

Abstract
A substantial proportion of influenza-related childhood deaths are due to infection with influenza B viruses, which co-circulate in the human population as two antigenically distinct lineages defined by the immunodominant receptor binding protein, hemagglutinin. While broadly cross-reactive, protective monoclonal antibodies against the hemagglutinin of influenza B viruses have been described, none targeting the neuraminidase, the second most abundant viral glycoprotein, have been reported. Here, we analyze a panel of five murine anti-neuraminidase monoclonal antibodies which demonstrate broad binding, neuraminidase inhibition, in vitro antibody-dependent cell-mediated cytotoxicity, and in vivo protection against influenza B viruses belonging to both HA lineages and spanning over 70 years of antigenic drift. Electron microscopic analysis of two neuraminidase-antibody complexes shows that the conserved neuraminidase epitopes are located on the head of the molecule and that they are distinct from the enzymatic active site. In the mouse model, one therapeutic dose of antibody 1F2 was more protective than the current standard of treatment, oseltamivir, given twice daily for six days.
Introduction
Influenza B viruses (IBVs) co-circulate in humans as two lineages based on the genetic and antigenic differences of the hemagglutinin (HA) glycoprotein. The two lineages – Yamagata (named after the B/Yamagata/16/88 strain) and Victoria (named after the B/Victoria/2/87 strain) – are thought to have diverged from a common ancestor strain in the 1970s (1, 2). While IBVs are responsible for 20–30% of influenza cases per year on average, IBV is the predominant cause of influenza disease in some years (3–6). Current studies challenge the notion that influenza B cases are clinically milder than those of influenza A, with the finding of no difference between influenza B and influenza A in terms of the length of hospital stay, intensive care unit admission frequency, or rate of death among hospitalized influenza patients (7). Additionally, epidemiologic data suggest IBVs disproportionally afflict children. During the 2010–2011 influenza season in the United States, IBVs accounted for 25% of all influenza infections but caused 38% of influenza-related pediatric deaths, and nearly half of these children had no pre-existing health conditions (8).
Neuraminidase (NA) inhibitors are the only antivirals officially recommended by the Advisory Committee on Immunization Practices (ACIP) for the treatment of influenza virus infection (9). This is particularly problematic for IBV infections since oseltamivir has been shown to be less effective when treating influenza B than when treating influenza A in both pediatric and adult outpatient populations (10–12); furthermore, zanamavir (an alternative NA inhibitor) is not approved for children under the age of seven (9). Given the substantial disease burden attributable to IBV despite the availability of vaccines and antivirals, development of therapeutics, such as the monoclonal antibodies (mAbs) presented here, is crucial.
There have been reports of murine and human mAbs against the IBV HA (13–15), but no broadly cross-reactive, protective mAbs binding the IBV NA have been reported thus far. The potential of the IBV NA globular head domain to harbor highly conserved epitopes has been recognized for some time (16). MAbs against the IBV NA were previously isolated, yet the antibodies were not assessed for in vivo protection, and structures of antibody bound to NA were not solved (16–18). Although the importance of anti-NA immunity in protection from viral infection has been extensively demonstrated (19–28), far less is known about NA epitopes compared to HA epitopes. While NA does not serve as the receptor binding protein, it is critically responsible for freeing nascent virus from host cells and virus in the airway from mucins (29–31); thus, antibodies that bind to the NA and interfere with its activity may confer protection through several mechanisms. Here, for the first time, we extensively characterize a panel of broadly cross-reactive mouse mAbs against the IBV NA in vitro and in vivo. We also present structures of recombinant IBV NA in complex with the fragment antigen-binding (Fab) portion of antibody, allowing for the direct comparison of structural binding footprints with critical binding residues (mapped using escape mutagenesis). Furthermore, such studies highlight the potential benefits of targeting the conserved regions of the NA when designing innovative influenza virus vaccines.
Results
Using hybridoma technology, we identified a panel of five broadly cross-reactive IBV NA-binding mAbs from the spleen of a single female BALB/c mouse that was serially immunized with IBVs from both the Victoria (V) and Yamagata (Y) lineages. All IgG-producing hybridoma clones were initially screened for binding to the ancestral B/Lee/40 strain (purified, whole virus), binding to recombinant, baculovirus-expressed NA (rNA) from B/Yamagata/16/88, and inhibiting neuraminidase activity of B/Wisconsin/1/2010 (Y) virus. Five hybridomas – 1F2 (IgG2a), 1F4 (IgG2a), 3G1 (IgG2a), 4B2 (IgG2a), and 4F11 (IgG2b) – were selected based on robust reactivity and were further tested for binding and neuraminidase inhibition (NI) activity to a wide array of IBVs covering both lineages and spanning 73 years of antigenic drift. The five mAbs were encoded by different light chain/heavy chain combinations with various degrees of mutations as compared to the germline sequences (Supplemental Data Set 1). All five mAbs displayed broad cross-reactivity to purified whole virus and rNA in enzyme-linked immunosorbent assays (ELISAs) and functionally inhibited NA enzymatic activity in enzyme-linked lectin assays (ELLAs) (Figs. 1A,B). Most mAbs displayed minimal binding concentrations to rNA in the sub-micromolar range, with binding to certain rNA substrates reaching minimal binding concentrations in the nanomolar range (between 1.5–150 nM). These findings were corroborated by affinity measurements via bio-layer interferometry where all but one mAb (4B2) reached sub-nanomolar binding affinities (Supplementary Figure 1). Since whole virus was coated based on amount of total protein in the preparation, the minimal binding concentrations in these ELISAs tended to be greater, on average, than those in which rNA was used as a substrate; other differences in binding between whole virus and rNA may be attributed to variation in protein folding and epitope availability. As expected, if a mAb did not bind the NA from a particular strain at a concentration of 10 μg/ml, it did not display NI activity against that strain. However, for some mAb/NA combinations binding was observed at higher antibody concentrations. Binding and NI activity against the ancestral B/Lee/40 strain was weak, but still present, compared to the negative control. By displaying a subsample of IBV NA amino acid sequences as a phylogenetic tree, we observe that the strains used in this study are representative of the amino acid sequence diversity of IBV NAs, strengthening the conclusion that these anti-NA mAbs are broadly cross-reactive (Fig. 1C).
Fig. 1. In vitro binding of IBV anti-NA mAbs.
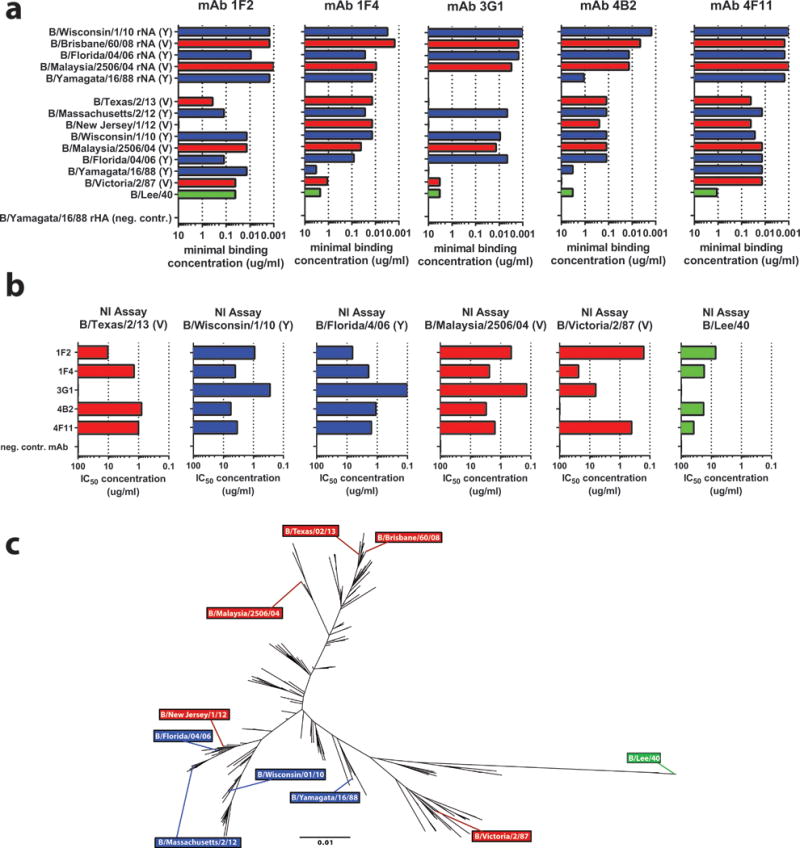
(a) Bar graphs represent the minimal binding concentrations of anti-NA mAbs to either rNA (top, coated at 2 ug/mL), or purified whole virus (bottom, coated at 5 ug/mL) as measured by ELISA. rHA from B/Yamagata/16/88 was used as a negative control substrate (b) MAbs were tested via ELLA to assess NI activity; bar graphs represent IC50 values. Technical duplicates were performed in a and b, with the mean displayed graphically. Victoria lineage strains (with lineage referring to the HA) are shown in red, Yamagata lineage strains in blue, and the ancestral B/Lee strain in green. MAb 8H9 (anti-H6, murine IgG) was used as a negative control. (c) Phylogenetic tree of 280 randomly subsampled IBV strains spanning all years since IBV was first isolated (1940-present). Strains used to assess mAb binding are labeled using the same color scheme as that in panels (a) and (b). The scale bar represents a 1% difference in amino acid sequence.
Protective epitopes on the IBV NA have yet to be described. To map the relevant epitopes on the surface of the NA protein, we carried out electron microscopic analysis of the complex between rNA and the Fab portions of antibodies 1F2 and 4F11. We focused our analysis on 4F11 (because it displayed the widest binding breadth and greatest binding affinities overall), and 1F2 (because it displayed the strongest binding affinity to the ancestral B/Lee/40 strain) Fab fragments. The resultant density maps (resolution ~ 25 Å) were interpreted (Figs 2A,B) using coordinates from the X-ray structures of NA from B/Brisbane/60/2008 (PDB ID: 4CPL) and a Fab that binds an IBV HA (PDB ID: 4FQL). Oblique and top views of the complex (Figs. 2A, B), enable visualization of the locations of the bound antibodies at the periphery of the NA tetramer, indicating that 1F2 has an orientation that is tilted relative to the plane of the NA tetramer, while 4F11 binds in an orientation in which the Fab is in the same plane as that of the tetramer. In Fig. 2C, the top view of antibody binding is shown with respect to the NA enzymatic active site. Detailed inspection of the footprints of the bound Fab molecules shows that the structural footprints of 4F11 and 1F2 are adjacent to each other, but separate (Fig. 2D). The amino acid residues of the binding footprints are highly conserved across all IBVs, consistent with the broad binding profiles of mAbs 4F11 and 1F2 (Fig. 2E). Interestingly, the binding sites of both Fabs appear to not directly overlap the enzymatic active site of NA (Fig. 2C). Our reconstructions thus show that antibodies that inhibit NA activity need not contact the catalytic site directly and instead may function by binding and sterically hindering access of the NA to substrate.
Fig. 2. Negative-stain electron microscopy analysis of NA structures reveals binding footprints for 1F2 and 4F11.
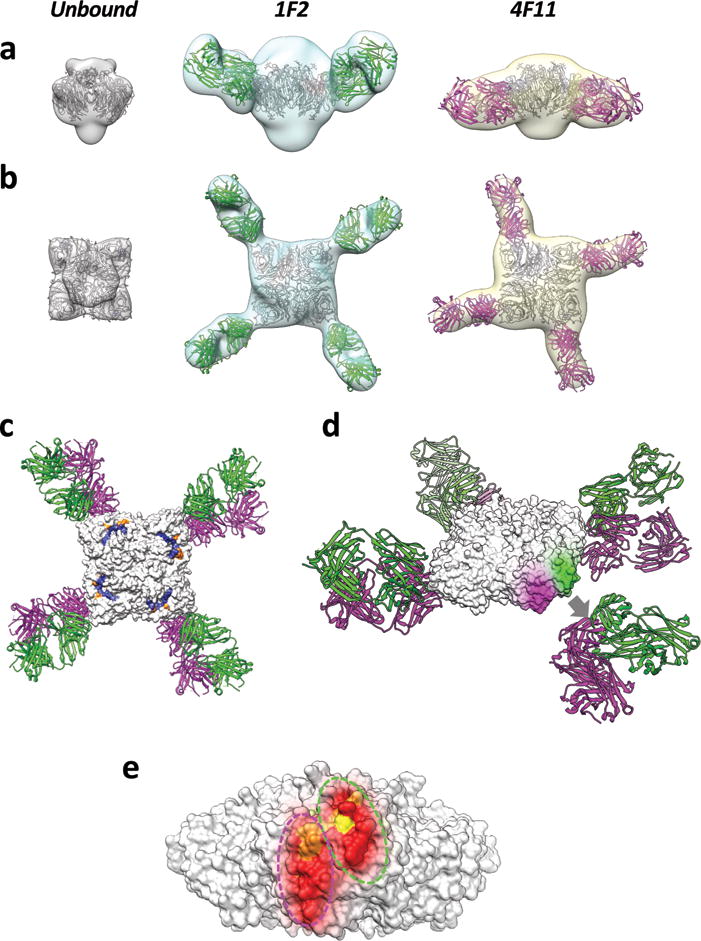
Side (a) and top (b) view isosurface representations of unbound, 1F2, and 4F11 bound NA density maps (grey, cyan, and yellow, respectively) fitted with coordinates for the NA tetramer and Fab (1F2 and 4F11 in green and magenta, respectively) x-ray coordinates. (a) and (b) are superimposed in top (c) and oblique (d) views of the IBV NA-Fab complexes. The top view also highlights the location of active site and framework residues in blue and orange, respectively. In the oblique view, the 1F2 and 4F11 binding footprints (green and magenta, respectively) are highlighted on the surface of the NA tetramer, with the corresponding Fab coordinates displaced away from the highlighted epitope region for purposes of visualization (grey arrow, d). Binding footprints of both Fabs are shown on a single NA tetramer to show their non-overlapping, but spatially adjacent locations (e). Residues within the binding footprint are colored as a heat map based on percent amino acid conservation (red: 98–100% conserved; orange: 75–98% conserved; yellow: 50–75% conserved).
Residues critical for antibody binding were identified through the generation of escape mutant IBVs. Escape mutants generated against 4 out of the 5 mAbs showed drastic loss of binding to mAbs via immunofluorescence staining of infected Madin-Darby canine kidney (MDCK) cells (Supplementary Figures 2A,B). Interestingly, the 4B2 escape mutant retained robust binding to antibody, but is nevertheless a true escape mutant, as demonstrated by its ability to grow to high titers compared to wild type (wt) virus in the presence of antibody (Supplementary Figure 2C). In this instance, mutations in other gene segments may have contributed to the ability of the mutant virus to escape antibody pressure. The critical residues identified in 1F2 and 4F11 escape mutants (E338K and G385R, respectively) are located either within or at the periphery of the binding footprints determined by electron microscopy (Supplementary Figure 2D). The critical residue identified in the 3G1 escape mutant (G346R) maps to a location close to the active site, consistent with the finding that this was the only one of the analyzed mAbs that could inhibit NA enzymatic activity to levels comparable to oseltamivir when using a small molecule substrate (Supplementary Figure 3F). Interestingly, mAb 1F2, which has a footprint adjacent to the 3G1 escape mutation, competed with mAb 3G1 in an ELISA assay (Supplementary Figure 4). This was an asymmetric interaction since mAb 3G1 was unable to block 1F2 binding. The critical residue identified in the 1F4 escape mutant (Q453R) mapped close to the monomer-monomer interface of the NA tetramer. Of relevance, a quaternary, protective epitope spanning two monomers of the NA from pandemic H1N1 (A/California/07/2009) has been previously reported as the target of a human mAb (32). It is conceivable that 1F4 binds to IBV NA in a similar manner. MAb 1F4 asymmetrically competed with mAb 4F11, suggesting that it binds closer to the 4F11 footprint than to the 1F2 footprint (Supplementary Figures 2 and 4). Finally, deep sequencing of the 4B2 escape mutant did indeed reveal a non-synonymous mutation in NA (G344E), although the mutation does not strongly alter the binding of mAb 4B2 as assessed by immunofluorescence (Supplementary Figure 2D). Competition ELISA analysis corroborated these findings with asymmetric competition between 4B2 with both 1F2 and 4F11 (Supplementary Figure 4). The mutation found in the 4B2 escape mutant is located right above both the 1F2 and the 4F11 footprint. All generated escape mutants lost sensitivity to the respective mAbs in an NI assay with the exception of the 4B2 escape mutant, which was still inhibited by mAb 4B2 (Supplementary Figure 6). However, this virus does not seem to escape by abolishing antibody binding to the NA as discussed above (Supplementary Figure 2C).
As previous reports have demonstrated the potential of influenza A virus NA-directed antibodies to protect in vivo (25, 27), we decided to test the in vivo efficacy of our panel of mAbs in a well-established influenza virus challenge model, using female BALB/c mice. In the case of all five mAbs, mice were fully protected from morbidity (Supplementary Figures 5A,D) and mortality (Figs. 3A,D) when administered antibody prophylactically at the highest tested dose (5 mg/kg) and challenged with 5 murine lethal doses (mLD50) of either B/Malaysia/2506/04 (Victoria lineage) or B/Florida/04/06 (Yamagata lineage) virus strains. At lower prophylactic doses of 1 mg/kg and 0.5 mg/kg, the mAbs did not prevent morbidity (Supplementary Figures 5B,C), but nevertheless prevented mortality against B/Malaysia/2506/04 virus, with 1F2 demonstrating 100% protection at both of the lower doses tested (Figs. 3B,C). All five mAbs were 100% protective when administered to mice 24 hours post infection (hpi) with 5mLD50 B/Malaysia/2506/04 virus, and 3 out of 5 were 100% protective when administered 48 hpi (Figs. 3E,F; Supplementary Figures 5E,F).
Fig. 3. In vivo efficacy of IBV anti-NA mAbs.

To test prophylactic efficacy, female BALB/c mice (5 per group) were administered either 5, 1, or 0.5 mg/kg of mAb intraperitoneally 2 h prior to a 5 mLD50 challenge with B/Malaysia/2506/04 virus (a–c) or administered 5 mg/kg of mAb intraperitoneally 2 h prior to a 5 mLD50 challenge with B/Florida/04/06 virus (d). To test therapeutic efficacy, mice were administered 5 mg/kg of each antibody either 24 (e) or 48 (f) h after challenge with 5 mLD50 B/Malaysia/2506/04 virus. Murine mAb 8H9 was used as a negative control in all experiments. Differences in survival were analyzed using a Mantel-Cox log rank test. Statistical significance is indicated where tested as follows: n.s. is p > 0.05, * is p ≤ 0.05, ** is p ≤ 0.01, *** is p ≤ 0.001 and **** is p ≤ 0.0001.
To investigate the in vivo protective breadth of the mAb panel, mice were prophylactically administered with antibodies as in Fig. 3, but were sacrificed on days 3 or 6 for lung titer analysis. When mice were challenged with B/Malaysia/2506/04 virus, lung titers were significantly reduced on day 6 – but not 3 – post infection relative to the negative control group (P ≤ 0.0001), suggesting enhanced viral clearance as a possible mechanism of protection (Fig. 4A). This pattern was also seen when mice were challenged with B/Yamagata/16/88, B/Victoria/2/87, and B/Lee/40 viruses, respectively (Supplementary Figures 3A–C).
Fig. 4. Non-neutralizing IBV anti-NA mAbs reduce viral lung titers in mice, activate ADCC, inhibit activity of a drug-resistant IBV, and demonstrate superior effectiveness to oseltamivir.
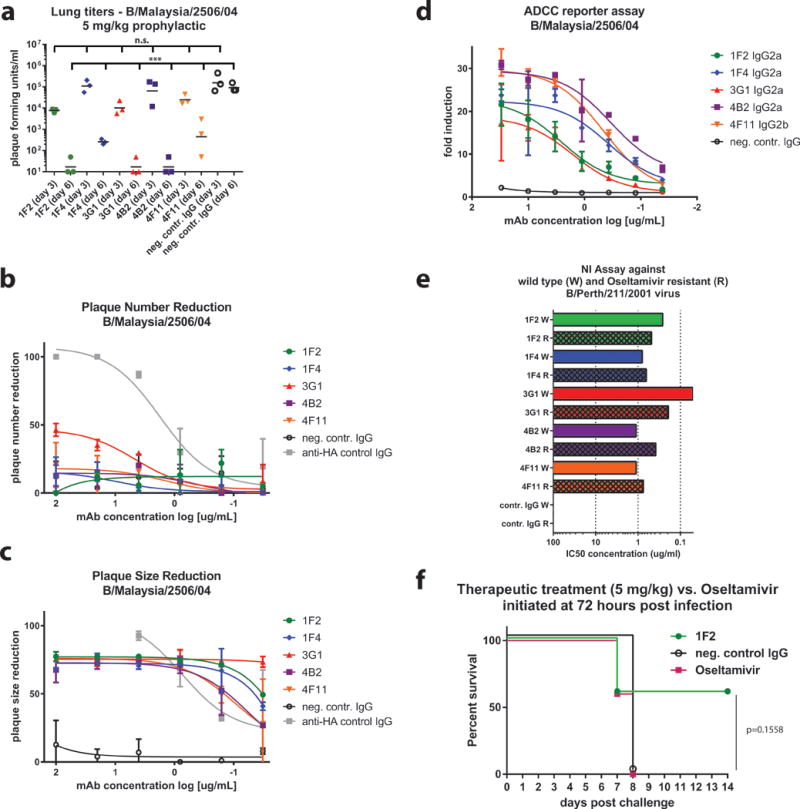
(a) Female BALB/c mice (3 per group) were administered 5 mg/kg antibody prophylactically, challenged with B/Malaysia/2506/04 virus in identical fashion to Figure 3A, and sacrificed on day 3 or 6 post-infection for lung titer analysis. Lung titers in groups treated with anti-NA mAbs are significantly reduced on day 6 post-infection compared to negative control mAb 8H9. When added to both the infectious inoculum and the solid agar overlay in a PRNA, IBV anti-NA mAbs did not reduce plaque number (b) – but reduced plaque size (c), of B/Malaysia/2504/06 virus in a titratable fashion compared to negative control mAb 8H9. The exception was 3G1, which in addition to reducing plaque size, was able to also reduce plaque number up to approximately 50%. A neutralizing murine mAb against the IBV HA was used as a positive control. (d) Anti-NA mAbs incubated with MDCK cells infected with B/Malaysia/2504/06 virus (MOI = 3) were able to engage Fc receptors and activate ADCC in vitro. Fold induction is defined as RLU (induced by antibody)/RLU (no antibody control background). Murine mAb 2G12 (anti-Ebolavirus Gp) is used as a negative control. (e) NI assay against wild type (W) and oseltamivir-resistant (R) B/Perth/211/2001 virus. Bar graphs represent IC50 values. Technical duplicates were performed in (b, c, and e). Technical triplicates were performed in (d). (f) Female BALB/c mice (5 per group) were administered either 5 mg/kg of mAb 1F2 intraperitoneally, 5 mg/kg negative control mAb 8H9 intraperitoneally, or placed on a twice daily, 20 mg/kg, 6 day-long regimen of oseltamivir delivered via oral gavage and initiated at 72 hpi. Percent survival is shown. Lung virus titers were compared using a one-way ANOVA corrected for multiple comparisons. Differences in survival were analyzed using a Mantel-Cox log rank test. Statistical significance is indicated where tested as follows: n.s. is p > 0.05, * is p ≤ 0.05, ** is p ≤ 0.01, *** is p ≤ 0.001 and **** is p ≤ 0.0001. Error bars shown in B, C and D represent the standard deviation.
NA antibodies, which are typically considered non-neutralizing, have been shown to decrease plaque size – but not plaque number – in plaque reduction neutralization assays (PRNAs), an in vitro phenomenon that stems from their ability to inhibit viral egress (25, 27, 29, 32–34). Our anti-NA mAbs displayed this phenotype in a PRNA when tested against B/Malaysia/2506/04 virus (Figs. 4B,C). Additionally, recent findings have shown that Fc-Fcϒ receptor interactions are necessary for broadly reactive HA head-, HA stalk-, and NA-directed antibodies to mediate protection in vivo (35–37). In light of the increasingly recognized implications of Fc receptor-mediated effector functions, such as antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), we decided to test the ability of our IBV NA-directed mAbs to engage with and activate Fc receptors in vitro. Using a commercially available ADCC Reporter Assay Core Kit (Promega), which utilizes Jurkat cells engineered to express firefly luciferase upon Fc receptor activation, we confirmed that all five mAbs displayed ADCC activity when incubated with MDCK cells infected with B/Malaysia/2506/04 or B/Florida/04/06 virus, respectively (Fig. 4D; Supplementary Figures 3D,E). Four of the five mAbs displayed ADCC activity when incubated with cells infected with B/Yamagata/16/88 virus, with the exception of 3G1, which is expected because 3G1 does not bind the NA of B/Yamagata/16/88.
As mentioned, 3G1 was the only mAb that possessed a critical binding residue located directly adjacent to the enzymatic active site (Supplementary Figure 2D). Yet, all mAbs displayed robust NI activity by ELLA, an assay that utilizes fetuin as a substrate (Fig. 1C). The ability of antibodies that bind epitopes outside of the active site to inhibit NA enzymatic activity by ELLA has been documented in the case of N1- and N2-binding mAbs (25, 38–40). Fetuin, a glycoprotein (molecular weight = 48.4 kDa), is immobilized to the 96-well plate during the ELLA coating process, so it is conceivable that antibodies that bind distal to the active site may inhibit access of the NA to fetuin by steric hindrance. To understand if this may account for the mechanism of inhibition in some of the other antibodies we studied, we assessed NI activity in an NA-Star assay, which uses a small, soluble chemiluminescent substrate (molecular weight = 684.5 Da). All five mAbs inhibited NA activity in the NA-Star assay to some extent, but only 3G1 was able to achieve 100% inhibition with an IC50 comparable to that of oseltamivir, a small molecule NA inhibitor (molecular weight = 312.4 Da) (Supplementary Figure 3F). Such data additionally supports the hypothesis that the epitope of 3G1 overlaps – or is in immediate proximity to – the enzymatic active site. In separate studies, we tested the role of steric hindrance by removal of the Fc region of the antibodies. Inhibition studies showed that 4F11 Fab, but not 1F2 Fab lost NI activity in the NA-Star assay, suggesting steric hindrance plays a role in the NI of this antibody.
Importantly, influenza viruses may develop resistance to oseltamivir and other NA inhibitors. As most of the mAbs from this study likely bind distal to the NA active site, it was hypothesized they could still effectively inhibit the enzymatic activity of drug-resistant IBV strains. Indeed, when tested against an IBV strain (B/Perth/211/2001) containing a known oseltamivir-resistance mutation (D197E) all five mAbs – even 3G1 – inhibited NA activity (Fig. 4E).
Finally, we compared treatment with mAb to treatment with oseltamivir in the mouse model. In the BALB/c mouse challenge model, a 20 mg/kg, twice-daily, 6-day long oral regimen of oseltamivir initiated 48 hpi with 5mLD50 B/Malaysia/2506/04 resulted in 100% survival (Supplementary Figure 7). However, timing of the onset of antiviral therapy with NA inhibitors is crucial, as the greatest reduction in morbidity has been observed when treatment is initiated 48 hours prior to symptom onset in both pediatric and adult cases of uncomplicated influenza (9, 41, 42). We thus chose to compare the therapeutic efficacy of mAb 1F2 to that of oseltamivir as late as 72 hpi. When administered at this time-point, a single bolus of 1F2 (5 mg/kg) delivered IP performed superiorly to the twice-daily regimen of oseltamivir, initiated at the same time point (72 hpi) and continued for six days. While not statistically significant, sixty percent of mice treated with mAb 1F2 survived, compared to 0% in the oseltamivir treatment group (Fig. 4G). Corresponding weight loss curves are shown in Supplementary Figure 3G.
Discussion
Here, for the first time, we elucidated broadly reactive epitopes located on the influenza B virus NA, using both structural and escape mutant mapping of murine monoclonal antibodies; the antibodies we generated bound to both Victoria and Yamagata lineage IBVs spanning 73 years of antigenic drift. While NA antibodies may inhibit enzymatic activity by binding, they also possess Fc regions capable of binding to – and activating – effector cells; indeed, we demonstrated the ability of our mAbs to robustly activate effector cells in vitro.
The critical importance of Fc effector functions – such as ADCP and ADCC – to the mechanism of protection of non-neutralizing antibodies against the influenza virus is becoming increasingly appreciated. It has been shown using Fc receptor knockout mice and mAbs deficient in Fc receptor binding (DA265-mutant Fc), for instance, that the protection mediated by broadly reactive HA-stalk antibodies in vivo is dependent on Fc-Fc receptor interactions (35, 36). Furthermore, Leon et al. demonstrated that the Fc activation mediated by HA-directed mAbs is epitope-dependent (43). Whether or not the NA mAbs described in this study depend on Fc receptor activation to mediate protection or whether or not the ADCC activation by NA-binding mAbs is epitope-dependent remains unknown, but may be elucidated using similar approaches. Of note, a human NA antibody activated ADCC to a lesser extent than an HA-stalk antibody in A549 cells infected with H3N2 influenza virus. However, when titrated into the assay, the NA-binding mAb was able to increase the level of ADCC achieved by the stalk-binding mAb in an additive fashion (44). Such a finding implies that NA antibodies may be able to cooperatively enhance the levels of ADCC achieved by HA antibodies during natural infection, most likely by providing increased contact points for Fc-Fc receptor engagement on the surface of virus or infected cells.
Finally, we demonstrated that mAb 1F2 exhibited superior efficacy to the standard of care, oseltamivir treatment, when administered 72 hpi in a mouse challenge model. Conceivably, the combined ability of an anti-NA antibody to interfere with NA enzymatic activity while engaging in effector functions that allow for enhanced, immune-mediated viral clearance may explain why mAb treatment is able to outperform treatment with NA inhibitor in vivo.
The mAbs in this study possess the potential to be developed into therapeutics for high-risk patient populations or against drug-resistant strains of IBV and highlight the benefits of targeting broadly protective NA epitopes in innovative influenza virus vaccine formulations. As the HA and NA drift independently, vaccines that elicit robust NA titers may prove especially useful in influenza seasons in which an HA drift event occurs.
Materials and Methods
Cells, viruses, and proteins
As described previously (27), Madin-Darby canine kidney (MDCK) cells (originated from MDCK (NBL-2); ATCC CCL-34) were grown in complete Dulbecco’s modified Eagle medium (DMEM; Life Technologies) supplemented with antibiotics (100 U/ml penicillin- 100 μg/ml streptomycin [Pen-Strep]; Gibco), 10% fetal bovine serum (FBS; HyClone), and 10 ml of 1 M HEPES (Life Technologies). Sf9 insect cells (originated from ATCC CRL-1711) were grown in TNM-FH insect medium (Gemini Bioproducts) supplemented with antibiotics (Pen-Strep) and 10% FBS, and High Five cells (BTI-TN-5B1-4 subclone; Vienna Institute of Biotechnology) (45) were grown in serum-free SFX-insect cell medium (HyClone). SP2/0 mouse myeloma cells (originated from SP2/0-Ag14; ATCC CRL-1581) were passaged and maintained in complete DMEM supplemented with antibiotics (Pen-Step) prior to fusion with primary mouse splenocytes. Monoclonal, immortalized B cells (obtained from the hybridoma fusion) were initially grown in Clonacell-HY Medium E (Stemcell Technologies) and gradually switched to serum-free hybridoma medium (Hybridoma-SFM; Life Technologies) for high-volume production. All cell lines used tested negative for mycoplasma contamination using the MycoAlert™ Mycoplasma Detection Kit (Lonza). No cell lines were further authenticated in this study.
The influenza viruses B/Lee/40, B/Victoria/2/87, B/Yamagata/16/88 B/Florida/04/06, B/Perth/211/2001 198D, B/Perth/211/2011 198E, B/Memphis/1B/2003, B/Malaysia/2506/04, B/Wisconsin/1/10, B/New Jersey/1/12, B/Massachusetts/2/12, B/Texas/2/13, and escape mutant viruses were grown in 8- to 10-day-old embryonated chicken eggs, and titers were determined on MDCK cells in the presence of TPCK (tosyl phenylalanyl chloromethyl ketone)-treated trypsin. To create purified virus preparations, allantoic fluid containing virus was harvested and subjected to low-speed centrifugation (at 3,000 × g for 30 min at 4°C) to remove cellular debris. Viruses were pelleted through a 30% sucrose cushion (30% sucrose in NTE buffer [100 mM NaCl, 10 mM Tris-HCl, 1 mM EDTA]; pH 7.4) by ultracentrifugation (Beckman L7-65 ultracentrifuge with SW-28 rotor at 25,000 rpm for 2 h). Once all of the supernatant was aspirated, virus pellets were resuspended in phosphate-buffered saline (PBS). B/Perth/211/2001 198D, B/Perth/211/2011 198E were kindly provided by Drs. Aeron Hurt and Elena Govorkova and are part of the ‘Panel of Influenza A and B Viruses for Assessment of Neuraminidase Inhibitor Susceptibility’ provided by the International Society for Influenza and other Respiratory Virus Diseases (ISIRV - https://isirv.org/site/images/stories/avg_documents/Resistance/avg%20leaflet%20nov12.pdf).
The recombinant proteins used – B/Yamagata/16/88, B/Malaysia/2506/04, B/Florida/04/06, B/Brisbane/60/08, B/Wisconsin/1/10 NAs – were expressed in High Five cells and purified from cell culture supernatants as described previously (46, 47). In brief, cultures were infected with recombinant baculoviruses at a multiplicity of infection of 10. Supernatants were then harvested by low-speed centrifugation at 72 h post infection and purified by using Ni-nitrilotriacetic acid resin (Qiagen) according to a published protocol (47).
Generation and screening of mAbs
Similarly to the protocol described previously (27, 48), six- to eight-week-old female BALB/c mice were first immunized with 40 ug of DNA (in 40 uL distilled H2O) encoding the open reading frame of the HA from the parental strain B/Yamagata/16/88 in the PCAGGS vector (DNA was delivered via intramuscular electroporation in the medial thigh using a TriGrid Delviery System [Ichor Medical Systems]) followed 4 weeks later by a second immunization with the HA from the parental strain B/Victoria/2/87 (this vaccination regimen was initially designed to elicit both HA- and NA-directed mAbs). Four weeks after the second immunization, mice were intranasally infected with a sublethal dose (104 plaque-forming units [PFU] in 50 uL PBS) of B/Florida/4/06, followed 6 weeks later by intranasal infection with a sublethal dose (104 PFU in 50 uL PBS) of B/Malaysia/2506/04. Approximately 6 weeks after the second infection, one mouse was boosted with a unilateral, intraperitoneal injection of 100 ug of formalin-inactivated, purified B/Wisconsin/1/2010 virus adjuvanted with 10 ug of poly(I·C).
Three days post-boost, one mouse was sacrificed, and its spleen was sterilely removed. The spleen was flushed forcefully with serum-free DMEM (with antibiotics [Pen-Strep]) using a 10-ml syringe with a 20-gauge needle, followed by repeated mashing with flat-ending forceps. Splenocytes and SP2/0 myeloma cells (in log phase) were combined in a 5:1 ratio, and cell fusion was mediated via slow, drop-wise addition of 1 ml of polyethylene glycol (molecular weight, 4,000 g/mol). The splenocyte/SP2 mixture was resuspended in 25 ml of complete DMEM (supplemented with antibiotics [Pen-Strep], FBS, and HEPES) and left to incubate for 24 h. After this incubation, the cells were spun down, resuspended in 10 ml of complete DMEM, mixed with a proprietary bottle of 90 ml of semisolid Clonacell-HY Medium D (Stemcell Technologies), and dispensed onto tissue culture dishes (10 ml each) using a 10-ml syringe with a 15-gauge Luer Stub adapter (Becton Dickinson). Individual colonies were picked 10 days later and transferred into 96-well plates containing Clonacell-HY Medium E. Five days after transfer to 96-well plates, hybridoma supernatants were screened by ELISA for binding reactivity to B/Lee/40 (purified, whole virus), B/Yamagata/16/88 (rNA), and NI activity to B/Wisconsin/1/2010 virus. Positive clones were isotyped using a Pierce rapid antibody isotyping kit (Life Technologies); only the mAbs isotyped to the IgG heavy-chain subclasses were selected for further expansion and purification. All animal procedures were performed in accordance with the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee (IACUC).
Expansion and purification of mAbs
Mabs were produced and purified as described previously (27). Fab fragments for mAbs 1F2 and 4F11 were generated by papain digestion by Southern Biotech (Birmingham, AL).
ELISA
ELISAs were performed as described previously (27). An endpoint titer was defined as the final concentration at which the antibody signal remained greater than 3 standard deviations above the average of the blank wells, as described in (49).
Enzyme-linked lectin assay
Enzyme-linked lectin assays (ELLAs), used to determine NA inhibition (NI) activity, were performed as described in detail in previous reports (26, 27).
Sequencing variable sequences of B NA antibodies
5×106 cells from hybridoma cell lines were used to purify the total mRNA using a Direct-zol RNA Miniprep kit (Zymo Research) according to the manufacturer’s instructions. Purified RNA was reverse transcribed using SuperScript™ III Reverse Transcriptase (Thermo Fisher) and the cDNA was stabilized in a Terminal Deoxynucleotidyl Transferase (TdT) tailing reaction (Invitrogen). The product of TdT tailing was used as a template in a PCR reaction using high fidelity Taq Platinum polymerase (Invitrogen) with a variable region specific 5′ consensus anchor primer (5′ GGCCACGCGTCGACTAGTACGGGIIGGGIIGGGIIG 3′) and a constant region specific reverse primer (5′ CCTTGACCAGGCATCCTAGAGTC 3′ or 5′ GGAGGTGTGCACACTGCTGGACAG 3′, for IgG2a and IgG2b respectively). PCR reaction products were sequenced and the obtained sequences were entered into the IMGT/V-QUEST database tool (http://www.imgt.org/IMGT_vquest/share/textes/) to determine complete variable region sequence as well as individual germline genes. In cases where the quality of sequencing read was not sufficient, the PCR product was first cloned into Strataclone vectors (Agilent Technologies) which were then amplified in StrataClone competent cells, purified using QIAPrep Spin Miniprep Kit (Qiagen) and then sequenced before entering them into the IMGT/V-Quest.
Antibody/antigen biotinylation
Antibodies and rNA were biotinylated using an EZ-Link NHS-PEG4-Biotin kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
KD determination using biolayer interferometry
Antibody dissociation constants (KD values) were determined by biolayer interferometry using an Octet Red96 instrument (ForteBio, Inc.), as described previously (37). Biosensors were loaded with rNA from B/Malaysia/2506/04.
Phylogenetic tree generation
A subsample of 280 sequences were chosen to form the phylogenic tree. For years in which 10 or less sequences were available, all sequences were used; for years with greater than 10 sequences, a random selection of 10 sequences was chosen to minimize the bias of more recent isolates. Sequences were aligned using MUSCLE and manually edited using the MEGA 6.0 software when applicable. Phylogenetic tree was assembled using Clustal Omega web server with a Neighbor-joining clustering method and default setting. The tree was cleaned and edited using FigTree.
Negative stain electron microscopy
Recombinant NA and Fabs were diluted with buffer (5mM HEPES, 150 mM NaCl pH 7.3) to approximately 0.02 mg/mL and 0.04 mg/mL respectively. To prepare Fab bound samples, equal volumes of NA and Fabs 1F2 or 4F11 were mixed and incubated for 5–10 minutes. The samples were adsorbed to plasma cleaned (Solarus Model 950 cleaner, Gatan Inc., Pleasanton, CA) EM grids coated with continuous carbon film that were subsequently washed with buffer and stained with 0.75% uranyl formate. Images were collected using EPU software (FEI Company, Hillsboro, OR) on a Tecnai T12 electron microscope (FEI Company) fitted with a 4K CCD camera (Gatan Inc.) at an effective pixel size of 0.18 nm in the specimen plane. The software package RELION 1.4 (50) was used to obtain 3D reconstructions. The maps for unbound NA, and for the complexes with 1F2, and 4F11 were constructed using 47,592, 4,326, and 13,665 particles, respectively and visualized using UCSF Chimera software (51).
Modeling Fab binding footprints
NA and Fab (Influenza B HA Fab CR8033) X-ray coordinates (PDB IDs: 4CPL and 4FQL, respectively) were fitted to density maps using UCSF Chimera software. To highlight the binding footprints of the Fabs, the regions of NA that most closely interacted with each Fab were identified by manual inspection.
Multiple sequence alignment
Sequences were obtained from the Global Initiative on Sharing All Influenza Data (GISAID, http://platform.gisaid.org/) with any laboratory-associated strains or truncated sequences removed from analysis. There were a total of 2409 sequences in the final file that was used for alignments. Sequence alignments were performed using MEGA 6.0 software (MUSCLE alignment).
Percent conservation calculation
Escape residues were isolated from the whole NA protein sequence alignment using sequence editing tools in MEGA 6.06. A subsample of 944 sequences was used for calculations. For years in which 50 or less sequences were available, all sequences were used; for years with greater than 50 sequences, a random selection of 50 sequences was chosen to minimize the bias of more recent isolates. To give the percentage of the number of sequences that contained a specific amino acid at each escape reside location, the amino acid phenotypes at the site were sorted and then divided by the total number of sequences.
Generation of IBV anti-NA mAb escape mutant viruses
MAb escape mutant variants of B/Malaysia/2506/04 virus were generated based on the methods described in (25). MAb (250 ug) and virus (106 PFU) were combined (total volume, 800 ul), incubated for 1 h at RT, split evenly into thirds and injected into three 8-day old embryonated chicken eggs. After incubating for 72 hours at 33°C, allantoic fluid was harvested and plaqued in the presence of mAb (100 ug/ml in both inoculum and overlay). After incubating for 72 hours at 33°C, plaque assays were inspected for escape variants (as evidenced by large plaque size). Large plaques were picked and inoculated into 10-day old embryonated chicken eggs for amplification. All escape mutant variants, excluding that of mAb 4B2, were generated in this way. In the case of 4B2, B/Malaysia/2506/04 virus was serially passaged on an MDCK cell monolayer in the presence of increasing amounts of mAb, with a starting concentration of 0.25×IC50 (as calculated from the NI assay against B/Malaysia/2506/04 virus). Initially, MDCK cells in 1 well of a 6-well plate were infected with B/Malaysia/2506/04 virus at an MOI of 0.1 in the presence of 0.5×IC50 of mAb. After incubating for 72 hours at 33°C, 10 ul of supernatant was collected and used to directly inoculate a fresh monolayer of MDCK cells in the presence of increased mAb concentration. This process was repeated for 15 passages, until the final antibody concentration was ~1 mg/ml. Throughout serial passaging, successful infection was confirmed by the presence of cytopathic effect (CPE) or – if CPE was not clear – positive staining with polyclonal anti-IBV mouse serum (detailed immunostaining procedure is described below in the “evaluation of the prophylactic and therapeutic efficacy in mice” section). Both CPE and positive immunostaining were present in the last passage. Virus was additionally passaged in the presence of an irrelevant mouse mAb (3C12, anti-N8, IgG, characterized previously in (27)) throughout all experiments to control for mutational variants obtained from passaging alone. Viruses were plaque purified once serial passaging was completed to create monoclonal stocks for deep sequencing and growth curve analysis.
Deep sequencing of escape mutant variants
RNA from the escape mutant variants was obtained using the Direct-zol RNA kit (Zymo Research). The samples were processed using the Illumina TruSeq RNA Sample Preparation Kit according to the manufacturers instructions and sequenced using a MiSeq Illumina instrument. Reads were consolidated and aligned to B/Malaysia/2506/04 using Bowtie2. The assembled genomes and minority variants were visualized with the Integrative Genomic Viewer (IGV) (Broad Institute).
Immunofluorescence
To screen for escape mutants, MDCK cells were plated in 96-well, sterile, flat-bottom tissue culture plates (Sigma) and subsequently infected with either wt B/Malaysia/2506/04 or mutant viruses at an MOI of 10. After incubating for 18 hours at 33°C in MEM lacking TPCK-trypsin (to limit viral growth to 1 infectious cycle), media was removed, and cells were fixed with 3.7% formaldehyde for at least 1 h at 4°C. Next, the formaldehyde was discarded and the cell monolayer was blocked with 3% milk in PBS for at least 1 h. For the primary antibody step, plates were incubated with either the respective IBV anti-NA mAb (30 ug/ml), a positive infection control (a polyclonal cocktail of purified mouse mAb IgGs against the IBV HA [1:1000 dilution], or irrelevant negative control mouse mAb 8H9 in PBS, 1% milk (100 ul/well) for 1 h at RT, while shaking. Next, plates were washed 3 times with PBS and incubated with Alexa Fluor® 488 goat anti-mouse secondary antibody in PBS, 1% milk (100 ul/well) for 1 h at RT in the dark. Finally, after washing 3 additional times, cells were visualized via fluorescent microscopy.
Growth curve analysis
To compare viral fitness in the presence of mAb, growth curves were performed in MDCK cells. Cells were plated as a confluent monolayer in 12-well tissue culture plates (Sigma) and infected with virus at an MOI of 0.01 (final volume of 1 ml/well). The experiment was performed in triplicate for each time point and each antibody condition. MAb was added to infection media at a concentration of 10 ug/ml. Cells were incubated at 33°C, and supernatant was collected at 12, 24, 48, or 72 hpi. Collected supernatant was clarified by centrifugation (at a relative centrifugal force of 3,000 for 10 min at 4°C) and immediately stored at −80°C. For simplicity, only titers at 72 hpi are reported here. Viral titers were assessed via hemagglutination assays, as described previously (52–54).
3D mapping of escape mutations
Escape mutations were represented on a 3D structure of the NA of B/Brisbane/60/2008 (PDB ID: 4CPL) using PyMOL™ version 1.8.4.2 (Schrodinger, LLC).
Competition ELISAs
Microtiter 96-well plates (Immulon 4 HBX, Thermo Fisher Scientific) were coated with 2 μg/mL (50 μL/well) of rNA from B/Florida/04/2006 diluted in coating solution (KPL). The plates were incubated at 4°C overnight. The next day, plates were washed three times with PBS containing 0.1% Tween-20 (PBS-T) and then incubated for 1 h at 20°C with 225 μL/well of blocking solution (PBS-T with 3% goat serum (Life Technologies, Inc.) and 0.5% milk powder)). After the blocking solution was removed, the competing monoclonal antibodies were diluted in blocking solution at 20 ug/ml and transferred to the plate. Blocking solution with no antibodies was used as a no-competition control. The final volume in each well after dilution was 100 μL. After a 2 h incubation period at 20°C, the plates were washed three times with PBS-T. Following the wash, the biotinylated target antibodies were diluted in 1:3 steps with a starting concentration of 30 ug/ml in blocking solution and then incubated for 2 h at 20°C. Subsequently, the plates were washed three times with PBS-T and then incubated with Streptavidin conjugated to HRP (1:3000, 50 μL/well, Thermo Fisher Scientific). After 1 h at 20°C the plates were washed four times with PBS-T and then developed with SigmaFast o-phenylenediamine dichloride (OPD, 100 μl/well) (Sigma) for 10 min. The reaction was stopped with the addition of 3 M hydrochloric acid (50μl/well). The plates were immediately read using a Synergy H1 hybrid multimode microplate reader (BioTek) at an optical density of 490 nm.
Animals
All animal procedures were performed in accordance with the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee (IACUC). Female mice (species: Mus musculus; strain: BALB/c) aged 4–6 weeks (The Jackson Laboratory) were used for all studies. Researchers performing animal experiments were not blinded. Mice were randomly assigned to infection and treatment groups without the use of a specific algorithm. A sample size of 3 mice per group was chosen for lung titer analyses and 5 mice per group for challenge studies according to the general practices in the influenza field. Sample sizes were not determined using power analyses.
Evaluation of the prophylactic and therapeutic efficacy in mice
Prophylactic and therapeutic protection studies and quantification of viral lung titers in mice were performed as described previously in (26, 27).
PRNAs
Plaque reduction neutralization assays (PRNAs) were performed according to the protocols described by Tan et al. (57) and Wohlbold et al. (27), with some modifications. In duplicate, six 5-fold dilutions of mAbs (highest concentration: 100 ug/ml; lowest concentration: 3.2 × 10−1 ug/ml) were prepared in serum free, 1 × MEM and each dilution was incubated with 100 PFU of virus for 1 h 30 min at RT, on a shaker. The inocula were then plaqued on MDCK cell monolayers in either 12 (B/Victoria/2/87, B/Yamagata/16/88, B/Victoria/2/87 viruses) or 6-well (B/Malaysia/2506/04 virus) plates, similar to the protocol used to plaque lung titers (described earlier). After 3 days of incubation at 33°C, the cells were fixed with 3.7% formaldehyde for at least 1 h at 4°C and blocked with 3% milk in PBS for at least 1 h. For the primary antibody step, plates were then incubated with a cocktail of broadly-reactive, anti-IBV HA mouse mAbs (1:5000 dilution in PBS, 1% milk) for 1 h at RT, while shaking. Next, plates were washed with PBS and incubated with an anti-mouse secondary antibody conjugated to HRP (Sigma) for 30 min at 37°C. Finally, plates were washed and stained with True-Blue (KPL) so plaques could be visualized and counted. The plaques were counted in each mAb dilution, and the percent inhibition for each mAb at each dilution was calculated based on a no-antibody control. An irrelevant murine IgG (8H9) was used as a negative control. The data were analyzed by using Prism software (GraphPad). We also assessed the decrease in plaque size upon incubation with antibody. To analyze average plaque diameter, 10 plaques were randomly selected in each well using a technique described previously (27).
Mouse ADCC assays
Assessment of the ability of mAbs to trigger ADCC was performed using a commercial ADCC kit (Promega) and according to the manufacturer’s instructions. Briefly, MDCK cells were seeded into white, flat bottom, 96-well cell culture plates (Costar) at a density of 3.0 × 104 cells/well and incubated overnight at 37°C and 5% CO2. The following day, the cells were infected with B/Yamagata/16/88, B/Malaysia/2506/04 or B/Florida/04/06 virus at a multiplicity of infection of 3 and incubated at 33°C, 5 % CO2. Sixteen hours later, cell medium was exchanged for 3-fold serial dilutions of antibody in assay buffer, starting at 30 ug/mL. Effector cells were added and after another 6 hours of incubation (37 °C, 5 % CO2), Bio-Glo™ luminescence reagent and substrate (Promega) were added and luminescence was measured on a Synergy H1 microplate reader (BioTek). Data were analyzed using Prism 6 software (GraphPad).
NA-Star Assay
The NA-Star Influenza Neuraminidase Inhibitor Resistance Detection Kit (Applied Biosystems) was used to assess mAb (or Fab) inhibition of the neuraminidase’s ability to cleave a small, soluble, chemiluminescent substrate (sodium (2-chloro-5-(4-methoxyspiro{1,2-dioxetane-3,2′-(5-chloro)tricyclo[3.3.1.13,7]decan}-4-yl-phenyl 5-acetamido-3,5-dideoxy-α-D-glycero-D-galacto-2-nonulopyranoside)onate). To measure antibody-mediated inhibition, mAb (or Fab) was diluted 1:3 in NA-Star Assay Buffer (starting concentration, 100 ug/ml; final volume per well, 25 ul) in white, flat bottom, 96-well cell culture plates. Twenty-five ul of virus at the determined 3×EC50 concentration was added to each well and plates were shaken and incubated for 30 min at 37°C. The remainder of the assay was performed identically to the method described above beginning with the addition of NA-Star Substrate. Data points were expressed as percent inhibition of maximal NA enzymatic activity, which was determined by the activity of virus without the addition of antibody. Curves were plotted using Prism software (GraphPad).
Oseltamivir treatment studies
Oseltamivir phosphate (Fischer Scientific, United States Pharmacopeia [USP] Reference Standard) was administered to mice via oral gavage twice daily (every 12 hours) at a dose of 20 mg/kg (in a total volume of 100 ul water for injection [Gibco]) for 6 days following treatment commencement. This dosing (40 mg/kg/day) was based off of recently published dosing regimens used in the BALB/c mouse model (55, 56) as well as the standard dosing recommended for therapeutic treatment in adult humans by the Advisory Committee on Immunization Practices (ACIP) (9).
Statistical analysis
Statistical analysis was performed using Prism software (GraphPad). Lung virus titers were compared using a one-way ANOVA corrected for multiple comparisons. Differences in survival were analyzed using a Mantel-Cox log rank test. Statistical significance is indicated where tested as follows: n.s. is p > 0.05, * is p ≤ 0.05, ** is p ≤ 0.01, *** is p ≤ 0.001 and **** is p ≤ 0.0001.
Supplementary Material
Acknowledgments
We thank Irina Margine for pilot studies, Madhusudan Rajendran Marilyne Panis and Raffael Nachbagauer for their assistance in the deep sequencing analysis of IBV mutants and competition ELISAs, Ariana Hirsh for producing recombinant neuraminidase proteins and Dr. Nicole Bouvier for her instructions in oral gavaging. This work was funded by NIAID grant R01 AI117287 (FK) and U19 AI109946 (PP, FK).
Footnotes
Author Contributions: T.J.W., K.A.P., S.S., and F.K. designed experiments and wrote the manuscript. T.J.W., K.A.P., V.C., and P.M. performed experiments. J.T. and F.A. assisted with experiments. F.A. and G.S.T. generated reagents. T.J.W., K.A.P., V.C., V.F., J.T., E.K., B.R.T., P.P., S.S., and F.K. analyzed and interpreted data.
Competing Financial Interests: The Icahn School of Medicine at Mount Sinai has filed patents regarding use of the described mAbs as therapeutics (application # 62/483,262). TJW, PP and FK are named as inventors on the application.
Data availability: The data that support the findings of this study are contained in the manuscript or available from the corresponding author upon request. The GEO reference for the RNA-Seq data pertaining to the viruses used in this paper is GSE96091 (S. Fig. 2). EM maps have been deposited in the EMDataBank under the following accession numbers: negative stain of influenza B virus recombinant neuraminidase bound to 1F2 Fab (EMD-8768), negative stain of influenza B virus recombinant neuraminidase bound to 4F11 Fab (EMD-8769), and negative stain of influenza B virus recombinant neuraminidase (EMD-8770).
References and Notes
- 1.Shaw M, Palese P. Orthomyxoviridae: The Viruses and Their Replication. Fields Virol. 2013;2:1648–1689. [Google Scholar]
- 2.Chen JM, Guo YJ, Wu KY, Guo JF, Wang M, Dong J, Zhang Y, Li Z, Shu YL. Exploration of the emergence of the Victoria lineage of influenza B virus. Arch Virol. 2007;152:415–422. doi: 10.1007/s00705-006-0852-6. [DOI] [PubMed] [Google Scholar]
- 3.Molinari NA, Ortega-Sanchez IR, Messonnier ML, Thompson WW, Wortley PM, Weintraub E, Bridges CB. The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine. 2007;25:5086–5096. doi: 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 4.Dijkstra F, Donker GA, Wilbrink B, Van Gageldonk-Lafeber AB, Van Der Sande MAB. Long time trends in influenza-like illness and associated determinants in The Netherlands. Epidemiol Infect. 2009;137:473–9. doi: 10.1017/S095026880800126X. [DOI] [PubMed] [Google Scholar]
- 5.Heikkinen T, Ikonen N, Ziegler T. Impact of Influenza B Lineage-level Mismatch between Trivalent Seasonal Influenza Vaccines and Circulating Viruses, 1999–2012. Clin Infect Dis. 2014:1–6. doi: 10.1093/cid/ciu664. [DOI] [PubMed] [Google Scholar]
- 6.Brottet E, Vandroux D, Gauzere BA, Antok E, Jafar-Bandjee MC, Michault A, Filleul L. Influenza season in Reunion dominated by infuenza B virus circulation associated with numerous cases of severe disease, France, 2014. Eurosurveillance. 2014;19:1–4. doi: 10.2807/1560-7917.es2014.19.39.20916. [DOI] [PubMed] [Google Scholar]
- 7.Su S, Chaves SS, Perez A, D’Mello T, Kirley PD, Yousey-Hindes K, Farley MM, Harris M, Sharangpani R, Lynfield R, Morin C, Hancock EB, Zansky S, Hollick GE, Fowler B, McDonald-Hamm C, Thomas A, Horan V, Lou Lindegren M, Schaffner W, Price A, Bandyopadhyay A, Fry AM. Comparing clinical characteristics between hospitalized adults with laboratory-confirmed influenza A and B virus infection. Clin Infect Dis. 2014;59:252–5. doi: 10.1093/cid/ciu269. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control. Influenza-Associated Pediatric Deaths — United States, September 2010 – August 2011. MMWR Morb Mortal Wkly Rep. 2011;60 [PubMed] [Google Scholar]
- 9.Fiore AE, Fry A, Shay D, Gubareva L, Bresee JS, Uyeki TM. Antiviral agents for the treatment and chemoprophylaxis of influenza — recommendations of the Advisory Committee on Immunization Practices (ACIP) MMWR Recomm Rep. 2011;60:1–24. [PubMed] [Google Scholar]
- 10.Kawai N, Ikematsu H, Iwaki N, Maeda T, Satoh I, Hirotsu N, Kashiwagi S. A comparison of the effectiveness of oseltamivir for the treatment of influenza A and influenza B: a Japanese multicenter study of the 2003–2004 and 2004–2005 influenza seasons. Clin Infect Dis. 2006;43:439–444. doi: 10.1086/505868. [DOI] [PubMed] [Google Scholar]
- 11.Kawai N, Ikematsu H, Iwaki N, Kawashima T, Maeda T, Mitsuoka S, Kondou K, Satoh I, Miyachi K, Yamaga S, Shigematsu T, Hirotsu N, Kashiwagi S. Longer virus shedding in influenza B than in influenza A among outpatients treated with oseltamivir. J Infect. 2007;55:267–272. doi: 10.1016/j.jinf.2007.05.176. [DOI] [PubMed] [Google Scholar]
- 12.Sugaya N, Mitamura K, Yamazaki M, Tamura D, Ichikawa M, Kimura K, Kawakami C, Kiso M, Ito M, Hatakeyama S, Kawaoka Y. Lower clinical effectiveness of oseltamivir against influenza B contrasted with influenza A infection in children. Clin Infect Dis. 2007;44:197–202. doi: 10.1086/509925. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Cheng F, Lu M, Tian X, Ma J. Crystal structure of unliganded influenza B virus hemagglutinin. J Virol. 2008;82:3011–20. doi: 10.1128/JVI.02477-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dreyfus C, Laursen NS, Kwaks T, Zuijdgeest D, Khayat R, Ekiert DC, Lee JH, Metlagel Z, Bujny MV, Jongeneelen M, van der Vlugt R, Lamrani M, Korse HJWM, Geelen E, Sahin O, Sieuwerts M, Brakenhoff JPJ, Vogels R, Li OTW, Poon LLM, Peiris M, Koudstaal W, Ward AB, Wilson IA, Goudsmit J, Friesen RHE. Highly Conserved Protective Epitopes on Influenza B Viruses. Science (80-) 2012;337:1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yasugi M, Kubota-Koketsu R, Yamashita A, Kawashita N, Du A, Sasaki T, Nishimura M, Misaki R, Kuhara M, Boonsathorn N, Fujiyama K, Okuno Y, Nakaya T, Ikuta K. Human Monoclonal Antibodies Broadly Neutralizing against Influenza B Virus. PLoS Pathog. 2013;9:1–12. doi: 10.1371/journal.ppat.1003150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Air GM, Laver WG, Luo M, Stray SJ, Legrone G, Webster RG. Antigenic, Sequence, and Crystal Variation in Influenza B Neuraminidase. Virology. 1990;177:578–587. doi: 10.1016/0042-6822(90)90523-t. [DOI] [PubMed] [Google Scholar]
- 17.Laver WG, Luo M, Bossart TJ, Babu YS, Smith C, Accavitti MA, Tulloch PA, Air GM. Crystallization and Preliminary X-Ray Analysis of Type B Influenza Virus Neuraminidase Complexed with Antibody Fab Fragments. Virology. 1988;167:621–624. [PubMed] [Google Scholar]
- 18.Doyle TM, Li C, Bucher DJ, Hashem AM, Van Domselaar G, Wang J, Farnsworth A, She YM, Cyr T, He R, Brown EG, Hurt AC, Li X. A monoclonal antibody targeting a highly conserved epitope in influenza B neuraminidase provides protection against drug resistant strains. Biochem Biophys Res Commun. 2013;441:226–229. doi: 10.1016/j.bbrc.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 19.Schulman JL, Khakpour M, Kilbourne E. Protective effects of specific immunity to viral neuraminidase on influenza virus infection in mice. J Virol. 1968;2:776–778. doi: 10.1128/jvi.2.8.778-786.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dowdle WR, Coleman MT, Mostow SR, Kaye HS, Schoenbaum SC. Inactivated influenza vaccines. 2. Laboratory indices of protection. Postgrad Med J. 1973;49:159–63. doi: 10.1136/pgmj.49.569.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Couch RB, Kasel JA, Gerin JL, Schulman JL, Kilbourne ED, Lanier WS, Warden A, Taylor B. Induction of Partial Immunity to Influenza by a Neuraminidase-Specific Vaccine. J Infect Dis. 1974;129 doi: 10.1093/infdis/129.4.411. [DOI] [PubMed] [Google Scholar]
- 22.Johansson BE, Kilbourne ED. Immunization with purified N1 and N2 influenza virus neuraminidases demonstrates cross-reactivity without antigenic competition. Proc Natl Acad Sci U S A. 1994;91:2358–2361. doi: 10.1073/pnas.91.6.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rockman S, Brown LE, Barr IG, Gilbertson B, Lowther S, Kachurin A, Kachurina O, Klippel J, Bodle J, Pearse M, Middleton D. Neuraminidase-inhibiting antibody is a correlate of cross-protection against lethal H5N1 influenza virus in ferrets immunized with seasonal influenza vaccine. J Virol. 2013;87:3053–3061. doi: 10.1128/JVI.02434-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Easterbrook JD, Schwartzman LM, Gao J, Kash JC, Morens DM, Couzens L, Wan H, Eichelberger MC, Taubenberger JK. Protection against a lethal H5N1 influenza challenge by intranasal immunization with virus-like particles containing 2009 pandemic H1N1 neuraminidase in mice. Virology. 2012;432:39–44. doi: 10.1016/j.virol.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wan H, Gao J, Xu K, Chen H, Couzens LK, Rivers KH, Easterbrook JD, Yang K, Zhong L, Rajabi M, Ye J, Sultana I, Wan XF, Liu X, Perez DR, Taubenberger JK, Eichelberger MC. Molecular basis for broad neuraminidase immunity: conserved epitopes in seasonal and pandemic H1N1 as well as H5N1 influenza viruses. J Virol. 2013;87:9290–9300. doi: 10.1128/JVI.01203-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wohlbold TJ, Nachbagauer R, Xu H, Tan GS, Hirsh A, Brokstad KA, Cox RJ, Palese P, Krammer F. Vaccination with Adjuvanted Recombinant Neuraminidase Induces Broad Heterologous, but Not Heterosubtypic, Cross-Protection against Influenza Virus Infection in Mice. MBio. 2015;6:1–13. doi: 10.1128/mBio.02556-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wohlbold TJ, Chromikova V, Tan G, Meade P, Amanat F, Comella P, Hirsh A, Krammer F. Hemagglutinin stalk- and neuraminidase-specific monoclonal antibodies protect against lethal H10N8 influenza virus infection in mice. J Virol. 2015;90:851–861. doi: 10.1128/JVI.02275-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Memoli MJ, Shaw PA, Han A, Czajkowski L, Reed S, Athota R, Bristol T, Fargis S, Risos K, Powers JH, Davey RT, Taubenberger JK. Evaluation of Antihemagglutinin and Antineuraminidase Antibodies as Correlates of Protection in an Influenza A/H1N1 Virus Healthy Human Challenge Model. MBio. 2016;7:e00417–16. doi: 10.1128/mBio.00417-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palese P, Tobita K, Ueda M, Compans RW. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology. 1974;61:397–410. doi: 10.1016/0042-6822(74)90276-1. [DOI] [PubMed] [Google Scholar]
- 30.Matrosovich MN, Matrosovich TY, Roberts NA, Klenk H, Gray T. Neuraminidase Is Important for the Initiation of Influenza Virus Infection in Human Airway Epithelium Neuraminidase Is Important for the Initiation of Influenza Virus Infection in Human Airway Epithelium. 2004 doi: 10.1128/JVI.78.22.12665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen M, Zhang XQ, Senaati HP, Chen HW, Varki NM, Schooley RT, Gagneux P. Influenza A penetrates host mucus by cleaving sialic acids with neuraminidase. Virol J. 2013;10:321. doi: 10.1186/1743-422X-10-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wan H, Yang H, Shore DA, Garten RJ, Couzens L, Gao J, Jiang L, Carney PJ, Villanueva J, Stevens J, Eichelberger MC. Structural characterization of a protective epitope spanning A(H1N1)pdm09 influenza virus neuraminidase monomers. Nat Commun. 2015;6:6114. doi: 10.1038/ncomms7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kilbourne ED. Comparative Efficacy of Neuraminidase-Specific and Conventional Influenza Virus Vaccines in Induction of Antibody to Neuraminidase in Humans. J Infect Dis. 1976;134:384–394. doi: 10.1093/infdis/134.4.384. [DOI] [PubMed] [Google Scholar]
- 34.Webster RG, Laver WG, Kilbourne ED. Reactions of antibodies with surface antigens of influenza virus. J Gen Virol. 1968;3:315–326. doi: 10.1099/0022-1317-3-3-315. [DOI] [PubMed] [Google Scholar]
- 35.DiLillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat Med. 2014;20:143–51. doi: 10.1038/nm.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DiLillo DJ, Palese P, Wilson PC, Ravetch JV. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J Clin Invest. 2016;126:605–610. doi: 10.1172/JCI84428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henry Dunand CJ, Leon PE, Huang M, Choi A, Chromikova V, Ho IY, Tan GS, Cruz J, Hirsh A, Zheng N-Y, Mullarkey CE, Ennis FA, Terajima M, Treanor JJ, Topham DJ, Subbarao K, Palese P, Krammer F, Wilson PC. Both Neutralizing and Non-Neutralizing Human H7N9 Influenza Vaccine-Induced Monoclonal Antibodies Confer Protection. Cell Host Microbe. 2016;19:800–813. doi: 10.1016/j.chom.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Webster RG, Brown LE, Laver WG. Antigenic and biological characterization of influenza virus neuraminidase (N2) with monoclonal antibodies. Virology. 1984;135:30–42. doi: 10.1016/0042-6822(84)90114-4. [DOI] [PubMed] [Google Scholar]
- 39.Air GM, Els MC, Brown LE, Laver WG, Webster RG. Location of Antigenic Sites on the Three-Dimensional Structure of the Influenza N2 Virus Neuraminidase. Virology. 1985;145:337–348. doi: 10.1016/0042-6822(85)90157-6. [DOI] [PubMed] [Google Scholar]
- 40.Gulati U, Hwang C, Venkatramani L, Gulati S, Stray SJ, Lee JT, Laver WG, Bochkarev A, Zlotnick A, Air GM. Antibody Epitopes on the Neuraminidase of a Recent H3N2 Influenza Virus (A/Memphis/31/98) J Virol. 2002;76:12274–12280. doi: 10.1128/JVI.76.23.12274-12280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicholson KG, Aoki FY, Osterhaus A, Trottier S, Carewicz O, Mercier CH, Rode A, Kinnersley N, Ward P. Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. Lancet. 2000;355:1845–1850. doi: 10.1016/s0140-6736(00)02288-1. [DOI] [PubMed] [Google Scholar]
- 42.Heinonen S, Silvennoinen H, Lehtinen P, Vainionpää R, Vahlberg T, Ziegler T, Ikonen N, Puhakka T, Heikkinen T. Early oseltamivir treatment of influenza in children 1-3 years of age: a randomized controlled trial. Clin Infect Dis. 2010;51:887–894. doi: 10.1086/656408. [DOI] [PubMed] [Google Scholar]
- 43.Leon PE, He W, Mullarkey CE, Bailey MJ, Miller MS, Krammer F, Palese P, Tan GS. Optimal activation of Fc-mediated effector functions by influenza virus hemagglutinin antibodies requires two points of contact. Proc Natl Acad Sci U S A. 2016;113:E5944–E5951. doi: 10.1073/pnas.1613225113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He W, Tan GS, Mullarkey CE, Lee AJ, Lam MMW, Krammer F, Henry C, Wilson PC, Ashkar AA, Palese P, Miller MS. Epitope specificity plays a critical role in regulating antibody-dependent cell-mediated cytotoxicity against influenza A virus. Proc Natl Acad Sci U S A. 2016;113:11931–11936. doi: 10.1073/pnas.1609316113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krammer F, Schinko T, Palmberger D, Tauer C, Messner P, Grabherr R. Trichoplusia ni cells (High Five™) are highly efficient for the production of influenza A virus-like particles: A comparison of two insect cell lines as production platforms for influenza vaccines. Mol Biotechnol. 2010;45:226–234. doi: 10.1007/s12033-010-9268-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krammer F, Margine I, Tan GS, Pica N, Krause JC, Palese P. A carboxy-terminal trimerization domain stabilizes conformational epitopes on the stalk domain of soluble recombinant hemagglutinin substrates. PLoS One. 2012;7:e43603. doi: 10.1371/journal.pone.0043603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Margine I, Palese P, Krammer F. Expression of functional recombinant hemagglutinin and neuraminidase proteins from the novel H7N9 influenza virus using the baculovirus expression system. J Vis Exp. 2013:e51112. doi: 10.3791/51112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang TT, Tan GS, Hai R, Pica N, Petersen E, Moran TM, Palese P. Broadly protective monoclonal antibodies against H3 influenza viruses following sequential immunization with different hemagglutinins. PLoS Pathog. 2010;6:e1000796. doi: 10.1371/journal.ppat.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nachbagauer R, Wohlbold TJ, Hirsh A, Hai R, Sjursen H, Palese P, Cox RJ, Krammer F. Induction of broadly-reactive anti-hemagglutinin stalk antibodies by an H5N1 vaccine in humans. J Virol. 2014;88:13260–13268. doi: 10.1128/JVI.02133-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scheres SHW. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera - A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Wohlbold TJ, Hirsh A, Krammer F. An H10N8 influenza virus vaccine strain and mouse challenge model based on the human isolate A/Jiangxi-Donghu/346/13. Vaccine. 2015;33:1102–1106. doi: 10.1016/j.vaccine.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klausberger M, Wilde M, Palmberger D, Hai R, Albrecht RA, Margine I, Hirsh A, García-Sastre A, Grabherr R, Krammer F. One-shot vaccination with an insect cell-derived low-dose influenza A H7 virus-like particle preparation protects mice against H7N9 challenge. Vaccine. 2014;32:355–62. doi: 10.1016/j.vaccine.2013.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krammer F, Albrecht RA, Tan GS, Margine I, Hai R, Schmolke M, Runstadler J, Andrews SF, Wilson PC, Cox RJ, Treanor JJ, García-Sastre A, Palese P. Divergent H7 Immunogens Offer Protection from H7N9 Virus Challenge. J Virol. 2014;88:3976–85. doi: 10.1128/JVI.03095-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hai R, Schmolke M, Leyva-Grado VH, Thangavel RR, Margine I, Jaffe EL, Krammer F, Solórzano A, García-Sastre A, Palese P, Bouvier NM. Influenza A(H7N9) virus gains neuraminidase inhibitor resistance without loss of in vivo virulence or transmissibility. Nat Commun. 2013;4:1–16. doi: 10.1038/ncomms3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marathe BM, Wong S, Vogel P, Garcia-Alcalde F, Webster RG, Webby RJ, Najera I, Govorkova EA. Combinations of Oseltamivir and T-705 Extend the Treatment Window for Highly Pathogenic Influenza A(H5N1) Virus Infection in Mice. Sci Rep. 2016;6:1–14. doi: 10.1038/srep26742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan GS, Krammer F, Eggink D, Kongchanagul A, Moran TM, Palese P. A Pan-H1 Anti-Hemagglutinin Monoclonal Antibody with Potent Broad-Spectrum Efficacy In Vivo. J Virol. 2012;86:6179–6188. doi: 10.1128/JVI.00469-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.