG signaling in pulmonary fibrosis in vivo. AREG stimulation also induces airway smooth muscle cell proliferation, which leads to airway remodeling in vivo [180].
Figure 6.

Classic and trans IL-6R signaling. In “classic” signaling membrane-bound IL-6R stimulated by secreted IL-6 associates with a homodimer of signal transducing glycoprotein (gp130) to activate downstream signaling molecules, including the ubiquitous transcription factor STAT-3. In trans-signaling, the soluble form of IL-6R (sIL-6R,) generated either by ADAM17 or by alternative mRNA splicing, binds to IL-6 and this IL-6/IL-6R complex activates gp130 on the airway epithelial cells (autocrine IL-6R trans-signaling) or underlying fibroblasts, myoblasts, or smooth muscle cells that do not express IL-6R (paracrine IL-6R trans-signaling), but do express gp130 to evoke activation of STAT-3 signaling.
These data together suggest that exaggerated and chronic AREG release may contribute to mucus plugging, excessive inflammation, and tissue remodeling in CF and COPD. Therefore, the EGFR/ADAM17/AREG signaling pathway is a potential therapeutic target to regulate both inflammation and lung cell proliferation in COPD and CF. However, AREG is only one of the many players involved in the autocrine and paracrine signaling leading to lung pathology. There is no available literature presenting intervention in AREG function in clinical trials.
8. Role of IL-6R in Human Lung Inflammation and Tissue Regeneration
Another prominent ADAM17 substrate involved in lung disease is the IL-6 coreceptor IL-6R. In “classic” signaling, this transmembrane receptor type I binds to IL-6, which evokes an association with a homodimer of signal transducing glycoprotein, gp130. This trimer dimerizes to form a hexameric complex composed of IL-6, IL-6R, and gp130 [187] (Figure 6). Alternatively, the extracellular domain of IL-6R can also evoke a trans-signaling cascade, when it is shed by ADAM17, producing a soluble form (sIL-6R) (Figure 6) [188] or ADAM10 [188–190]. sIL-6R can be also generated by alternative splicing of IL-6R mRNA [191–193]. However, data suggest that shedding rather than the alternative splicing takes part in trigger-induced generation of the sIL-6R [128]. Indeed, we observed that in primary bronchial epithelial cells in air liquid culture, cigarette smoke enhanced ADAM17-dependent sIL-6R shedding but not the production of the alternatively spliced mRNA [87]. Both the alternatively spliced form and the shed form of sIL-6R can create functional complexes with IL-6 (IL-6/sIL-6R) [194], mediating a transfer of stress responses from epithelial cells to underlying mesenchymal and myeloid cells, involved in tissue remodeling and inflammation (trans-signaling, Figure 6).
IL-6 activated membrane bound IL-6R has anti-inflammatory, antiapoptotic, and regenerative properties [128, 195–198]. In contrast, trans-signaling mediated by IL-6/sIL-6R complexes binding to gp130 is thought to maintain inflammation and promote inflammation-associated cancer [128, 195, 199, 200] (Figure 6).
Importantly, IL-6R expression is not ubiquitous, but restricted to specific cell types, such as lymphoid cells, hepatocytes, and airway epithelial cells [4, 128, 201, 202]. However, cells that do not express IL-6R but do express gp130 can still transduce IL-6 signals via binding of IL-6/sIL-6R complexes in trans. Thus, smooth muscle cells and endothelial cells are responsive to IL-6 through IL-6/sIL-6R trans-signaling provided by epithelial cells [203, 204] (Figure 6).
The role of IL-6R in CF and COPD has not been studied in detail. In COPD patients, elevated levels of IL-6R have been observed in peripheral blood leukocytes [205] and sputum samples [206]. Recently, genetic variants of IL-6R have been linked with lung function [207] and COPD severity [208]. In contrast to elevated IL-6R levels in sputum of COPD patients [207], the levels of sIL-6R in BALF from CF patients were not different in comparison to control, possibly due to enhanced degradation of sIL-6R by serine proteases [209]. Furthermore, it is not evident what the impact of luminal IL-6R on the activity of the subepithelial tissue is, which is separated from the lumen by tight junctions. Shedding of IL-6R from epithelial cells occurs mainly towards the basal side where it can engage in paracrine and autocrine signaling (Figure 6) [4, 87]. Inhibition of sIL-6R by intraperitoneal injection of a recombinant decoy receptor (gp130Fc) attenuates pulmonary fibrosis, whereas activation of IL-6 trans-signaling in cell lines enhances fibroblast proliferation and extracellular matrix protein production [210], which are the hallmarks of the pulmonary fibrosis progression.
Together, these data show the importance of IL-6R trans-signaling in inflammation and lung remodeling and offer possibilities for therapeutic interventions [14].
8.1. Species Specificity of IL-6R Signaling
Murine models have been invaluable in the study of inflammatory disease, but should be analyzed with caution. Species differences in IL-6R mediated signaling have been observed. Human IL-6 stimulates human and murine cells, whereas murine IL-6 only stimulates murine IL-6R signaling [211]. Because of this species specificity, transgenic mice expressing human sIL-6R from a liver promoter did not bind the endogenous murine IL-6, and as a consequence the transgenic animals do not have a transgene specific phenotype [211, 212]. Garbers et al. reported that human IL-6R is a substrate of human ADAM17, but murine IL-6R is a substrate of murine ADAM10 [213]. However, Schumacher et al. subsequently revealed that trigger-induced shedding of both human and mouse IL-6R is mediated by murine ADAM17, but constitutive release of IL-6R is largely mediated by mADAM10 in mice [190]. Additionally, in humans, but not in mice, the sIL-6R can be generated by translation from an alternatively spliced mRNA [190, 214]. Of note, the soluble gp130 form, which circulates in human plasma, blocks IL-6R trans-signaling responses and does not show species specificity, meaning that it interacts with human and mouse sIL-6R complexes [215]. All of this has implications in the extrapolation of IL-6R data obtained in mouse models to human, but is consistent with a prominent role of EGFR/ADAM17 sIL-6R trans-signaling in human lung pathology.
8.2. Dual Control of the STAT3 Transcription Pathway by the EGFR/ADAM17 Axis
IL-6R-mediated signaling leads to activation of the transcription factor STAT3 [216], ERK [217], and PI3K [201], linking this signaling pathway with the EGFR/ADAM17 axis. STAT3 is also activated through a parallel pathway in trans-signaling, which involves shed EGFR ligands [218](Figure 7). The STAT3 pathway is involved in tissue repair [198], carcinogenesis [9, 219], and immune responses [220]. Notably, STAT3 was found to be a modifier gene of cystic fibrosis lung disease [221], and enhanced STAT3 phosphorylation was observed in lung tissue from smokers and COPD patients [222]. Consequently, a disturbance in the control of the complex EGFR/ADAM17/STAT3 pathway would likely play a role in Cystic Fibrosis and COPD chronic lung disease.
Figure 7.

ADAM17/EGFR signaling an important player in CF and COPD lung disease. In airway epithelial cells in culture, both CFTR deficiency [61] and cigarette smoke exposure [87] cause enhanced activity of the EGFR/ADAM17 signaling cascade, and thus enhanced release of proinflammatory cytokines and growth factors that are ADAM17 substrates and signaling molecules that are downstream of the EGFR/MAPK pathway (Figure 1) including the proinflammatory CXCL8 (IL-8). In chronic lung disease, this could contribute to mucous metaplasia, inflammation, and tissue remodeling. A potential link between CFTR deficiency, COPD, and EGFR/ADAM17 activity (dashed arrows) is the extracellular redox potential, which is in part dependent on CFTR-related glutathione transport [30, 165, 166], and which in turn regulates ADAM17 activity (Figure 5). In COPD, CS induced long-term downregulation of CFTR expression [16, 47, 80] can have the same long-term effect, in addition to the acute oxidative stress caused by cigarette smoke. In this figure, we focus on two canonical ADAM17 substrates: proinflammatory IL-6 receptor (IL-6R) and growth factor amphiregulin (AREG). AREG is proteolytically cleaved by ADAM17 from airway epithelial cells and activates EGFR in the airway epithelial cells and on the underlying fibroblasts. IL-6R trans-signaling requires ADAM17-mediated shedding of the extracellular domain or alternative mRNA splicing. The soluble sIL-6R binds to IL-6 creating IL-6/sIL-6R complexes that trans-activate gp130 on the underlying myofibroblasts and myeloid cells. Signals transduced by AREG and IL-6R shed from airway epithelial cells converge in combined activation of the transcription factor STAT3 in myofibroblasts and smooth muscle cells. STAT3 is involved in fibrotic responses and inflammatory lung disease, and a modifier of CF lung disease [221]. Based on available literature cited in text, enhanced EGFR/ADAM17 signaling may contribute to hyperinflammation and epithelial metaplasia, fibroblast and smooth muscle cell activation, angiogenesis, and net deposition of extracellular matrix (tissue remodeling) observed in COPD and CF lung disease.
9. ADAM17 Phosphorylation and Trafficking
The serine- and threonine-rich ADAM17 cytoplasmic tail [112] has three phosphorylation sites that have been proposed to activate ADAM17: Thr735, Ser791, and Ser819 (Figure 2). Pro-ADAM17 and mature ADAM17 are phosphorylated at Thr735 under resting conditions, but phorbol ester (PMA) treatment further increases phosphorylation at this site [223]. ERK 1/2 and p38 MAP kinase phosphorylate ADAM17 at Thr735 [126: Diaz-Rodriguez, 2002 #237] [224], and activate ADAM17 proteolytic activity, leading to shedding of IL-6R and TGF-alpha [126]. Stimulation of ADAM17 increases its phosphorylation without changing the total protein level, suggesting that phosphorylation plays a role in the regulation of the proteolytic activity of ADAM17. However, mutation of the phosphorylation sites individually or in combination and even removal of the whole cytoplasmic tail has no significant effect on stimulated shedding in cell models, suggesting that phosphorylation may be a minor regulatory mechanism of ADAM17 proteolytic activity, or that it plays a context and cell type dependent role in intracellular transport, processing, and maturation [112, 121, 225–227].
9.1. The Role of ADAM17, AREG/IL-6R, and EGFR Trafficking in Cellular Signaling
The majority of EGFR signaling is believed to occur at the plasma membrane. However, many studies show that upon ligand-dependent activation, EGFR is rapidly internalized into endosomes, where hypothetically it may continue to signal [228, 229]. Up till now, the triggered endocytosis of EGFR has been interpreted as signal attenuation, due to lysosomal degradation [230]. However, recent reviews elaborate on the role of EGFR endocytosis and recycling in active signal transduction [10, 230–233]. How triggered intracellular trafficking of EGFR relates to the regulation of the EGFR/ADAM17 axis has not been explained in detail. So far, these studies largely focus on deregulated EGFR trafficking in the context of carcinogenesis. However, EGFR trafficking in CFTR deficiency and its effect on the signaling cascade has not been investigated.
Most reports assume that ADAM17 cleaves and sheds it substrates at the extracellular membrane. However, ADAMs and its substrates have been localized in various subcellular compartments like lysosomes [234], endosomes [175, 235], and exosomes [233]; thus, it is difficult to identify an exact site of ADAM17 mediated shedding and how it interacts with EGFR [236].
Doedens et al. proposed that endocytosis from the cell surface is a pre-requisite for ADAM17 catalytic activity [237]. Lorenzen et al. suggested that ADAM17 and its substrate meet in the ER/Golgi pathway and that the prodomain does not interfere with the enzyme-substrate interaction [100]. Xu et al. showed that ADAM17 is localized at the cell surface as a dimer and associates with tissue inhibitor of metalloproteinase-3 (TIMP-3) [126]. Upon activation with PMA or anisomycin, the amount of ADAM17 at the cell surface increases, followed by dissociation of ADAM17 from TIMP-3, and ADAM17 dimers convert to monomers. According to the authors, these monomers correspond to the active form of ADAM17 and are predominantly found in cytoplasm [147], suggesting that the monomers are internalized, and shedding may occur intracellularly. Also, recent reports suggest that ADAM-mediated cleavage occurs from an intracellular vesicular pool in several cell types. Triggered shedding of FasL by ADAM10 and ADAM17 by T-lymphocytes involves trafficking of proteases and substrates to an intracellular membrane raft compartment [234]. Moreover, two proteins that regulate ADAM17-dependent shedding of EGFR ligands, annexins, and a phosphofurin acidic cluster sorting protein 2 are close to ADAM17 in the intracellular vesicular compartment [235] as shown by proximity ligation assay (PLA). Consistent with this, we have shown by PLA that upon CS exposure ADAM17 and ADAM17-P appear in close proximity with its substrates AREG and IL-6R in the intracellular compartment in ALI-HBEC, whereas under basal condition ADAM17 or ADAM17-P substrate complexes were infrequent [87]. Together, reports from various cellular models suggest that the shedding process may occur in an intracellular compartment and that triggered activity involves active trafficking of ADAM17 and its substrates. It remains to be established whether ADAM17 sheds its substrate in intracellular vesicles followed by secretion or whether the ADAM17/substrate complexes in these vesicles need to be transported to the membrane first in order to deliver ADAM17 and its substrate for cleavage. In both cases, a vesicle trafficking and membrane fusion event is involved, which may be subject to regulation, adding another level of complexity.
A further aspect of the relationship between EGFR/ADAM17 signaling and membrane trafficking concerns the production and intercellular exchange of exosomes. Higginbotham et al. showed the importance of paracrine exosomal AREG-mediated signaling in breast cancer cells [233]. Recipient LM2-4175 cells rapidly take up AREG-containing exosomes in an EGFR-dependent manner and enhanced invasion of LM2-4175 cells through matrigel [233]. Also, EGFR- and ADAM17-containing exosomes have been described [238, 239]. Such exosomes are considered important in the resolution of tissue injury and inflammation, presumably because they allow delivery of functional signaling complexes from triggered cells to neighboring cells. While most available studies address the role of exosomes in the progression of cancer, their role in chronic lung disease and possible implications for future treatment is under study [240].
10. Summary and Conclusions
Several lines of evidence, discussed above, suggest that the ADAM17/EGFR axis and downstream regulatory pathways are hyperactive in CF and COPD chronic lung disease, promoting inflammation and tissue remodeling by shedding EGFR binding growth factors and proinflammatory agonists from airway epithelial cells. This may contribute to inflammation, epithelial metaplasia, fibroblast and smooth muscle cell activation, and net deposition of extracellular matrix. Taken together, we propose that pathology-driven trans-signaling at least in part depends on airway epithelial AREG and IL-6R shedding. Importantly, signals transduced by shed AREG and IL-6R from airway epithelial cells may converge in activation of the transcription factor STAT3 in lung fibroblasts, myofibroblasts, and smooth muscle cells (Figure 7). Moreover, unbalanced ligand shedding towards the submucosa in CF and COPD and likely other chronic lung disease will affect the activity of resident and infiltrating myeloid cells, including dendritic cells, macrophages, and neutrophils.
While evidence in cellular and animals models is compelling, direct evidence for EGFR/ADAM17 hyperactivity from patient lung tissue in situ is scarce. This is in part due to a lack of appropriate airway material for analysis, especially from early disease and healthy controls. In a recent study, Zuo et al. show enhanced AREG expression in biopsies from smokers lungs, associated with remodeling lesions [181], consistent with enhanced EGFR activity as shown in primary cells in culture [87, 181]. However, successful pharmacological interventions in the EGFR/ADAM17 pathway have not been reported to our knowledge. Activation of CFTR activity may help to reduce EGFR/ADAM17 activity and resolve COPD pathology [85], but no clinical evidence for this is yet available. Targeting EGFR/ADAM17 in CF patients has not been attempted. Conversely, it would be important to study patients treated with CFTR-targeted medication for evidence of reduced activity of the EFGR/ADAM17 axis in biomarker and biopsy studies.
Genetic analysis of the COPD and CF populations, aiming at identification of genes that determine the pathology (“modifier genes”) would also provide important evidence for the involvement of the EGFR/ADAM17 axis. So far, as already cited above, STAT3, controlled by the EGFR/ADAM17 axis (Figure 7), is a modifier of CF lung disease [221]. IL-6R, a prominent ADAM17 substrate linking the axis to STAT3 in trans-signaling, is a modifier of COPD as well as asthma [207, 208]. So far, polymorphisms associated with the ADAM17, AREG, or EGFR genes have not been directly linked to chronic lung disease, but that may be due to limitations of the studies.
A possible mechanistic link between EGFR/ADAM17 activity, CF, and COPD is suggested by the observation that CFTR activity is diminished in COPD airways. Reduced CFTR activity interferes with glutathione transport in CF airways, adding to oxidative stress, which would activate the EGFR/ADAM17 axis both on CF and COPD. Further studies in vivo and in vitro are required to establish this.
Interventions in the EGFR/ADAM17 pathway may reduce CF and COPD lung pathology. However, since long-term systemic delivery of available EGFR and ADAM17 inhibitors likely causes undesirable side effects, novel methods to control this pathways will likely be required. Studies of stress-induced dynamic trafficking of the membrane proteins involved in the EGFR/ADAM17 axis in advanced 3D airway culture models may allow the development of novel modulators that will allow a more targeted approach to suit the requirements for treatment of CF and COPD lung disease.
Disclosure
Marta Stolarczyk present address is Department of Medicine V, Translational Oncology, University of Heidelberg, Heidelberg, Germany.
Conflicts of Interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
Acknowledgements
Marta Stolarczyk was supported by the Dutch Lung Foundation (Longfonds, Grant no. 3.3.10.027). The authors thank Susanne Bogers MSc for her help in editing the final manuscript and literature list.
References
- 1.Hiemstra P. S., McCray P. B., Jr, Bals R. The innate immune function of airway epithelial cells in inflammatory lung disease. The European Respiratory Journal. 2015;45(4):1150–1162. doi: 10.1183/09031936.00141514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gras D., Chanez P., Vachier I., Petit A., Bourdin A. Bronchial epithelium as a target for innovative treatments in asthma. Pharmacology & Therapeutics. 2013;140(3):290–305. doi: 10.1016/j.pharmthera.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Zhu L., Lee P. K., Lee W. M., Zhao Y., Yu D., Chen Y. Rhinovirus-induced major airway mucin production involves a novel TLR3-EGFR-dependent pathway. American Journal of Respiratory Cell and Molecular Biology. 2009;40(5):610–619. doi: 10.1165/rcmb.2008-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomez M. I., Sokol S. H., Muir A. B., Soong G., Bastien J., Prince A. S. Bacterial induction of TNF-alpha converting enzyme expression and IL-6 receptor alpha shedding regulates airway inflammatory signaling. The Journal of Immunology. 2005;175(3):1930–1936. doi: 10.4049/jimmunol.175.3.1930. [DOI] [PubMed] [Google Scholar]
- 5.Hudy M. H., Proud D. Cigarette smoke enhances human rhinovirus-induced CXCL8 production via HuR-mediated mRNA stabilization in human airway epithelial cells. Respiratory Research. 2013;14(1):p. 88. doi: 10.1186/1465-9921-14-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parnia S., Hamilton L. M., Puddicombe S. M., Holgate S. T., Frew A. J., Davies D. E. Autocrine ligands of the epithelial growth factor receptor mediate inflammatory responses to diesel exhaust particles. Respiratory Research. 2014;15(1):p. 22. doi: 10.1186/1465-9921-15-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amatngalim G. D., van Wijck Y., de Mooij-Eijk Y., et al. Basal cells contribute to innate immunity of the airway epithelium through production of the antimicrobial protein RNase 7. The Journal of Immunology. 2015;194(7):3340–3350. doi: 10.4049/jimmunol.1402169. [DOI] [PubMed] [Google Scholar]
- 8.Robb C. T., Regan K. H., Dorward D. A., Rossi A. G. Key mechanisms governing resolution of lung inflammation. Seminars in Immunopathology. 2016;38(4):425–448. doi: 10.1007/s00281-016-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wendt M. K., Balanis N., Carlin C. R., Schiemann W. P. STAT3 and epithelial–mesenchymal transitions in carcinomas. JAK-STAT. 2014;3(2, article e28975) doi: 10.4161/jkst.28975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh B., Coffey R. J. Trafficking of epidermal growth factor receptor ligands in polarized epithelial cells. Annual Review of Physiology. 2014;76(1):275–300. doi: 10.1146/annurev-physiol-021113-170406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lacroix M., Rousseau F., Guilhot F., et al. Novel insights into interleukin 6 (IL-6) Cis- and trans-signaling pathways by differentially manipulating the assembly of the IL-6 signaling complex. The Journal of Biological Chemistry. 2015;290(45):26943–26953. doi: 10.1074/jbc.M115.682138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adam D., Roux-Delrieu J., Luczka E., et al. Cystic fibrosis airway epithelium remodelling: involvement of inflammation. The Journal of Pathology. 2015;235(3):408–419. doi: 10.1002/path.4471. [DOI] [PubMed] [Google Scholar]
- 13.Pini L., Pinelli V., Modina D., Bezzi M., Tiberio L., Tantucci C. Central airways remodeling in COPD patients. International Journal of Chronic Obstructive Pulmonary Disease. 2014;9:927–933. doi: 10.2147/copd.s52478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boon M., Verleden S. E., Bosch B., et al. Morphometric analysis of explant lungs in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 2016;193(5):516–526. doi: 10.1164/rccm.201507-1281OC. [DOI] [PubMed] [Google Scholar]
- 15.Johannesson B., Hirtz S., Schatterny J., Schultz C., Mall M. A. CFTR regulates early pathogenesis of chronic obstructive lung disease in βENaC-Overexpressing mice. PLoS ONE. 2012;7(8, article e44059) doi: 10.1371/journal.pone.0044059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rab A., Rowe S. M., Raju S. V., Bebok Z., Matalon S., Collawn J. F. Cigarette smoke and CFTR: implications in the pathogenesis of COPD. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2013;305(8):L530–L541. doi: 10.1152/ajplung.00039.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mall M. A., Hartl D. CFTR: cystic fibrosis and beyond. The European Respiratory Journal. 2014;44(4):1042–1054. doi: 10.1183/09031936.00228013. [DOI] [PubMed] [Google Scholar]
- 18.Scheller J., Chalaris A., Garbers C., Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends in Immunology. 2011;32(8):380–387. doi: 10.1016/j.it.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Xu W., Liu C., Kaartinen V., et al. TACE in perinatal mouse lung epithelial cells promotes lung saccular formation. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2013;305(12):L953–L963. doi: 10.1152/ajplung.00189.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horiuchi K., Kimura T., Miyamoto T., et al. Cutting edge: tnf-α-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. The Journal of Immunology. 2007;179(5):2686–2689. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- 21.The Clinical and Functional TRanslation of CFTR (CFTR2) US CF Foundation: Johns Hopkins University; 2011. [Google Scholar]
- 22.Cheng S. H., Gregory R. J., Marshall J., et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63(4):827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 23.Kreda S. M., Mall M., Mengos A., et al. Characterization of wild-type and δf508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Molecular Biology of the Cell. 2005;16(5):2154–2167. doi: 10.1091/mbc.E04-11-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edelman A., Sallenave J. M. Cystic fibrosis, a multi-systemic mucosal disease: 25 years after the discovery of CFTR. The International Journal of Biochemistry & Cell Biology. 2014;52:2–4. doi: 10.1016/j.biocel.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Elborn J. S. Cystic fibrosis. Lancet. 2016;388(10059):2519–2531. doi: 10.1016/S0140-6736(16)00576-6. [DOI] [PubMed] [Google Scholar]
- 26.Poulsen J. H., Fischer H., Illek B., Machen T. E. Bicarbonate conductance and pH regulatory capability of cystic fibrosis transmembrane conductance regulator. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(12):5340–5344. doi: 10.1073/pnas.91.12.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linsdell P., Hanrahan J. W. Glutathione permeability of CFTR. The American Journal of Physiology. 1998;275(1, Part 1):C323–C326. doi: 10.1152/ajpcell.1998.275.1.C323. [DOI] [PubMed] [Google Scholar]
- 28.Gao L., Kim K. J., Yankaskas J. R., Forman H. J. Abnormal glutathione transport in cystic fibrosis airway epithelia. The American Gournal of Physiology. 1999;277(1, Part 1):L113–L118. doi: 10.1152/ajplung.1999.277.1.L113. [DOI] [PubMed] [Google Scholar]
- 29.Quinton P. M. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet. 2008;372(9636):415–417. doi: 10.1016/S0140-6736(08)61162-9. [DOI] [PubMed] [Google Scholar]
- 30.Ballatori N., Krance S. M., Notenboom S., Shi S., Tieu K., Hammond C. L. Glutathione dysregulation and the etiology and progression of human diseases. Biological Chemistry. 2009;390(3):191–214. doi: 10.1515/BC.2009.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuchs S. I., Gappa M., Eder J., Unsinn K. M., Steinkamp G., Ellemunter H. Tracking Lung Clearance Index and chest CT in mild cystic fibrosis lung disease over a period of three years. Respiratory Medicine. 2014;108(6):865–874. doi: 10.1016/j.rmed.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 32.Stick S., Tiddens H., Aurora P., et al. Early intervention studies in infants and preschool children with cystic fibrosis: are we ready? The European Respiratory Journal. 2013;42(2):527–538. doi: 10.1183/09031936.00108212. [DOI] [PubMed] [Google Scholar]
- 33.Ramsey K. A., Ranganathan S. Interpretation of lung function in infants and young children with cystic fibrosis. Respirology. 2014;19(6):792–799. doi: 10.1111/resp.12329. [DOI] [PubMed] [Google Scholar]
- 34.Mott L. S., Park J., Murray C. P., et al. Progression of early structural lung disease in young children with cystic fibrosis assessed using CT. Thorax. 2012;67(6):509–516. doi: 10.1136/thoraxjnl-2011-200912. [DOI] [PubMed] [Google Scholar]
- 35.Long F. R., Williams R. S., Adler B. H., Castile R. G. Comparison of quiet breathing and controlled ventilation in the high-resolution CT assessment of airway disease in infants with cystic fibrosis. Pediatric Radiology. 2005;35(11):1075–1080. doi: 10.1007/s00247-005-1541-4. [DOI] [PubMed] [Google Scholar]
- 36.Rossi U., Owens C. The radiology of chronic lung disease in children. Archives of Disease in Childhood. 2005;90(6):601–607. doi: 10.1136/adc.2004.051383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mott L. S., Graniel K. G., Park J., et al. Assessment of early bronchiectasis in young children with cystic fibrosis is dependent on lung volume. Chest. 2013;144(4):1193–1198. doi: 10.1378/chest.12-2589. [DOI] [PubMed] [Google Scholar]
- 38.Rosenow T., Oudraad M. C., Murray C. P., et al. Quantitative structural lung disease computed tomography outcome in young children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 2015;191(10):1158–1165. doi: 10.1164/rccm.201501-0061OC. [DOI] [PubMed] [Google Scholar]
- 39.Wilke M., Buijs-Offerman R. M., Aarbiou J., et al. Mouse models of cystic fibrosis: phenotypic analysis and research applications. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2011;10(Supplement 2):S152–S171. doi: 10.1016/S1569-1993(11)60020-9. [DOI] [PubMed] [Google Scholar]
- 40.Meyerholz D. K. Lessons learned from the cystic fibrosis pig. Theriogenology. 2016;86(1):427–432. doi: 10.1016/j.theriogenology.2016.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeong J. H., Joo N. S., Hwang P. H., Wine J. J. Mucociliary clearance and submucosal gland secretion in the ex vivo ferret trachea. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2014;307(1):L83–L93. doi: 10.1152/ajplung.00009.2014. [DOI] [PubMed] [Google Scholar]
- 42.Quinton P. M. Role of epithelial HCO3(-) transport in mucin secretion: lessons from cystic fibrosis. American journal of physiology. Cell physiology. 2010;299(6):C1222–C1233. doi: 10.1152/ajpcell.00362.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shamsuddin A. K. M., Quinton P. M. Native small airways secrete bicarbonate. American Journal of Respiratory Cell and Molecular Biology. 2014;50(4):796–804. doi: 10.1165/rcmb.2013-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmid A., Meili D., Salathe M. Soluble adenylyl cyclase in health and disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2014;1842(12):2584–2592. doi: 10.1016/j.bbadis.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang X. X., Ostedgaard L. S., Hoegger M. J., et al. Acidic pH increases airway surface liquid viscosity in cystic fibrosis. The Journal of Clinical Investigation. 2016;126(3):879–891. doi: 10.1172/JCI83922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Derichs N., Jin B. J., Song Y., Finkbeiner W. E., Verkman A. S. Hyperviscous airway periciliary and mucous liquid layers in cystic fibrosis measured by confocal fluorescence photobleaching. FASEB journal : Official Publication of the Federation of American Societies for Experimental Biology. 2011;25(7):2325–2332. doi: 10.1096/fj.10-179549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghosh A., Boucher R. C., Tarran R. Airway hydration and COPD. Cellular and Molecular Life Sciences. 2015;72(19):3637–3652. doi: 10.1007/s00018-015-1946-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tarran R., Button B., Picher M., et al. Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. The Journal of Biological Chemistry. 2005;280(42):35751–35759. doi: 10.1074/jbc.M505832200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schmid A., Sutto Z., Schmid N., et al. Decreased soluble adenylyl cyclase activity in cystic fibrosis is related to defective apical bicarbonate exchange and affects ciliary beat frequency regulation. The Journal of Biological Chemistry. 2010;285(39):29998–30007. doi: 10.1074/jbc.M110.113621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pezzulo A. A., Tang X. X., Hoegger M. J., et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487(7405):109–113. doi: 10.1038/nature11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shah V. S., Meyerholz D. K., Tang X. X., et al. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science. 2016;351(6272):503–507. doi: 10.1126/science.aad5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abou Alaiwa M. H., Reznikov L. R., Gansemer N. D., et al. pH modulates the activity and synergism of the airway surface liquid antimicrobials β-defensin-3 and LL-37. Proceedings of the National Academy of Sciences. 2014;111(52):18703–18708. doi: 10.1073/pnas.1422091112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Linnane B., Vaish S., Clarke D., O'Sullivan N., McNally P. The findings of a clinical surveillance bronchoalveolar lavage programme in pre-school patients with cystic fibrosis. Pediatric Pulmonology. 2015;50(4):327–332. doi: 10.1002/ppul.23118. [DOI] [PubMed] [Google Scholar]
- 54.Armstrong D. S., Grimwood K., Carlin J. B., et al. Lower airway inflammation in infants and young children with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 1997;156(4):1197–1204. doi: 10.1164/ajrccm.156.4.96-11058. [DOI] [PubMed] [Google Scholar]
- 55.Stoltz D. A., Meyerholz D. K., Pezzulo A. A., et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Science Translational Medicine. 2010;2(29):p. 29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Veltman M., Stolarczyk M., Radzioch D., et al. Correction of lung inflammation in a F508del CFTR murine cystic fibrosis model by the sphingosine-1-phosphate lyase inhibitor LX2931. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2016;311(5):L1000–L1014. doi: 10.1152/ajplung.00298.2016. [DOI] [PubMed] [Google Scholar]
- 57.Rosen B. H., Evans T., Keiser N. W., et al. P 151 Muco-inflammatory bronchiectasis in cystic fibrosis ferrets does not require infection. Pediatric Pulmonology. 2016;51:p. 249. [Google Scholar]
- 58.Huaux F., Noel S., Dhooghe B., et al. Dysregulated proinflammatory and fibrogenic phenotype of fibroblasts in cystic fibrosis. PLoS One. 2013;8(5, article e64341) doi: 10.1371/journal.pone.0064341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruscia E. M., Bonfield T. L. Cystic fibrosis lung immunity: the role of the macrophage. Journal of Innate Immunity. 2016;8(6):550–563. doi: 10.1159/000446825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Margaroli C., Tirouvanziam R. Neutrophil plasticity enables the development of pathological microenvironments: implications for cystic fibrosis airway disease. Molecular and Cellular Pediatrics. 2016;3(1):p. 38. doi: 10.1186/s40348-016-0066-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim S., Beyer B. A., Lewis C., Nadel J. A. Normal CFTR inhibits epidermal growth factor receptor-dependent pro-inflammatory chemokine production in human airway epithelial cells. PLoS One. 2013;8(8, article e72981) doi: 10.1371/journal.pone.0072981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsuchiya M., Kumar P., Bhattacharyya S., et al. Differential regulation of inflammation by inflammatory mediators in cystic fibrosis lung epithelial cells. Journal of Interferon & Cytokine Research. 2013;33(3):121–129. doi: 10.1089/jir.2012.0074. [DOI] [PubMed] [Google Scholar]
- 63.Veit G., Bossard F., Goepp J., et al. Proinflammatory cytokine secretion is suppressed by TMEM16A or CFTR channel activity in human cystic fibrosis bronchial epithelia. Molecular Biology of the Cell. 2012;23(21):4188–4202. doi: 10.1091/mbc.E12-06-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Torphy T. J., Allen J., Cantin A. M., et al. Considerations for the conduct of clinical trials with antiinflammatory agents in cystic fibrosis. A cystic fibrosis foundation workshop report. Annals of the American Thoracic Society. 2015;12(9):1398–1406. doi: 10.1513/AnnalsATS.201506-361OT. [DOI] [PubMed] [Google Scholar]
- 65.Lands L. C., Stanojevic S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database of Systematic Reviews. 2016;4, article Cd001505 doi: 10.1002/14651858.CD001505.pub4. [DOI] [PubMed] [Google Scholar]
- 66.Cheng K., Ashby D., Smyth R. L. Oral steroids for long-term use in cystic fibrosis. Cochrane Database of Systematic Reviews. 2013;Cd000407 doi: 10.1002/14651858.CD000407.pub3. [DOI] [PubMed] [Google Scholar]
- 67.Littlewood K. J., Higashi K., Jansen J. P., et al. A network meta-analysis of the efficacy of inhaled antibiotics for chronic Pseudomonas infections in cystic fibrosis. Journal of Cystic Fibrosis. 2012;11(5):419–426. doi: 10.1016/j.jcf.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 68.Yang C., Chilvers M., Montgomery M., Nolan S. J. Dornase alfa for cystic fibrosis. Cochrane Database of Systematic Reviews. 2016;4, article Cd001127 doi: 10.1002/14651858.CD001127.pub3. [DOI] [PubMed] [Google Scholar]
- 69.Cai Z. W., Liu J., Li H. Y., Sheppard D. N. Targeting F508del-CFTR to develop rational new therapies for cystic fibrosis. Acta pharmacologica Sinica. 2011;32(6):693–701. doi: 10.1038/aps.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cholon D. M., Esther C. R., Jr, Gentzsch M. Efficacy of lumacaftor-ivacaftor for the treatment of cystic fibrosis patients homozygous for the F508del-CFTR mutation. Expert Review of Precision Medicine and Drug Development. 2016;1(3):235–243. doi: 10.1080/23808993.2016.1175299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boyle M. P., Bell S. C., Konstan M. W., et al. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. The Lancet Respiratory Medicine. 2014;2(7):527–538. doi: 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- 72.Wainwright C. E., Elborn J. S., Ramsey B. W., et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for phe508del cftr. The New England Journal of Medicine. 2015;373(3):220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rowe S. M., Heltshe S. L., Gonska T., et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. American journal of respiratory and critical care medicine. 2014;190(2):175–184. doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schnur A., Hegyi P., Rousseau S., Lukacs G. L., Veit G. Epithelial anion transport as modulator of chemokine signaling. Mediators of Inflammation. 2016;2016:20. doi: 10.1155/2016/7596531.7596531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hogg J. C., Timens W. The pathology of chronic obstructive pulmonary disease. Annual Review of Pathology. 2009;4(1):435–459. doi: 10.1146/annurev.pathol.4.110807.092145. [DOI] [PubMed] [Google Scholar]
- 76.Du Q., Jin J., Liu X., Sun Y. Bronchiectasis as a comorbidity of chronic obstructive pulmonary disease: a systematic review and meta-analysis. PLoS One. 2016;11(3, article e0150532) doi: 10.1371/journal.pone.0150532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.da Silva S. M., Paschoal I. A., De Capitani E. M., Moreira M. M., Palhares L. C., Pereira M. C. COPD phenotypes on computed tomography and its correlation with selected lung function variables in severe patients. International Journal of Chronic Obstructive Pulmonary Disease. 2016;11:503–513. doi: 10.2147/COPD.S90638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mehta A. J., Miedinger D., Keidel D., et al. Occupational exposure to dusts, gases, and fumes and incidence of chronic obstructive pulmonary disease in the Swiss Cohort Study on Air Pollution and Lung and Heart Diseases in Adults. American Journal of Respiratory and Critical Care Medicine. 2012;185(12):1292–1300. doi: 10.1164/rccm.201110-1917OC. [DOI] [PubMed] [Google Scholar]
- 79.Varga J. J., Barbier M., Mulet X., et al. Genotypic and phenotypic analyses of a Pseudomonas aeruginosa chronic bronchiectasis isolate reveal differences from cystic fibrosis and laboratory strains. BMC Genomics. 2015;16(1):p. 883. doi: 10.1186/s12864-015-2069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raju S. V., Solomon G. M., Dransfield M. T., Rowe S. M. Acquired cystic fibrosis transmembrane conductance regulator dysfunction in chronic bronchitis and other diseases of mucus clearance. Clinics in Chest Medicine. 2016;37(1):147–158. doi: 10.1016/j.ccm.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cantin A. M. Cystic fibrosis transmembrane conductance regulator. Implications in cystic fibrosis and chronic obstructive pulmonary disease. Annals of the American Thoracic Society. 2016;13(Supplement 2):S150–S155. doi: 10.1513/AnnalsATS.201509-588KV. [DOI] [PubMed] [Google Scholar]
- 82.Clunes L. A., Davies C. M., Coakley R. D., et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. The FASEB Journal. 2012;26(2):533–545. doi: 10.1096/fj.11-192377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sloane P. A., Shastry S., Wilhelm A., et al. A pharmacologic approach to acquired cystic fibrosis transmembrane conductance regulator dysfunction in smoking related lung disease. PLoS One. 2012;7(6, article e39809) doi: 10.1371/journal.pone.0039809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bodas M., Min T., Vij N. Critical role of CFTR-dependent lipid rafts in cigarette smoke-induced lung epithelial injury. AJP: Lung Cellular and Molecular Physiology. 2011;300(6):L811–L820. doi: 10.1152/ajplung.00408.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Raju S. V., Rasmussen L., Sloane P. A., Tang L. P., Libby E. F., Rowe S. M. Roflumilast reverses CFTR-mediated ion transport dysfunction in cigarette smoke-exposed mice. Respiratory Research. 2017;18(1):p. 173. doi: 10.1186/s12931-017-0656-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ni I., Ji C., Vij N. Second-hand cigarette smoke impairs bacterial phagocytosis in macrophages by modulating CFTR dependent lipid-rafts. PLoS One. 2015;10(3, article e0121200) doi: 10.1371/journal.pone.0121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stolarczyk M., Amatngalim G. D., Yu X., Veltman M., Hiemstra P. S., Scholte B. J. ADAM17 and EGFR regulate IL-6 receptor and amphiregulin mRNA expression and release in cigarette smoke-exposed primary bronchial epithelial cells from patients with chronic obstructive pulmonary disease (COPD) Physiological Reports. 2016;4(16, article e12878) doi: 10.14814/phy2.12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cholon D. M., Quinney N. L., Fulcher M. L., et al. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Science Translational Medicine. 2014;6(246, article 246ra296) doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schamberger A. C., Mise N., Meiners S., Eickelberg O. Epigenetic mechanisms in COPD: implications for pathogenesis and drug discovery. Expert Opinion on Drug Discovery. 2014;9(6):609–628. doi: 10.1517/17460441.2014.913020. [DOI] [PubMed] [Google Scholar]
- 90.Comer B. S., Ba M., Singer C. A., Gerthoffer W. T. Epigenetic targets for novel therapies of lung diseases. Pharmacology & Therapeutics. 2015;147:91–110. doi: 10.1016/j.pharmthera.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lambert J. A., Raju S. V., Tang L. P., et al. Cystic fibrosis transmembrane conductance regulator activation by roflumilast contributes to therapeutic benefit in chronic bronchitis. American Journal of Respiratory Cell and Molecular Biology. 2014;50(3):549–558. doi: 10.1165/rcmb.2013-0228OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Durham A. L., Caramori G., Chung K. F., Adcock I. M. Targeted anti-inflammatory therapeutics in asthma and chronic obstructive lung disease. Translational Research. 2016;167(1):192–203. doi: 10.1016/j.trsl.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Merkert S., Bednarski C., Gohring G., Cathomen T., Martin U. Generation of a gene-corrected isogenic control iPSC line from cystic fibrosis patient-specific iPSCs homozygous for p.Phe508del mutation mediated by TALENs and ssODN. Stem Cell Research. 2017;23:95–97. doi: 10.1016/j.scr.2017.07.010. [DOI] [PubMed] [Google Scholar]
- 94.Benam K. H., Mazur M., Choe Y., Ferrante T. C., Novak R., Ingber D. E. Human lung small airway-on-a-chip protocol. Methods in Molecular Biology. 2017;1612:345–365. doi: 10.1007/978-1-4939-7021-6_25. [DOI] [PubMed] [Google Scholar]
- 95.Gohy S. T., Hupin C., Pilette C., Ladjemi M. Z. Chronic inflammatory airway diseases: the central role of the epithelium revisited. Clinical & Experimental Allergy. 2016;46(4):529–542. doi: 10.1111/cea.12712. [DOI] [PubMed] [Google Scholar]
- 96.Thorley A. J., Tetley T. D. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. International Journal of Chronic Obstructive Pulmonary Disease. 2007;2(4):409–428. [PMC free article] [PubMed] [Google Scholar]
- 97.Janes P. W., Saha N., Barton W. A., et al. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123(2):291–304. doi: 10.1016/j.cell.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 98.Takeda S., Igarashi T., Mori H., Araki S. Crystal structures of VAP1 reveal ADAMs’ MDC domain architecture and its unique C-shaped scaffold. The EMBO Journal. 2006;25(11):2388–2396. doi: 10.1038/sj.emboj.7601131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takeda S. Three-dimensional domain architecture of the ADAM family proteinases. Seminars in Cell & Developmental Biology. 2009;20(2):146–152. doi: 10.1016/j.semcdb.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 100.Lorenzen I., Lokau J., Dusterhoft S., et al. The membrane-proximal domain of A Disintegrin and Metalloprotease 17 (ADAM17) is responsible for recognition of the interleukin-6 receptor and interleukin-1 receptor II. FEBS Letters. 2012;586(8):1093–1100. doi: 10.1016/j.febslet.2012.03.012. [DOI] [PubMed] [Google Scholar]
- 101.Dusterhoft S., Michalek M., Kordowski F., et al. Extracellular juxtamembrane segment of adam17 interacts with membranes and is essential for its shedding activity. Biochemistry. 2015;54(38):5791–5801. doi: 10.1021/acs.biochem.5b00497. [DOI] [PubMed] [Google Scholar]
- 102.Deshmukh H. S., Case L. M., Wesselkamper S. C., et al. Metalloproteinases mediate mucin 5AC expression by epidermal growth factor receptor activation. American Journal of Respiratory and Critical Care Medicine. 2005;171(4):305–314. doi: 10.1164/rccm.200408-1003OC. [DOI] [PubMed] [Google Scholar]
- 103.Shao M. X., Nakanaga T., Nadel J. A. Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-α-converting enzyme in human airway epithelial (NCI-H292) cells. AJP: Lung Cellular and Molecular Physiology. 2004;287(2):L420–L427. doi: 10.1152/ajplung.00019.2004. [DOI] [PubMed] [Google Scholar]
- 104.Shao M. X., Nadel J. A. Dual oxidase 1-dependent MUC5AC mucin expression in cultured human airway epithelial cells. Proceedings of the National Academy of Sciences. 2005;102(3):767–772. doi: 10.1073/pnas.0408932102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Richter A., O'Donnell R. A., Powell R. M., et al. Autocrine ligands for the epidermal growth factor receptor mediate interleukin-8 release from bronchial epithelial cells in response to cigarette smoke. American Journal of Respiratory Cell and Molecular Biology. 2002;27(1):85–90. doi: 10.1165/ajrcmb.27.1.4789. [DOI] [PubMed] [Google Scholar]
- 106.Pasqualon T., Pruessmeyer J., Weidenfeld S., et al. A transmembrane C-terminal fragment of syndecan-1 is generated by the metalloproteinase ADAM17 and promotes lung epithelial tumor cell migration and lung metastasis formation. Cellular and Molecular Life Sciences. 2015;72(19):3783–3801. doi: 10.1007/s00018-015-1912-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Broekman W., Amatngalim G. D., de Mooij-Eijk Y., et al. TNF-α and IL-1β-activated human mesenchymal stromal cells increase airway epithelial wound healing in vitro via activation of the epidermal growth factor receptor. Respiratory Research. 2016;17(1):p. 3. doi: 10.1186/s12931-015-0316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lemjabbar H., Li D., Gallup M., Sidhu S., Drori E., Basbaum C. Tobacco smoke-induced lung cell proliferation mediated by tumor necrosis factor α-converting enzyme and amphiregulin. Journal of Biological Chemistry. 2003;278(28):26202–26207. doi: 10.1074/jbc.M207018200. [DOI] [PubMed] [Google Scholar]
- 109.Vallath S., Hynds R. E., Succony L., Janes S. M., Giangreco A. Targeting EGFR signalling in chronic lung disease: therapeutic challenges and opportunities. European Respiratory Journal. 2014;44(2):513–522. doi: 10.1183/09031936.00146413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Breshears L. M., Schlievert P. M., Peterson M. L. A disintegrin and metalloproteinase 17 (adam17) and epidermal growth factor receptor (egfr) signaling drive the epithelial response to staphylococcus aureus toxic shock syndrome toxin-1 (tsst-1) Journal of Biological Chemistry. 2012;287(39):32578–32587. doi: 10.1074/jbc.M112.352534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vermeer P. D., Panko L., Karp P., Lee J. H., Zabner J. Differentiation of human airway epithelia is dependent on erbB2. AJP: Lung Cellular and Molecular Physiology. 2006;291(2):L175–L180. doi: 10.1152/ajplung.00547.2005. [DOI] [PubMed] [Google Scholar]
- 112.Fan H., Turck C. W., Derynck R. Characterization of growth factor-induced serine phosphorylation of tumor necrosis factor-alpha converting enzyme and of an alternatively translated polypeptide. Journal of Biological Chemistry. 2003;278(20):18617–18627. doi: 10.1074/jbc.M300331200. [DOI] [PubMed] [Google Scholar]
- 113.Chen Y.-T., Gallup M., Nikulina K., et al. Cigarette smoke induces epidermal growth factor receptor-dependent redistribution of apical muc1 and junctional β-catenin in polarized human airway epithelial cells. The American Journal of Pathology. 2010;177(3):1255–1264. doi: 10.2353/ajpath.2010.091129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Finigan J. H., Faress J. A., Wilkinson E., et al. Neuregulin-1-human epidermal receptor-2 signaling is a central regulator of pulmonary epithelial permeability and acute lung injury. Journal of Biological Chemistry. 2011;286(12):10660–10670. doi: 10.1074/jbc.M110.208041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao J., Chen H., Peschon J. J., et al. Pulmonary hypoplasia in mice lacking tumor necrosis factor-α converting enzyme indicates an indispensable role for cell surface protein shedding during embryonic lung branching morphogenesis. Developmental Biology. 2001;232(1):204–218. doi: 10.1006/dbio.2001.0176. [DOI] [PubMed] [Google Scholar]
- 116.Dreymueller D., Uhlig S., Ludwig A. ADAM-family metalloproteinases in lung inflammation: potential therapeutic targets. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2015;308(4):L325–L343. doi: 10.1152/ajplung.00294.2014. [DOI] [PubMed] [Google Scholar]
- 117.Chalaris A., Adam N., Sina C., et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. The Journal of Experimental Medicine. 2010;207(8):1617–1624. doi: 10.1084/jem.20092366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Park J. A., Sharif A. S., Shiomi T., et al. Human neutrophil elastase-mediated goblet cell metaplasia is attenuated in TACE-deficient mice. AJP: Lung Cellular and Molecular Physiology. 2013;304(10):L701–L707. doi: 10.1152/ajplung.00259.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Miettinen P. J., Berger J. E., Meneses J., et al. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376(6538):337–341. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 120.Le Gall S. M., Bobé P., Reiss K., et al. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor α, l-selectin, and tumor necrosis factor α. Molecular Biology of the Cell. 2009;20(6):1785–1794. doi: 10.1091/mbc.E08-11-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Le Gall S. M., Maretzky T., Issuree P. D., et al. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. Journal of Cell Science. 2010;123(22):3913–3922. doi: 10.1242/jcs.069997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sham D., Wesley U. V., Hristova M., van der Vliet A. ATP-mediated transactivation of the epidermal growth factor receptor in airway epithelial cells involves DUOX1-dependent oxidation of Src and ADAM17. PLoS One. 2013;8(1, article e54391) doi: 10.1371/journal.pone.0054391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Baumgart A., Seidl S., Vlachou P., et al. ADAM17 regulates epidermal growth factor receptor expression through the activation of Notch1 in non-small cell lung cancer. Cancer Research. 2010;70(13):5368–5378. doi: 10.1158/0008-5472.CAN-09-3763. [DOI] [PubMed] [Google Scholar]
- 124.Ermert M., Pantazis C., Duncker H. R., Grimminger F., Seeger W., Ermert L. In situ localization of TNFα/β, TACE and TNF receptors TNF-R1 and TNF-R2 in control and LPS-treated lung tissue. Cytokine. 2003;22(3-4):89–100. doi: 10.1016/S1043-4666(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 125.Dijkstra A., Postma D. S., Noordhoek J. A., et al. Expression of ADAMs (“a disintegrin and metalloprotease”) in the human lung. Virchows Archiv. 2009;454(4):441–449. doi: 10.1007/s00428-009-0748-4. [DOI] [PubMed] [Google Scholar]
- 126.Xu P., Derynck R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Molecular Cell. 2010;37(4):551–566. doi: 10.1016/j.molcel.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Booth B. W., Sandifer T., Martin E. L., Martin L. D. IL-13-induced proliferation of airway epithelial cells: mediation by intracellular growth factor mobilization and ADAM17. Respiratory Research. 2007;8(1):p. 51. doi: 10.1186/1465-9921-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rose-John S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. International Journal of Biological Sciences. 2012;8(9):1237–1247. doi: 10.7150/ijbs.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nam H.-S., Lee S. Y., Kim S. J., et al. The soluble tumor necrosis factor-alpha receptor suppresses airway inflammation in a murine model of acute asthma. Yonsei Medical Journal. 2009;50(4):569–575. doi: 10.3349/ymj.2009.50.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dang M., Dubbin K., D'Aiello A., Hartmann M., Lodish H., Herrlich A. Epidermal growth factor (EGF) ligand release by substrate-specific a disintegrin and metalloproteases (ADAMs) involves different protein kinase C (PKC) isoenzymes depending on the stimulus. Journal of Biological Chemistry. 2011;286(20):17704–17713. doi: 10.1074/jbc.M110.187823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Horiuchi K., Le Gall S., Schulte M., et al. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Molecular Biology of the Cell. 2007;18(1):176–188. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Killock D. J., Ivetic A. The cytoplasmic domains of TNFalpha-converting enzyme (TACE/ADAM17) and L-selectin are regulated differently by p38 MAPK and PKC to promote ectodomain shedding. Biochemical Journal. 2010;428(2):293–304. doi: 10.1042/BJ20091611. [DOI] [PubMed] [Google Scholar]
- 133.Wang X. J., Feng C. W., Li M. ADAM17 mediates hypoxia-induced drug resistance in hepatocellular carcinoma cells through activation of EGFR/PI3K/Akt pathway. Molecular and Cellular Biochemistry. 2013;380(1-2):57–66. doi: 10.1007/s11010-013-1657-z. [DOI] [PubMed] [Google Scholar]
- 134.Maretzky T., Evers A., Zhou W., et al. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nature Communications. 2011;2:p. 229. doi: 10.1038/ncomms1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Koff J. L., Shao M. X., Ueki I. F., Nadel J. A. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. AJP: Lung Cellular and Molecular Physiology. 2008;294(6):L1068–L1075. doi: 10.1152/ajplung.00025.2008. [DOI] [PubMed] [Google Scholar]
- 136.Yu H., Zhou X., Wen S., Xiao Q. Flagellin/TLR5 responses induce mucus hypersecretion by activating EGFR via an epithelial cell signaling cascades. Experimental Cell Research. 2012;318(6):723–731. doi: 10.1016/j.yexcr.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 137.Wang R., Ahmed J., Wang G., et al. Airway epithelial expression of TLR5 is downregulated in healthy smokers and smokers with chronic obstructive pulmonary disease. Journal of Immunology. 2012;189(5):2217–2225. doi: 10.4049/jimmunol.1101895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gschwind A., Hart S., Fischer O. M., Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. The EMBO Journal. 2003;22(10):2411–2421. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Heijink I. H., Brandenburg S. M., Noordhoek J. A., Slebos D. J., Postma D. S., van Oosterhout A. J. Role of aberrant metalloproteinase activity in the pro-inflammatory phenotype of bronchial epithelium in COPD. Respiratory Research. 2011;12(1):p. 110. doi: 10.1186/1465-9921-12-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Baginski T. K., Dabbagh K., Satjawatcharaphong C., Swinney D. C. Cigarette smoke synergistically enhances respiratory mucin induction by proinflammatory stimuli. American Journal of Respiratory Cell and Molecular Biology. 2006;35(2):165–174. doi: 10.1165/rcmb.2005-0259OC. [DOI] [PubMed] [Google Scholar]
- 141.Kim S., Lewis C., Nadel J. A. CCL20/CCR6 feedback exaggerates epidermal growth factor receptor-dependent MUC5AC mucin production in human airway epithelial (NCI-H292) cells. Journal of Immunology. 2011;186(6):3392–3400. doi: 10.4049/jimmunol.1003377. [DOI] [PubMed] [Google Scholar]
- 142.Liu K., Gualano R. C., Hibbs M. L., Anderson G. P., Bozinovski S. Epidermal growth factor receptor signaling to Erk1/2 and STATs control the intensity of the epithelial inflammatory responses to rhinovirus infection. Journal of Biological Chemistry. 2008;283(15):9977–9985. doi: 10.1074/jbc.M710257200. [DOI] [PubMed] [Google Scholar]
- 143.Mahmoodi M., Sahebjam S., Smookler D., Khokha R., Mort J. S. Lack of tissue inhibitor of metalloproteinases-3 results in an enhanced inflammatory response in antigen-induced arthritis. The American Journal of Pathology. 2005;166(6):1733–1740. doi: 10.1016/S0002-9440(10)62483-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Smookler D. S., Mohammed F. F., Kassiri Z., Duncan G. S., Mak T. W., Khokha R. Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. Journal of Immunology. 2006;176(2):721–725. doi: 10.4049/jimmunol.176.2.721. [DOI] [PubMed] [Google Scholar]
- 145.Wisniewska M., Goettig P., Maskos K., et al. Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. Journal of Molecular Biology. 2008;381(5):1307–1319. doi: 10.1016/j.jmb.2008.06.088. [DOI] [PubMed] [Google Scholar]
- 146.Zheng D. Y., Zhao J., Yang J. M., Wang M., Zhang X. T. Enhanced ADAM17 expression is associated with cardiac remodeling in rats with acute myocardial infarction. Life Sciences. 2016;151:61–69. doi: 10.1016/j.lfs.2016.02.097. [DOI] [PubMed] [Google Scholar]
- 147.Xu P., Liu J., Sakaki-Yumoto M., Derynck R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Science Signaling. 2012;5(222):p. ra34. doi: 10.1126/scisignal.2002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Carvalho-Oliveira I. M., Charro N., Aarbiou J., et al. Proteomic analysis of naphthalene-induced airway epithelial injury and repair in a cystic fibrosis mouse model. Journal of Proteome Research. 2009;8(7):3606–3616. doi: 10.1021/pr900021m. [DOI] [PubMed] [Google Scholar]
- 149.Buijs-Offerman R. M., Aarbiou J., Ooms M., Dik W., Wilke M., Scholte B. J. Sustained activaiton of EGFR and IL6 pathways in f508del CFTR mutant mice after airway injury. Pediatric Pulmonology. 2010;(Supplement 33):p. 260. [Google Scholar]
- 150.Nadel J. A. The CFTR and EGFR relationship in airway vascular growth, and its importance in cystic fibrosis. European Respiratory Journal. 2013;42(6):1438–1440. doi: 10.1183/09031936.00107513. [DOI] [PubMed] [Google Scholar]
- 151.Martel G., Bérubé J., Rousseau S. The protein kinase TPL2 Is essential for ERK1/ERK2 activation and cytokine gene expression in airway epithelial cells exposed to pathogen-associated molecular patterns (PAMPs) PLoS ONE. 2013;8(3, article e59116) doi: 10.1371/journal.pone.0059116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Martin C., Coolen N., Wu Y., et al. CFTR dysfunction induces vascular endothelial growth factor synthesis in airway epithelium. European Respiratory Journal. 2013;42(6):1553–1562. doi: 10.1183/09031936.00164212. [DOI] [PubMed] [Google Scholar]
- 153.Berube J., Roussel L., Nattagh L., Rousseau S. Loss of cystic fibrosis transmembrane conductance regulator function enhances activation of p38 and ERK MAPKs, increasing interleukin-6 synthesis in airway epithelial cells exposed to Pseudomonas aeruginosa. The Journal of Biological Chemistry. 2010;285(29):22299–22307. doi: 10.1074/jbc.M109.098566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Trinh N. T., Prive A., Maille E., Noel J., Brochiero E. EGF and K+ channel activity control normal and cystic fibrosis bronchial epithelia repair. AJP: Lung Cellular and Molecular Physiology. 2008;295(5):L866–L880. doi: 10.1152/ajplung.90224.2008. [DOI] [PubMed] [Google Scholar]
- 155.Kelly M., Trudel S., Brouillard F., et al. Cystic fibrosis transmembrane regulator inhibitors CFTR(inh)-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition. Journal of Pharmacology and Experimental Therapeutics. 2010;333(1):60–69. doi: 10.1124/jpet.109.162032. [DOI] [PubMed] [Google Scholar]
- 156.Gutscher M., Pauleau A. L., Marty L., et al. Real-time imaging of the intracellular glutathione redox potential. Nature Methods. 2008;5(6):553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 157.Boots A. W., Hristova M., Kasahara D. I., Haenen G. R., Bast A., van der Vliet A. ATP-mediated activation of the NADPH oxidase DUOX1 mediates airway epithelial responses to bacterial stimuli. Journal of Biological Chemistry. 2009;284(26):17858–17867. doi: 10.1074/jbc.M809761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hristova M., Habibovic A., Veith C., et al. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. Journal of Allergy and Clinical Immunology. 2016;137(5):1545–1556.e11. doi: 10.1016/j.jaci.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dusterhoft S., Jung S., Hung C. W., et al. Membrane-proximal domain of a disintegrin and metalloprotease-17 represents the putative molecular switch of its shedding activity operated by protein-disulfide isomerase. Journal of the American Chemical Society. 2013;135(15):5776–5781. doi: 10.1021/ja400340u. [DOI] [PubMed] [Google Scholar]
- 160.Paulsen C. E., Truong T. H., Garcia F. J., et al. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nature Chemical Biology. 2011;8(1):57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Willems S. H., Tape C. J., Stanley P. L., et al. Thiol isomerases negatively regulate the cellular shedding activity of ADAM17. Biochemical Journal. 2010;428(3):439–450. doi: 10.1042/BJ20100179. [DOI] [PubMed] [Google Scholar]
- 162.Dusterhoft S., Hobel K., Oldefest M., et al. A disintegrin and metalloprotease 17 dynamic interaction sequence, the sweet tooth for the human interleukin 6 receptor. Journal of Biological Chemistry. 2014;289(23):16336–16348. doi: 10.1074/jbc.M114.557322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ziady A. G., Hansen J. Redox balance in cystic fibrosis. The International Journal of Biochemistry & Cell Biology. 2014;52:113–123. doi: 10.1016/j.biocel.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Juarez J. C., Manuia M., Burnett M. E., et al. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proceedings of the National Academy of Sciences. 2008;105(20):7147–7152. doi: 10.1073/pnas.0709451105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gould N. S., Min E., Martin R. J., Day B. J. CFTR is the primary known apical glutathione transporter involved in cigarette smoke-induced adaptive responses in the lung. Free Radical Biology & Medicine. 2012;52(7):1201–1206. doi: 10.1016/j.freeradbiomed.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Marson F. A., Bertuzzo C. S., Ribeiro A. F., Ribeiro J. D. Polymorphisms in the glutathione pathway modulate cystic fibrosis severity: a cross-sectional study. BMC Medical Genetics. 2014;15(1):p. 27. doi: 10.1186/1471-2350-15-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hirota N., Risse P. A., Novali M., et al. Histamine may induce airway remodeling through release of epidermal growth factor receptor ligands from bronchial epithelial cells. The FASEB Journal. 2012;26(4):1704–1716. doi: 10.1096/fj.11-197061. [DOI] [PubMed] [Google Scholar]
- 168.Deacon K., Knox A. J. Human airway smooth muscle cells secrete amphiregulin via bradykinin/COX-2/PGE2, inducing COX-2, CXCL8, and VEGF expression in airway epithelial cells. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2015;309(3):L237–L249. doi: 10.1152/ajplung.00390.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Meng C., Liu G., Mu H., Zhou M., Zhang S., Xu Y. Amphiregulin may be a new biomarker of classically activated macrophages. Biochemical and Biophysical Research Communications. 2015;466(3):393–399. doi: 10.1016/j.bbrc.2015.09.037. [DOI] [PubMed] [Google Scholar]
- 170.Matsumoto K., Fukuda S., Nakamura Y., Saito H. Amphiregulin production by human eosinophils. International Archives of Allergy and Immunology. 2009;149(1) Supplement 1:39–44. doi: 10.1159/000210652. [DOI] [PubMed] [Google Scholar]
- 171.Bles N., Di Pietrantonio L., Boeynaems J. M., Communi D. ATP confers tumorigenic properties to dendritic cells by inducing amphiregulin secretion. Blood. 2010;116(17):3219–3226. doi: 10.1182/blood-2010-01-265611. [DOI] [PubMed] [Google Scholar]
- 172.Adib-Conquy M., Pedron T., Petit-Bertron A.-F., et al. Neutrophils in cystic fibrosis display a distinct gene expression pattern. Molecular Medicine. 2008;14(1-2):36–44. doi: 10.2119/2007-00081.Adib-Conquy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang S. W., Oh C. K., Cho S. H., et al. Amphiregulin expression in human mast cells and its effect on the primary human lung fibroblasts. Journal of Allergy and Clinical Immunology. 2005;115(2):287–294. doi: 10.1016/j.jaci.2004.11.037. [DOI] [PubMed] [Google Scholar]
- 174.Brown C. L., Meise K. S., Plowman G. D., Coffey R. J., Dempsey P. J. Cell surface ectodomain cleavage of human amphiregulin precursor is sensitive to a metalloprotease inhibitor. Release of a predominant N-glycosylated 43-kDa soluble form. Journal of Biological Chemistry. 1998;273(27):17258–17268. doi: 10.1074/jbc.273.27.17258. [DOI] [PubMed] [Google Scholar]
- 175.Gephart J. D., Singh B., Higginbotham J. N., et al. Identification of a novel mono-leucine basolateral sorting motif within the cytoplasmic domain of amphiregulin. Traffic. 2011;12(12):1793–1804. doi: 10.1111/j.1600-0854.2011.01282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zaiss D. M. W., Gause W. C., Osborne L. C., Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. 2015;42(2):216–226. doi: 10.1016/j.immuni.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Enomoto Y., Orihara K., Takamasu T., et al. Tissue remodeling induced by hypersecreted epidermal growth factor and amphiregulin in the airway after an acute asthma attack. The Journal of Allergy and Clinical Immunology. 2009;124(5):913–920.e7. doi: 10.1016/j.jaci.2009.08.044. [DOI] [PubMed] [Google Scholar]
- 178.Kim K. W., Jee H. M., Park Y. H., Choi B. S., Sohn M. H., Kim K. E. Relationship between amphiregulin and airway inflammation in children with asthma and eosinophilic bronchitis. Chest. 2009;136(3):805–810. doi: 10.1378/chest.08-2972. [DOI] [PubMed] [Google Scholar]
- 179.Shimizu S., Kouzaki H., Ogawa T., Takezawa K., Tojima I., Shimizu T. Eosinophil-epithelial cell interactions stimulate the production of MUC5AC mucin and profibrotic cytokines involved in airway tissue remodeling. American Journal of Rhinology & Allergy. 2014;28(2):103–109. doi: 10.2500/ajra.2014.28.4018. [DOI] [PubMed] [Google Scholar]
- 180.Shim J. Y., Park S. W., Kim D. S., Shim J. W., Jung H. L., Park M. S. The effect of interleukin-4 and amphiregulin on the proliferation of human airway smooth muscle cells and cytokine release. Journal of Korean Medical Science. 2008;23(5):857–863. doi: 10.3346/jkms.2008.23.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zuo W. L., Yang J., Gomi K., Chao I., Crystal R. G., Shaykhiev R. EGF-Amphiregulin interplay in airway stem/progenitor cells links the pathogenesis of smoking-induced lesions in the human airway epithelium. Stem Cells. 2017;35(3):824–837. doi: 10.1002/stem.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lemjabbar-Alaoui H., Sidhu S. S., Mengistab A., Gallup M., Basbaum C. TACE/ADAM-17 phosphorylation by PKC-epsilon mediates premalignant changes in tobacco smoke-exposed lung cells. PLoS One. 2011;6(3, article e17489) doi: 10.1371/journal.pone.0017489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Blanchet S., Ramgolam K., Baulig A., Marano F., Baeza-Squiban A. Fine particulate matter induces amphiregulin secretion by bronchial epithelial cells. American Journal of Respiratory Cell and Molecular Biology. 2004;30(4):421–427. doi: 10.1165/rcmb.2003-0281RC. [DOI] [PubMed] [Google Scholar]
- 184.Okumura S., Sagara H., Fukuda T., Saito H., Okayama Y. FcepsilonRI-mediated amphiregulin production by human mast cells increases mucin gene expression in epithelial cells. The Journal of Allergy and Clinical Immunology. 2005;115(2):272–279. doi: 10.1016/j.jaci.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 185.Val S., Belade E., George I., Boczkowski J., Baeza-Squiban A. Fine PM induce airway MUC5AC expression through the autocrine effect of amphiregulin. Archives of Toxicology. 2012;86(12):1851–1859. doi: 10.1007/s00204-012-0903-6. [DOI] [PubMed] [Google Scholar]
- 186.Zhou Y., Lee J. Y., Lee C. M., et al. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-β-induced pulmonary fibrosis. Journal of Biological Chemistry. 2012;287(50):41991–42000. doi: 10.1074/jbc.M112.356824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Boulanger M. J., Chow D. C., Brevnova E. E., Garcia K. C. Hexameric structure and assembly of the interleukin-6/IL-6 α-receptor/gp130 complex. Science. 2003;300(5628):2101–2104. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- 188.Mullberg J., Oberthur W., Lottspeich F., et al. The soluble human IL-6 receptor. Mutational characterization of the proteolytic cleavage site. Journal of Immunology. 1994;152:4958–4968. [PubMed] [Google Scholar]
- 189.Baran P., Nitz R., Grotzinger J., Scheller J., Garbers C. Minimal interleukin 6 (IL-6) receptor stalk composition for IL-6 receptor shedding and IL-6 classic signaling. Journal of Biological Chemistry. 2013;288(21):14756–14768. doi: 10.1074/jbc.M113.466169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Schumacher N., Meyer D., Mauermann A., et al. Shedding of endogenous interleukin-6 receptor (IL-6R) is governed by a disintegrin and metalloproteinase (ADAM) proteases while a full-length IL-6R isoform localizes to circulating microvesicles. Journal of Biological Chemistry. 2015;290(43):26059–26071. doi: 10.1074/jbc.M115.649509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lust J. A., Donovan K. A., Kline M. P., Greipp P. R., Kyle R. A., Maihle N. J. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine. 1992;4(2):96–100. doi: 10.1016/1043-4666(92)90043-Q. [DOI] [PubMed] [Google Scholar]
- 192.Rath K. S., Funk H. M., Bowling M. C., Richards W. E., Drew A. F. Expression of soluble interleukin-6 receptor in malignant ovarian tissue. American Journal of Obstetrics and Gynecology. 2010;203(3):230.e1–230.e8. doi: 10.1016/j.ajog.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 193.Lamas J. R., Rodriguez-Rodriguez L., Tornero-Esteban P., et al. Alternative splicing and proteolytic rupture contribute to the generation of soluble IL-6 receptors (sIL-6R) in rheumatoid arthritis. Cytokine. 2013;61(3):720–723. doi: 10.1016/j.cyto.2012.12.025. [DOI] [PubMed] [Google Scholar]
- 194.Lee S. Y., Buhimschi I. A., Dulay A. T., et al. IL-6 trans-signaling system in intra-amniotic inflammation, preterm birth, and preterm premature rupture of the membranes. Journal of Immunology. 2011;186(5):3226–3236. doi: 10.4049/jimmunol.1003587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rose-John S., Waetzig G. H., Scheller J., Grotzinger J., Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opinion on Therapeutic Targets. 2007;11(5):613–624. doi: 10.1517/14728222.11.5.613. [DOI] [PubMed] [Google Scholar]
- 196.Grivennikov S., Karin E., Terzic J., et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Barkhausen T., Tschernig T., Rosenstiel P., et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Critical Care Medicine. 2011;39(6):1407–1413. doi: 10.1097/CCM.0b013e318211ff56. [DOI] [PubMed] [Google Scholar]
- 198.Tadokoro T., Wang Y., Barak L. S., Bai Y., Randell S. H., Hogan B. L. IL-6/STAT3 promotes regeneration of airway ciliated cells from basal stem cells. Proceedings of the National Academy of Sciences. 2014;111(35):E3641–E3649. doi: 10.1073/pnas.1409781111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nowell M. A., Williams A. S., Carty S. A., et al. Therapeutic targeting of IL-6 trans signaling counteracts STAT3 control of experimental inflammatory arthritis. Journal of Immunology. 2009;182(1):613–622. doi: 10.4049/jimmunol.182.1.613. [DOI] [PubMed] [Google Scholar]
- 200.Brooks G. D., McLeod L., Alhayyani S., et al. IL6 trans-signaling promotes KRAS-Driven lung carcinogenesis. Cancer Research. 2016;76(4):866–876. doi: 10.1158/0008-5472.CAN-15-2388. [DOI] [PubMed] [Google Scholar]
- 201.Chalaris A., Garbers C., Rabe B., Rose-John S., Scheller J. The soluble Interleukin 6 receptor: generation and role in inflammation and cancer. European Journal of Cell Biology. 2011;90(6-7):484–494. doi: 10.1016/j.ejcb.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 202.Takizawa H., Ohtoshi T., Yamashita N., Oka T., Ito K. Interleukin 6-receptor expression on human bronchial epithelial cells: regulation by IL-1 and IL-6. The American Journal of Physiology. 1996;270, 3 Part 1:L346–L352. doi: 10.1152/ajplung.1996.270.3.L346. [DOI] [PubMed] [Google Scholar]
- 203.Klouche M., Bhakdi S., Hemmes M., Rose-John S. Novel path to activation of vascular smooth muscle cells: up-regulation of gp130 creates an autocrine activation loop by IL-6 and its soluble receptor. Journal of Immunology. 1999;163:4583–4589. [PubMed] [Google Scholar]
- 204.Romano M., Sironi M., Toniatti C., et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6(3):315–325. doi: 10.1016/S1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 205.Edmiston J. S., Archer K. J., Scian M. J., Joyce A. R., Zedler B. K., Murrelle E. L. Gene expression profiling of peripheral blood leukocytes identifies potential novel biomarkers of chronic obstructive pulmonary disease in current and former smokers. Biomarkers. 2010;15(8):715–730. doi: 10.3109/1354750X.2010.512091. [DOI] [PubMed] [Google Scholar]
- 206.Ravi A. K., Khurana S., Lemon J., et al. Increased levels of soluble interleukin-6 receptor and CCL3 in COPD sputum. Respiratory Research. 2014;15(1):p. 103. doi: 10.1186/s12931-014-0103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wang Y., Hu H., Wu J., et al. The IL6R gene polymorphisms are associated with sIL-6R, IgE and lung function in Chinese patients with asthma. Gene. 2016;585(1):51–57. doi: 10.1016/j.gene.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 208.Perez-Rubio G., Silva-Zolezzi I., Fernandez-Lopez J. C., et al. Genetic variants in il6r and adam19 are associated with copd severity in a Mexican mestizo population. COPD: Journal of Chronic Obstructive Pulmonary Disease. 2016;13(5):610–615. doi: 10.3109/15412555.2016.1161017. [DOI] [PubMed] [Google Scholar]
- 209.McGreal E. P., Davies P. L., Powell W., et al. Inactivation of IL-6 and soluble IL-6 receptor by neutrophil derived serine proteases in cystic fibrosis. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2010;1802(7-8):649–658. doi: 10.1016/j.bbadis.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 210.Le T. T., Karmouty-Quintana H., Melicoff E., et al. Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. Journal of Immunology. 2014;193(7):3755–3768. doi: 10.4049/jimmunol.1302470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Peters M., Jacobs S., Ehlers M., et al. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. Journal of Experimental Medicine. 1996;183(4):1399–1406. doi: 10.1084/jem.183.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.van Dam M., Mullberg J., Schooltink H., et al. Structure-function analysis of interleukin-6 utilizing human/murine chimeric molecules. Involvement of two separate domains in receptor binding. The Journal of Biological Chemistry. 1993;268:15285–15290. [PubMed] [Google Scholar]
- 213.Garbers C., Janner N., Chalaris A., et al. Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. The Journal of Biological Chemistry. 2011;286(17):14804–14811. doi: 10.1074/jbc.M111.229393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Scheller J., Chalaris A., Schmidt-Arras D., Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2011;1813(5):878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 215.Jostock T., Mullberg J., Ozbek S., et al. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. European Journal of Biochemistry. 2001;268(1):160–167. doi: 10.1046/j.1432-1327.2001.01867.x. [DOI] [PubMed] [Google Scholar]
- 216.Oberg H. H., Wesch D., Grussel S., Rose-John S., Kabelitz D. Differential expression of CD126 and CD130 mediates different STAT-3 phosphorylation in CD4+CD25- and CD25high regulatory T cells. International Immunology. 2006;18(4):555–563. doi: 10.1093/intimm/dxh396. [DOI] [PubMed] [Google Scholar]
- 217.Mihara M., Hashizume M., Yoshida H., Suzuki M., Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clinical Science. 2012;122(4):143–159. doi: 10.1042/CS20110340. [DOI] [PubMed] [Google Scholar]
- 218.Johnston P. A., Grandis J. R. STAT3 signaling: anticancer strategies and challenges. Molecular Interventions. 2011;11(1):18–26. doi: 10.1124/mi.11.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Geiger J. L., Grandis J. R., Bauman J. E. The STAT3 pathway as a therapeutic target in head and neck cancer: Barriers and innovations. Oral Oncology. 2016;56:84–92. doi: 10.1016/j.oraloncology.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hillmer E. J., Zhang H., Li H. S., Watowich S. S. STAT3 signaling in immunity. Cytokine & Growth Factor Reviews. 2016;31:1–15. doi: 10.1016/j.cytogfr.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Labenski H., Hedtfeld S., Becker T., Tummler B., Stanke F. Initial interrogation, confirmation and fine mapping of modifying genes: STAT3, IL1B and IFNGR1 determine cystic fibrosis disease manifestation. European Journal of Human Genetics. 2011;19(12):1281–1288. doi: 10.1038/ejhg.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Yew-Booth L., Birrell M. A., Lau M. S., et al. JAK-STAT pathway activation in COPD. European Respiratory Journal. 2015;46(3):843–845. doi: 10.1183/09031936.00228414. [DOI] [PubMed] [Google Scholar]
- 223.Diaz-Rodriguez E., Montero J. C., Esparis-Ogando A., Yuste L., Pandiella A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor α-converting enzyme at threonine 735: a potential role in regulated shedding. Molecular Biology of the Cell. 2002;13(6):2031–2044. doi: 10.1091/mbc.01-11-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Soond S. M., Everson B., Riches D. W., Murphy G. ERK-mediated phosphorylation of Thr735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. Journal of Cell Science. 2005;118(11):2371–2380. doi: 10.1242/jcs.02357. [DOI] [PubMed] [Google Scholar]
- 225.Wang Y., Herrera A. H., Li Y., Belani K. K., Walcheck B. Regulation of mature ADAM17 by redox agents for L-selectin shedding. Journal of Immunology. 2009;182(4):2449–2457. doi: 10.4049/jimmunol.0802770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Reddy P., Slack J. L., Davis R., et al. Functional analysis of the domain structure of tumor necrosis factor-α converting enzyme. Journal of Biological Chemistry. 2000;275(19):14608–14614. doi: 10.1074/jbc.275.19.14608. [DOI] [PubMed] [Google Scholar]
- 227.Li C., Hao M., Cao Z., et al. Naked2 acts as a cargo recognition and targeting protein to ensure proper delivery and fusion of TGF-α containing exocytic vesicles at the lower lateral membrane of polarized MDCK cells. Molecular Biology of the Cell. 2007;18(8):3081–3093. doi: 10.1091/mbc.E07-02-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Sigismund S., Argenzio E., Tosoni D., Cavallaro E., Polo S., Di Fiore P. P. Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Developmental cell. 2008;15(2):209–219. doi: 10.1016/j.devcel.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 229.Goh L. K., Huang F., Kim W., Gygi S., Sorkin A. Multiple mechanisms collectively regulate clathrin-mediated endocytosis of the epidermal growth factor receptor. The Journal of Cell Biology. 2010;189(5):871–883. doi: 10.1083/jcb.201001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Tan X., Lambert P. F., Rapraeger A. C., Anderson R. A., Stress-Induced E. G. F. R. Trafficking: mechanisms, functions, and therapeutic implications. Trends in Cell Biology. 2016;26(5):352–366. doi: 10.1016/j.tcb.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tomas A., Futter C. E., Eden E. R. EGF receptor trafficking: consequences for signaling and cancer. Trends in Cell Biology. 2014;24(1):26–34. doi: 10.1016/j.tcb.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Jones S., Cunningham D. L., Rappoport J. Z., Heath J. K. The non-receptor tyrosine kinase Ack1 regulates the fate of activated EGFR by inducing trafficking to the p62/NBR1 pre-autophagosome. Journal of Cell Science. 2014;127(5):994–1006. doi: 10.1242/jcs.136895. [DOI] [PubMed] [Google Scholar]
- 233.Higginbotham J. N., Demory Beckler M., Gephart J. D., et al. Amphiregulin exosomes increase cancer cell invasion. Current Biology. 2011;21(9):779–786. doi: 10.1016/j.cub.2011.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ebsen H., Lettau M., Kabelitz D., Janssen O. Subcellular localization and activation of ADAM proteases in the context of FasL shedding in T lymphocytes. Molecular Immunology. 2015;65(2):416–428. doi: 10.1016/j.molimm.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 235.Dombernowsky S. L., Samsoe-Petersen J., Petersen C. H., et al. The sorting protein PACS-2 promotes ErbB signalling by regulating recycling of the metalloproteinase ADAM17. Nature Communications. 2015;6:p. 7518. doi: 10.1038/ncomms8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Murphy G. Regulation of the proteolytic disintegrin metalloproteinases, the ‘Sheddases’. Seminars in Cell & Developmental Biology. 2009;20(2):138–145. doi: 10.1016/j.semcdb.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 237.Doedens J. R., Black R. A. Stimulation-induced down-regulation of tumor necrosis factor-α converting enzyme. Journal of Biological Chemistry. 2000;275(19):14598–14607. doi: 10.1074/jbc.275.19.14598. [DOI] [PubMed] [Google Scholar]
- 238.Zhang H., Deng T., Liu R., et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nature Communications. 2017;8, article 15016 doi: 10.1038/ncomms15016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Groth E., Pruessmeyer J., Babendreyer A., et al. Stimulated release and functional activity of surface expressed metalloproteinase ADAM17 in exosomes. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2016;1863(11):2795–2808. doi: 10.1016/j.bbamcr.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 240.Wahlund C. J. E., Eklund A., Grunewald J., Gabrielsson S. Pulmonary extracellular vesicles as mediators of local and systemic inflammation. Frontiers in Cell and Developmental Biology. 2017;5:p. 39. doi: 10.3389/fcell.2017.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]