


Abstract
A methodology to achieve high-throughput de novo sequencing of synthetic peptide mixtures is reported. The approach leverages shotgun nanoliquid chromatography coupled with tandem mass spectrometry-based de novo sequencing of library mixtures (up to 2000 peptides) as well as automated data analysis protocols to filter away incorrect assignments, noise, and synthetic side-products. For increasing the confidence in the sequencing results, mass spectrometry-friendly library designs were developed that enabled unambiguous decoding of up to 600 peptide sequences per hour while maintaining greater than 85% sequence identification rates in most cases. The reliability of the reported decoding strategy was additionally confirmed by matching fragmentation spectra for select authentic peptides identified from library sequencing samples. The methods reported here are directly applicable to screening techniques that yield mixtures of active compounds, including particle sorting of one-bead one-compound libraries and affinity enrichment of synthetic library mixtures performed in solution.
Keywords: de novo sequencing, synthetic peptide mixtures, shotgun nanoliquid chromatography, tandem mass spectrometry, one-bead one-compound libraries
Graphical abstract
INTRODUCTION
Decoding the structure of identified active compounds is a necessary step in most embodiments of the combinatorial discovery process. With regard to peptide libraries, several approaches to sequence determination of identified peptides are established. DNA/RNA encoding is one of the most commonly used strategies because the DNA sequencing technology is reliable, fast, and cheap.1 This approach is commonly utilized in phage,2,3 yeast,4,5 and mRNA6,7 display strategies. Genetic encoding of peptide libraries is limited, however, in its ability to successfully incorporate a variety of “privileged” nonproteogenic amino acids and their analogues (β-, γ-, d-amino acids and peptoids),8–11 although this issue can be circumvented to a certain degree in mRNA display.12,13 The application of DNA encoding to synthetic peptide libraries was reported more than 20 years ago.14 However, the strategy has not caught on until recently.15–18
Synthetic peptide libraries are commonly decoded by either Edman degradation19–22 or MALDI-coupled23–25 tandem mass spectrometry-based (MS/MS) de novo peptide sequencing. Edman degradation, although reliable, is slow,26 not easily amenable to multiplexing, and, like genetic encoding, is generally limited to α-amino acids.27 Mass spectrometry-based strategies are able to decode amino acids of great structural diversity28–30 but require that a peptide forms a complete fragmentation ladder for unambiguous de novo sequence assignment. Unfortunately, many peptides do not generate fragmentation spectra of this quality.31–34 In proteomics, partially correct sequence assignments can be sufficient to identify a peptide by the use of database matching. This approach is not generally applicable to the sequencing of synthetic peptide libraries when each of a series of candidate assignments is possible according to the library design.
In this work, we investigate the use of a priori library design considerations to improve the reliability with which synthetic peptide mixtures can be sequenced by automated de novo assignment of MS/MS spectra. By constraining library designs (as described below), candidate de novo assignments can be “matched” to library design features in a fashion reminiscent of database matching in proteomics. A priori constraints imposed on peptide sequence features enabled us to reject candidate assignments inconsistent with the library design and in some cases to replace them with a design-consistent assignment deemed lower quality by de novo sequencing software. In this way, we were able to improve the reliability of automated sequence assignments made to peptides exhibiting suboptimal fragmentation spectra.
This strategy is in principle applicable to any MS/MS-based peptide sequencing approach, including those employing MALDI-TOF/TOF instrumentation. However, we believe its real value lies in high-throughput sequencing of MS/MS data generated by liquid chromatography/tandem mass spectrometry (LC/MS/MS) analysis of synthetic peptide mixtures. MS/MS-based decoding of synthetic peptide libraries is generally performed on individual compounds isolated from screens of one-bead one-compound (OBOC) libraries, and MALDI-TOF/TOF instrumentation is well suited to such a workflow. In contrast, an MS/MS-based decoding strategy applicable to complex peptide mixtures has not been realized despite longstanding interest in such a method.35,36 LC/MS/MS is capable of separating mixtures containing thousands of peptides on the time scale of hours, as recent advances in proteomics indicate.37–39 Analogous analysis of synthetic peptide mixtures is complicated by the presence of synthetic side-products, unknown number of input peptides in the sample, and the absence of useful databases to help discarding incorrect sequence assignments and noise. Improving the reliability of automated de novo sequence assignments mitigates these issues and potentially enables the realization of a high-throughput LC/MS/MS-based decoding strategy. At least two powerful screening techniques, particle sorting of OBOC libraries14,17 and affinity enrichment performed in solution,40–42 generate mixtures of active compounds and could benefit from the development of such a strategy.
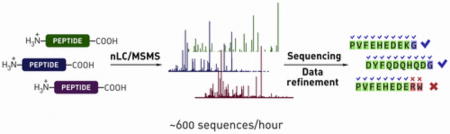
To address this need, we investigate the feasibility of a high-throughput LC/MS/MS-based approach to the decoding of synthetic peptide mixtures (Figure 1). First, we study the utility of library design rules for improving the accuracy of automated de novo sequence assignments and demonstrate the benefits of sequence-constrained libraries in the context of high-throughput de novo sequencing. Second, we describe automated data analysis routines to identify peptides of interest and filter them away from noise, incorrect assignments, and synthetic side-products. Finally, we establish conditions for high-throughput nLC/MS/MS analysis of peptide libraries and demonstrate that decoding can be performed at a rate of 600 peptides/hour without substantial losses in the sequence identification rate.
Figure 1.

Five-step experimental workflow for the proposed library decoding strategy enables automated sequence assignment and data analysis.
RESULTS
Constraining the Library Design Rules
We began by identifying the features of a library peptide sequence that might be used to distinguish a potentially correct assignment from an incorrect one (that is, one inconsistent with library design rules). In most cases, all intended library members possess some common features that allow the discrimination of actual peptides from noise, nonsense de novo assignments, and synthetic impurities such as deletions and truncations. Two such features are peptide length and the presence of a fixed subsequence (“constant region”). For instance, in our case, the use of glycyl-OCH2-phenylacetamidomethyl (PAM) ester as a TFA-stable/base-labile cleavable linker43,44 for use in OBOC libraries resulted in a fixed C-terminal Gly residue for all library peptides.
In addition to these features, we introduced an alternating monomer subset (AMS) design in an attempt to (a) increase de novo sequencing assignment confidence in the peptide variable region and (b) resolve assignment ambiguities for fragmentation spectra with incomplete ion ladders, especially for those cases where both complementary b and y ions are missing. Briefly, in this approach, a library is constructed using standard solid-phase peptide synthesis (SPPS) procedures using two nonintersecting amino acid subsets, alternating them according to position, i.e., subset1-subset2-subset1-subset2-etc. Monomer subsets are constructed by dividing the total monomer pool into subsets of equal size such that the resulting molecular weight of each subset1-subset2 dipeptide is unique (details in Supporting Information (SI) 3.3).
We anticipated that the AMS approach would improve the overall decoding confidence by decreasing the probability of randomly assigning a subsequence allowed by the library design for spectra where definitive assignments may not be possible. More importantly, we hoped to resolve any potential ambiguous isomeric dipeptide arrangements because every subset1-subset2 dipeptide has a unique molecular weight (the list of all possible dipeptide molecular weights for the AMS design we utilized in this work is provided in SI 3.3). Additionally, this approach is straightforward in its experimental implementation as it does not require any extra chemical manipulations during or after library assembly, which can become an important consideration for libraries of great size. One obvious disadvantage of working with AMS peptide libraries is, of course, their artificially limited diversity, both in terms of the theoretical library size and limited structural-positional variance of library members. We expect that this drawback can be mitigated by increasing the size and structural diversity of the parent monomer set, such that structural differences in alternating subsets become less prominent. Critical evaluation of the performance of AMS libraries in screening applications will be required to substantiate this claim.
Automated Decoding of Synthetic Peptide Mixtures
With these considerations in mind, we turned to constructing a model 10-mer OBOC library to study our hypotheses. For a total amino acid set of 16 amino acids, a number of feasible monomer subsets exist. We chose the combination where amino acids Asp, Phe, His, Lys, Met, Pro, Trp, and Leu are combined in subset 1 (ss1) and Ala, Glu, Gly, Gln, Ser, Thr, Val, and Tyr constitute subset 2 (ss2), trying to separate monomers of similar functionality into different subsets where possible. Cys, Ile, Asn, and Arg were excluded for various reasons (SI 2.2). The library of the general design ss1-ss2-ss1-ss2-ss1-ss2-ss1-ss2-ss1-Gly (library 1) was constructed using established Fmoc SPPS procedures45 on Tentagel S-NH2 30 µm resin (Rapp Polymere, 0.20 mmol/g amine loading) functionalized with PAM linker for TFA-orthogonal release of peptides from the solid support prior to nLC/MS/MS analysis. Next, we established a standard protocol for nLC/MS/MS analysis of mixtures containing peptides from a few hundred beads and found that nLC can successfully detect and separate peptides for the subsequent MS/MS analysis running a 70 min-long gradient (SI 3.5) even when only 2% of the total sample volume (~80 fmol/peptide) was submitted for analysis. Collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) peptide fragmentation spectra were obtained for each precursor ion automatically in a data-dependent fashion on a Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer. De novo peptide sequencing of the acquired data was performed in PEAKS 7 or PEAKS 8 (BioInformatics Solutions Inc.),46,47 which is widely regarded as one of the most reliable de novo sequencing software.34,48,49 Using PEAKS, CID and HCD spectra were merged, prefiltered to remove noise, and sequenced allowing Met-oxide as a variable post-translational modification. Fifteen candidate sequence assignments were created for each merged secondary scan.
Finally, we created automated Python-based routines for postprocessing data analysis to eliminate noise, synthetic impurities, duplicates, resolve certain sequencing ambiguities, and to select the best candidate sequence assignment for each merged MS/MS scan. Briefly, in the first step of this process, a priori library design rules are used to eliminate all sequence candidates of length other than 10 not bearing a C-terminal Gly residue or not having a correct monomer in each of the allocated positions. During this step, peptide candidates with incorrect amino acid ordering and incorrect dipeptide assignments are removed from further consideration as a result of the inherent properties of the AMS. For example, one sequence candidate for a peptide containing Asp-Ala subsequence can be interpreted by the sequencing software as Gly-Glu if no fragmentation is observed between these two residues, but because Gly-Glu is not a valid dipeptide in the AMS design, this sequence is rejected during the data analysis step (Figure 2). Next, for each remaining spectrum, a single candidate is kept, discarding all other peptides with lower sequencing scores from PEAKS (average local confidence (ALC) scores, from 0 to 99), and duplicate sequences are labeled as nonunique. Finally, the resulting unique sequence assignments are refined further by excluding prominent synthetic impurities that were not eliminated in the previous steps. If two unique sequences have an identifiable main product/side product relationship, the side product is eliminated. In this way, peptides containing oxidized Met residues, deamidation of Gln to Glu, which occasionally happens during saponification of PAM ester, sodium adducts, and a few less prominent side-reactions are identified, and their corresponding sequences are discarded. The list of remaining sequence assignments (or “peptides”) is considered to be the final result of the workflow and can be utilized further to perform appropriate statistical analysis.
Figure 2.

Spectra with incomplete b/y-fragmentation ladders can be unambiguously assigned using the AMS principle. Displayed are merged, preprocessed CID and HCD spectra for 491.243 Da/e precursor ions. The first candidate sequence does not match the library design pattern due to the fact that the pair of b3/y7 ions is not observed in the spectrum, and consequently, an unreliable assignment is made. The molecular weight difference of 186.064 Da between the observed fragment ions corresponds to four different dipeptides (Gly-Glu, Glu-Gly, Ala-Asp, and Asp-Ala), but only one of them (Asp-Ala) matches the parent design, which makes an unambiguous assignment possible. ss1: monomer subset 1, ss2: monomer subset 2, cr: constant region.
Evaluating the Performance of the Method
Having established the workflow for high-throughput decoding of peptide libraries, we sought to evaluate the performance of the method. To this end, we analyzed a naïve, i.e., not preselected in any way, aliquot containing 660 beads bearing library 1 peptides by nLC/MS/MS and performed the downstream data analysis as described above. As summarized in Table 1, each of the filtration steps played a significant role in filtering out incorrect assignments, and the final sequence list consisted of 587 unique peptides (0.89 sequence identification rate; full sequence list is provided in SI Appendix I). Manual analysis of the peptides rejected in each filtration step verified that the automated process leads to an identical outcome. As shown in Figure 3a, the resulting data set has positional amino acid frequency distribution close to uniform (χ2-test; P-value = 0.42, details in SI 3.4), suggesting that the workflow did not bias the results based on amino acid composition, at least not in an immediately obvious way. Additionally, as demonstrated in Figure 3B and C, the post de novo noise filtration process primarily removed peptides with low sequencing scores and high assignment mass errors while retaining high confidence and low mass error ones. More specifically, the unprocessed PEAKS output data set had average ALC of 57 ± 27 (one standard deviation) and assignment mass error of −0.8 ± 5.7 ppm, whereas in the final peptide data set, these parameters were 82 ± 14 and −0.7 ± 1.0 ppm, respectively, reinforcing the notion that low confidence assignments and noise are selected against during the filtration process.
Table 1.
Three Data Analysis Steps (Matching, Finding Unique Peptides, and Sequence Refinement) Are Necessary to Eliminate Noise and Incorrect Assignments: Data Analysis Summary for a Sample Consisting of 660 Beads Bearing Library 1 Peptides
| data analysis | stage no. of sequences |
|---|---|
| unfiltered PEAKS output | 5526 |
| matched | 2396 |
| matched, unique | 913 |
| matched, unique, refined | 587 |
Figure 3.

Alternating monomer set (AMS) increases confidence in sequencing results. All data are from the 660-bead sample of library 1 peptides (587 unique sequences identified). (A) Color-coded positional amino acid frequency is shown on the left, and the mean amino acid frequencies are plotted on the right. Each nonzero cell in the matrix has the expected value of 0.125, and the observed values map closely to it. (B) Sequencing quality scatterplot for the unfiltered PEAKS output. (C) Sequencing quality scatterplot for the final filtered data set. Most peptides with low sequencing score and/or large assignment errors are removed during the postsequencing filtration. (D) Precision-recall curves for different data filtration methods. ALC thresholds (0–99) are applied to the reference and method 1 and 2 data sets, and the corresponding precision and recall values are calculated for each data set. Method 1 is inferior to AMS in both precision and recall. (E) Total number of sequences recovered as a function of sequencing score for different data filtration methods. Results diverge in the region of medium (50–85) sequencing scores.
Evaluating the Importance of the AMS Library Design
Next, we turned to assessing the utility of the AMS design for gaining confidence in sequencing results. The above parsed data set (containing 587 unique peptides) was taken as a reference, operating under the assumption that the AMS does not bias de novo sequencing. Then, in method 1, the PEAKS output was parsed without imposing the AMS design rules; that is, assuming that the library monomer composition is uniform with respect to all nine positions, and the monomer set consists of the 16 amino acids comprising the sum of two alternating subsets. The resulting data set was expected to contain some erroneous assignments, inconsistent with the AMS library design. Analysis of sequences present in the method 1 output but absent from the reference data set could be used to estimate the frequency at which AMS eliminates a priori incorrect assignments. Processing the data set using method 1 led to 756 unique sequences, of which only 502 were found in the reference data set. On the basis of this outcome, we concluded that the use of the AMS in combination with postsequencing filtration routines improves sequencing accuracy by rejecting 254 incorrectly assigned peptides (38% of 660 analyzed beads).
A portion of the 254 spectra given inaccurate assignments was rescued by use of the AMS design. In method 2, we parsed the PEAKS output assuming the AMS design but considering only the single most confident peptide candidate per spectrum, i.e., the candidate with the highest ALC score. For some spectra, the highest ALC score candidate will contain dipeptide assignments inconsistent with the AMS design, but a lower ALC score candidate will be AMS-consistent. Assignments to such spectra will be eliminated by method 2 but present in the reference data set. Comparison of the method 2 set to the AMS set allowed us to estimate the frequency at which the AMS approach recovers sequences containing incorrect dipeptide assignments. We found that method 2 yielded 505 unique peptides, 500 of which were from the reference data set. Therefore, the AMS design helped to recover 87 peptides (13% of analyzed beads), which would not have been considered top candidates by the software.
A precision-recall curve comparing the reference, method 1, and method 2 data sets is shown in Figure 3D. “Precision” here is the fraction of sequences in a data set that are also found in the reference set. “Recall” is the ratio of sequences in a data set that match the reference to the total number of peptides in the sample, in theory (in this case, 660). By these metrics, both methods 1 and 2 compare unfavorably against the reference data set. Method 1 suffers from lower recall (0.76 vs 0.89) and lower precision (0.66 vs 1.00), and method 2 leads to lower recall (0.76 vs 0.89).
A closer look reveals that the data subsets consisting of de novo peptides with high sequencing confidence (ALC≳ 83) overlapped almost exactly, whereas subsets of medium sequencing scores (50 ≲ ALC ≲ 83) diverged significantly. This behavior is particularly evident from the analysis of the absolute sequence recall as a function of a sequencing score: recall between the data sets diverged in the ALC range of ~50 to ~83; almost no sequences were found at ALC values lower than 50, and no significant divergence was observed at high ALC values (Figure 3E).
These results suggest an alternative strategy for reliable MS/MS sequencing of peptide libraries, namely, discarding peptides with sequencing scores lower than ~85. Such a strategy obviates the need for AMS at the expense of a much lower sequence recall. For instance, parsing the data described above using method 1 and discarding sequences with ALC scores less than 85 yields 310 unique peptides (recall: 0.47, precision: 0.97). A fully analogous experiment performed for a separate 306 bead sample of library 1 peptides (details are in SI 3.1, Appendix II) corroborated the conclusions listed here.
To further probe the reliability of our approach, we resynthesized individual peptides identified as described above and subjected the resulting peptidyl resins to the library analysis conditions. We found that in all cases MS/MS spectra of resynthesized peptides agreed well with those observed in a corresponding library sequencing experiment (Figure 4, SI 3.2), reinforcing our belief in the accuracy of PEAKS assignments obtained by our approach.
Figure 4.

Proposed library decoding workflow yields reliable results. Sequences assigned to spectra from a library analysis were resynthesized and subjected to analogous analytical conditions. Original library-derived spectra are shown in blue; spectra of authentic peptides are displayed in red. (A) Overlaid raw CID fragmentation spectra (collision energy = 20.6 eV, precursor ion: 710.82 Da/e) for GCβFLDEVEFPHG peptide (β = β-alanine). (B) Overlaid raw CID fragmentation spectra (collision energy = 21.1 eV, precursor ion: 654.77 Da/e) for GCβFADASEFPHG peptide.
Evaluating the Analysis Throughput
Next, we studied the potential throughput of our approach using library 1 as a model AMS library. In particular, we were interested in investigating the sequence identification rate as a function of sample complexity. To this end, we prepared bead aliquots and manually counted the exact number of beads for samples comprised of less than 1000 beads. For more complex analytes, the number of beads was estimated in three ways, and the average value was assumed. The targeted complexity of the prepared aliquots was 50, 150, 300, 600, or 1200 beads, but the actual bead counts deviated from these numbers. All aliquots were analyzed by nano-LC/MS/MS (CID and HCD) running a linear 70 min long 2 → 48% acetonitrile in water gradient. All downstream data processing was performed as described above. As summarized in Figure 5, we found that the simplest bead mixtures yielded the highest sequence identification rates: all samples comprised of approximately 50 beads recovered more than 95% of analyzed peptides. For samples comprised of between 50 and 660 beads, we observed a gradual decrease in sequence recall with increasing sample complexity and a steep drop in sequence identification rate for more complex samples. We attributed these observations to the phenomenon of “peptide interference”: too many peptides eluting off an nLC column per unit time may cause the mass spectrometer to omit analysis of some and prevent the isolation of individual precursor ions for others. Indeed, when we reanalyzed one of the more complex samples comprised of approximately 1600 beads and extended the LC gradient to either 2 or 3 h, we observed a uniform increase of the sequence identification rates from 0.75 to 0.87, which corresponded to an extra 185 peptides identified for the 3 h gradient. These results suggest, assuming that the overall sequence recovery of 85% is deemed satisfactory, that AMS library samples can be routinely analyzed at a rate of at least 600 peptides/hour and that samples comprised of thousands of peptides can also be successfully decoded by extending LC gradients accordingly.
Figure 5.

Approximately 600 peptides/hour can be decoded while keeping the sequence identification rate above 0.85. (A) Sequence identification rate as a function of sample complexity. Gradual reduction of recall values is observed as samples become more complex. (B) Analysis of a 1600 bead library sample (marked red in panel A) under different nLC conditions. Extending the gradient time improves sequence recall.
Analyzing Libraries Containing Multiple Nonproteogenic Amino Acid Residues
The ability to sequence peptides comprised of structurally diverse unnatural amino acids is one of the major advantages of mass spectrometry over other decoding techniques. Accordingly, we sought to demonstrate the applicability of our approach toward sequencing libraries comprised of nonproteogenic amino acids. For this study, we prepared another model library (library 2) comprised of proteogenic and nonproteogenic α-, β-, and δ-amino acids (details in the SI 2.2.2) and analyzed it using the standard protocol, encoding unnatural amino acids as fixed post-translational modifications on unused proteogenic amino acids during the de novo sequencing step in PEAKS. Analysis of three samples containing 183, 212, and 220 beads revealed that library 2 can be decoded nearly as efficiently as library 1 with sequence identification rates reaching 86%. Additionally, the overall quality of fragmentation spectra was comparable to those observed for libraries of proteogenic peptides, as evaluated by manual inspection. Two representative high-quality MS/MS spectra and their respective sequence assignments featuring multiple unnatural amino acids are demonstrated in Figure 6. Taken together, these observations indicate that our approach can be utilized to decode structurally diverse libraries of polypeptides and peptidomimetics. Further work is required to demonstrate the applicability of our approach to the sequencing of non-natural polymers comprised of tertiary amides (peptoids) or other linkages.
Figure 6.

Libraries of peptides with multiple unnatural amino acids can be successfully decoded using the proposed strategy. (A, B) Merged, postprocessed CID and HCD spectra for a 615.84 Da/e precursor ion and a 499.78 Da/e precursor ions with corresponding assignments and decoded sequences. One-letter encoding of unnatural amino acids highlighted in green are shown on the right.
DISCUSSION
In this work, we investigated the use of constraining library designs to increase the reliability and throughput of de novo sequencing of synthetic peptide mixtures. This involved the development of new MS/MS-friendly library designs and associated data analysis procedures to take advantage of these designs for the interpretation of de novo sequencing output from commercial software. Our results indicate that the proposed library design principle improves both sequencing accuracy and peptide recovery rate. Using nLC/MS/MS for high-throughput analysis of synthetic peptide mixtures, at least 600 peptides/hour can be decoded by the use of this strategy while keeping the peptide identification rate above 85%. Combined with the fact that our approach is experimentally straightforward, no extra chemical manipulations or specialty reagents are required at any stage during library synthesis or analysis, our results suggest that the approach described herein may be a feasible high-throughput method for decoding mixtures of synthetic peptides. Application of the described methods to the decoding of compound mixtures obtained from library screens will be required to test their true utility for this purpose.
One potential application of this methodology is the decoding of compound mixtures obtained from particle sorting of OBOC libraries. Particle sorting is well suited for the automated analysis of on-bead screens of OBOC libraries prepared on small monosized resins (10 or 30 micron),50,51 which are compatible with commercial flow cytometers. In contrast to manual screening methods in which individual beads displaying active compounds are selected for decoding, particle sorting generally yields mixtures of beads. Historically, DNA encoding has been required to decode such libraries, as the amount of material on a single bead (~100 fmol for 0.2 mmol/g resin) was insufficient for MS/MS analysis. The unique sensitivity provided by the nLC strategy described here enables the analysis of ~80 fmol of individual peptides, in principle enabling the decoding of libraries prepared on 10 µm diameter resin.
A second use case of our strategy is the decoding of mixtures obtained by affinity enrichment performed in solution. Affinity enrichment is a powerful screening technique that has become widely used in the pharmaceutical industry for the identification of drug leads. However, the challenge of identifying active compounds (often small molecules identified by their exact mass) by mass spectrometry generally limits the complexity of library pools that can be screened using this strategy.40 A strategy for de novo sequencing of peptide mixtures should increase the size of synthetic peptide libraries amenable to the affinity selection approach.
For any conceivable screening application, we believe that the described method will be most valuable in analyzing sparse libraries of great diversity (both in terms of member size and variable region length). We consider libraries of theoretical diversity on the order of 107 members, consisting of six variable positions occupied by one of 16 possible amino acids, to be “low diversity”. MS/MS sequencing of hexapeptides from such a library should be more robust than the sequencing of analogous decapeptides, and consequently, the value of AMS can be expected to decrease for libraries of low diversity. Analyzing OBOC peptide libraries of high redundancy52 using our approach may lead to a challenge in identifying the number of beads on which a given sequence was displayed; this challenge may become prominent during decoding of selected peptides where some sort of sequence convergence has occurred. Although technically possible, such identification was not attempted in this work.
We anticipate that the presented experimentation and the reasoning behind it are not inherently limited to decoding active compounds obtained from screens for binders. Some of the potential applications of such a methodology may include direct, selection-free reactivity library profiling to identify unique chemical reactivities displayed by short peptide sequences, rapid profiling of enzyme specificity for various enzymes acting on peptidic substrates, or the generation of customized (defined only by the library design) mass spectral databases to extend our knowledge of peptide fragmentation pathways and improve the accuracy of modern de novo sequencing algorithms. The development of some of these techniques is the focus of our ongoing investigations.
Supplementary Material
Acknowledgments
This work was supported by the Defense Advanced Research Projects Agency (Award No. 023504-001, B.L.P.). C.Z. is a recipient of the Bristol-Myers Squibb Fellowship in Synthetic Organic Chemistry. The authors would like to thank Prof. JoAnne Stubbe, Prof. Elizabeth Nolan, Dr. Mark D. Simon, Mr. Ethan D. Evans, Dr. Surin K. Mong, and Mr. Alexander J. Mijalis for insightful discussions and helpful suggestions.
Footnotes
ASSOCIATED CONTENT
- Experimental procedures, characterization of synthesized compounds, supplementary tables, and additional data; parts of the data analysis code are available to interested academic users upon direct request to authors (PDF)
The authors declare no competing financial interest.
References
- 1.Goodwin S, Mcpherson J, Mccombie R. Coming of Age: Ten Years of next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith G. Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 3.Smith G, Petrenko V. Phage Display. Chem. Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 4.Boder E, Wittrup D. Yeast Surface Display for Screening Combinatorial Polypeptide Libraries. Nat. Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 5.Wittrup D, Boder E. Yeast Surface Display for Directed Evolution of Protein Expression, Affinity, and Stability. Methods Enzymol. 2000;328:430–444. doi: 10.1016/s0076-6879(00)28410-3. [DOI] [PubMed] [Google Scholar]
- 6.Mattheakis L, Bhatt R, Dower W. An in Vitro Polysome Display System for Identifying Ligands from Very Large Peptide Libraries. Proc. Natl. Acad. Sci. U. S. A. 1994;91:9022–9026. doi: 10.1073/pnas.91.19.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson D, Keefe A, Szostak J. The Use of mRNA Display to Select High-Affinity Protein-Binding Peptides. Proc. Natl. Acad. Sci. U. S. A. 2001;98:3750–3755. doi: 10.1073/pnas.061028198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuckermann R, Kodadek T. Peptoids as Potential Therapeutics. Curr. Opin. Mol. Ther. 2009;11:299–307. [PubMed] [Google Scholar]
- 9.Fisher B, Gellman S. Impact of γ-Amino Acid Residue Preorganization on A/γ-peptide Foldamer Helicity in Aqueous Solution. J. Am. Chem. Soc. 2016;138:10766–10769. doi: 10.1021/jacs.6b06177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng R, Gellman S, DeGrado W. Beta-Peptides: From Structure to Function. Chem. Rev. 2001;101:3219–3232. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- 11.Fowler S, Blackwell H. Structure–function Relationships in Peptoids: Recent Advances toward Deciphering the Structural Requirements for Biological Function. Org. Biomol. Chem. 2009;7:1508–1524. doi: 10.1039/b817980h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlippe Y, Hartman M, Josephson K, Szostak J. In Vitro Selection of Highly Modified Cyclic Peptides That Act as Tight Binding Inhibitors. J. Am. Chem. Soc. 2012;134:10469–10477. doi: 10.1021/ja301017y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hipolito C, Suga H. Ribosomal Production and in Vitro Selection of Natural Product-like Peptidomimetics: The FIT and RaPID Systems. Curr. Opin. Chem. Biol. 2012;16:196–203. doi: 10.1016/j.cbpa.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 14.Needels M, Jones D, Tate E, Heinkel G, Kochersperger L, Dower W, Barrett R, Gallop M. Generation and Screening of an Oligonucleotide-Encoded Synthetic Peptide Library. Proc. Natl. Acad. Sci. U. S. A. 1993;90:10700–10704. doi: 10.1073/pnas.90.22.10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macconnell A, Mcenaney P, Cavett V, Paegel B. DNA-Encoded Solid-Phase Synthesis: Encoding Language Design and Complex Oligomer Library Synthesis. ACS Comb. Sci. 2015;17:518–534. doi: 10.1021/acscombsci.5b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Malone M, Paegel B. What Is a “DNA-Compatible” Reaction? ACS Comb. Sci. 2016;18:182–187. doi: 10.1021/acscombsci.5b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendes K, Malone M, Ndungu J, Suponitsky-Kroyter I, Cavett V, Mcenaney P, Macconnell A, Doran T, Ronacher K, Stanley K, Utset O, Walzl G, Paegel B, Kodadek T. High-Throughput Identification of DNA-Encoded IgG Ligands That Distinguish Active and Latent Mycobacterium Tuberculosis Infections. ACS Chem. Biol. 2017;12:234–243. doi: 10.1021/acschembio.6b00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macconnell A, Price A, Paegel B. An Integrated Micro Fluidic Processor for DNA-Encoded Combinatorial Library Functional Screening. ACS Comb. Sci. 2017;19:181–192. doi: 10.1021/acscombsci.6b00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam K, Lehman A, Song A, Doan N, Enstrom A, Maxwell J, Liu R. Synthesis and Screening of “‘One-Bead One-Compound’” Combinatorial Peptide Libraries. Methods Enzymol. 2003;369:298–322. doi: 10.1016/S0076-6879(03)69017-8. [DOI] [PubMed] [Google Scholar]
- 20.Boeijen A, Liskamp R. Sequencing of Peptoid Peptidomimetics by Edman Degradation. Tetrahedron Lett. 1998;39:3589–3592. [Google Scholar]
- 21.Joo SH, Xiao Q, Ling Y, Gopishetty B, Pei D. High-Throughput Sequence Determination of Cyclic Peptide Library Members by Partial Edman Degradation/Mass Spectrometry. J. Am. Chem. Soc. 2006;128:13000–13009. doi: 10.1021/ja063722k. [DOI] [PubMed] [Google Scholar]
- 22.Thakkar A, Cohen A, Connolly M, Zuckermann R, Pei D. High-Throughput Sequencing of Peptoids and Peptide-Peptoid Hybrids by Partial Edman Degradation and Mass Spectrometry. J. Comb. Chem. 2009;11:294–302. doi: 10.1021/cc8001734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Semmler A, Weber R, Przybylski M. De Novo Sequencing of Peptides on Single Resin Beads by MALDI-FTICR Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2010;21:215–219. doi: 10.1016/j.jasms.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Lee S, Lim J, Tan S, Cha J, Yeo S, Agnew H, Heath J. Accurate MALDI-TOF/TOF Sequencing of One-Bead - One-Compound Peptide Libraries with Application to the Identification of Multiligand Protein Affinity Agents Using in Situ Click Chemistry Screening. Anal. Chem. 2010;82:672–679. doi: 10.1021/ac902195y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cha J, Lim J, Zheng Y, Tan S, Ang Y, Oon J, Ang M, Ling J, Bode M, Lee S. Process Automation toward Ultra-High-Throughput Screening of Combinatorial One-Bead-One-Compound (OBOC) Peptide Libraries. J. Lab. Autom. 2012;17:186–200. doi: 10.1177/2211068211433503. [DOI] [PubMed] [Google Scholar]
- 26.Thakkar A, Wavreille A-S, Pei D. Traceless Capping Agent for Peptide Sequencing by Partial Edman Degradation and Mass Spectrometry. Anal. Chem. 2006;78:5935–5939. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]
- 27.Grant G, Crankshaw M, Gorka J. Edman Sequencing as Tool for Characterization of Synthetic Peptides. Methods Enzymol. 1997;289:395–419. doi: 10.1016/s0076-6879(97)89056-8. [DOI] [PubMed] [Google Scholar]
- 28.Bathany K, Owens N, Guichard G, Schmitter J-M. equencing of Oligourea Foldamers by Tandem Mass. J. Am. Soc. Mass Spectrom. 2013;24:458–462. doi: 10.1007/s13361-012-0546-0. [DOI] [PubMed] [Google Scholar]
- 29.Schreiber J, Quadroni M, Seebach D. Sequencing of Beta-Peptides by Mass Spectrometry. Chimia (Aarau) 1999;53:621–626. [Google Scholar]
- 30.Heerma W, Versluis C, de Koster C, Kruijtzer J, Zigrovic I, Liskamp R. Comparing Mass Spectrometric Characteristics of Peptides and Peptoids. Rapid Commun. Mass Spectrom. 1996;10:459–464. doi: 10.1002/(SICI)1097-0231(19960315)10:4<459::AID-RCM501>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 31.Ma B, Johnson R. De Novo Sequencing and Homology Searching. Mol. Cell. Proteomics. 2012:1–16. doi: 10.1074/mcp.O111.014902. O111.01490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steen H, Mann M. The ABC’s (and XYZ’s) of Peptide Sequencing. Nat. Rev. Mol. Cell Biol. 2004;5:699–711. doi: 10.1038/nrm1468. [DOI] [PubMed] [Google Scholar]
- 33.Fischer B, Roth V, Roos F, Grossmann J, Baginsky S, Widmayer P, Gruissem W, Buhmann J. NovoHMM: A Hidden Markov Model for de Novo Peptide Sequencing. Anal. Chem. 2005;77:7265–7273. doi: 10.1021/ac0508853. [DOI] [PubMed] [Google Scholar]
- 34.Betancourt H, Garay HE, Cabrales A, Albericio F, Yang H, Zubarev R, Besada V, Osvaldo A. Introducing an Asp-Pro Linker in the Synthesis of Random One-Bead-One-Compound Hexapeptide Libraries Compatible with ESI-MS Analysis. ACS Comb. Sci. 2012;14:145–149. doi: 10.1021/co200159r. [DOI] [PubMed] [Google Scholar]
- 35.Metzger J, Wiesmuller K-H, Gnau V, Brunjes J, Jung G. Ion-Spray Mass Spectrometry and High-Performance Liquid Chromatography-Mass Spectrometry of Synthetic Peptide Libraries. Angew. Chem. Int. Ed. Engl. 1993;32:894–896. [Google Scholar]
- 36.Sußmuth R, Jung G. Impact of Mass Spectrometry on Combinatorial Chemistry. J. Chromatogr., Biomed. Appl. 1999;725:49–65. doi: 10.1016/s0378-4347(98)00513-1. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Fonslow B, Shan B, Baek M-C, Yates J. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013;113:2343–2394. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bantscheff M, Lemeer S, Savitski M, Kuster B. Quantitative Mass Spectrometry in Proteomics: Critical Review Update from 2007 to the Present. Anal. Bioanal. Chem. 2012;404:939–965. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- 39.Altelaar M, Munoz J, Heck A. Next-Generation Proteomics: Towards an Integrative View of Proteome Dynamics. Nat. Rev. Genet. 2012;14:35–48. doi: 10.1038/nrg3356. [DOI] [PubMed] [Google Scholar]
- 40.O'Connell T, Ramsay J, Rieth S, Shapiro M, Stroh J. Solution-Based Indirect Affi Nity Selection Mass Spectrometry - A General Tool For High-Throughput Screening Of Pharmaceutical Compound Libraries. Anal. Chem. 2014;86:7413–7420. doi: 10.1021/ac500938y. [DOI] [PubMed] [Google Scholar]
- 41.Kaur S, Mcguire L, Tang D, Dollinger G, Huebner V. Affinity Selection and Mass Spectrometry-Based Strategies to Identify Lead Compounds in Combinatorial Libraries. J. Protein Chem. 1997;16:505–511. doi: 10.1023/a:1026369729393. [DOI] [PubMed] [Google Scholar]
- 42.Zuckermann R, Kerr J, Siani M, Banville S, Santi D. Identification of Highest-Affinity Ligands by Affinity Selection from Equimolar Peptide Mixtures Generated by Robotic Synthesis. Proc. Natl. Acad. Sci. U. S. A. 1992;89:4505–4509. doi: 10.1073/pnas.89.10.4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitchell A, Kent S, Engelhard M, Merrifield R. A New Synthetic Route to Tert-Butyloxycarbonylaminoacyl-4-(Oxymethyl)-phenylacetamidomethyl-Resin, as Improved Support for Solid-Phase Peptide Synthesis. J. Org. Chem. 1978;43:2845–2852. [Google Scholar]
- 44.Pulley S, Hegedus L. Solid-Phase, Solution, and Segment Condensation Peptide Syntheses Incorporating Chromium Carbene Complex-Derived Nonproteinogenic (“Unnatural”) Amino Acid Fragments. J. Am. Chem. Soc. 1993;115:9037–9047. [Google Scholar]
- 45.Coin I, Beyermann M, Bienert M. Solid-Phase Peptide Synthesis: From Standard Procedures to the Synthesis of Difficult Sequences. Nat. Protoc. 2007;2:3247–3256. doi: 10.1038/nprot.2007.454. [DOI] [PubMed] [Google Scholar]
- 46.Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, Lajoie G. PEAKS: Powerful Software for Peptide de Novo Sequencing by Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom. 2003;17:2337–2342. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- 47.Ma B, Zhang K, Liang C. An Effective Algorithm for Peptide de Novo Sequencing from MS/MS Spectra. J. Comput. Syst. Sci. 2005;70:418–430. [Google Scholar]
- 48.Bringans S, Kendrick T, Lui J, Lipscombe R. A Comparative Study of the Accuracy of Several de Novo Sequencing Software Packages for Datasets Derived by Matrix-Assisted Desorption/ionization and Electrospray. Rapid Commun. Mass Spectrom. 2008;22:3450–3454. doi: 10.1002/rcm.3752. [DOI] [PubMed] [Google Scholar]
- 49.Ma B. Novor: Real-Time Peptide de Novo Sequencing Software. J. Am. Soc. Mass Spectrom. 2015;26:1885–1894. doi: 10.1007/s13361-015-1204-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rapp W, Fritz H, Bayer E. Monosized 15 Micron Grafted Microspheres for Ultra High Speed Peptide Synthesis. Proc. 12th Am. Pept. Symp. 1991:529–530. [Google Scholar]
- 51.Bayer E. Towards the Chemical Synthesis of Proteins. Angew. Chem., Int. Ed. Engl. 1991;30:113–129. [Google Scholar]
- 52.Doran T, Gao Y, Mendes K, Dean S, Simanski S, Kodadek T. Utility of Redundant Combinatorial Libraries in Distinguishing High and Low Quality Screening Hits. ACS Comb. Sci. 2014;16:259–270. doi: 10.1021/co500030f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.