

Abstract
Anti-angiogenic and vascular disrupting therapies rely on the dependence of tumors on new blood vessels to sustain tumor growth. We previously reported a potent vascular disrupting agent, a theranostic nanosystem consisting of a tumor vasculature-homing peptide (CGKRK) fused to a pro-apoptotic peptide [D(KLAKLAK)2] coated on iron oxide nanoparticles. This nanosystem showed promising therapeutic efficacy in glioblastoma (GBM) and breast cancer models. However, complete control of the tumors was not achieved, and some tumors became non-responsive to the treatment. Here we examined the non-responder phenomenon in an aggressive MCF10-CA1a breast tumor model. In the treatment-resistant tumors we noted the emergence of CD31-negative patent neovessels and a concomitant loss of tumor homing of the nanosystem. In vivo phage library screening in mice bearing non-responder tumors showed that compared to untreated and treatment-sensitive tumors, treatment sensitive tumors yield a distinct landscape of vascular homing peptides characterized by over-representation of peptides that target αv integrins. Our approach may be generally applicable to the development of targeted therapies for tumors that have failed treatment.
Keywords: Tumor angiogenesis, tumor homing peptide, phage display, drug resistance, integrins
Graphical abstract
INTRODUCTION
All vertebrate tissues depend on blood circulation, and that includes tumors, which require angiogenesis as an essential step in tumor initiation and progression [1,2]. In the past decades, several therapies have been developed that specifically block tumor angiogenesis [3–6]. Some of the anti-angiogenic drugs have already been approved for the treatment of solid tumors, such as breast cancer [7–9]. The blood vessel-directed therapies can act synergistically with conventional chemotherapies and tend to have non-overlapping toxicities. However, like all anticancer therapies, anti-angiogenic agents and vascular disrupting agents (VDAs) trigger compensatory changes that undermine the efficacy. For example, current anti-angiogenic and VDA therapies preferentially eliminate immature tumor blood vessels, leaving mature vessels intact and even improving their function [10–12]. Some of the molecular markers that distinguish tumor vessels from normal vessels disappear as a result of the therapy, making it difficult to target the remaining vessels for destruction [13]. However, development of affinity probes to target tumors that have failed treatment and become resistant has received little attention.
Recently we reported a novel tumor vessel-disrupting nanosystem consisting of three elements: (i) a tumor-homing 5-amino acid peptide (CGKRK) that specifically delivers its payload to the mitochondria of tumor endothelial cells and tumor cells, (ii) a pro-apoptotic peptide [D(KLAKLAK)2] that disrupts mitochondria, and (iii) multivalent presentation of the peptides on iron oxide nanoparticles, which greatly enhances the activities of the peptides [14,15]. The nanoparticles, dubbed nanoworms (NWs) because of their elongated shape [14], showed therapeutic efficacy against lentiviral-induced glioblastoma models and a MCF10-CA1 breast cancer model [15,16]. Nearly complete control was achieved in one glioblastoma model, but in a more aggressive glioma and in the breast cancer model, some tumors escaped the control and become resistant to the therapy.
In vivo phage display of peptides is used to discover for agnostic discovery of homing peptides specific for different pathologies including tumors, atherosclerotic plaques, wounds, and severe brain injuries [17–21].
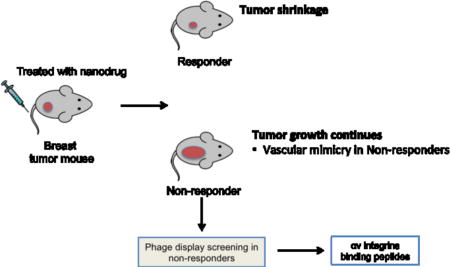
Here, we compare the vasculature of MCF10-CA1a breast cancers that are either sensitive or resistant to the nanosystem. We then use in vivo phage display to identify peptides that selectively recognize the vasculature of the treatment-resistant tumors.
RESULTS
Development of therapeutic resistance in breast tumors treated with CGKRK-D[KLAKLAK]2-NWs
The CGKRK-D[KLAKLAK]2-NW-based nanosystem used in this study, and the treatment schedule are schematically depicted in Figure 1A and B. As has been reported [15,16], the nanosystem therapy resulted in significant overall reduction of tumor volume compared to control-treated mice. The treated mice appeared to fall into two categories, one with very small tumors (and occasional cures), termed the responders, and another with large tumors, the non-responders (Fig 1C). The tumor volume in these sub-groups correlated with tumor cell proliferation and apoptosis; the responder group showed significantly reduced cell proliferation as measured by Ki67 staining (Fig. 1D), whereas the non-responder group was similar to the PBS-treated group. Conversely, the responder tumors displayed more TUNEL staining than the PBS and non-responder groups (Fig. 1E). This dichotomy in the tumor responses suggested that comparison of the two groups may provide clues to designing strategies that overcome the treatment resistance.
Figure 1. Design of the theranostic nanosystem and activity in MCF10Ca1a breast tumor model.
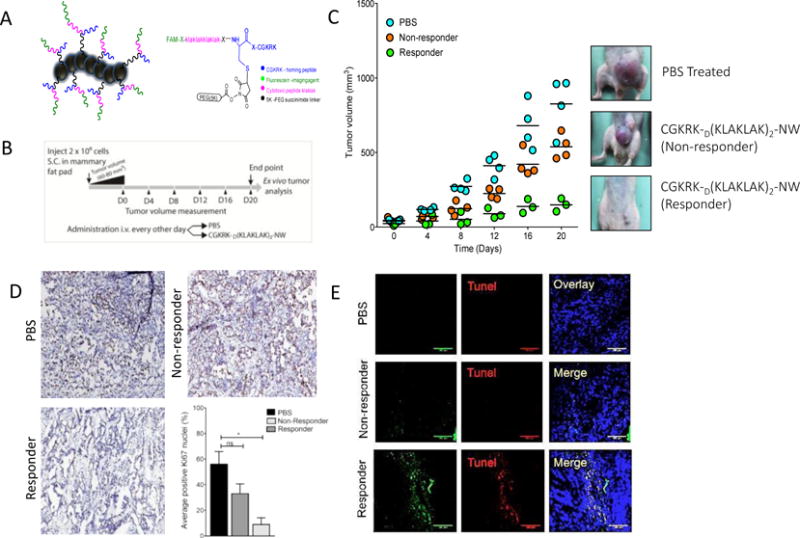
(A) A chimeric peptide combining a tumor-homing peptide (CGKRK) and a pro-apoptotic peptide is covalently coupled to iron oxide NWs (length 40-50 nm; Park et al., 2008). An extra cysteine was added to the N-terminus of the CGKRK peptide for the NW coupling. The drug peptide and the fluorophor were attached to the free N-terminus of the same cysteine residue. (B) Schematic representation of the treatment regimen. (C) Mice bearing MCF10CA1a orthotopic tumor xenografts were intravenously injected with peptide-coated NWs or PBS every other day for 3 weeks at a dose of 5 mg/kg. PBS, n = 5; CGKRKD[KLAKLAK]2-NWs, n = 7. The tumors were grouped into responder and non-responder tumors. One of three independent experiments with similar results is shown. (D) Sections from the treated tumors were stained with an antibody against proliferating cell nuclear antigen (Ki67). The graph shows the percentage of Ki67 positive nuclei. The results are expressed as a mean ±SD (*P < 0.01, one way ANOVA, Kruskal-Wallis test n = 3 mice per group, scale bar = 200μm). (E) TUNEL staining of treated tumors. Merged image: NWs, green; TUNEL-positive nuclei, red; DAPI-stained nuclei. blue. Scale bars, 100 μm.
Vascular changes in therapy-resistant tumors
The nanosystem therapy used here was designed to target and disrupt the tumor vasculature based on the homing properties of the CGKRK peptide. Analysis of the vascular changes after completion of the treatment showed a striking 75% reduction of blood vessels detectable with CD31 staining in the responder tumors relative to the PBS control group (Fig. 2A). The non-responder vascular density was intermediate, and the vessels appeared larger than in the other groups. Perfusion with labeled tomato lectin to map patent vascular structures revealed a different picture. In the PBS-treated and responder groups, most of the CD31+ vessels were also positive for lectin staining (Fig. 2B). In contrast, over 20% of the non-responder vessels were positive for the lectin, but not for CD31 (Fig. 2B, bar graph). In agreement with the lectin results, ultrasound analysis of the non-responding tumors revealed efficient tumor circulation in the non-responder tumors (Fig. 2C).
Figure 2. Vascular changes in responder and non-responder tumors.
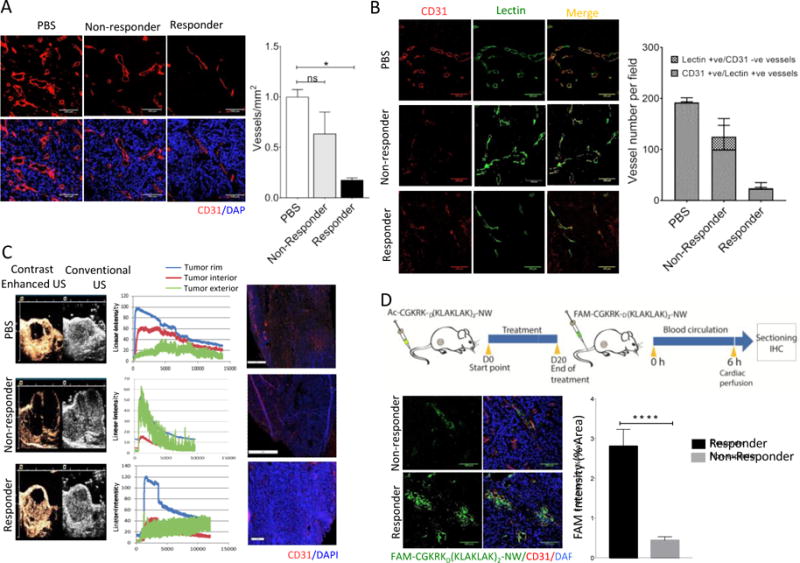
(A) Representative confocal images of tumor vasculature stained with anti-CD31 (red). CD31-positive blood vessels were quantified by counting 10 random fields at 20× magnification, and the results are presented as number of vessels per field (***P < 0.001, one way ANOVA, Tukey’s posthoc test, n = 3 mice per group, scale bar = 100μm). (B) Blood vessel density estimated by anti-mouse CD31 staining (red) and lectin (green) perfusion. Representative images from 3 mice analyzed in each group are shown. Blood vessels were quantified and analyzed as described for panel A, n = 3 mice per group, scale bar = 100 μm). (C) Tumor-bearing mice were treated as in Figure 1, and at the end of the treatment, blood flow in control and treated tumors was measured by contrast-enhanced ultrasound. The grey images outline the entire tumor; the brown color images show the blood flow. Graph is showing enhancement-analysis curves of blood flow in different tumor regions and the surrounding tissue. (D) Schematic representation of a homing experiment. Tumor-bearing mice were treated with the nanosystem for three weeks, and at the end of the treatment, FAM-labeled peptide-NWs were intravenously injected (5mg iron/kg) and allowed to circulate for 5 hours. The mice were perfused through the heart with PBS, and tumors and organs were collected and processed for fluorescence microscopy. Tumor sections were stained with anti-CD31 and examined by confocal microscopy. NWs, green; blood vessels (CD31), red; DAPI-stained nuclei, blue. (Scale bars, 100μm). Bar diagram, quantification of NW accumulation in tumors. Slides were scanned using Scanscope. NW fluorescence in 10 random areas of each tumor section (n = 3/group) was quantified using ImageJ software. (****P < 0.0001, paired t test, n = 3 mice per group, Error bars = mean ±SD).
The presence of lectin-positive, CD31-negative vessels in the non-responder tumors suggested the presence of an alternative circulatory system not based on endothelially-lined blood vessels of mouse origin. We next stained the sections with an anti-human CD31 antibody to examine the possibility that the alternative circulation was derived from the human tumor cells through trans-differentiation into endothelial cells. Such trans-differentiation has been observed in other tumor types [22], including glioblastomas [23, 24]. We observed no staining for human CD31 (not shown), but some of the lectin-positive vessels in non-responder tumors stained with human-specific anti-CD105, a marker for immature blood vessels (Suppl. Fig. 1). This finding suggests that a subset of the alternative vessels in the non-responder tumors originate from the human tumor cells.
Homing studies with CGKRK-D[KLAKLAK]2-NWs at the completion of the treatment revealed a significant decrease in NW accumulation in the non-responder tumors (Fig. 2D). The reduced NW homing did not seem to be due to lower receptor expression because the main receptor for the CGKRK peptide, p32, was expressed at similar levels in the vessels and tumor cells of the two types of tumors (Suppl Fig 2). These results show that even the non-responder vessels that do stain positive for mouse CD31 are altered, as they could not be targeted by the nanosystem.
Phage screening using treated tumors
To probe vascular changes after the NW treatment, and to identify peptides that recognize the altered vessels in the resistant tumors, we screened phage-displayed peptide libraries in mice bearing treated tumors (Fig. 3A). High-throughput sequencing of the peptide-encoding segment in the genomes of the recovered phages revealed a strikingly high frequency of the integrin-binding RGD motif among the most commonly represented peptide sequences from non-responder tumors (20 of the 200 most abundant peptide sequences contained the RGD motif). In contrast, the responder tumor pool yielded a single RGD peptide among the top 200. The top 20 motifs from each tumor type are listed in Fig. 3B.
Figure 3. Phage screening on nanosystem-treated tumors.
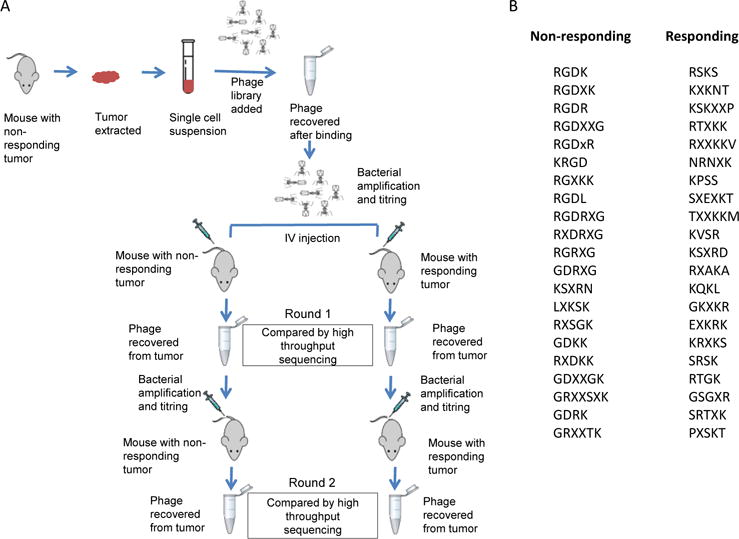
(A) Schematic showing steps in the phage screening. (B) A sampling of then top 20 motifs among the most frequent 200 4-amino acid motifs in responding and non-responding tumors. X-denotes any amino acid. Only full-length (cyclic) inserts were considered.
Peptides containing an RGD motif are widely used for tumor targeting [25]. Our phage screening results suggest that a RGD peptide could restore effective tumor targeting for tumors that have become resistant to a CGKRK-targeted vascular disrupting treatment. Indeed, two RGD peptides (a cyclic and a linear peptide) effectively directed NWs to both CD31+/lectin+ and CD31−/lectin+ vascular structures in non-responder tumors (Fig. 4).
Figure 4. Homing of NWs coated with RGD peptides to non-responder tumors.
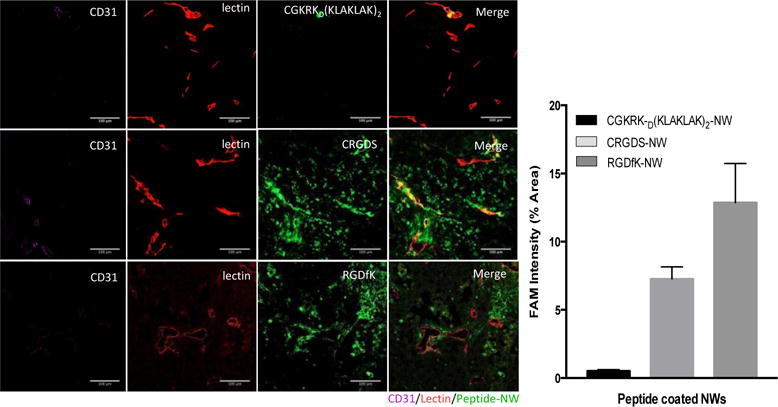
(A) Tumor-bearing mice were treated with the nanosystem for 3 weeks, and at the end of the treatment, FAM-labeled peptide-NWs (green) were intravenously injected (5mg iron/kg) and allowed to circulate for 5 hours. The mice were further injected with 100μg of DyLight 488 labeled Lycopersicon esculentum lectin, (green). After 5 minutes, the mice were perfused with PBS through the left ventricle of the heart, followed by 4% formaldehyde in PBS. Tumors and organs were collected and processed for fluorescence microscopy. (B) Quantification of NW accumulation in tumors. Slides were scanned using Scanscope. NW fluorescence in 10 random areas of each tumor section (n = 3/group) was quantified using ImageJ software. (****P < 0.0001, paired t test, n = 3 mice per group. Error bars = mean ±SD).
Integrin immunochemistry and microarray analysis
Given the abundance of RGD-containing phage peptides recovered from non-responder tumors and the strong homing of RGD functionalized nanoparticles to these tumors, we analyzed expression of the main target of RGD peptides, integrin, αvβ3, in the treated tumors. A robust expression of β3 integrin subunit in non-responder tumors was apparent, whereas the signal was much lower in responder tumors (Fig. 5). The PBS group also showed β3 staining, although the staining was weaker and its pattern different from the non-responders.
Figure 5. Integrin β3 expression in treated tumors.
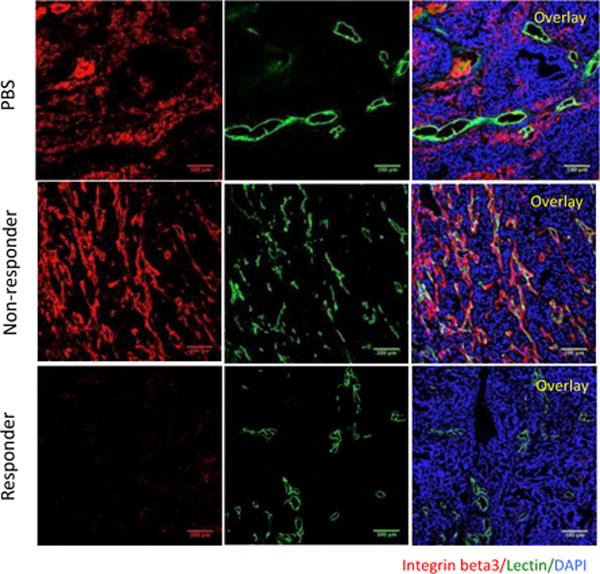
(A) Tumor sections were stained with anti-β3 integrin antibodies (red) and examined by confocal microscopy. Lectin, green; β3, red; DAPI-stained nuclei, blue. (Scale bars, 100 μm).
In agreement with the immunostaining data, microarrray analysis revealed elevated expression of β3 mRNA in the non-responder tumors, and the mRNA for another αv-associated integrin β subunit, β6, was also elevated (Table S1). Significantly, a number of other angiogenesis-associated genes (FGF2, stefin A, CD300, Igfbp2, Pim1, Cxcl10, MMP13 and ITGB3) were also highly expressed in these tumors. We also noted another factor potentially involved in the formation of the non-responder neovasculature, high expression of a set of genes involved in stemness and epithelial-to-mesenchymal transition. These genes included putative tumor-stemness marker CDH2 (N-cadherin), SNAI2, and Zinc-finger transcription factors, p63 and ITGA6 (CD49f).
These results suggest that that significant biological information of potential importance to therapy can be gleaned from the analysis of the vasculature of tumors that have become resistant to a treatment.
DISCUSSION
In the current study we explored the changes in the breast tumor vasculature that are associated with the emergence of resistance to a vascular disrupting treatment. We observed an abundant and efficient vasculature in the treatment resistant tumors that was partially composed of tumor cell-derived vessels. We also identified a highly expressed marker in these vessels that allows effective recognition of vessels in the non-responder tumors.
The vascular disrupting nanosystem we have developed is remarkably effective in glioblastoma and breast cancer models [15–16], even eradicating some of the tumors. However, some tumors grow nearly as fast as the controls. This dichotomy in the response provided a model for the study of the molecular and cellular basis of the resistance to the therapy.
The CGKRK peptide, which is the homing device in the nanosystem is capable of binding to tumor vessels and tumor cells [17], but unless combined with a tumor-penetrating peptide [15], this peptide primarily takes its payload only to tumor vessels [16]. That treatment with the CGKRK-guided therapeutic nanosystem targets blood vessels was apparent from the greatly reduced number of vessels in the tumors that responded to the treatment. Another indication to that effect was the altered vasculature of the non-responder tumors.
The most striking change in the non-responder vessels was the emergence of blood vessels that appeared to be of human origin. The non-responder vessels also appeared generally immature in that they not stain for mouse or human CD31, but about 20% of the vessels expressed a marker of immature blood vessels, CD105. The CD105 staining was with an antibody that recognizes human, but not mouse CD105, suggesting tumor cell origin of the vessels. Despite their immaturity and apparent origin by trans-differentiation from the human tumor cells, these vessels were functional as evidenced by lectin perfusion. Trans-differentiation of human tumor cells into cells capable of forming functional blood vessels has been demonstrated in melanoma [22] and glioblastoma [23,24]. These studies used tumors that were not subjected to any treatment. Our results indicate that breast cancer cells can also possess this capacity to trans-differentiate, although it may require a tumor vessel-directed therapy to manifest itself.
Another important change we observed in the non-responder vessels was that they could no longer be targeted with the CGKRK nanosystem. This was despite the fact that overexpression of p32, a putative receptor for CGKRK, was maintained in these tumors after the treatment. Possible reasons for this discrepancy include selective loss of cell surface p32 in tumor endothelial cells. Alternatively, endothelial cells could have lost their ability to internalize the peptide (and its payload) through p32 binding.
Phage screening identified peptides containing the integrin binding RGD motif as potentially effective targeting elements for tumors that have developed a vasculature resistant to CGKRK targeting. RGD peptides are among the most commonly used ligands for synaphic tumor targeting [18]. Their target receptors, the αv integrins (primarily αvβ3 and αvβ5), and α5β1 integrin, are strongly expressed in blood vessels undergoing angiogenesis, such as tumor vessels [26,27], but not in normal vessels. Tumor cells also commonly express these integrins. In agreement with the prevalence of RGD peptides in the non-responder phage screen, the β3 integrin was particularly highly expressed in the vasculature of these tumors. Thus, targeted treatment systems suitable for complementing CGKRK targeting are already available. Also, using both CGKRK and RGD targeting at the same time might prevent the development of resistance to either therapy in analogy with other combination therapies for cancer. [28,29]. A number of clinical and pre-clinical studies have shown role of integrins in driving a stem phenotype, drug resistance, and metastasis [30–32]. Damiano et al, showed that cell adhesion-mediated drug resistance, a pro-survival and anti-apoptotic program, is dictated by integrins/ECM interactions [33]. Furthermore, αvβ3 integrin expression has been reported on a subpopulation of breast cancer stem cells and its expression has been associated with poor outcome and high incidence of metastasis in a variety of epithelial cancers [34–36].
Our results also provide information on the mechanisms underlying the development of resistance to a vascular disrupting agent. In addition to being a marker of blood vessels undergoing angiogenesis, β3 integrin has been shown to promote tumor stemness, including tumor initiation and tumor stem cell self-renewal [30], and it is also involved in drug resistance [31]. In the non-responding tumors, β3 was mostly expressed in the blood vessel, but these tumors expressed elevated levels of other stem cell markers, such as N-cadherin, p63, and CD49f, suggesting stem cell-like properties may be important in the vascular trans-differentiation.
Further studies in spontaneous tumor models are needed to determine how broadly applicable this phenomena is and to determine to what extent the features we have established for the CGKRK-D[KLAKLAK]2-NW system are shared by other vascular disrupting agents or anti-angiogenic therapies. However, our data show that multiple pathways are at play in acquired resistance to our vascular therapy, including blunting the targeted homing of the agent, vascular trans-differentiation, and stemness. A combination therapy addressing these pathways may overcome such therapy resistance.
MATERIALS AND METHODS
Peptide synthesis and NW conjugates
Peptides were synthesized with a 5(6)-fluorescein carboxylate (FAM) label and amide-blocked C-terminus. An extra cysteine with a free sulfhydryl group was added to cyclic peptides for coupling purposes [37]. Linear peptides contained an N-terminal cysteine residue. The synthesis and subsequent conjugation of aminated dextran coated iron oxide NWs have been described [14–16]. For peptide coupling to NWs, aminated NWs were PEGylated with maleimide-5KPEG-NHS (JenKem Technology) and peptides were conjugated to the functionalized particles through a thioether bond between the cysteine thiol in the peptide and the maleimide on the particles.
Cell lines and tumors
The MCF10CA1a human breast tumor cell line was obtained from the Barbara Ann Karmanos Cancer Institute (Detroit, MI). Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 1% Glutamine-Pen-Strep and 100 ng/ml (Sigma-Aldrich, St. Louis, MI). The cell line was validated at the DNA facility of this institute.
To produce orthotopic MCF10CA1a tumors, BALB/c nude mice were injected into the mammary fat pad with 2 × 106 cells suspended in 100 μl of PBS. Animal experimentation was performed according to procedures approved by the Sanford Burnham Prebys IACUC.
Tumor treatment and peptide-NW homing
Tumor treatment was started when the tumors were about 50-60 mm3 in diameter. The tumor-bearing mice were intravenously injected with peptide-coated NWs at a dose of 7.5 mg/kg in 100 μl of PBS on alternate days for three weeks. A control group was similarly injected with 100 μl of PBS. Tumor volume was calculated by using the following formula: volume = (d2 × D)/2, where d and D are the smallest and the largest tumor diameters, respectively (REF).
To test homing of peptide-conjugated NWs in treated mice, NWs were injected into the tail vein (7.5 mg of iron/kg of body weight in 100 μl of PBS). After 5-6 hours of NW circulation the mice were euthanized by cardiac perfusion with PBS under anesthesia, and tumors and organs were dissected and analyzed for NW homing.
Histological and immunostaining of tumors
Tissues were fixed in 4% paraformaldehyde overnight at 4°C, cryoprotected in 30% sucrose overnight, and frozen in optimum cutting temperature embedding medium. Tissue sections (7 μm) were cut and stained with hematoxylin and eosin or processed for immunostaining. The sections were first incubated for 1 hour at room temperature with 10% serum from the species in which the secondary antibody was generated, followed by incubation with the primary antibody. Monoclonal anti-rat CD31 (BD Pharmingen, San Jose, CA) was detected with Alexa 594 goat anti-rat secondary antibody (Molecular Probes, Eugene, OR). Rabbit antibodies against human and mouse p32 [38], anti-human CD105 (Abcam Antibodies), anti-mouse CD105 (Abcam Antibodies), rabbit antibodies against human and mouse integrin β3 (Abcam Antibodies), Anti-mouse PDGRR-β (Abcam Antibodies) were detected with Alexa 594 goat anti-rabbit secondary antibody (Molecular Probes). Each staining experiment included sections stained with the secondary antibody only as a negative control. Nuclei were stained with DAPI (4,6-diamidino-2-phenylindole; 5 μg/mL; Molecular Probes). The sections were mounted in gel/mount mounting medium (Biomeda) and viewed under a LSM 710 NLO Multiphoton Laser Point Scanning Confocal Microscope (Zeiss). For quantification of staining and peptide-NW accumulation in tumors, stained slides were scanned using Scanscope. 10 random areas of each tumor section (n = 3/group) was quantified using ImageJ software.
Immunoperoxidase staining with anti Ki67 was performed using DAB (MP Biomedicals) reaction, and the sections were counterstained with hematoxylin (Sigma Aldrich). Species specificity of antibodies, when relevant, was tested by staining mouse and human tissues with the antibody.
Click-iT TUNEL 647 Alexa Fluor imaging assay (Invitrogen) was carried out according to the manufacturer’s instructions. Nuclei were counterstained with DAPI. The sections were mounted in Gel/Mount mounting medium (Biomeda, Foster City, CA) and viewed under a Fluoview 500 confocal microscope (Olympus America, Center Valley, PA).
Contrast-enhanced ultrasound (CEUS) and lectin perfusion
DEFINITY contrast agent (Lantheus Medical Imaging; 5 μL in 45 μL of saline) was injected into the tail vein of mice with a 28-gauge insulin syringe. A Philips iU22 ultrasound system with an L12-5 transducer was used for imaging with a low-mechanical-index, contrast-specific imaging mode (power-modulation mode). The imaging parameters were set as follows (and kept identical throughout the study): depth 2 cm, focus zone 2 cm, mechanical index 0.06, 5 frames per second. A 2-minute cine loop was saved for time-intensity curve analysis with Philips QLab. Contrast-enhanced ultrasound (CEUS) was performed after three weeks of treatment with the nanosystem or PBS. The tumor rim and center and the surrounding tissue were examined separately to quantify the efficiency of tumor circulation. Image analysis was performed with Photoshop with a threshold method in which the pixels with enhancement and those without were counted (the threshold was set arbitrarily, because there was no fixed number). The percentage area with enhancement was equal to the pixel number of the enhanced area divided by the pixel number of the entire tumor ×100.
Mice were perfused with lycopersicon esculentum agglutinin (LEA, tomato lectin; Vector Labs, Burlingame, CA) to label tumor vasculature. A bolus of 100 μg of tomato lectin in 100 μl volume injected intravenously, followed by 5 minutes of circulation, after which the mouse was perfused through the left ventricle of the heart with 10 ml PBS, followed by 20 ml PBS 4 % paraformaldehyde.
Following the perfusion fixation, tissues were collected and placed into 4 % paraformaldehyde for overnight fixation at 4 °C. The following day, the tissues were transferred to a solution of 30 % sucrose in PBS for cryoprotection and embedded in optimum cutting temperature embedding medium. Tissue sections (7 μm) were cut and processed for immunostaining.
Phage screening
The first step of the phage screening was an ex vivo enrichment by incubating 1010 pfu of a CX7C naïve phage library with tumor cell suspension made from 50 mg of tumor tissue. The tumor cell suspension was prepared from non-responder tumors excised from mice treated with the nanosystem for three weeks. The tumors were cut with a razor blade into small pieces and cell suspensions were generated in Medimachine (BD Biosciences). The cells were incubated with phage overnight at 4 °C, followed by 3 washes with 15 ml of DMEM containing 1% bovine serum albumin (DMEM–BSA) in a 50ml Falcon tube. The cells were pelleted by centrifugation at 400 g for 5 min. After the last wash, the cell pellet was lysed in 1 ml LB containing 1% Nonidet P-40 (LB-NP40), and the phage titrated [39]. The phage pool recovered from the cell suspension was amplified in E. coli, and 4× 109 pfu were injected intravenously in mice with responding and non-responding tumors. The phages were allowed to circulate for 30 min, after which the mice were anaesthetized with 2.5% avertin and perfused with PBS through the heart. Tumors were excised and homogenized in 1 ml LB-NP40 using a handheld homogenizer with disposable plastic hard-tissue probes (Omni International Inc.). The phage pool was titrated, amplified, and peptide-encoding portion of the phage genome was sequenced using Ion Torrent high-throughput sequencing. The in vivo round was repeated once by injected the phage pool from the first round into tumor bearing mice and repeating the steps outlined above.
RNA isolation and microarray analysis
To isolate RNA from tumors, previously snap frozen tissues were homogenized in TRIzol® reagent (Ambion®). Total RNA was extracted from the tumors using an RNeasy Mini Kit (Qiagen, Valencia, CA). Labeled cRNA was prepared from 500 ng RNA using the Illumina® RNA Amplification Kit from Ambion (Invitrogen, CA, USA). The labeled cRNA (1500 ng for mouse and 750 for human) was hybridized overnight at 58°C to the Sentrix MouseWG-6 Expression BeadChip or humanHT-12 (>46,000 gene transcripts; Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. BeadChips were subsequently washed and developed with fluorolink streptavidin-Cy3 (GE Healthcare). BeadChips were scanned with an Illumina BeadArray Reader.
Supplementary Material
Acknowledgments
This work was supported by grants R01CA152327 (ER), and Cancer Center Support grant CA30199 to Sanford Burnham Prebys from the National Cancer Institute of NIH. TT was supported by Susan Komen for Cure Foundation Award KG110704, European Research Council Starting Grant GliomaDDS from European Regional Development Fund, and Wellcome Trust International Fellowship (WT095077MA).
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 3.Ferrara N, Alitalo K. Clinical applications of angiogenic growth factors and their inhibitors. Nat Med. 1999;5:1359–1364. doi: 10.1038/70928. [DOI] [PubMed] [Google Scholar]
- 4.Singh M, Ferrara N. Modeling sand predicting clinical efficacy for drugs targeting the tumor milieu. Nat Biotechnol. 2012;30:648–657. doi: 10.1038/nbt.2286. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 6.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hein A, Lambrechts D, von MinckwitzG, Häberle L, Eidtmann H, Tesch H, Untch M, Hilfrich J, Schem C, Rezai M, Gerber B, Costa SDan, Blohmer JU, Schwedler K, Kittel K, Fehm T, Kunz G, Beckmann MW, Ekici AB, Hanusch C, Huober J, Liedtke C, Mau C, Moisse M, Müller V, Nekljudova V, Peuteman G, Rack B, Rübner M, Brussel TVan, Wang L, Weinshilboum RM, Loibl S, Fasching PA. Genetic variants in VEGF pathway genes in neoadjuvant breast cancer patients receiving bevacizumab: Results from the randomized phase III GeparQuinto study. Int J Cancer. 2015;15:2981–2988. doi: 10.1002/ijc.29656. [DOI] [PubMed] [Google Scholar]
- 8.Penson RT, Huang HQ, Wenzel LB, Monk BJ, Stockman S, Long HJ. Bevacizumab for advanced cervical cancer: patient-reported outcomes of a randomised, phase 3 trial (NRG Oncology-Gynecologic Oncology Group protocol 240) Lancet Oncol. 2015;16:301–311. doi: 10.1016/S1470-2045(15)70004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Dabrosin C, Yin X, Fuster MM, Arreola A, Rathmell WK, Generali D, Nagaraju GP, El-Rayes B, Ribatti D, Chen YC, Honoki K, Fujii H, Georgakilas AG, Nowsheen S, Amedei A, Niccolai E, Amin A, Ashraf SS, Helferich B, Yang X, Guha G, Bhakta D, Ciriolo MR, Aquilano K, Chen S, Halicka D, Mohammed SI, Azmi AS, Bilsland A, Keith WN, Jensen LD. Broad targeting of angiogenesis for cancer prevention and therapy. Semin Cancer Biol. 2015;35(Suppl):S224–243. doi: 10.1016/j.semcancer.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubenstein JL, Kim J, Ozawa T, Westpal M, Deen DF, Shuman MA. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia. 2000;2:306–314. doi: 10.1038/sj.neo.7900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, Kostaras E, Nathan MR, Wotherspoon A, Gao ZH, Shi Y, Eynden GVanden, Daley F, Peckitt C, Tan X, Salman A, Lazaris A, Gazinska P, Berg TJ, Eltahir Z, Ritsma L, Rheenen Jvan, Khashper A, Brown G, Nyström H, Sund M, Laere SVan, Loyer E, Dirix L, Cunningham D, Metrakos P, Reynolds AR. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med. 2016;22:1294–1302. doi: 10.1038/nm.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye W. The complexity of translating anti-angiogenesis therapy from basic science to clinics. Dev Cell. 2016;18:114–125. doi: 10.1016/j.devcel.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Jain RK, Duda DG, Willett CG, Sahani DV, Zhu AX, Loeffler JS, Batchelor TT, Sorensen AG. Biomarkers of response and resistance to antiangiogenic therapy. Nat Rev Clin Oncol. 2009;6:327–338. doi: 10.1038/nrclinonc.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park JH, Von Maltzahn G, Zhang L, Schwartz MP, Bhatia SN, Ruoslahti E, Sailor MJ. Magnetic Iron Oxide Nanoworms for Tumor Targeting and Imaging. Adv Mater. 2008;20:1630–1635. doi: 10.1002/adma.200800004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agemy L, Friedmann-Morvinski D, Kotamraju VR, Roth L, Sugahara KN, Girard OM, Mattrey RF, Verma IM, Ruoslahti E. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc Natl Acad Sci U S A. 2011;108:17450–17455. doi: 10.1073/pnas.1114518108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agemy L, Friedmann-Morvinski D, Kotamraju VR, Sharma S, Sugahara KN, Ruoslahti E. Proapoptotic Peptide-Mediated Cancer Therapy to Cell Surface p32. Molecular Therapy. 2013;21:2195–2204. doi: 10.1038/mt.2013.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joyce JA, Laakkonen P, Bernasconi M, Bergers G, Ruoslahti E, Hanahan D. Stage-specific vascular markers revealed by phage display in a mouse model of pancreatic islet tumorigenesis. Cancer Cell. 2003;4:393–403. doi: 10.1016/s1535-6108(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 18.Ruoslahti E. Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv Mater. 2012;24:3747–3756. doi: 10.1002/adma.201200454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.She ZG, Hamzah J, Kotamraju VR, Pang HB, Jansen S, Ruoslahti E. Plaque-penetrating peptide inhibits development of hypoxic atherosclerotic plaque. J Control Release. 2016;28:212–220. doi: 10.1016/j.jconrel.2016.07.020. [DOI] [PubMed] [Google Scholar]
- 20.Järvinen TA, Ruoslahti E. Target-seeking antifibrotic compound enhances wound healing and suppresses scar formation in mice. Proc Natl Acad Sci U S A. 2010;14:21671–21676. doi: 10.1073/pnas.1016233107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mann AP, Scodeller P, Hussain S, Joo J, Kwon E, Braun GB, Mölder T, She ZG, Kotamraju VR, Ranscht B, Krajewski S, Teesalu T, Bhatia SN, Sailor MJ, Ruoslahti E. A peptide for targeted, systemic delivery of imaging and therapeutic compounds into acute brain injuries. Nat Commun. 2016;28:1–11. doi: 10.1038/ncomms11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;6:411–421. doi: 10.1038/nrc1092. [DOI] [PubMed] [Google Scholar]
- 23.Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, De Maria R. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;9:824–828. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 24.Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, Pastorino S, Yang M, Hoffman RM, Kesari S, Verma IM. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A. 2011;11:4274–4280. doi: 10.1073/pnas.1016030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruoslahti E. Tumor penetrating peptides for improved drug delivery. Adv Drug Deliv Rev. 2016 Apr; doi: 10.1016/j.addr.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin αvβ3 for angiogenesis. Science. 1994a;264:569–571. doi: 10.1126/science.7512751. [DOI] [PubMed] [Google Scholar]
- 27.Kim S, Bell K, Mousa SA, Varner JA. Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am J Pathol. 2000;156:1345–1362. doi: 10.1016/s0002-9440(10)65005-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cesca M, Bizzaro F, Zucchetti M, Giavazzi R. Tumor delivery of chemotherapy combined with inhibitors of angiogenesis and vascular targeting agents. Front Oncol. 2013;1:259. doi: 10.3389/fonc.2013.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ocaña A, Amir E, Vera F, Eisenhauer EA, Tannock IF. Addition of bevacizumab to chemotherapy for treatment of solid tumors: similar results but different conclusions. J Clin Oncol. 2011;20:254–256. doi: 10.1200/JCO.2010.32.0275. [DOI] [PubMed] [Google Scholar]
- 30.Laetitia S, Shumei K, Franovic MAleksandra, Fernanda C, Lesperance J, Kathryn C, Cheresh DA. An integrin β3–KRAS–RalB complex drives tumour stemness and resistance to EGFR inhibition. Nature Cell Biology. 2014;16:457–468. doi: 10.1038/ncb2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laetitia S, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25:234–240. doi: 10.1016/j.tcb.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeLay M, Jahangiri A, Carbonell WS. Microarray Analysis Verifies Two Distinct Phenotypes of Glioblastomas Resistant to Antiangiogenic Therapy. Clin Cancer Res. 2012;10:2930–2942. doi: 10.1158/1078-0432.CCR-11-2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Damiano JS. Integrins as novel drug targets for overcoming innate drug resistance. Current Cancer Drug targets. 2002;2:37–43. doi: 10.2174/1568009023334033. [DOI] [PubMed] [Google Scholar]
- 34.Lo PK, Kanojia D, Liu X. CD49f and CD61 identify Her2/neu-induced mammary tumour-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFbeta signaling. Oncogene. 2011;31:2614–2626. doi: 10.1038/onc.2011.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. 2008;68:7711–7717. doi: 10.1158/0008-5472.CAN-08-1949. [DOI] [PubMed] [Google Scholar]
- 36.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 37.Kotamraju VR, Sharma S, Kolhar P, Agemy L, Pavlovich J, Ruoslahti E. Increasing tumor accessibility with conjugatable disulfide bridged tumor-penetrating peptides for cancer diagnosis and treatment. Breast Cancer (Auckl) 2015;9(Suppl 2):79–87. doi: 10.4137/BCBCR.S29426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fogal V, Zhang L, Krajewski S, Ruoslahti E. Cancer Res. 2008;68:7210–7218. doi: 10.1158/0008-5472.CAN-07-6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teesalu T, Sugahara KN, Ruoslahti E. Mapping of vascular ZIP codes by phage display. Methods Enzymol. 2012;503:35–56. doi: 10.1016/B978-0-12-396962-0.00002-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.