

Preface
Biologists have long been intrigued by the possibility that cells can change identity, a phenomenon known as cellular plasticity. The discovery that terminally differentiated cells can be coaxed experimentally to become pluripotent has invigorated the field, and recent studies have demonstrated that changes in cell identity are not limited to the laboratory. Specifically, certain adult cells retain the capacity to de-differentiate or trans-differentiate under physiological conditions as part of an organ’s normal injury response. Recent studies have highlighted the extent to which cell plasticity contributes to tissue homeostasis, findings that have implications for cell-based therapy.
Introduction
Under most circumstances, cellular identity – the product of normal differentiation – is a stable feature within tissues. Maintaining cellular identity is crucial for normal tissue function, as chaos would result if changes in cell differentiation states led cardiomyocytes to stop contracting or adult neurons to cease generating action potentials. Such stability is achieved through epigenetic regulation – modifications to chromatin or DNA – that result in heritable patterns of tissue-specific gene expression1.
But it is clear that under experimental conditions cell identity can be altered. The potential of fully mature adult cells to dramatically change their identity was first exposed by John Gurdon, who showed that terminally differentiated cells could – under the extreme experimental conditions of nuclear transplantation – be converted into cells with the properties of a fertilized egg2. Since those pioneering experiments, cellular plasticity has been the focus of intense investigation, with cellular conversions falling into two major categories: de-differentiation and trans-differentiation3. De-differentiation refers to the reversion of a differentiated cell into one with greater developmental potential, such as a stem cell or a progenitor cell4 (Figure 1). The most dramatic example of this is the phenomenon of induced pluripotent stem cells (iPSCs), in which overexpression of a limited number of transcription factors can induce terminally differentiated cells to become pluripotent in vitro5. Trans-differentiation, by contrast, refers to the conversion of one mature cell type into another (Figure 1). Such interconversions may involve a de-differentiation step, whereby cells go through a primitive stem-like state, or may involve a more direct route, bypassing such stem-like intermediates. The activity of MyoD – a basic helix-loop-helix transcription factor that can convert fibroblasts, chondrocytes, retinal pigmented epithelial cells and other cell types into muscle when ectopically expressed–is an example of the latter process6, 7. While these experimental methods for manipulating cell identity have been the focus of most mammalian plasticity studies, it also appears that cell identity can also change under “natural” conditions4 (See Box 1 for a discussion of in vivo and in vitro cellular plasticity). In marine animals, cellular de-differentiation and trans-differentiation constitute part of the organism’s normal response to injury8, 9. Recently, however, has it become apparent that mammalian cells share this property and can change their identity in response to physiological stresses independent of any experimental efforts to redirect fate. While the precise role of adult cell plasticity remains to be determined on a case-by-case basis, the existence of multiple examples of the phenomenon throughout the animal kingdom implies a conserved role in tissue homeostasis and repair.
Figure 1. Models of differentiation, de-differentiation, and trans-differentiation.
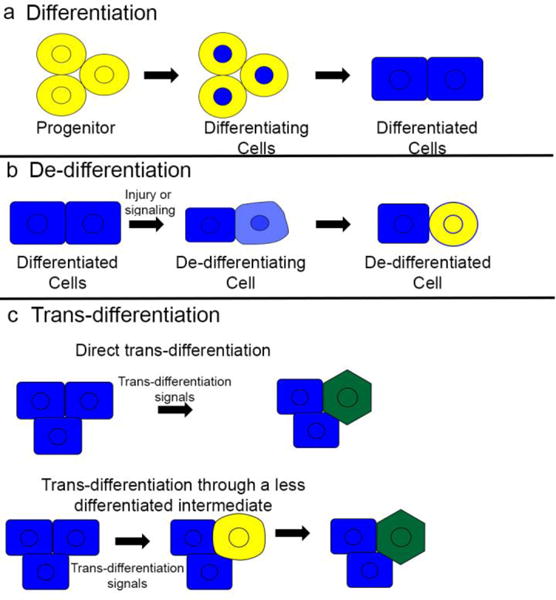
(a) During development and regeneration, progenitors and stem cells differentiate to form mature, differentiated cells. (b) In some cases of injury or stem cell ablation differentiated cells may de-differentiate, returning to an earlier fate and becoming progenitor cells again. (c) Injury and ablation may also induce differentiated cells to take on a different mature cell fate, a process known as trans-differentiation. Trans-differentiation may either occur directly, without any reversion to immature phenotypes, or through a de-differentiation step before cells re-differentiate to a new mature phenotype.
Box 1. Criteria for evaluating cellular plasticity.
In the initial enthusiasm over cell plasticity studies at the beginning of this century, many studies reported that cells could make extreme changes in identity, crossing developmental germ layers as neurons became blood cells102 and bone marrow cells produced hepatocytes103. These remarkable claims were based largely on studies conducted in vitro or which utilized transplantation under extreme experimental conditions. As these studies came under criticism for problems with reproducibility or their relevance to physiological in vivo situation3, it became clear that better criteria were needed to support claims of de- or trans-differentiation48. Amongst these were the requirements that cells be clearly identified before and after fate change, that cells be functional in their new fate, and that cells be properly integrated in the tissue48. Thus, while it is true in principle that there are no absolute limits to cellular plasticity (since any cell can be reprogrammed to a pluripotent state, which in turn can give rise to any other cell type), claims of cellular plasticity – like any scientific assertion – needs to be assessed with caution: Is the conversion taking place in vitro or in vivo? What assays are being used to assess cell identity? Are de-differentiation and/or trans-differentiation occurring under experimental or physiological conditions? While experimentally-mediated trans-differentiation may have important applications in the future (see Box 3), such findings do not necessarily represent normal paths to plasticity in vivo.
In this review, we focus on such “physiological” reprogramming, using examples from mammalian and non-mammalian systems to understand how and why cells change their identity during a normal injury response. Implicit in this is the belief that understanding such processes at the cellular and molecular level will foster greater insight into diseases where cellular plasticity plays a role – including cholestatic liver injury, diabetes, and neural injuries – and thus facilitate the development of novel therapies that employ cell replacement.
Epimorphosis and de-differentiation
More than a century ago, Thomas Hunt Morgan made a distinction between regenerative processes that utilized cellular proliferation (which he termed “epimorphosis”) and those which did not (which he termed “morphyllaxis”)10. Over time, epimorphosis has come to refer to those regenerative processes involving cellular de-differentiation and re-differentiation (typically involving proliferation), in which cells return to their original identity, while morphyllaxis is used to refer to regeneration involving the trans-differentiation of cells from one identity to another (Figure 1). With the advent of refined microscopy and lineage tracing tools, it has become apparent that both forms of cellular plasticity – de-differentiation and trans-differentiation – can contribute to tissue regeneration in a variety of settings. A third type of regeneration – not discussed here as it involves compensatory reconstitution of mass without changes in cell identity – is referred to as hypertrophy or hyperplasia, and involves the growth or replication of existing differentiated cells, respectively11.
De-differentiation in invertebrates
One of the best-studied examples of cellular plasticity is the Drosophila melanogaster testis. During de-differentiation, cells with a more specialized differentiation state revert back to a more progenitor or stem cell identity characterized by the expression of immature cell markers and stem-like functional properties, including selfrenewal and the capacity to produce differentiated spermatids4. In the fly testis, germline stem cells (GSCs) reside near a specialized niche known as the “hub” (Figure 2), which provides the environment and signals necessary for stem cell maintenance. In this niche, the GSCs divide to give rise to more GSCs or differentiate to give rise to a gonialblasts, which differentiate into spermatogonia and the spermatocyte lineage12. Because GSCs rely on STAT signaling for maintenance13, 14, it was possible to deplete the niche of GSCs by genetically removing STAT signaling15. Remarkably, when this was done new GSCs emerged via de-differentiation of gonialblasts and spermatogonia following restoration of STAT signaling15. De-differentiation, as marked by repopulation of the GSC niche and remnants from spermatogonia cysts breaking up to form single cells, was also seen following other methods of GSC ablation. Loss of GSCs by forced differentiation through ectopic expression of differentiation factor bag-of-marbles (Bam)16 induced de-differentiation, suggesting it was triggered by a decrease in the size of the stem cell pool rather than as a consequence of altered STAT signaling. While gonialblasts and spermatogonia can become GSCs following this transient ablation, spermatocytes cannot15, a finding that probably reflects changes in chromatin that impose a barrier to de-differentiation. Moreover, de-differentiation also appears to be conserved in mammals, as spermatogonia cultured in the presence of GDNF and FGF2 can regenerate the mammalian GSC niche.17, 18. A similar phenomenon takes place in the D. melanogaster ovary following forced differentiation of the GSCs19.
Figure 2. Examples of de-differentiation.
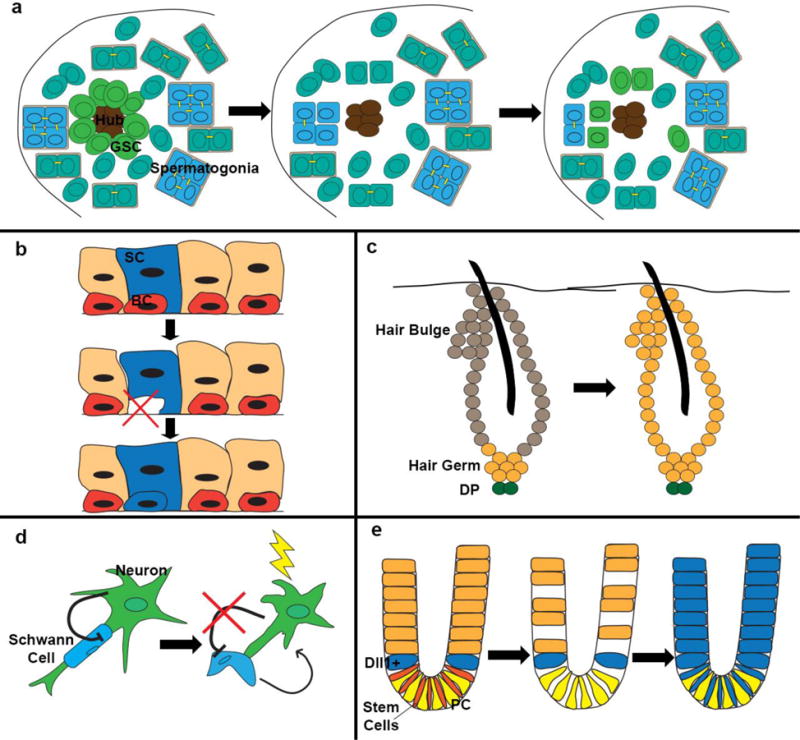
(a) Following loss of germ stem cells, differentiating spermatogonia may break apart from their multi-cell cysts to de-differentiate and form new germ stem cells around the hub. (b) In the lung airway, ablation of basal stem cells (BC) induces differentiated secretory cells (SC) to de-differentiate and form new BC. (c) During the hair cycle, hair germ cells give rise to transiently amplifying cells that make up the hair. Bulge cells contribute to the outer root sheath and to some cells of the hair germ for the next cycle. Following ablation of bulge cells, hair germ cells can de-differentiate and give rise to new bulge cells. DP: dermal papilla. (d) Following injury to the neuron, Schwann cells disassemble from the neuron and de-differentiate (a process normally inhibited by signals from the neuron), secreting factors to help with neuron regeneration. (e) In the intestine, radiation injury, which ablates crypt stem cells and cells further up in the crypt, exposes Dll1+ secretory cells to new signals from the paneth cells (PC) in the crypt which induce de-differentiation and expansion of the cell’s progeny up the crypt, as shown by lineage tracing.
De-differentiation in amphibians and teleosts
Amphibian limb regeneration is the archetypal example of epimorphic regeneration. Following limb amputation, cells near the amputation site undergo de-differentiation or stem cell activation to produce a pool of proliferating progenitors called a blastema. The blastema will ultimately produce all of the tissues in a fully regenerated limb. Originally the blastema was thought to consist of pluripotent cells that could give rise to many limb tissues20, 21, but more recent studies have suggested this apparent plasticity of the stem cell pool may have been due to sample contamination. In particular, one study using lineage-specific labeling found that the blastema is not composed of a homogenous pool of pluripotent cells, but instead contains a heterogeneous mix of lineage-restricted progenitors; for example, muscle-derived blastemal cells only produced muscle and Schwann cells only gave rise to new Schwann cells22. Although the dermis exhibited some adaptability in cell fate, these new cell fates were restricted to cells that also originate from the lateral plate mesoderm developmentally, suggesting that any de-differentiation which occurs remains partially lineage restricted within lateral plate mesoderm22.
Genetic lineage-tracing techniques have also demonstrated distinct species-specific mechanisms of regeneration. For example, applying the technique in urodele salamanders revealed that limb amputation in the newt Notophthalmus viridescens induced de-differentiation of multinucleated myofibers into proliferating mononuclear cells to generate new muscle in the regenerating limb. However, the axolotl Ambystoma mexicanum does not de-differentiate myofibers but instead relies on Pax7+ satellite cells, which are quiescent muscle stem cells, to regenerate limb muscle23. Importantly, this study found that the newt blastema did not contain Pax7+ cells, demonstrating that even within amphibians, different species can use different mechanisms to regenerate lost tissue.
De-differentiation also contributes to heart regeneration in the zebrafish Danio rerio. Zebrafish can regrow heart tissue following amputation of up to 20% of the left ventricle24. In response to amputation, differentiated cardiomyocytes disassemble their sarcomere apparatus, repress sarcomeric genes, and induce expression of genes involved in cell proliferation25, 26.
De-differentiation in mammals
Tissue regeneration occurs to a more limited extent in mammals than it does in amphibians and invertebrates. Nevertheless, several examples of de-differentiation during mammalian regeneration have recently come to light, including skin, intestine, lung, and nervous system (Additional information on plasticity in the skin and intestine can be found in two recent reviews27, 28).
During hair growth, the hair-producing cells within the follicle are derived from two progenitor populations – the bulge stem cells and their associated hair germ cells (Figure 3). The cells of the hair germ are the dominant progenitors within the hair follicle, giving rise to the majority of differentiated hair cells during a normal growth cycle. Specifically, hair germ cells undergo more divisions than bulge cells29 and give rise to the rapidly dividing “transient amplifying” cells that make the hair30. However, with each cycle a small number of bulge stem cells repopulate the bulge and the hair germ, in preparation for a new cycle. Thus, the hair follicle appears to contain a stem cell hierarchy – akin to GSCs and spermatogonia in the D. melanogaster testis – whereby bulge cells constitute a reserve source of stem cells that can give rise to the hair germ. The environment around the stem cells – the stem cell “niche” – appears to be critical in determining their fate, as location dictates fate in a stereotypical fashion31. Although the signals inducing these stem cell fates are unclear, the slow cycling upper cells in the outer root sheath typically repopulate the bulge, while the lower outer root sheath cells typically lose their stem cell characteristics30. Similar to the fly testis, hair germ cells are able to repopulate the niche following bulge cell ablation via de-differentiation31.
Figure 3. Signaling from surrounding cells and the environment induce de-differentiation.
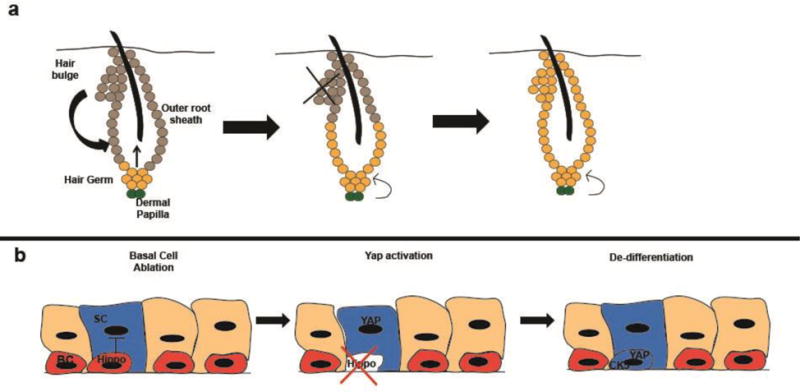
De-differentiation is induced as cells are exposed to new de-differentiation signals or have inhibitory signals silenced. (a), During the normal cycle of hair growth and regeneration, the hair germ produces transient amplifying cells that make up the hair while the hair bulge contributes to the outer root sheath. Following experimental ablation of the bulge, hair germ cells can de-differentiate and give rise to new bulge cells. This process relies on signals from the dermal papillae, as mechanical separation of the hair germ and dermal papilla prevents de-differentiation. (b), Basal cells adjacent to secretory cells (with one secretory cell genetically labeled in the diagram) secrete signals, possibly Hippo, preventing de-differentiation of secretory cells. Following ablation of basal cells, these signals are lost and secretory cell de-differentiation is no longer inhibited, allowing accumulation of the YAP transcription factor, expression of basal cell genes such as CK5, and de-differentiation of secretory cells into basal cells. These dedifferentiated basal cells can then differentiate to form new secretory cells. BC= basal cell, SC= secretory cell
De-differentiation also occurs in the intestine following injury (Figure 2). As with prior examples, the intestinal crypts, which contain stem cells that self-renew and produce the intestinal epithelium, appear to be organized as stem cell hierarchies whereby slowly dividing Bmi1+ cells give rise to rapidly dividing Lgr5+ cells, both of which are multipotent and can give rise to all intestinal cells, including transit amplifying, absorptive, and secretory cell types32, 33. These stem cells, in turn, give rise to committed progenitor cells, including Delta-like 1 (Dll1)+ crypt cells that differentiate exclusively along the secretory lineage34. However, after exposure to radiation – which eliminates the Lgr5+ stem cells – Dll1+ secretory precursor cells have been reported to de-differentiate into Lgr5+ stem cells which can subsequently generate all the cell types of the intestinal epithelium34.
Following this paradigm, the lung also utilizes de-differentiation to generate new stem cells in the airway epithelium following stem cell ablation. The tracheal airway is a pseudostratified epithelium, with cytokeratin 5 (CK5+) basal cells located in close contact with the basal lamina35, 36. These basal cells act as stem cells, as they can self-renew and give rise to both secretory and ciliated lineages37, and basal cell proliferation and differentiation is dramatically increased following airway injury38. If, however, CK5+ basal cells are ablated, cells from the secretory lineage can de-differentiate to give rise to new basal cells; such cells have the full phenotypic and functional characteristics of airway stem cells39 (Figure 2).
Finally, de-differentiation is a component of the injury response in the nervous system. Following nerve injury and the ensuing inflammatory response, mature Schwann cells take on an immature phenotype, downregulating myelin-associated factors and expressing growth factors and pro-regenerative factors to support axonal repair (Figure 2). Once de-differentiated Schwann cells make contact with neurons, the cells re-differentiate and myelinate axons again40. Blocking Schwann cell de-differentiation, either experimentally or as a result of aging, impairs neural regeneration41.
Mechanisms of de-differentiation
In the examples discussed above, de-differentiation is a physiological response to tissue injury or cell ablation. Because this does not occur under homeostatic conditions but only following perturbation, differentiated cells must either be exposed to new signals that cause de-differentiation or be released from signals that normally inhibit their reversion. It appears that distinct molecular mechanisms – involving both cell autonomous and non-cell autonomous factors – control cell identity in different systems.
Competence to de-differentiate
Competence, the intrinsic ability of a cell to respond to certain signals, is a prerequisite for cellular de-differentiation. In newt limb regeneration, de-differentiation involves the segregation of syncytial muscle fibers into individual muscle progenitor cells, and recent work has suggested that this might occur through initiation, but not completion, of apoptosis and caspase 3 activation within muscle fibers42. These progenitor cells proliferate before they re-differentiate23, a process that involves inactivation of the tumor suppressor retinoblastoma (Rb) protein. As mammalian skeletal muscle fails to de-differentiate following injury, it has been proposed that cell cycle regulators – like Rb – may constrain mammalian regeneration. Consistent with this notion, simultaneous deleting Rb and the tumor suppressor ARF (another inhibitor of cell proliferation) from mouse muscle permits de-differentiation43. Interestingly, ARF is absent from the newt genome, which may render newt muscle intrinsically more responsive to de-differentiation signals than mammalian muscle.
Microenvironment as a de-differentiation driver or obstacle
In addition to such cell-intrinsic factors, signals from the microenvironment drive de-differentiation. In the hair follicle, for example, de-differentiation of hair germ cells following ablation of the bulge requires the dermal papillae, a cluster of cells located beneath the hair germ44 (Figure 2). If the dermal papillae is physically separated from the epithelium, hair germ cells are unable repopulate the bulge niche and the hair follicle does not regenerate31, suggesting that the dermal papillae (and its surrounding mesenchyme) provide signalsthat promote de-differentiation. At present, the identity of these signals is unknown. Similarly, de-differentiation of Dll1+ secretory precursor cells into intestinal stem cells relies on signals from the crypt. Normally, intestinal stem cells are maintained by Wnt signals provided by the niche, and when Dll1+ cells (grown in organ culture) are exposed to soluble Wnt ligand they de-differentiate and give rise to Lgr5+ stem cells34. Thus, damage to the intestinal crypts may cause secretory progenitor cells to be newly exposed to Wnt signals, leading to expression of the Wnt target Lgr545 and de-differentiation.
Finally, de-differentiation requires the repression of inhibitory signals. Following neuronal injury, Schwann cells are displaced from neurons, thus relieving an inhibitory signal and allowing them to de-differentiate to a more immature regeneration-promoting state. Although the signals preventing de-differentiation are unknown, the process is likely to involve cAMP, as high cAMP levels in Schwann cells inhibit de-differentiation and promote re-differentiation, possibly through induction of the pro-myelination transcription factorKrox2046.
Similarly, loss of an inhibitory signal appears to underlie secretory cell de-differentiation following basal cell ablation in the airway (Figure 3)39. The Hippo pathway, and its downstream mediator YAP, could be one such inhibitory signal; YAP is normally absent from secretory cells, but its overexpression within secretory cells induces basal cell de-differentiation47. This de-differentiation likely occurs through transcriptional changes, as YAP binds upstream of several genes, including CK5, FGFR2, EGFR, Integrin α6, and Integrin β4, which are more highly expressed in basal stem cells than secretory cells. Hence, it is enticing to imagine a feedback loop in which signals from stem cells repress de-differentiation in adjacent cells, thus ensuring the proper balance between stem cells and their differentiated progeny. Under such a scenario, stem cell ablation would eliminate this inhibitory signal, resulting in the subsequent de-differentiation of adjacent differentiated cells into stem cells.
Morphyllaxis and trans-differentiation
Trans-differentiation is defined as the conversion of a cell from one specialized state to another. The process may be direct, involving a direct conversion, or involve a de-differentiation event prior to re-differentiation (Figure 1). Importantly, trans-differentiation requires both repressive and activating effects on gene expression, as genes specific to the starting state must be turned off while genes specific to the new state must be turned on. In addition, corresponding changes in functional properties must also occur48. It should be stressed that there is a difference between what cells do normally and what they are capable of doing under experimental conditions, including cell culture (Box 1). Thus, while many examples of trans-differentiation have been reported in vitro, including some that may have important practical applications for therapies49–51 there are only a few examples of bona fide trans-differentiation occurring in vivo, and only a small subset of these take place under physiological conditions. For this review, we focus on these in vivo examples of trans-differentiation, particularly those that constitute a natural (physiological) response to injury.
Trans-differentiation in invertebrates
In C. elegans, in which all cell lineages have been defined, trans-differentiation has been reported under both experimental and natural (developmental) conditions. Experimentally, a fifteen minute pulse of ectopic GATA transcription factor ELT-7 is sufficient to induce pharyngeal margin cells to transdifferentiate into intestinal cells52. Trans-differentiation in this setting is direct, as no morphological or functional intermediates have been observed. Instead, cells activate intestinal gene expression and adopt an intestinal phenotype directly, before the pharyngeal gene expression program is repressed.
Furthermore, trans-differentiation also occurs naturally during C. elegans normal development. Specifically, a single hindgut cell crosses germ layers during the second larval stage and trans-differentiates into a neuron in 100% of developing worms53. In this example, the hindgut cell first de-differentiates without proliferating, turning off hindgut-specific genes and undergoing an epithelial to mesenchymal transition. This cell then migrates to its new position before re-differentiating into a neuron with its corresponding gene expression pattern. As most studies of vertebrate trans-differentiation have been done in the context of injury (and in adult animals), it is unknown whether other organisms might employ a similar process of trans-differentiation during normal development.
Trans-differentiation in amphibians
Regeneration of the adult newt eye, which involves trans-differentiation, has been studied since the late 19th century54. Following surgical removal of the adult lens, pigmented epithelial cells of the iris de-differentiate, proliferate, and then re-differentiate as lens cells. During de-differentiation, these cells lose pigmentation and begin to express the pluripotency reprogramming factors Sox2 and Klf455 and the linker histone B4, which is normally found in oocytes or in nuclei of differentiated cells used for somatic cell nuclear transfer56. During re-differentiation, these cells express γ-crystallin and other genes specific to the lens cell fate57. Although the de-differentiated cells express pluripotency factors, they remain committed to a lens fate, as regenerating iris transplanted into other regions of the newt still results in the generation of lens tissue58, 59.
Trans-differentiation in mammals
The best examples of mammalian trans-differentiation come from two developmentally-related tissues: the liver and the pancreas. The liver is composed primarily of hepatocytes, which perform most of its metabolic and synthetic functions, and biliary epithelial cells (BECs), which line the bile ducts and are critical for carrying bile out of liver60. Following injury to the liver or ectopic activation of signaling pathways (as detailed below), hepatocytes can change fate, turning off hepatocyte-specific genes and turning on markers of BECs (Figure 4a)61–63. Analysis of cell size, ultrastructural appearance, and mRNA expression indicate that the trans-differentiated cells more closely resemble BECs than hepatocytes62. Because early progenitor markers of hepatoblasts were not observed61, it seems that these cells are directly reprogrammed into BECs without going through an intermediate de-differentiation step. The process occurs over a prolonged period of time (weeks) and involves step-wise changes in gene expression (Figure 4a). Thus, while many cells undergo a complete and stable conversion to BECs, some hepatocytes initiate but do not complete the trans-differentiation process, reverting back to a hepatocyte fate when the reprogramming stimulus is removed62.
Figure 4. Trans-differentiation in the liver and pancreas leads to tissue repair.
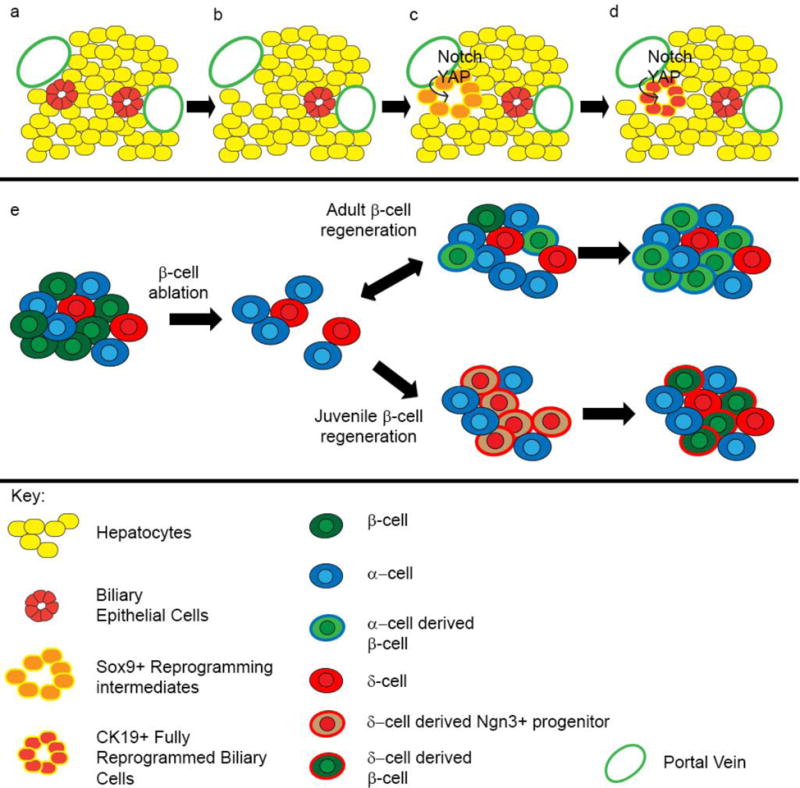
Hepatocytes trans-differentiate into biliary cells following liver injury. (a) Hepatic tissue prior to injury, with hepatocytes in yellow and biliary epithelial cells (bile ducts) in red. (b) Following injury and bile duct damage, (c) hepatocytes receive signals including Notch and activation of the YAP transcription factor that induce their trans-differentiation into biliary cells (shown in orange). During the trans-differentiation process, hepatocytes turn on a subset of “early” biliary genes, such as Sox9, which are specific to the new cell fate. (d) Over time trans-differentiated hepatocytes become mature bile ducts, expressing “late” bile duct markers such as CK19. (e) Normal pancreatic islets contain β, α, and δ cells. Following experimental ablation of β-cells, adult and juvenile islets follow different pathways to regenerate the missing tissue. In adult islets, α cells proliferate and begin to trans-differentiate directly in insulin-producing cells. Although these trans-differentiated cells are functional and produce insulin, they still maintain some α-gene expression, suggesting they are not fully trans-differentiated. In juvenile islets, δ cells proliferate and turn on δ cell-specific genes. These cells de-differentiate to Ngn3+ progenitor cells, then re-differentiate as β-cells. In this example, different pathways may lead to restoration of islet cells.
Because of the central role of pancreatic β cells in the pathogenesis of diabetes, the pancreas has also been a major focus of trans-differentiation research. In addition to β cells the pancreatic islets of Langerhans contain glucagon-producing α cells and somatostatin-producing δ cells64. In type I diabetes mellitus (T1DM), β cells are attacked by the immune system, leaving patients with an insufficient number of insulin-producing β cells65. The finding that T1DM can be successfully treated by transplanting patients with cadaver-derived islets has inspired many researchers to try to generate new β cells in vitro66. With this goal in mind, an in vivo approach was taken to make new β cells, delivering three transcription factors known to play an important role in β cell development – Pdx1, MafA, and Ngn3 (PMN) – into pancreatic exocrine cells67. Remarkably, transduced cells adopted many of the specialized features of true β cells – without an apparent de-differentiation intermediate. Furthermore, the cells secreted sufficient quantities of insulin to significantly improve blood glucose levels in animals that had undergone β cell ablation67. Exocrine cells were also found to trans-differentiate into β-like cells following administration of epidermal growth factor (EGF) and ciliary neurotropic factor (CNTF) to hypoglycemic animals, a result which may have strong therapeutic potential68. Similar findings of partial β cell trans-differentiation have been reported following mis-expression of the PMN transcription factors in the proximal intestine – a region of the gut tube that is embryonically related to the pancreas69, or when subsets of these factors have been expressed in various tissues70–72. Hence, it appears that there are multiple ways to prompt cells within endoderm-derived organs to adopt features of β cells.
But what about trans-differentiation under normal conditions? Beyond these examples of experimentally-prompted trans-differentiation, it is also clear that endocrine cell plasticity is evoked as a physiological response to β cell damage. Specifically, ablation of greater than 99% of β cells causes a substantial number of glucagon-producing α cells to trans-differentiate into insulin-producing β-like cells73. This change in cell identity has profound physiological consequences, as those mice with robust trans-differentiation no longer required exogenous insulin 6 months after β cell ablation. Interestingly, and in contrast to some other examples of trans-differentiation, the reprogrammed β cells still expressed a number of α-cell specific genes, indicating that functionally relevant trans-differentiation does not require complete repression of the transcriptional program of the starting cell type.
More recent work found that juvenile mice use a different mechanism of trans-differentiation following massive β cell loss in which somatostatin-producing δ cells become β-like cells74. Unlike adult α cell trans-differentiation, juvenile δ cell trans-differentiation engages a de-differentiation and re-differentiation pathway that requires re-expression of the Neurogenin 3 transcription factor, a marker of embryonic endocrine progenitor cells74. These results suggest that cells reach similar endpoints via divergent sources and paths depending on cellular context (Figure 4b). In addition, it is likely that these trans-differentiation “routes” are bi-directional, as several studies have shown that β cells can be converted to α cells in vivo through a number of experimental approaches75, 76 (Figure 4b).
Finally, whereas cellular plasticity may be a part of the solution when it comes to T1DM, it may be part of the problem when it comes to type 2 diabetes mellitus (T2DM). Type 2 diabetes is the more common form of the disease and reflects the combined effects of insulin resistance (whereby target tissues no longer respond to the same levels of insulin) and reduced β cell function. It has long been thought that decreased β cell function was due to β cells lost over time due to chronic stress. But several recent studies have suggested that β cell trans-differentiation, rather than death, contributes the losses in β cell function that drive T2DM. Specifically, it was reported that in mouse models of T2DM, β cells took on features of other endocrine cell types or stem cells, implicating cell plasticity as a source for β cell dysfunction77. Evidence supporting this view has recently been obtained from studies of human T2DM patients78.
Mechanisms of trans-differentiation
Trans-differentiation involves massive cellular reorganization at the transcriptional, post-transcriptional, and cell biological level. Moreover, the extracellular signals that drive trans-differentiation must be tightly regulated in a spatiotemporal-specific manner; without such tight control, alterations in cell identity could lead to marked disruptions in tissue function. Although the intracellular and extracellular factors required for cells to change identity have been identified in a few specific cases, most of the molecular drivers of cellular plasticity remain unknown. Nevertheless, chromatin remodeling – the process whereby new transcriptional programs are activated and old ones are silenced – is likely to lie at the center of this process.
Chromatin modifications in invertebrates and amphibians
The intestinal-to-neuronal cell identity switch in C. elegans provides one example of the importance of chromatin modification in trans-differentiation. Genetic screens for genes that interrupted this event revealed jmjd-3.1 – a demethylase that removes a transcriptionally repressive methyl mark from histone 3 lysine 27 (H3K27) – as essential for activating the motor neuron transcriptional program. Similarly, mutations in set1, which activates transcription by methylating histone 3 lysine 4 (H3K4), also inhibit trans-differentiation79.
For trans-differentiation to occur, multiple regions of chromatin must adopt an “open” configuration which is more accessible to transcription factors and chromatin-modifying enzymes, permitting global changes in transcription. In this respect, it is perhaps not surprising that Sox2 and Klf4 are expressed during newt lens regeneration55. Furthermore, the linker histone B4, which facilitates chromatin remodeling, is required for iris-to-lens trans-differentiation56. These chromatin changes are likely essential for the cells to repress iris-specific genes and activate lens-specific genes. One such gene is γ-crystallin, whose expression is almost absent when histone B4 is knocked down.
Signalling pathways in the mammalian liver and pancreas
Developmentally-important signaling pathways are crucial for trans-differentiation. In the embryonic liver, Notch signaling regulates bile duct development, where it coordinates differentiation and morphogenesis80, while in the adult liver ectopic Notch signaling converts hepatocytes to a biliary fate61. The trans-differentiation process requires the Notch-dependent DNA binding protein RBP-J, and occurs in a stepwise fashion that takes days to weeks, thus resembling reprogramming to pluripotency81. The signals that activate Notch during hepatocyte trans-differentiation remain unknown, but recent work has implicated the Hippo pathway, and its downstream transcriptional activator YAP. Like Notch, the Hippo pathway is involved in embryonic liver development, where YAP is required for biliary development82. Ectopic YAP expression in adult hepatocytes leads to biliary trans-differentiation that is partially Notch-dependent, as YAP is bound to the promoters of both Notch2 and Sox9, an earlier marker of biliary cells, in de-differentiated cells83. Together, these findings suggest that Notch and Hippo act in concert as “master regulators” of biliary identity in the liver (Figure 4b).
As noted above, studies investigating trans-differentiation in the pancreas suggest that developing and adult mice use different cellular substrates to generate new β cells following injury73, 74. These studies leave open several intriguing questions regarding cell plasticity during maturation and aging. It is currently unclear how pancreatic α or δ cells sense the decreased quantity of β cells and why trans-differentiation from α cells is direct while trans-differentiation from δ cells involves a de-differentiation intermediate. By analogy with the basal cells of the lung, it is possible that β cells send an inhibitory signal that prevents trans-differentiation under normal circumstances and which is disrupted following β cell ablation. Alternatively, α and δ cells could be differentially sensitive to decreasing levels of insulin.
Initiation versus maintenance of new cellular identities
Cellular reprogramming, whether by de-differentiation or trans-differentiation, requires dramatic changes in gene expression. Such changes are needed to activate genes associated with the new fate and to inhibit genes associated with the previous fate. In some cases, master regulators that control the expression of many genes, such as MyoD, may control the process6, while in other cases specific changes in chromatin may serve to establish a new fate. Regardless of how they are established, changes in gene expression are most likely maintained over the long term by alterations in chromatin structure56, 79, 84, such as histone modifications that can turn genes of the old fate off and genes of the new fate on.
In this regard, it is worth noting that trans-differentiation need not be an “all or nothing” phenomenon. For example, cellular phenotype may change as a result of transcriptional or post-transcriptional changes, such as overexpression of the YAP transcription factor in the de-differentiation of secretory cells in the lung, but then revert back in the absence of subsequent epigenetic modifications once the stimulus is removed. Another case in point is hepatocyte trans-differentiation, where some trans-differentiating cells revert back to the hepatocyte fate when the injury is resolved. It seems theoretically likely that cells that have undergone stable trans-differentiation are maintained in their new (biliary) identity, while cells that are in the midst of the trans-differentiation process can return back to their original hepatocyte identity62.
Cellular plasticity, metaplasia and cancer
Metaplasia describes changes within tissues whereby one cell type is replaced with another85. Although the phenomenon may involve trans-differentiation, it has also been proposed that metaplasia can also result from cell migration or altered cell fate decisions during normal differentiation – so-called “trans-fating”. Tissue metaplasia is frequently associated with a predisposition to cancer (Table 1)86, 87. One of the most common examples of metaplasia is “Barrett’s esophagus,” in which the normal squamous epithelium of the esophagus is replaced by columnar cells with features of the intestinal epithelium88. Barrett’s metaplasia is strongly associated with the development of esophageal adenocarcinoma89, 90, which has led to the notion that trans-differentiation is associated with oncogenic risk. However, the extent to which the Barrett’s-associated risk of cancer is due to trans-differentiation remains unclear. Whether or not Barrett’s is a true metaplastic event, uncontrolled cell plasticity has been hypothesized to be carcinogenic91. Specifically, mouse models of cancer – particularly pancreas and liver cancer – provide strong evidence that trans-differentiation plays a central role in cancer-initiating events.
Table 1.
Examples of metaplasia in well-known cancers
| Type of Cancer | Affected tissue | Cell types undergoing metaplasia | References |
|---|---|---|---|
| Intrahepatic Cholangiocarcinoma | Liver | Hepatocytes to biliary cells | 97, 98 |
| Pancreatic Ductal Adenocarcinoma | Pancreas | Exocrine cells to ductal cells | 92–95, 104 |
| Barrett’s metaplasia/Esophageal adenocarcinoma | Esophagus | Esophageal cells to intestinal-like cells | 89, 90 |
| Bladder squamous cell carcinoma | Bladder | Transitional epithelium to squamous cells | 105 |
| Intestinal metaplasia/Gastric cancer | Stomach | Gastric squamous cells to intestinal cells | 106 |
| Cervical cancer | Cervix | Metaplasia to squamous cells | 107 |
| Non-Small Cell Lung cancer | Lung | Metaplasia to squamous cells | 108 |
Most pancreatic cancers – and their neoplastic precursors known as pancreatic intraepithelial neoplasias (PanINs) – have the histological appearance of ductal cells92. However, several studies suggest that PanINs arise from phenotypically-distinct acinar cells through a process termed “acinar-to-ductal metaplasia”92–95. For instance, a recent study found that deletion of Ptf1a, a gene controlling acinar cell fate, increased PanIN development and subsequently increased the rate of pancreatic cancer96. It is unclear why acinar cell trans-differentiation predisposes cells to cancer, but it may be that only particular cell types are susceptible to the growth-promoting effects of certain oncogenic mutations (that is, the right mutations occurring in the right cells at the right time).
Similarly, trans-differentiation event may precede further oncogenic steps during the development of intrahepatic cholangiocarcinoma (ICC), a cancer of the small bile ducts of the liver. Although ICCs have a ductal (biliary) appearance histologically, several studies have suggested that they originate from phenotypically-distinct hepatocytes97, 98. Although these studies did not investigate whether changes in cell identity come before or after other oncogenic events, it is enticing to imagine that the same dramatic changes in gene expression (and chromatin structure) that underlie cellular plasticity could also lead to dysregulated expression of oncogenes or repression of tumor suppressor genes.
Interestingly, some of the same signals that produce cellular reprogramming can also be oncogenic. As discussed above, Notch signaling induces hepatocytes to trans-differentiate into biliary cells, and activation of Notch in hepatic cells results in hepatocellular carcinomas with biliary features99 or frank intrahepatic cholangiocarcinoma97. YAP, which also induces hepatocyte-to-biliary trans-differentiation in the liver, is also associated with the formation of liver cancers100. These findings suggest that the signals which allow adult cells to change their identity may put tissues at increased risk of malignant transformation. Such an association could provide an additional explanation for the well-appreciated association between tissue inflammation following chronic injury (the in vivo stimulus for trans-differentiation) and cancer.
Conclusions and perspectives
It may be useful to think of cells in a tissue as players on a soccer pitch – each having a specific position and role. In the hierarchy of the team, the goalkeeper has a particularly crucial responsibility, protecting the goal and calling out strategy to his or her teammates. As long as the goalkeeper is tending the goal, all the other players remain in position. But if the goalkeeper is incapacitated, someone else must take on the job, at least temporarily. This, of course, is “de-differentiation.” Likewise, players must be able to “trans-differentiate” into an offensive or defensive role if a teammate goes down, even if is not their normal task. Even if one or two players is temporarily disabled, plasticity allows the team to function without falling apart.
At the cellular level, changes in identity can occur experimentally through the activation of specific signaling pathways or physiologically as a response to signals released during injury and inflammation. These processes are tightly regulated, as uncontrolled plasticity could destabilize tissues and/or lead to cancer. The fact that cells exhibit plasticity in the setting of injury indicates that de-differentiation and trans-differentiation are important for organ physiology and regeneration. However, the extent and precise role of cellular plasticity remains to be defined, and the widespread existence of the phenomenon raises a number of important questions.
To begin, one might ask why tissues, particularly those that harbor stem cells, exploit de-differentiation? While the answer to this question is unknown, we speculate that tissues utilize de-differentiation as a backup system, to generate reserve stem cells should the original stem cell pool be lost. In the examples of lung, intestine and skin discussed in this review, de-differentiation may result in a restoration of the stem cell pool after injury or ablation. Indeed, there is evidence that stem cells passively or actively inhibit de-differentiation, the result of an ongoing dialogue between stem cells and their progeny.
One can also ask the converse question: if cells can undergo de-differentiation and trans-differentiation, then why maintain a stem cell pool in the first place? Again, the answer is unclear, but it may indicate that plasticity comes at a cost. In particular, the epigenetic changes required for a cell to change its identity may also predispose it to cancer101, and hence reducing the frequency with which cells change identity might reduce the risk of malignant transformation. Alternatively, differentiation from stem cells might provide a more efficient or spatially defined way to control cellular identity. If this is the case, then de-differentiation and trans-differentiation might be the preferred mechanism of maintaining tissue homeostasis only when normal spatial signals for differentiation are disrupted by injury.
The ability to exploit cellular plasticity to generate needed cell types in vivo has tremendous therapeutic potential (Table 2). Although most examples of plasticity discussed in this review were conducted in model organisms, it is likely that the cellular relationships identified in these studies are conserved in humans. For example, one study found that human hepatocytes transplanted into mouse livers undergo trans-differentiation into biliary cells62, indicating that human hepatocytes also undergo trans-differentiation as a physiological response to injury. Generating new β cells in the pancreas as a treatment for diabetes has high priority in the field of regenerative medicine, and trans-differentiation – from either α cells or other endoderm derivatives – is an attractive approach.
Table 2.
Possible therapeutics targets of cellular plasticity
| Organ/Tissue | Disease or injury | Transdifferentiation Target | Current research |
|---|---|---|---|
Cochlea
|
Cochlear hair cell loss | Induce Supporting cells → Hair cells | New hair cells can be generated from neighboring supporting cells by inhibitin ephrin-B2 signaling109. |
| Heart |
Regenerating cardiac muscle following injury | Induce Fibroblasts → Cardiomyocyte | Cardiomyctes can be generated from cardiac fibroblasts in vivo by treatment with exogenous factors after injury110. |
Lung alveoli
|
Lung repair following injury | Alveolar Type I and Type II cells can each give rise to both Type I and II cells | Using lineage tracing, Type I and Type II alveolar cells have bidirectional cellular plasticity111 |
Nervous system
|
Parkinson’s disease | Induce fibroblasts → Dopaminergic neurons, transplanted | Fibroblasts can be converted directly to dopaminergic neurons in vitro112. |
Kidney
|
Chronic Kidney Disease | Inhibit pathological transdifferentiation of pericytes → fibrotic myofibroblasts | Inhibiting trans-differentiation of pericytes prevents them from becoming myofibroblasts and inducing fibrosis113. |
Pancreatic Islets of Langerhans
|
Type 1 Diabetes | α or δ cell transdifferentiation to β cells | Following ablation of β cells, remaining islet cells may trans-differentiate to generate new β cells73, 74 |
Before such strategies can be applied, a deeper mechanistic understanding of cellular plasticity is needed. Are there generalized principles of epigenetic remodeling that underlie all changes in adult cellular identity, or is every case different? What are the spatial cues that establish functional cellular arrangements following de-differentiation and trans-differentiation? And finally, what are the mechanisms by which cellular plasticity might predispose tissues to cancer? Hopefully, answering these questions will open up new opportunities for cell-based therapies of disease, approaches that will rely on cells already present in the body.
Glossary terms
- Epigenesis
Morgan’s term for regeneration using cellular proliferation
- Glucagon
A hormone secreted by pancreatic α cells that increases serum glucose levels
- Lateral plate mesoderm
A developmental division of mesoderm that gives rise to tendon, bone, connective tissue, and dermis within the vertebrate limb
- Lineage tracing
Permanently labeling a population of cells in order to trace their progeny and fate
- Linker histone
Histones that are responsible for stabilizing the complex of DNA wrapped around histones that forms nucleosomes
- Metaplasia
Changes in tissue whereby one cell type is replaced by another, often associated with increased cancer risk
- Morphyllaxis
Morgan’s term for regeneration using existing material in the animal, without relying on proliferation
- Multinucleated myofibers
Syncytial muscle fibers form from many muscle progenitors that fuse together to generate a single fiber with many nuclei
- Myelin
An electrically insulating sheath provided by Schwann cells membranes that surrounds axons
- Pancreatic islets of Langerhans
Endocrine cells in the pancreas responsible for producing the hormones used for glucose management
- Pioneer factor
A DNA binding factor that directly binds closed, condensed chromatin and can open the chromatin to make it more accessible for other factors to bind
- Pluripotency factor
Also known as Yamanaka factors, the factors are Sox2, Oct3/4, Myc, and Klf4 transcription factors that can induce differentiated cells to reprogram into induced pluripotent stem cells
- Progenitors
Immature cells, often lineage restricted, that can proliferate and give rise to differentiated cells. This is often a short-term state, compared to stem cell populations which may be maintained for a lifetime
- Reprogramming
Change in the identity of a differentiated cell. Usage often overlaps with de- and trans-differentiation, although reprogramming generally refers to a complete and stable shift. The most extreme example is reprogramming of a differentiated cell to a pluripotent state
- Sarcomere apparatus
Actin, myosin, and associated proteins within mature muscle fibers that are organized in such a way that they may move relative to each other to produce muscle contractions
- Satellite cells
Pax7+ muscle stem cells that reside next to muscle fibers and mediate muscle regeneration in many vertebrate species
- Schwann cells
Cells that surround and envelope neurons in myelin sheaths, allowing for proper conduction along the nerve
- Somatic cell nuclear transfer
A technique whereby nuclei from differentiated cells are transplanted into oocytes. These nuclei are reprogrammed to a germ cell state, and ultimately can generate a new organism
- Somatostatin
A hormone secreted by pancreatic δ cells that inhibits secretion of other pancreatic hormones
References
- 1.Holliday R. Epigenetics: a historical overview. Epigenetics. 2006;1:76–80. doi: 10.4161/epi.1.2.2762. [DOI] [PubMed] [Google Scholar]
- 2.Gurdon JB. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J Embryol Exp Morphol. 1962;10:622–40. [PubMed] [Google Scholar]
- 3.Raff M. Adult stem cell plasticity: fact or artifact? Annu Rev Cell Dev Biol. 2003;19:1–22. doi: 10.1146/annurev.cellbio.19.111301.143037. [DOI] [PubMed] [Google Scholar]
- 4.Jopling C, Boue S, Izpisua Belmonte JC. Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nat Rev Mol Cell Biol. 2011;12:79–89. doi: 10.1038/nrm3043. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 6.Tapscott SJ, et al. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science. 1988;242:405–11. doi: 10.1126/science.3175662. [DOI] [PubMed] [Google Scholar]
- 7.Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- 8.Galliot B. Hydra, a fruitful model system for 270 years. Int J Dev Biol. 2012;56:411–23. doi: 10.1387/ijdb.120086bg. [DOI] [PubMed] [Google Scholar]
- 9.Baguna J. The planarian neoblast: the rambling history of its origin and some current black boxes. Int J Dev Biol. 2012;56:19–37. doi: 10.1387/ijdb.113463jb. [DOI] [PubMed] [Google Scholar]
- 10.Morgan TH. Growth and regeneration in Planaria lugubris. 1901 [Google Scholar]
- 11.Goss RJ. Kinetics of Compensatory Growth. Q Rev Biol. 1965;40:123–46. doi: 10.1086/404538. [DOI] [PubMed] [Google Scholar]
- 12.de Cuevas M, Matunis EL. The stem cell niche: lessons from the Drosophila testis. Development. 2011;138:2861–9. doi: 10.1242/dev.056242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tulina N, Matunis E. Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science. 2001;294:2546–9. doi: 10.1126/science.1066700. [DOI] [PubMed] [Google Scholar]
- 14.Kiger AA, Jones DL, Schulz C, Rogers MB, Fuller MT. Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science. 2001;294:2542–5. doi: 10.1126/science.1066707. [DOI] [PubMed] [Google Scholar]
- 15.Brawley C, Matunis E. Regeneration of male germline stem cells by spermatogonial dedifferentiation in vivo. Science. 2004;304:1331–4. doi: 10.1126/science.1097676. This paper discovered evidence of de-differentiation by demonstrating that differentiated spermatogonia could de-differentiate into germline stem cells in drosophila mutants after extreme germline stem cell loss. [DOI] [PubMed] [Google Scholar]
- 16.Sheng XR, Brawley CM, Matunis EL. Dedifferentiating spermatogonia outcompete somatic stem cells for niche occupancy in the Drosophila testis. Cell Stem Cell. 2009;5:191–203. doi: 10.1016/j.stem.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barroca V, et al. Mouse differentiating spermatogonia can generate germinal stem cells in vivo. Nat Cell Biol. 2009;11:190–6. doi: 10.1038/ncb1826. [DOI] [PubMed] [Google Scholar]
- 18.Nakagawa T, Sharma M, Nabeshima Y, Braun RE, Yoshida S. Functional hierarchy and reversibility within the murine spermatogenic stem cell compartment. Science. 2010;328:62–7. doi: 10.1126/science.1182868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kai T, Spradling A. Differentiating germ cells can revert into functional stem cells in Drosophila melanogaster ovaries. Nature. 2004;428:564–9. doi: 10.1038/nature02436. This study shows that differentiating cyst cells in the drosophila ovary can de-differentiate into germline stem cells when the germline stem cells are lost to excessive differentiation. [DOI] [PubMed] [Google Scholar]
- 20.Steen TP. Stability of chondrocyte differentiation and contribution of muscle to cartilage during limb regeneration in the axolotl (Siredon mexicanum) J Exp Zool. 1968;167:49–78. doi: 10.1002/jez.1401670105. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka EM, Reddien PW. The cellular basis for animal regeneration. Dev Cell. 2011;21:172–85. doi: 10.1016/j.devcel.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kragl M, et al. Cells keep a memory of their tissue origin during axolotl limb regeneration. Nature. 2009;460:60–5. doi: 10.1038/nature08152. [DOI] [PubMed] [Google Scholar]
- 23.Sandoval-Guzman T, et al. Fundamental differences in dedifferentiation and stem cell recruitment during skeletal muscle regeneration in two salamander species. Cell Stem Cell. 2014;14:174–87. doi: 10.1016/j.stem.2013.11.007. Sandoval-Guzman, et al. found that after limb amputation certain species of salamanders use de-differentiation of post-mitotic muscle fibers to produce more muscle progenitors for limb regeneration. [DOI] [PubMed] [Google Scholar]
- 24.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–90. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 25.Kikuchi K, et al. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464:601–5. doi: 10.1038/nature08804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jopling C, et al. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–9. doi: 10.1038/nature08899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science. 2014;344:1242281. doi: 10.1126/science.1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodell MA, Nguyen H, Shroyer N. Somatic stem cell heterogeneity: diversity in the blood, skin and intestinal stem cell compartments. Nat Rev Mol Cell Biol. 2015;16:299–309. doi: 10.1038/nrm3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greco V, et al. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell. 2009;4:155–69. doi: 10.1016/j.stem.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu YC, Pasolli HA, Fuchs E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell. 2011;144:92–105. doi: 10.1016/j.cell.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rompolas P, Mesa KR, Greco V. Spatial organization within a niche as a determinant of stem-cell fate. Nature. 2013;502:513–8. doi: 10.1038/nature12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barker N, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–7. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 33.Tian H, et al. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478:255–9. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Es JH, et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol. 2012;14:1099–104. doi: 10.1038/ncb2581. This study found that committed secretory precursors de-differentiate into new crypt stem cells following intestinal crypt injury and that these de-differentiated stem cells can ultimately produce all the cell types of the intestine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans MJ, Van Winkle LS, Fanucchi MV, Plopper CG. Cellular and molecular characteristics of basal cells in airway epithelium. Exp Lung Res. 2001;27:401–15. doi: 10.1080/019021401300317125. [DOI] [PubMed] [Google Scholar]
- 36.Schoch KG, et al. A subset of mouse tracheal epithelial basal cells generates large colonies in vitro. Am J Physiol Lung Cell Mol Physiol. 2004;286:L631–42. doi: 10.1152/ajplung.00112.2003. [DOI] [PubMed] [Google Scholar]
- 37.Rock JR, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A. 2009;106:12771–5. doi: 10.1073/pnas.0906850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hong KU, Reynolds SD, Watkins S, Fuchs E, Stripp BR. In vivo differentiation potential of tracheal basal cells: evidence for multipotent and unipotent subpopulations. Am J Physiol Lung Cell Mol Physiol. 2004;286:L643–9. doi: 10.1152/ajplung.00155.2003. [DOI] [PubMed] [Google Scholar]
- 39.Tata PR, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–23. doi: 10.1038/nature12777. This study demonstrates that differentiated cells are able to de-differentiate in response to stem cell ablation to replenish the stem cell population and that this de-differentiation is likely inhibited and regulated by contact with existing stem cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mirsky R, et al. Novel signals controlling embryonic Schwann cell development, myelination and dedifferentiation. J Peripher Nerv Syst. 2008;13:122–35. doi: 10.1111/j.1529-8027.2008.00168.x. [DOI] [PubMed] [Google Scholar]
- 41.Painter MW, et al. Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron. 2014;83:331–43. doi: 10.1016/j.neuron.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, et al. Turning terminally differentiated skeletal muscle cells into regenerative progenitors. Nat Commun. 2015;6:7916. doi: 10.1038/ncomms8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pajcini KV, Corbel SY, Sage J, Pomerantz JH, Blau HM. Transient inactivation of Rb and ARF yields regenerative cells from postmitotic mammalian muscle. Cell Stem Cell. 2010;7:198–213. doi: 10.1016/j.stem.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsu YC, Li L, Fuchs E. Emerging interactions between skin stem cells and their niches. Nat Med. 2014;20:847–56. doi: 10.1038/nm.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Lau W, Peng WC, Gros P, Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014;28:305–16. doi: 10.1101/gad.235473.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monje PV, Soto J, Bacallao K, Wood PM. Schwann cell dedifferentiation is independent of mitogenic signaling and uncoupled to proliferation: role of cAMP and JNK in the maintenance of the differentiated state. J Biol Chem. 2010;285:31024–36. doi: 10.1074/jbc.M110.116970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao R, et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self-renewal of stem cells. Dev Cell. 2014;30:151–65. doi: 10.1016/j.devcel.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagers AJ, Weissman IL. Plasticity of adult stem cells. Cell. 2004;116:639–48. doi: 10.1016/s0092-8674(04)00208-9. [DOI] [PubMed] [Google Scholar]
- 49.Huang P, et al. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature. 2011;475:386–9. doi: 10.1038/nature10116. [DOI] [PubMed] [Google Scholar]
- 50.Khurana S, Mukhopadhyay A. In vitro transdifferentiation of adult hematopoietic stem cells: an alternative source of engraftable hepatocytes. J Hepatol. 2008;49:998–1007. doi: 10.1016/j.jhep.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 51.Vierbuchen T, et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–41. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Riddle MR, Weintraub A, Nguyen KC, Hall DH, Rothman JH. Transdifferentiation and remodeling of post-embryonic C. elegans cells by a single transcription factor. Development. 2013;140:4844–9. doi: 10.1242/dev.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jarriault S, Schwab Y, Greenwald I. A Caenorhabditis elegans model for epithelial-neuronal transdifferentiation. Proc Natl Acad Sci U S A. 2008;105:3790–5. doi: 10.1073/pnas.0712159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Del Rio-Tsonis K, Tsonis PA. Eye regeneration at the molecular age. Dev Dyn. 2003;226:211–24. doi: 10.1002/dvdy.10224. [DOI] [PubMed] [Google Scholar]
- 55.Maki N, et al. Expression of stem cell pluripotency factors during regeneration in newts. Dev Dyn. 2009;238:1613–6. doi: 10.1002/dvdy.21959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maki N, et al. Oocyte-type linker histone B4 is required for transdifferentiation of somatic cells in vivo. FASEB J. 2010;24:3462–7. doi: 10.1096/fj.10-159285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mizuno N, Agata K, Sawada K, Mochii M, Eguchi G. Expression of crystallin genes in embryonic and regenerating newt lenses. Dev Growth Differ. 2002;44:251–6. doi: 10.1046/j.1440-169x.2002.00639.x. [DOI] [PubMed] [Google Scholar]
- 58.Reyer RW, Woolfitt RA, Withersty LT. Stimulation of lens regeneration from the newt dorsal iris when implanted into the blastema of the regenerating limb. Dev Biol. 1973;32:258–81. doi: 10.1016/0012-1606(73)90240-6. [DOI] [PubMed] [Google Scholar]
- 59.Ito M, Hayashi T, Kuroiwa A, Okamoto M. Lens formation by pigmented epithelial cell reaggregate from dorsal iris implanted into limb blastema in the adult newt. Dev Growth Differ. 1999;41:429–40. doi: 10.1046/j.1440-169x.1999.00447.x. [DOI] [PubMed] [Google Scholar]
- 60.Stanger BZ. Cellular homeostasis and repair in the mammalian liver. Annu Rev Physiol. 2015;77:179–200. doi: 10.1146/annurev-physiol-021113-170255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yanger K, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27:719–24. doi: 10.1101/gad.207803.112. Using genetic lineage tracing, Yanger, et al found that forced Notch signaling or injury are sufficient to induce hepatocytes to transdifferentiate into biliary cells in a stepwise process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tarlow BD, et al. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell. 2014;15:605–18. doi: 10.1016/j.stem.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Michalopoulos GK, Barua L, Bowen WC. Transdifferentiation of rat hepatocytes into biliary cells after bile duct ligation and toxic biliary injury. Hepatology. 2005;41:535–44. doi: 10.1002/hep.20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koh DS, Cho JH, Chen L. Paracrine interactions within islets of Langerhans. J Mol Neurosci. 2012;48:429–40. doi: 10.1007/s12031-012-9752-2. [DOI] [PubMed] [Google Scholar]
- 65.Cogger K, Nostro MC. Recent advances in cell replacement therapies for the treatment of type 1 diabetes. Endocrinology. 2015;156:8–15. doi: 10.1210/en.2014-1691. [DOI] [PubMed] [Google Scholar]
- 66.Kushner JA, MacDonald PE, Atkinson MA. Stem cells to insulin secreting cells: two steps forward and now a time to pause? Cell Stem Cell. 2014;15:535–6. doi: 10.1016/j.stem.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 67.Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–32. doi: 10.1038/nature07314. This study found that forced expression of beta cell-specific transcription factors in pancreatic acinar cells leads to transdifferentiation of acinar cells directly into functional beta-cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Baeyens L, et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol. 2014;32:76–83. doi: 10.1038/nbt.2747. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Chen YJ, et al. De novo formation of insulin-producing “neo-beta cell islets” from intestinal crypts. Cell Rep. 2014;6:1046–58. doi: 10.1016/j.celrep.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferber S, et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes in liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6:568–72. doi: 10.1038/75050. [DOI] [PubMed] [Google Scholar]
- 71.Shternhall-Ron K, et al. Ectopic PDX-1 expression in liver ameliorates type 1 diabetes. J Autoimmun. 2007;28:134–42. doi: 10.1016/j.jaut.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 72.Horb ME, Shen CN, Tosh D, Slack JM. Experimental conversion of liver to pancreas. Curr Biol. 2003;13:105–15. doi: 10.1016/s0960-9822(02)01434-3. [DOI] [PubMed] [Google Scholar]
- 73.Thorel F, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149–54. doi: 10.1038/nature08894. Thorel, et al determined that following beta cell ablation in the pancreas, alpha cells could transdifferentiate into functional beta cells without needing exogenous factors to initiate transdifferentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chera S, et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature. 2014;514:503–7. doi: 10.1038/nature13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao T, et al. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 2014;19:259–71. doi: 10.1016/j.cmet.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A. Pancreatic beta cell identity is maintained by DNA methylation-mediated repression of Arx. Dev Cell. 2011;20:419–29. doi: 10.1016/j.devcel.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–34. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo S, et al. Inactivation of specific beta cell transcription factors in type 2 diabetes. J Clin Invest. 2013;123:3305–16. doi: 10.1172/JCI65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zuryn S, et al. Transdifferentiation. Sequential histone-modifying activities determine the robustness of transdifferentiation. Science. 2014;345:826–9. doi: 10.1126/science.1255885. [DOI] [PubMed] [Google Scholar]
- 80.Zong Y, et al. Notch signaling controls liver development by regulating biliary differentiation. Development. 2009;136:1727–39. doi: 10.1242/dev.029140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yanger K, Stanger BZ. Liver cell reprogramming: parallels with iPSC biology. Cell Cycle. 2014;13:1211–2. doi: 10.4161/cc.28381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang N, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yimlamai D, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157:1324–38. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Katsuyama T, Paro R. Epigenetic reprogramming during tissue regeneration. FEBS Lett. 2011;585:1617–24. doi: 10.1016/j.febslet.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 85.Slack JM. Metaplasia and transdifferentiation: from pure biology to the clinic. Nat Rev Mol Cell Biol. 2007;8:369–78. doi: 10.1038/nrm2146. [DOI] [PubMed] [Google Scholar]
- 86.Tosh D, Slack JM. How cells change their phenotype. Nat Rev Mol Cell Biol. 2002;3:187–94. doi: 10.1038/nrm761. [DOI] [PubMed] [Google Scholar]
- 87.Corbett JL, Tosh D. Conversion of one cell type into another: implications for understanding organ development, pathogenesis of cancer and generating cells for therapy. Biochem Soc Trans. 2014;42:609–16. doi: 10.1042/BST20140058. [DOI] [PubMed] [Google Scholar]
- 88.Shaheen NJ, Richter JE. Barrett’s oesophagus. Lancet. 2009;373:850–61. doi: 10.1016/S0140-6736(09)60487-6. [DOI] [PubMed] [Google Scholar]
- 89.Bhat S, et al. Risk of malignant progression in Barrett’s esophagus patients: results from a large population-based study. J Natl Cancer Inst. 2011;103:1049–57. doi: 10.1093/jnci/djr203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hvid-Jensen F, Pedersen L, Drewes AM, Sorensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N Engl J Med. 2011;365:1375–83. doi: 10.1056/NEJMoa1103042. [DOI] [PubMed] [Google Scholar]
- 91.Stanger BZ, Hebrok M. Control of cell identity in pancreas development and regeneration. Gastroenterology. 2013;144:1170–9. doi: 10.1053/j.gastro.2013.01.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63:2016–9. [PubMed] [Google Scholar]
- 93.De La OJ, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105:18907–12. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kopp JL, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:737–50. doi: 10.1016/j.ccr.2012.10.025. This study found that acinar-ductal-metaplasia and expression of ductal genes are critical for inducing acinar cells to give rise to pancreatic ductal adenocarcinoma and suggests that cellular reprogramming may be a critical step in tumor initiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu L, Shi G, Schmidt CM, Hruban RH, Konieczny SF. Acinar cells contribute to the molecular heterogeneity of pancreatic intraepithelial neoplasia. Am J Pathol. 2007;171:263–73. doi: 10.2353/ajpath.2007.061176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krah NM, et al. The acinar differentiation determinant PTF1A inhibits initiation of pancreatic ductal adenocarcinoma. Elife. 2015;4 doi: 10.7554/eLife.07125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fan B, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122:2911–5. doi: 10.1172/JCI63212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J Clin Invest. 2012;122:3914–8. doi: 10.1172/JCI63065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Villanueva A, et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 2012;143:1660–1669 e7. doi: 10.1053/j.gastro.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li H, et al. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int. 2012;32:38–47. doi: 10.1111/j.1478-3231.2011.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Abollo-Jimenez F, Jimenez R, Cobaleda C. Physiological cellular reprogramming and cancer. Semin Cancer Biol. 2010;20:98–106. doi: 10.1016/j.semcancer.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 102.Bjornson CR, Rietze RL, Reynolds BA, Magli MC, Vescovi AL. Turning brain into blood: a hematopoietic fate adopted by adult neural stem cells in vivo. Science. 1999;283:534–7. doi: 10.1126/science.283.5401.534. [DOI] [PubMed] [Google Scholar]
- 103.Petersen BE, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168–70. doi: 10.1126/science.284.5417.1168. [DOI] [PubMed] [Google Scholar]
- 104.Carriere C, Seeley ES, Goetze T, Longnecker DS, Korc M. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2007;104:4437–42. doi: 10.1073/pnas.0701117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Khan MS, Thornhill JA, Gaffney E, Loftus B, Butler MR. Keratinising squamous metaplasia of the bladder: natural history and rationalization of management based on review of 54 years experience. Eur Urol. 2002;42:469–74. doi: 10.1016/s0302-2838(02)00358-5. [DOI] [PubMed] [Google Scholar]
- 106.de Vries AC, Kuipers EJ. Epidemiology of premalignant gastric lesions: implications for the development of screening and surveillance strategies. Helicobacter. 2007;12(Suppl 2):22–31. doi: 10.1111/j.1523-5378.2007.00562.x. [DOI] [PubMed] [Google Scholar]
- 107.Elson DA, et al. Sensitivity of the cervical transformation zone to estrogen-induced squamous carcinogenesis. Cancer Res. 2000;60:1267–75. [PubMed] [Google Scholar]
- 108.Daniels JM, Sutedja TG. Detection and minimally invasive treatment of early squamous lung cancer. Ther Adv Med Oncol. 2013;5:235–48. doi: 10.1177/1758834013482345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Defourny J, et al. Cochlear supporting cell transdifferentiation and integration into hair cell layers by inhibition of ephrin-B2 signalling. Nat Commun. 2015;6:7017. doi: 10.1038/ncomms8017. [DOI] [PubMed] [Google Scholar]
- 110.Qian L, et al. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–8. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jain R, et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat Commun. 2015;6:6727. doi: 10.1038/ncomms7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pfisterer U, et al. Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci U S A. 2011;108:10343–8. doi: 10.1073/pnas.1105135108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nakagawa N, Duffield JS. Myofibroblasts in Fibrotic Kidneys. Curr Pathobiol Rep. 2013;1 doi: 10.1007/s40139-013-0025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]