

Abstract
Background & objectives:
A high incidence of hearing impairment is reported from the village of Dhadkai in the State of Jammu and Kashmir, India. Prevalence of endogamy in this community suggested a common genetic basis for the disorder. A genetic study was undertaken to ascertain the basis for the high incidence of hearing impairment in this region.
Methods:
In a two-step approach to identify the causative mutation/s, a whole-genome-based linkage analysis of an extended family of 45 members was carried out, which included 23 affected and 22 unaffected members. Mutational analysis for the candidate deafness genes helped reveal causative mutations in the family. In addition, seven deafness-causing genes, Cx26, SLC26A4, CLDN14, TMPRSS3, TMC1, TMIE and USH1C, were analyzed in smaller families with hearing impairment.
Results:
In the 45-member extended family, the critical chromosomal region mapped to 2p24-p22. The c.2122C>T (p.R708X) mutation in OTOF in 2p24-p22was identified as being the causal change. Linkage to 2p24-p22 locus was not observed in a particular branch of this extended family. Analysis of seven known deafness-causing genes in this branch revealed a mutation, c.254T>A (p.V85D), in CLDN14. Among seven small families unrelated to the 45-member extended family, hearing loss was attributable to p.R708X in OTOF in three families and to p.V85D in CLDN14 in one family; a new mutation c.1668T>A (p.Y556X) SLC26A4 was identified in two families and the causative change could not be identified in one family.
Interpretation & conclusions:
This study suggested considerable genetic heterogeneity in the causation of hearing loss in Dhadkai. Recessive mutations were observed in at least three genes causing hearing loss: OTOF (p.R708X), SLC26A4 (p.Y556X) and CLDN14 (p.V85D). Mutation p.R708X appeared to be the major cause of hearing impairment in Dhadkai.
Keywords: Autosomal recessive non-syndromic hearing loss, CLDN14, genetic heterogeneity, hearing impairment, OTOF, SLC26A4
Hearing loss constitutes the most common form of sensory defect, affecting one in 2000 newborns. Nearly 50 per cent of hearing impairment has a genetic basis to its aetiology. About 85 per cent of hereditary hearing loss is recessive in inheritance and non-syndromic in manifestation. To date, 88 genes and 145 loci implicated in non-syndromic hearing loss (NSHL) have been identified (hereditary hearing loss website: http://hereditaryhearingloss.org/). Mutations in a single gene connexin 26 (Cx26) are the most common cause of hearing loss worldwide1.
Dhadkai is a remote village located in Doda district of Jammu and Kashmir, India. According to the 2011 census, the village had a total population of 1774 inhabitants as 253 families (http://censusindia.gov.in). A majority of individuals in this village belong to the Gujjar tribe, which is an endogamous community. A high incidence of hearing loss has been reported from Dhadkai. In 2005, 72 cases of hearing-impaired individuals were reported in a survey conducted by the State's Social Welfare Department2. Other reports indicated an increase in the number of hearing-impaired individuals in this village2,3. To understand the genetic basis of this disorder, one extended family and seven small families were studied from this village.
Material & Methods
This study was conducted during 2004-2009 as a part of a deafness project carried out at Molecular Biology and Genetics Unit, Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bengaluru, India. Selection of families and collection of blood samples were carried out by scientists from University of Kashmir, Srinagar, and department of Audiology and Ali Yavar Jung National Institute for the Hearing Handicapped (AYJNIHH), Mumbai. Audiological assessment of members was carried out by AYJNIHH. This study was approved by the Institutional Human Bioethics and Biosafety Committee of JNCASR, the main coordinating centre for the study. Up to 10 ml of peripheral blood was collected after obtaining written informed consent for participation. DNA was extracted using the phenol-chloroform method.
A selected set of eight densely affected families comprising about 50 per cent of the affected individuals from Dhadkai village were examined. Families with one or more members exhibiting prelingual NSHL were selected. A large family (KSH01) was enrolled for genome-wide linkage analysis on the bases of its multi-generational nature and the presence of multiple affected and unaffected members. In case of singleton-affected families, samples of both parents and, in families with more than one affected member, sample of one parent were necessary. The mean age of hearing impaired members was 14 yr (range between 3 and 45 yr). The male/female ratio for affected members was 1.2:1. In KSH01 with 45 members, there were 23 affected and 22 unaffected members (Fig. 1A). A three-generation branch of KSH01 comprising 18 members (8 affected and 10 unaffected) was subjected to whole-genome linkage mapping (Fig. 1A). Seven small families, KSH02-08 (Fig. 1B) examined, included four families with at least two affected members (KSH02, KSH03, KSH04 and KSH08) and three families (KSH05, KSH06 and KSH07) each with a single affected member. Detailed clinical histories of the affected members were collected to evaluate age of onset and to examine the possibility of a non-genetic cause for hearing loss, such as a birth defect, acoustic trauma or ear infections. The degree of hearing loss was ascertained through pure-tone audiometric evaluations for air and bone conduction. Members exhibited hearing impairment of profound, bilateral and sensorineural type (Fig. 2C). No apparent additional clinical feature was noted segregating with the hearing loss, indicating its non-syndromic nature.
Fig. 1.

Pedigree analysis of KSH01-KSH08. (A) 2p24-p22 marker haplotypes and OTOF, c.2122C>T in KSH01. Members taken up for genome-wide scan (asterisk), additional affected members genotyped and their parents/ancestors are depicted. Of the 45 members of this family, all 24 affected members and 16 of 22 unaffected members (excluding 6 unaffected members from the extended family) are shown. Microsatellite markers (left-side), affected chromosomes (black bars), critical recombination boundaries (arrows), autozygous genotypes (bold italics) and inferred genotypes (parenthesis) are indicated. NSG (non-segregating) denotes the branch where linkage to 2p24-p22 was absent. (B) KSH02-KSH08 and KSH01.NSG showing the segregating mutations: c.2122C>T, c.254T>A and c.1668T>A. Squares, circles, filled and unfilled symbols denote males, females, affected and unaffected individuals, respectively.
Fig. 2.

Mutations OTOF p.R708X (c.2122C>T); CLDN14 p.V85D (c.254T>A) and SLC26A4 p.Y556X (c.1668T>A). (A) Representative sequence traces from the hearing impaired individuals exhibiting the mutations (lower panel) are shown. The corresponding wild-type alleles are shown in upper panel. (B) Protein schematics of OTOF, CLDN14 and SLC26A4 depicting approximate locations (asterisk) of mutations. (C) Audiograms of affected members KSH01-VI:14, KSH01.NSG-VI:2 and KSH04-II:3. Curves indicate the hearing thresholds for left (red) and right (blue) ears.
Genome-wide scan and linkage analysis: Genome-wide scan was carried out by genotyping 18 members of KSH01 using ABI PRISM Linkage mapping set v2.5 MD-10 (ThermoFisher Scientific Inc., USA). In the KSH01 extended family, 27 additional members were also examined for the seven microsatellite markers D2S162, D2S168, D2S305, D2S165, D2S367, D2S2259 and D2S391 from the mapping set, for a 52.5cM genetic region in 2p25-p21. Polymerase chain reactions (PCRs) were performed with fluorescence-labelled primers and True allele™ PCR premix on a Gene Amp PCR system 9700 (Thermo Fisher Scientific Inc.), according to the manufacturer's protocol. Aliquots from the fluorescence-labelled products were pooled and denatured at 94°C for five minutes, snap chilled and electrophoresed on an ABI3730 DNA Analyzer (Thermo Fisher Scientific Inc.). A reference individual CEPH 1347-02 with known genotypes was used as an internal control. Genotypes were called using ABI PRISM GeneMapper™ v3.7 (Thermo Fisher Scientific Inc.). Genotypes were checked for Mendelian inconsistencies if any, and haplotypes were generated manually, allowing for minimum number of possible recombination events. Two-point linkage analysis was carried out using MLINK v5.2 of linkage4. Disease allele frequency was taken as 0.001 and equal frequencies were assumed for the marker alleles. Logarithm of odds (LOD) scores were computed under a recessive model with consanguinity loops at 99 per cent penetrance value and 0 per cent phenocopy.
Mutation analysis: The proband, KSH01-VI:14, was examined for mutation in Cx26 by sequencing its coding and non-coding exons. Mutation in the otoferlin gene (OTOF) in VI:14 was detected by sequencing its 48 exons including the coding exons, 5’- and 3’-UTRs (untranslated regions) and intronic flanks (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/mapview/). Primer pairs were designed using Primer3 Input (http://primer3.ut.ee/). PCR amplification was carried out using 100 ng of genomic DNA, 10 pmol primers, 800 μM dNTPs (nucleoside triphosphates, New England Biolabs Inc., USA), 1.5 mM MgCl2 and 1 Uof Taq DNA polymerase (Thermo Fisher Scientific Inc., USA) in a 20 μl reaction volume. The amplified products were purified by Montage PCR96 clean-up reagents (EMD Millipore, USA). Cycle sequencing was performed using 20 ng of purified PCR products, 2.5 pmol of primer and ABI PRISM BigDye™ Terminator cycle sequencing reagents. The sequenced products were ethanol washed, denatured in formamide, snap chilled and electrophoresed using a 3730 DNA Analyzer. Each amplicon was sequenced in both directions and analyzed using SeqManII 5.01 (DNASTAR Inc., USA).
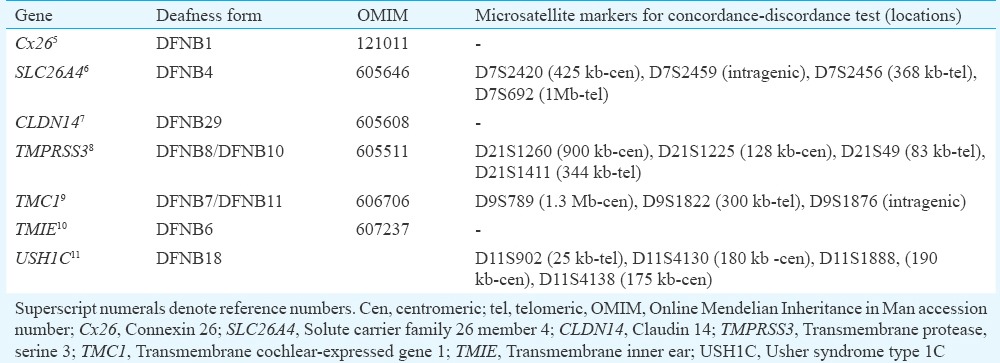
To examine the KSH01-non-segragating (NSG) branch and KSH04 for possible mutations in Cx26, transmembrane inner ear (TMIE) and claudin 14 (CLDN14), bidirectional sequencing of the exons and flanking intronic regions was done in an affected member. For analysis of solute carrier family 26, member 4 (SLC26A4), transmembrane protease, serine 3 (TMPRSS3), transmembrane cochlear-expressed gene 1(TMC1) and Usher syndrome type 1 C(USH1C) genes, a concordance/discordance test using polymorphic microsatellite markers closely linked to the gene of interest was performed in KSH04 and KSH01.NSG (Table I). PCR was carried out with 50 ng of genomic DNA, 2.5 pmol primers, 1.5 mM MgCl2 and 2.5 U Taq DNA polymerase on a GeneAmp PCR9700 and genotyping was done on a 3730 DNA Analyzer. Allele sizing was performed by GeneMapper v3.7. In those families which could not be excluded for a possible mutation in the gene on basis of concordance/discordance tests, complete SLC26A4 (21 exons), TMPRSS3 (13 exons), TMC1 (24 exons) and USH1C (28 exons) transcripts comprising exons and flanking intronic regions were analyzed by sequencing. All members included in the study were examined for the identified mutations. In KSH04 proband, heterozygous for mutation p.R708X, the possibility of a second OTOF mutation leading to compound heterozygous condition was examined by sequencing all the exons of OTOF.
Table I.
Autosomal recessive non-syndromic hearing loss genes analyzed in KSH01.non-segragating (NSG) and KSH04
Results
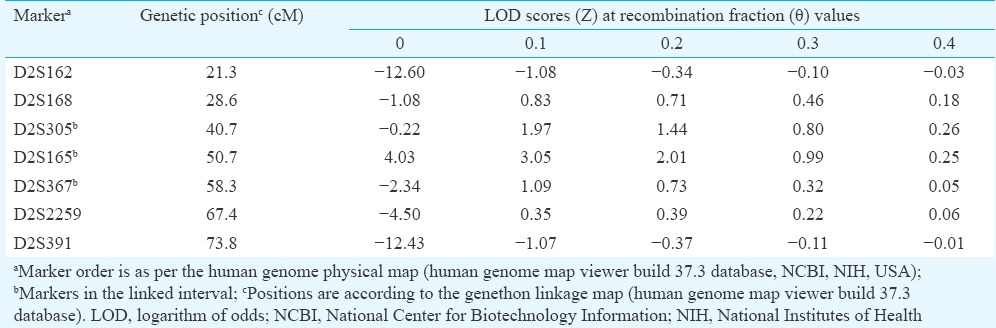
Major deafness locus in family KSH01 maps to chromosome 2p24-p22: KSH01 was an extended, six-generation family (Fig. 1A) exhibiting hearing loss transmitted in an apparently autosomal recessive manner. No Cx26 mutation was detected in the proband. In the genome-wide scan involving 18 members of KSH01, the highest two-point LOD score obtained was 4.03 (θ=0) for D2S165 (Fig. 1A and Table II).
Table II.
Logarithm of odds scores for certain markers in the 2p24-p22 region in KSH01
Haplotyping analysis identified a co-segregating marker, D2S165 amongst all the affected siblings. The recombination boundaries for the linked interval were defined by markers D2S305 and D2S367 (Fig. 1A). Genotyping the remaining members in the family for D2S165 and its neighbouring markers revealed autozygous regions in the affected siblings extending upto nearly 33.1cM (Fig. 1A: Individual V:20). The alleles shared by these siblings included D2S165 and D2S367. In affected sibs VI:5 and VI:6, though autozygosity was not noted, linkage to the same region was noted. Among the family members who underwent genome-wide scan, autozygous regions were observed for all the members except individual VI:9. In one of the branches of KSH01, named KSH01.NSG, comprising an available parent and four affected offsprings, linkage to D2S165 or its flanking region was not observed (Fig. 1A). This suggested the presence of a second deafness-causing locus elsewhere in the genome in the NSG branch.
p.R708X mutation in OTOF in KSH01: Sequence analysis of OTOF in the proband identified a homozygous mutation c.2122C>T (Fig. 2A and Table III). c.2122C>T transition created a premature stop codon at amino acid position 708 (p.R708X) of the long isoform of the protein (Fig. 2B). Except in the KSH01.NSG branch, p.R708X was found to co-segregate with hearing loss in the family (Figs 1A and 2C).
Table III.
Sequence variants observed in OTOF (KSH01 proband, VI: 14), CLDN14 (KSH01.non-segragating (NSG) member, VI: 2) and SLC26A4 (KSH04 proband, II: 3)
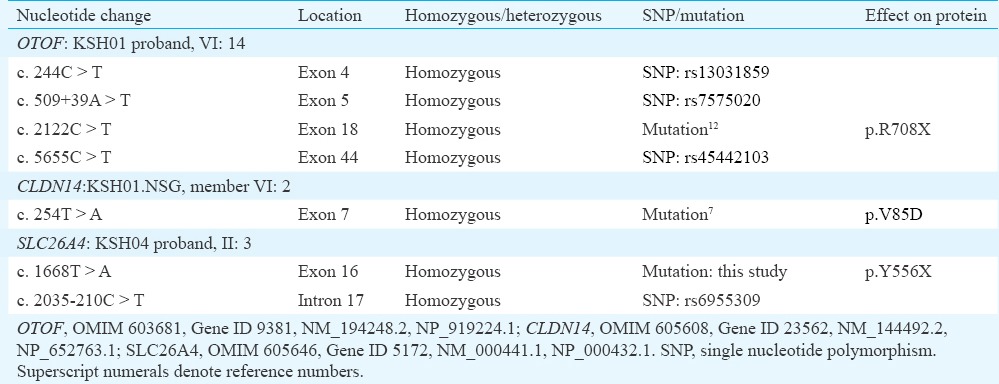
p.V85D mutation in CLDN14 in the KSH01.NSG branch: To identify the possible deafness-causing gene in KSH01.NSG, seven autosomal recessive NSHL (ARNSHL) genes, namely Cx26, SLC26A4, CLDN14, TMPRSS3, TMC1, USH1C and TMIE, were taken up for mutation analysis (Table I). These analyses revealed a homozygous mutation, c.254T>A (p.V85D), in CLDN147 (Table III and Fig. 2A). c.254T>A was found to segregate with hearing loss in KSH01.NSG (Figs 1B and 2C).
Presence of p.R708X, p.V85D as well as a new mutation p.Y556X in SLC26A4 in the Dhadkai families: Additional seven families (Fig. 1B, KSH02-08) were analyzed for p.R708X (OTOF) and p.V85D (CLDN14). Mutation p.R708X accounted for hearing loss in KSH02, KSH05 and KSH07 and p.V85D accounted for hearing loss in a single family, KSH03 (Fig. 1B and Table IV). However, in family KSH04, p.R708X was present in heterozygous condition in the two affected siblings (Fig. 1B). This suggested two possibilities: (i) KSH04 members had another OTOF mutation present elsewhere in the gene, or (ii) these members were carriers for p.R708X and were affected due to genetic defects elsewhere in the genome. To test the first possibility, the remaining exons of OTOF were sequenced in the KSH04 proband which revealed 11 known polymorphisms and a new intronic deletion, c.6011+66_67del. This deletion was a benign change as it did not segregate with the phenotype in the family. A haplotype analysis with c.6011+66_67del and three polymorphisms (c.2215-291G>C, c.2215-203_-204del and c.5655C>T) confirmed that the affected members did not share a second mutation in OTOF.
Table IV.
Summary of study on Dhadkai families: KSH01-KSH08
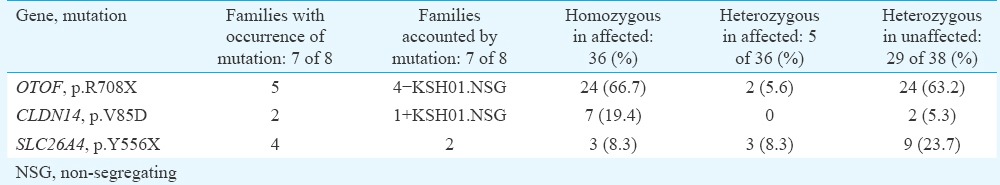
Hearing impairment in KSH04, KSH06 and KSH08 could not be attributed to either p.R708X or p.V85D (Fig. 1). In KSH04, analysis was done for the possibility of a mutation in Cx26, SLC26A4, TMPRSS3, TMC1, TMIE or USH1C. In SLC26A4, a new mutation c.1668T>A (p.Y556X) was found to segregate with hearing loss in this family (Table III and Figs 1B, 2A, C). c.1668T>A was absent in 96 chromosomes of normal hearing individuals examined. This mutation introduces a stop codon in place of tyrosine at the 556 position, thereby prematurely truncating the SLC26A4 protein in its C-terminal tail (Fig. 2B). Besides KSH04, p.Y556X also accounted for hearing loss in KSH06 (Fig. 1B). In KSH08, no mutation was detected.
Among the eight families (KSH01-08) of Dhadkai studied here, there was a predominance of OTOF, p.R708X mutation which was present in five families and could account for hearing impairment in 24 of the 36 (66.7%) affected individuals (Table IV). Among the unaffected members from these families, the carrier frequency for p.R708X was 0.61 (Table IV). In comparison, CLDN14, p.V85D mutation was not frequent. p.V85D was observed in two families (Table IV). SLC26A4, p.Y556X mutation accounted for hearing loss in two families, and in families KSH01 and KSH07, carriers of p.Y556X were observed (Fig. 1B and Table IV). Co-occurrence of SLC26A4, p.Y556X and OTOF, p.R708X mutations was observed in three families (KSH01, KSH02 and KSH04). Family members with both p.Y556X and p.R708X mutations included six unaffected individuals who were heterozygous for both the mutations and five affected individuals who were homozygous for one mutation and were coincidentally carrying a mutant allele of the second mutation.
Discussion
The Dhadkai clan with its endogamous culture was expected to be a genetically homogeneous population with a single founder mutation in a known or novel gene. However, this study involving one large and seven small families identified mutations in three known NSHL genes: OTOF, SLC26A4 and CLDN14.
OTOF mutations have been studied in deaf cohorts from Spain13,14, Turkey15, USA16,17, Colombia14, Argentina14, Pakistan18, Brazil19, China20,21, Taiwan22, Iran23, Japan24,25 and Korea26. OTOF is frequently implicated in ARNSHL and many of its pathogenic alleles include stop-codon changes and deletions or duplications that result in premature truncation of the protein. p.R708X, the mutation found in KSH01, results in an abnormal termination of the long isoform of OTOF protein exclusively. The prevalence of p.R708X mutation was about 0.36 per cent in 557 families from Pakistan exhibiting prelingual, recessive, severe-to-profound hearing loss18.
The contribution of OTOF mutations to the load of hereditary hearing loss in India remains largely unexplored. Previously, a splice-site mutation, IVS8-2A>G in OTOF, was identified in a family from Southwest India with three affected sibs27. In the present study, OTOF, p.R708X was found to be the most common mutation in Dhadkai. As evident from the KSH01 family structure, p.R708X was transmitted from a common ancestor that was at least six generations old.
Mutations in OTOF are frequently associated with the clinical phenotype of non-syndromic auditory neuropathy16 or more specifically auditory synaptopathy28. These hearing-impaired individuals do not have pure-tone audiometric and auditory brainstem response (ABR). However, transiently evoked otoacoustic emissions (TEOAEs), either unilateral or bilateral, and/or cochlear microphonics are present in them. Individuals have unusually poor reception of speech in proportion to the degree of hearing loss, and hearing aids may provide limited benefit16. Cochlear implants are found to enhance sound detection and improve communication skills in such cases29. Thus, patients with the OTOF, p.R708X in Dhadkai are likely to benefit from cochlear implantation.
CLDN14, p.V85D was the second mutation observed in the clan. Although mutations in CLDN14 have not been examined in any Indian cohort so far, p.V85D has been reported as a frequent CLDN14 mutation of a founder origin in Pakistan7,30. In a cohort of 800 NSHL families from Pakistan, 12 families exhibited p.V85D mutation (prevalence of 1.5%)30.
The third mutation noted in Dhadkai was a new mutation, p.Y556X in SLC26A4. Mutations in SLC26A4 are a frequent cause of hearing loss and are causally linked to a syndromic form of hearing impairment, Pendred syndrome31 (MIM 274600) and NSHL with enlarged vestibular aqueduct6 (MIM 600791).
The Dhadkai clan exemplified a case of genetic heterogeneity underlying hearing loss in a highly endogamous population. Presence of more than one mutation in a family/individual suggested assortative mating: all the three mutations p.R708X, p.Y556X and p.V85D occurred in KSH01extended family; and in smaller families KSH04 and KSH07, both p.Y556X and p.R708X were noted. The KSH08 family, wherein p.R708X (OTOF), p.V85D (CLDN14) and p.Y556X (SLC26A4) were absent, represented a fourth, as yet, unidentified genetic cause for hearing loss in Dhadkai.
From a sub-population perspective, allelic diversity and contribution of the NSHL genes to hearing impairment seem distinct in Dhadkai from the situation in the overall Indian population. In Dhadkai, OTOF, SLC26A4 and CLDN14 mutations accounted for hearing loss in 94 per cent of the cases examined. There are several possible approaches to reduce the incidence of hearing loss in Dhadkai: (i) appropriate genetic counselling about the risks involving deaf-deaf and intra-community marriages, and (ii) examining the carrier status in the Dhadkai population and where necessary offering prenatal screening for the mutations identified. Further, TEOAEs routinely conducted to evaluate hearing impairment in newborns are apparently misleading in cases of OTOF mutations. Genetic screens for neonates and use of suitable audiological tests (such as TEOAE coupled with ABR and tympanometry) should aid in early intervention and better management of the disability.
Acknowledgment
Authors thank members of families who participated in this study. Authors also thank Dr Aparna Ganapathy for help in mutational analysis of TMPRSS3, USH1C and TMC1, and Dr Sharat Chandra for critical reading of manuscript and helpful discussions. Financial support from DBT, New Delhi, ICMR, New Delhi and JNCASR, Bengaluru are gratefully acknowledged. NP acknowledges CSIR, New Delhi for junior and senior research fellowships.
Footnotes
Conflicts of Interest: None.
References
- 1.Kenneson A, Van Naarden Braun K, Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: AHuGE review. Genet Med. 2002;4:258–74. doi: 10.1097/00125817-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Malik MM. Dadkai Gandoh of Doda is a Village of Deaf and Dumb. Jammu and Kashmir Newspoint. 2012 Mar 4; [Google Scholar]
- 3.Kataria S. India's “silent” village of deaf-mutes. India: Reuters; 2009. [Google Scholar]
- 4.Lathrop GM, Lalouel JM. Easy calculations of LOD scores and genetic risks on small computers. Am J Hum Genet. 1984;36:460–5. [PMC free article] [PubMed] [Google Scholar]
- 5.Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, et al. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–3. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 6.Li XC, Everett LA, Lalwani AK, Desmukh D, Friedman TB, Green ED, et al. A mutation in PDS causes non-syndromic recessive deafness. Nat Genet. 1998;18:215–7. doi: 10.1038/ng0398-215. [DOI] [PubMed] [Google Scholar]
- 7.Wilcox ER, Burton QL, Naz S, Riazuddin S, Smith TN, Ploplis B, et al. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell. 2001;104:165–72. doi: 10.1016/s0092-8674(01)00200-8. [DOI] [PubMed] [Google Scholar]
- 8.Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, Chrast R, et al. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet. 2001;27:59–63. doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
- 9.Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet. 2002;30:277–84. doi: 10.1038/ng842. [DOI] [PubMed] [Google Scholar]
- 10.Naz S, Giguere CM, Kohrman DC, Mitchem KL, Riazuddin S, Morell RJ, et al. Mutations in a novel gene, TMIE, are associated with hearing loss linked to the DFNB6 locus. Am J Hum Genet. 2002;71:632–6. doi: 10.1086/342193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, et al. Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum Genet. 2002;110:527–31. doi: 10.1007/s00439-002-0732-4. [DOI] [PubMed] [Google Scholar]
- 12.Rodríguez-Ballesteros M, del Castillo FJ, Martín Y, Moreno-Pelayo MA, Morera C, Prieto F, et al. Auditory neuropathy in patients carrying mutations in the otoferlin gene (OTOF) Hum Mutat. 2003;22:451–6. doi: 10.1002/humu.10274. [DOI] [PubMed] [Google Scholar]
- 13.Migliosi V, Modamio-Høybjør S, Moreno-Pelayo MA, Rodríguez-Ballesteros M, Villamar M, Tellería D, et al. Q829X, a novel mutation in the gene encoding otoferlin (OTOF), is frequently found in Spanish patients with prelingual non-syndromic hearing loss. J Med Genet. 2002;39:502–6. doi: 10.1136/jmg.39.7.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodríguez-Ballesteros M, Reynoso R, Olarte M, Villamar M, Morera C, Santarelli R, et al. A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum Mutat. 2008;29:823–31. doi: 10.1002/humu.20708. [DOI] [PubMed] [Google Scholar]
- 15.Tekin M, Akcayoz D, Incesulu A. A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am J Med Genet A. 2005;138:6–10. doi: 10.1002/ajmg.a.30907. [DOI] [PubMed] [Google Scholar]
- 16.Varga R, Kelley PM, Keats BJ, Starr A, Leal SM, Cohn E, et al. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J Med Genet. 2003;40:45–50. doi: 10.1136/jmg.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varga R, Avenarius MR, Kelley PM, Keats BJ, Berlin CI, Hood LJ, et al. OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. J Med Genet. 2006;43:576–81. doi: 10.1136/jmg.2005.038612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi BY, Ahmed ZM, Riazuddin S, Bhinder MA, Shahzad M, Husnain T, et al. Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin Genet. 2009;75:237–43. doi: 10.1111/j.1399-0004.2008.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Romanos J, Kimura L, Fávero ML, Izarra FA, de Mello Auricchio MT, Batissoco AC, et al. Novel OTOF mutations in Brazilian patients with auditory neuropathy. J Hum Genet. 2009;54:382–5. doi: 10.1038/jhg.2009.45. [DOI] [PubMed] [Google Scholar]
- 20.Wang DY, Wang YC, Weil D, Zhao YL, Rao SQ, Zong L, et al. Screening mutations of OTOF gene in Chinese patients with auditory neuropathy, including a familial case of temperature-sensitive auditory neuropathy. BMC Med Genet. 2010;11:79. doi: 10.1186/1471-2350-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Fan YY, Wang SJ, Liang PF, Wang JL, Qiu JH, et al. Variants of OTOF and PJVK genes in Chinese patients with auditory neuropathy spectrum disorder. PLoS One. 2011;6:e24000. doi: 10.1371/journal.pone.0024000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu YH, Wu CC, Lu YC, Chen PJ, Lee WY, Liu AY, et al. Mutations in the OTOF gene in Taiwanese patients with auditory neuropathy. Audiol Neurootol. 2010;15:364–74. doi: 10.1159/000293992. [DOI] [PubMed] [Google Scholar]
- 23.Mahdieh N, Shirkavand A, Rabbani B, Tekin M, Akbari B, Akbari MT, et al. Screening of OTOF mutations in Iran: A novel mutation and review. Int J Pediatr Otorhinolaryngol. 2012;76:1610–5. doi: 10.1016/j.ijporl.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 24.Iwasa Y, Nishio SY, Yoshimura H, Kanda Y, Kumakawa K, Abe S, et al. OTOF mutation screening in Japanese severe to profound recessive hearing loss patients. BMC Med Genet. 2013;14:95. doi: 10.1186/1471-2350-14-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishio SY, Usami S. Deafness gene variations in a 1120 nonsyndromic hearing loss cohort: Molecular epidemiology and deafness mutation spectrum of patients in Japan. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):49S–60S. doi: 10.1177/0003489415575059. [DOI] [PubMed] [Google Scholar]
- 26.Bae SH, Baek JI, Lee JD, Song MH, Kwon TJ, Oh SK, et al. Genetic analysis of auditory neuropathy spectrum disorder in the Korean population. Gene. 2013;522:65–9. doi: 10.1016/j.gene.2013.02.057. [DOI] [PubMed] [Google Scholar]
- 27.Yasunaga S, Grati M, Chardenoux S, Smith TN, Friedman TB, Lalwani AK, et al. OTOF encodes multiple long and short isoforms: Genetic evidence that the long ones underlie recessive deafness DFNB9. Am J Hum Genet. 2000;67:591–600. doi: 10.1086/303049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–89. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 29.Rouillon I, Marcolla A, Roux I, Marlin S, Feldmann D, Couderc R, et al. Results of cochlear implantation in two children with mutations in the OTOF gene. Int J Pediatr Otorhinolaryngol. 2006;70:689–96. doi: 10.1016/j.ijporl.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Bashir ZE, Latief N, Belyantseva IA, Iqbal F, Riazuddin SA, Khan SN, et al. Phenotypic variability of CLDN14 mutations causing DFNB29 hearing loss in the Pakistani population. J Hum Genet. 2013;58:102–8. doi: 10.1038/jhg.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, et al. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–22. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]