

Abstract
BACKGROUND
Brentuximab vedotin is an anti-CD30 antibody–drug conjugate that has been approved for relapsed and refractory Hodgkin’s lymphoma.
METHODS
We conducted an open-label, multicenter, randomized phase 3 trial involving patients with previously untreated stage III or IV classic Hodgkin’s lymphoma, in which 664 were assigned to receive brentuximab vedotin, doxorubicin, vinblastine, and dacarbazine (A+AVD) and 670 were assigned to receive doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD). The primary end point was modified progression-free survival (the time to progression, death, or noncomplete response and use of subsequent anticancer therapy) as adjudicated by an independent review committee. The key secondary end point was overall survival.
RESULTS
At a median follow-up of 24.9 months, 2-year modified progression-free survival rates in the A+AVD and ABVD groups were 82.1% (95% confidence interval [CI], 78.7 to 85.0) and 77.2% (95% CI, 73.7 to 80.4), respectively, a difference of 4.9 percentage points (hazard ratio for an event of progression, death, or modified progression, 0.77; 95% CI, 0.60 to 0.98; P = 0.03). There were 28 deaths with A+AVD and 39 with ABVD (hazard ratio for interim overall survival, 0.72 [95% CI, 0.44 to 1.17]; P = 0.19). All secondary efficacy end points trended in favor of A+AVD. Neutropenia occurred in 58% of the patients receiving A+AVD and in 45% of those receiving ABVD; in the A+AVD group, the rate of febrile neutropenia was lower among the 83 patients who received primary prophylaxis with granulocyte colony-stimulating factor than among those who did not (11% vs. 21%). Peripheral neuropathy occurred in 67% of patients in the A+AVD group and in 43% of patients in the ABVD group; 67% of patients in the A+AVD group who had peripheral neuropathy had resolution or improvement at the last follow-up visit. Pulmonary toxicity of grade 3 or higher was reported in less than 1% of patients receiving A+AVD and in 3% of those receiving ABVD. Among the deaths that occurred during treatment, 7 of 9 in the A+AVD group were associated with neutropenia and 11 of 13 in the ABVD group were associated with pulmonary-related toxicity.
CONCLUSIONS
A+AVD had superior efficacy to ABVD in the treatment of patients with advanced-stage Hodgkin’s lymphoma, with a 4.9 percentage-point lower combined risk of progression, death, or noncomplete response and use of subsequent anticancer therapy at 2 years. (Funded by Millennium Pharmaceuticals and Seattle Genetics; ECHELON-1 ClinicalTrials.gov number, NCT01712490; EudraCT number, 2011-005450-60.)
Outcomes for patients with advanced-stage Hodgkin’s lymphoma have improved dramatically over the past half century.1 Although regional differences exist, the most commonly used frontline regimen — doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) — has not been modified since its original description in 1975.
Up to 30% of patients with stage III or IV Hodgkin’s lymphoma harbor refractory disease or relapse after frontline treatment with ABVD.2–4 Bleomycin is associated with unpredictable and sometimes fatal pulmonary toxicity and is often dropped from later cycles of chemotherapy owing to pulmonary symptoms.5,6 Recent studies suggest that response-adapted therapy guided by interim positron-emission tomography (PET) with 18F-fluorodeoxyglucose can provide a more individualized treatment approach, in which treatment intensity is de-escalated or intensified depending on the early response to treatment.7,8 Efforts are also being made to incorporate new drugs into established backbone regimens to improve efficacy and reduce toxicity.9
CD30 is a characteristic surface antigen expressed on Reed–Sternberg cells in classic Hodgkin’s lymphoma.10 Brentuximab vedotin is an antibody–drug conjugate composed of an anti-CD30 monoclonal antibody conjugated by a protease-cleavable linker to the microtubule-disrupting agent monomethyl auristatin E. Brentuximab vedotin has been approved for the treatment of classic Hodgkin’s lymphoma after failure of autologous stem-cell transplantation or after two or more multiagent chemotherapy regimens in patients who are not candidates for transplantation. The drug has also been approved as post-transplantation consolidation therapy for patients with Hodgkin’s lymphoma who are at increased risk for relapse or progression.11,12
A previous phase 1, dose-escalation trial involving patients with advanced Hodgkin’s lymphoma evaluated the use of frontline brentuximab vedotin combined with either ABVD or doxorubicin, vinblastine, and dacarbazine (AVD).13 Brentuximab vedotin plus AVD (A+AVD) had an acceptable side-effect profile and resulted in complete response in 24 of 25 patients (96%). Long-term follow-up showed a 5-year failure-free survival rate of 92% and an overall survival rate of 100% with A+AVD.14 On the basis of these findings, ECHELON-1, a large, international, open-label, randomized, multicenter, phase 3 trial was conducted to compare A+AVD with ABVD as frontline therapy in patients with stage III or IV classic Hodgkin’s lymphoma.
METHODS
TRIAL DESIGN
Patients were randomly assigned in a 1:1 ratio to receive A+AVD (1.2 mg of brentuximab vedotin per kilogram of body weight, 25 mg of doxorubicin per square meter of body-surface area, 6 mg of vinblastine per square meter, and 375 mg of dacarbazine per square meter) or ABVD (25 mg of doxorubicin per square meter, 10 units of bleomycin per square meter, 6 mg of vinblastine per square meter, and 375 mg of dacarbazine per square meter) intravenously on days 1 and 15 of each 28-day cycle for up to 6 cycles. Brentuximab vedotin was administered over 30 minutes, starting within approximately 1 hour after completion of AVD. Dose reductions and modifications are described in Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org. Patients were stratified according to region (Americas vs. Europe vs. Asia) and International Prognostic Score (IPS) risk group (low risk vs. intermediate risk vs. high risk). The IPS ranges from 0 to 7, with a score of 0 or 1 indicating low risk of treatment failure, a score of 2 or 3 intermediate risk, and a score of 4 to 7 high risk (see Table S2 in the Supplementary Appendix).15 The results of PET conducted at the end of the second 28-day cycle of treatment (hereafter referred to as PET2) guided an optional switch to alternative frontline therapy at the treating physician’s discretion for patients with a Deauville score of 5. The Deauville score is a 5-point scale on which higher scores indicate greater uptake of 18F-fluorodeoxyglucose at involved sites on PET. A score of 1 indicates no uptake, a score of 2 uptake at an initial site that is less than or equal to the uptake at the mediastinum, a score of 3 uptake at an initial site that is greater than uptake at the mediastinum but less than or equal to uptake at the liver, a score of 4 uptake at an initial site that is moderately increased as compared with the uptake at the liver, and a score of 5 markedly increased uptake at any site or uptake at a new site of disease.16
OVERSIGHT
The ECHELON-1 trial was conducted in accordance with regulatory requirements; the protocol (available at NEJM.org) was approved by institutional review boards and ethics committees at individual sites, and adhered to Good Clinical Practice guidelines (as defined by the International Conference on Harmonisation). A steering committee and an independent data and safety monitoring committee oversaw the conduct of the trial, and all the patients provided written informed consent.
The trial was designed by a committee consisting of six authors plus representatives of the sponsors, Millennium Pharmaceuticals and Seattle Genetics. Data were collected and trial procedures were overseen by trial investigators. Data were verified by the sponsors, analyzed by sponsor statisticians, and interpreted by academic authors and sponsor representatives. The manuscript was prepared by the authors with the assistance of a medical writer funded by the sponsors. All the authors had full access to the data, vouch for the completeness and accuracy of the data and adherence of the trial to the protocol, and had final responsibility for the manuscript content and the decision to submit the manuscript for publication.
PATIENTS
Patients were 18 years of age or older and had histologically confirmed advanced classic Hodgkin’s lymphoma (Ann Arbor stage III or IV, as determined on a 4-point scale, with higher stages indicating more widespread disease),17 according to the World Health Organization classification system.18 Patients who had not been previously treated with systemic chemotherapy or radiotherapy were eligible. Patients were required to have an Eastern Cooperative Oncology Group performance status of 0, 1, or 2 (on a scale of 0 to 5, with higher scores indicating greater disability)19; satisfactory absolute neutrophil counts (≥1500 per cubic millimeter), platelet counts (≥75,000 per cubic millimeter), and hemoglobin levels (≥8 g per deciliter) (with the exception of patients with involvement of the marrow); satisfactory levels of markers of liver function (total bilirubin level, <1.5 times the upper limit of normal [with the exception of patients with Gilbert’s syndrome] and alanine aminotransferase or aspartate aminotransferase levels, <3 times the upper limit of normal [with the exception of patients with involvement of the liver]); and satisfactory levels of markers of kidney function (serum creatinine level, <2.0 mg per deciliter [177 μmol per liter]; creatinine clearance or calculated creatinine clearance, >40 ml per minute; or both). Patients with nodular lymphocyte-predominant Hodgkin’s lymphoma were ineligible, as were those with peripheral sensory or motor neuropathy, a positive pregnancy test, known cerebral or meningeal disease, any evidence of residual disease from another cancer, diagnosis of another cancer within 3 years before the first dose, or any clinically relevant cardiovascular conditions.
END POINTS
The primary end point was modified progression-free survival, defined as time to disease progression, death, or modified progression (with the latter defined as evidence of noncomplete response after completion of frontline therapy according to review by an independent committee, followed by subsequent anticancer therapy). This end point was chosen specifically to evaluate the effectiveness of the primary chemotherapy and encompasses three possible outcomes, each of which represents a failure of the primary chemotherapy to eliminate Hodgkin’s lymphoma: documented progression20 at any time after initiation of primary chemotherapy, death from any cause, and detection of a response that was less than complete at the end of primary chemotherapy (Deauville score of 3, 4, or 5 on a PET scan), followed by the delivery of subsequent anticancer therapy. The latter outcome was considered to be an event only if noncomplete response was confirmed during review by an independent committee, whose members were unaware of group assignments, and was followed by the delivery of subsequent anticancer treatment that was not specified in the protocol. Additional justifications for, and explanation of, this choice of primary end point are provided in the Supplementary Appendix. Timing of the modified progression event was the date on which the first PET scan was obtained after completion of frontline therapy, showing the absence of complete response. In the absence of disease progression, a switch to an alternative frontline therapy before completion of primary chemotherapy with the randomized regimen was not considered to be an event.
The key secondary end point was overall survival, defined as the time from randomization to death from any cause. Other secondary and exploratory end points are described in the protocol.
ASSESSMENTS
Response and progression were evaluated in accordance with the Revised Response Criteria for Malignant Lymphoma.20 Computed tomographic scans were obtained at screening, at the end of cycle 2, after administration of the last dose of frontline therapy, and during the follow-up period (every 3 months for the first year and every 6 months thereafter). PET scans were obtained at screening, at the end of cycle 2, and at the end of treatment. Safety outcomes were the incidence of adverse events (defined according to the Medical Dictionary for Regulatory Activities [MedDRA], version 19.0, and the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03) and changes in vital signs and laboratory test results.
STATISTICAL ANALYSIS
According to statistical calculations, an estimated 260 modified progression-free survival events would give the trial 90% power to detect a hazard ratio for disease progression, death, or modified progression of 0.67 at a one-sided significance level of 0.025. The trial was powered on the following assumption: a 2-year modified progression-free survival of 81% for patients in the A+AVD group and 73% for patients in the ABVD group. Randomization of approximately 1240 patients was planned to achieve (with 95% probability) 260 modified progression-free survival events. The primary end point was summarized with the use of the Kaplan–Meier method and evaluated with the use of a stratified log-rank test. A stratified Cox regression model was used to estimate the hazard ratio and the 95% confidence interval for the treatment effect. The stratification factors included region and IPS risk group at baseline. The interim analysis for overall survival was to be performed if the result of the primary analysis was statistically significant. The final overall survival analysis will be performed after 112 deaths have occurred. Overall type I error for the overall survival analysis will be controlled with the use of the O’Brien–Fleming method with the Lan–DeMets alpha spending function.
All efficacy evaluations were performed in the intention-to-treat population unless otherwise specified. Safety was analyzed in patients who received at least one dose of the trial drug (the safety population).
RESULTS
PATIENTS
From November 19, 2012, through January 13, 2016, a total of 1334 patients at 218 sites in 21 countries were randomly assigned to receive A+AVD (664 patients) or ABVD (670 patients) (intention-to-treat population) (Fig. S1 in the Supplementary Appendix). Overall, 58% of the patients were men, 64% had stage IV disease, 62% had extranodal involvement at diagnosis, 58% had B symptoms (i.e., weight loss, night sweats, and fever), and the median age was 36 years (34% of patients were ≥45 years of age). Baseline characteristics were generally well balanced between the two groups (Table 1, and Table S3 in the Supplementary Appendix).
Table 1.
Patient Demographic and Clinical Characteristics at Baseline (Intention-to-Treat Population).*
| Characteristic | A+AVD (N = 664) |
ABVD (N = 670) |
Total (N = 1334) |
|---|---|---|---|
| Male sex — no. (%) | 378 (57) | 398 (59) | 776 (58) |
| Age — yr | |||
| Median | 35 | 37 | 36 |
| Range | 18–82 | 18–83 | 18–83 |
| Age categories — no. (%) | |||
| <45 yr | 451 (68) | 423 (63) | 874 (66) |
| 45–59 yr | 129 (19) | 145 (22) | 274 (21) |
| 60–64 yr | 24 (4) | 40 (6) | 64 (5) |
| ≥65 yr | 60 (9) | 62 (9) | 122 (9) |
| Regions — no. (%) | |||
| Americas | 261 (39) | 262 (39) | 523 (39) |
| Europe | 333 (50) | 336 (50) | 669 (50) |
| Asia | 70 (11) | 72 (11) | 142 (11) |
| Ann Arbor stage at initial diagnosis — no. (%)† | |||
| Stage II‡ | 1 (<1) | 0 | 1 (<1) |
| Stage III | 237 (36) | 246 (37) | 483 (36) |
| Stage IV | 425 (64) | 421 (63) | 846 (64) |
| Not applicable, unknown, or missing | 1 (<1) | 3 (<1) | 4 (<1) |
| International Prognostic Score — no. (%)§ | |||
| 0 or 1 | 141 (21) | 141 (21) | 282 (21) |
| 2 or 3 | 354 (53) | 351 (52) | 705 (53) |
| 4 to 7 | 169 (25) | 178 (27) | 347 (26) |
| ECOG performance status — no. (%)¶ | |||
| 0 | 376 (57) | 378 (57) | 754 (57) |
| 1 | 259 (39) | 262 (39) | 521 (39) |
| 2 | 28 (4) | 26 (4) | 54 (4) |
| Not obtained or missing | 1 (<1) | 4 (<1) | 5 (<1) |
| Extranodal involvement at diagnosis — no. (%) | |||
| Yes | 411 (62) | 416 (62) | 827 (62) |
| 1 extranodal site | 217 (33) | 223 (33) | 440 (33) |
| >1 extranodal sites | 194 (29) | 193 (29) | 387 (29) |
| No | 217 (33) | 228 (34) | 445 (33) |
| Unknown or missing | 36 (5) | 26 (4) | 62 (5) |
| Patients with any B symptom — no. (%)‖ | 399 (60) | 381 (57) | 780 (58) |
A full description of patient demographics and clinical characteristics at baseline can be found in Table S3 in the Supplementary Appendix. Percentages may not total 100 because of rounding. A+AVD denotes brentuximab vedotin plus doxorubicin, vinblastine, dacarbazine, and ABVD doxorubicin, bleomycin, vinblastine, and dacarbazine.
The Ann Arbor staging system ranges from I to IV, with higher stages indicating more widespread disease.
Patients in this category had a major protocol violation.
The International Prognostic Score ranges from 0 to 7, with higher scores indicating increased risk of treatment failure. Scores of 0 to 1 denote low risk, scores of 2 to 3 intermediate risk, and scores of 4 to 7 high risk.
Values for Eastern Cooperative Oncology Group (ECOG) performance status range from 0 to 5, with higher scores indicating greater disability.
B symptoms consist of night sweats, unexplained fever (temperature >38°C), or loss of more than 10% of body weight.
EFFICACY
After a median follow-up of 24.9 months (range, 0 to 49.3), the rate of the primary end point of independently determined modified progression-free survival was significantly higher in the A+AVD group than in the ABVD group (2-year modified progression-free survival rate, 82.1% [95% confidence interval {CI}, 78.7 to 85.0] vs. 77.2% [95% CI, 73.7 to 80.4]; hazard ratio for progression, death, or modified progression, 0.77 [95% CI, 0.60 to 0.98]; P = 0.03), corresponding to a 23% risk reduction (Fig. 1A). Events of progression, death, or modified progression occurred in 117 patients in the A+AVD group and in 146 patients in the ABVD group; disease progression occurred in 90 and 102 patients, respectively; death from any cause in 18 and 22 patients, respectively, and receipt of subsequent anticancer therapy after failure to achieve a complete response at the completion of frontline therapy (modified progression) in 9 and 22 patients, respectively (Table 2). The majority (71%) of these subsequent anticancer therapies consisted of salvage chemotherapy (7 of 9 patients in the A+AVD group and 15 of 22 patients in the ABVD group), with radiotherapy given to the remainder of patients in both groups (Table S4 in the Supplementary Appendix). Modified progression events assigned because of an end-of-treatment PET scan and subsequent treatment were predominantly associated with a Deauville score of 4 or 5 (a score of 3 was recorded in 7 of 31 patients [23%], a score of 4 in 10 of 31 patients [32%], and a score of 5 in 14 of 31 patients [45%]); these events also met the criteria for a progression event according to investigator assessment. Of note, only 7 of the 21 patients with a Deauville score of 3 on the end-of-treatment PET scan went on to receive additional therapy and were therefore determined to have had a modified progression event (2 patients in the A+AVD group and 5 patients in the ABVD group; Tables 2 and 3).
Figure 1. Modified Progression-free Survival in the Intention-to-Treat Population.
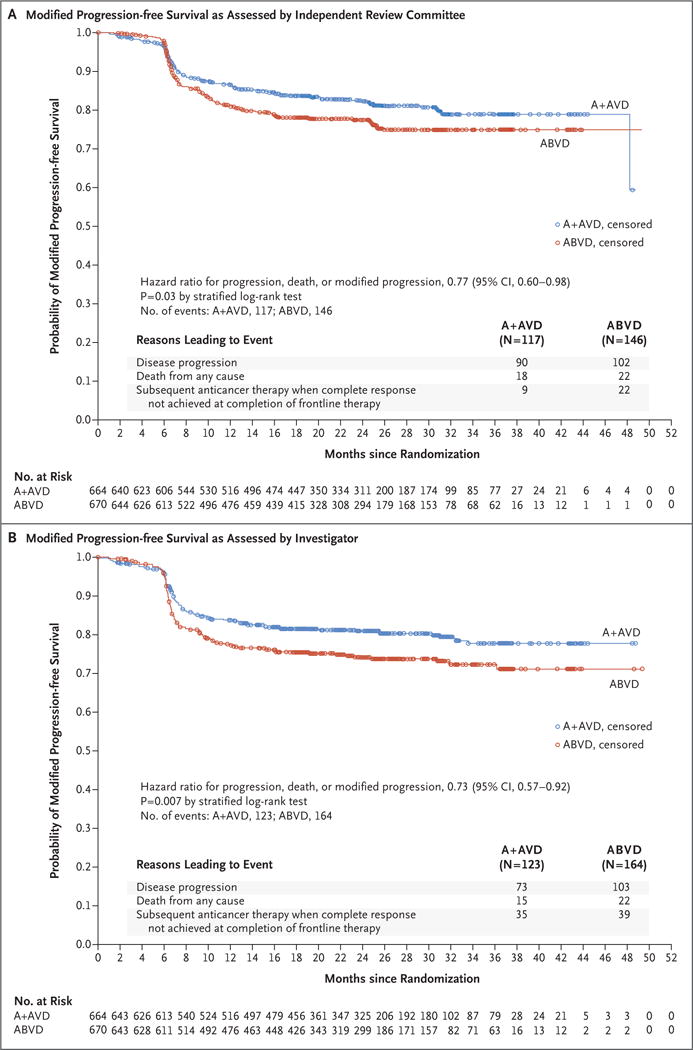
Panel A shows Kaplan–Meier estimates of modified progression-free survival, by treatment group, according to the independent review committee. The hazard ratio for treatment with A+AVD versus ABVD and the 95% confidence intervals (CIs) were based on a stratified Cox proportional-hazards regression model, with treatment as the explanatory variable. Stratification factors included region and International Prognostic Score risk group at baseline. Panel B shows Kaplan–Meier estimates of modified progression-free survival, by treatment group, according to investigators. In Panels A and B, circles indicate censored data. A+AVD denotes brentuximab vedotin plus doxorubicin, vinblastine, and dacarbazine, and ABVD doxorubicin, bleomycin, vinblastine, and dacarbazine.
Table 2.
Summary of Modified Progression-free Survival According to the Independent Review Committee and Concordance with Events Noted by Trial Investigators (Intention-to-Treat Population).
| Events | A+AVD (N = 664) |
ABVD (N = 670) |
Total (N = 1334) |
|---|---|---|---|
| Patients with events per independent review committee — no. | 117 | 146 | 263 |
| Progression — no./total no. (%) | 90/117 (77) | 102/146 (70) | 192/263 (73) |
| Death — no./total no. (%) | 18/117 (15) | 22/146 (15) | 40/263 (15) |
| Positive PET scan and subsequent treatment — no./total no. (%)* | 9/117 (8) | 22/146 (15) | 31/263 (12) |
| Patients with positive PET scan and subsequent treatment — no. | 9 | 22 | 31 |
| Salvage chemotherapy — no./total no. (%)† | 7/9 (78) | 15/22 (68) | 22/31 (71) |
| Met criteria for PFS event | |||
| PFS event or modified event reported by investigator — no. | 7 | 15 | 22 |
| PFS event reported by investigator — no./total no. (%) | 7/7 (100) | 13/15 (87) | 20/22 (91) |
| PFS event reported by independent review committee — no./total no. (%) | 2/7 (29) | 3/15 (20) | 5/22 (23) |
| Deauville score at end of treatment — no./total no. (%)‡ | |||
| 1 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 |
| 3 | 0 | 2/15 (13) | 2/22 (9) |
| 4 | 3/7 (43) | 4/15 (27) | 7/22 (32) |
| 5 | 4/7 (57) | 9/15 (60) | 13/22 (59) |
| Radiation — no./total no. (%) | 2/9 (22) | 7/22 (32) | 9/31 (29) |
| Met criteria for PFS event | |||
| PFS event or modified event reported by investigator — no. | 2 | 7 | 9 |
| PFS event reported by investigator — no./total no. (%) | 0 | 1/7 (14) | 1/9 (11) |
| PFS event reported by independent review committee — no./total no. (%) | 0 | 1/7 (14) | 1/9 (11) |
| Deauville score at end of treatment — no./total no. (%)‡ | |||
| 1 | 0 | 0 | 0 |
| 2 | 0 | 0 | 0 |
| 3 | 2/2 (100) | 3/7 (43) | 5/9 (56) |
| 4 | 0 | 3/7 (43) | 3/9 (33) |
| 5 | 0 | 1/7 (14) | 1/9 (11) |
There were 58 patients at risk for a modified progression event (end-of-treatment Deauville score ≥3 and no progressive disease at the end of treatment): 19 in the group receiving A+AVD versus 39 in the group receiving ABVD. However, only 9 patients in the A+AVD group and 22 patients in the ABVD group actually had a modified progression event because they received subsequent treatment. PET denotes positron-emission tomography, and PFS progression-free survival.
Salvage chemotherapy included the terms chemotherapy, high-dose chemotherapy plus transplantation, and immunotherapy according to medical review.
The Deauville score is a 5-point scale on which higher scores indicate greater uptake of 18F-fluorodeoxyglucose at involved sites on PET. A score of 1 indicates no uptake, a score of 2 uptake at an initial site that is less than or equal to the uptake at the mediastinum, a score of 3 uptake at an initial site that is greater than uptake at the mediastinum but less than or equal to uptake at the liver, a score of 4 uptake at an initial site that is moderately increased as compared with uptake at the liver, and a score of 5 markedly increased uptake at any site or uptake at a new site of disease. The absence of complete response at the end of primary chemotherapy was defined as a Deauville score of 3, 4, or 5.
Table 3.
Summary of Responses in the Intention-to-Treat Population.
| Measure | A+AVD (N = 664) |
ABVD (N = 670) |
Difference (95% CI)* |
|---|---|---|---|
| no. (%) | % | ||
| Complete response at end of randomized regimen† | 488 (73) | 472 (70) | 3.0 (−2.3 to 8.4) |
| Overall response at end of randomized regimen‡ | 569 (86) | 553 (83) | 3.2 (−2.2 to 8.6) |
| Complete response at end of frontline therapy§ | 488 (73) | 474 (71) | 2.7 (−2.6 to 8.1) |
| Deauville score¶ | |||
| ≤3 After completion of frontline therapy) | 570 (86) | 551 (82) | 3.6 (−1.8 to 9.0) |
| ≤2 After completion of frontline therapy | 563 (85) | 537 (80) | 4.6 (−0.8 to 10.0) |
| Summary at cycle 2 | |||
| 1 | 435 (66) | 414 (62) | |
| 2 | 131 (20) | 133 (20) | |
| 3 | 22 (3) | 30 (4) | |
| 4 | 26 (4) | 28 (4) | |
| 5 | 21 (3) | 30 (4) | |
| Unavailable | 29 (4) | 35 (5) | |
| Summary after completion of primary chemotherapy | |||
| 1 | 444 (67) | 425 (63) | |
| 2 | 119 (18) | 112 (17) | |
| 3 | 7 (1) | 14 (2) | |
| 4 | 12 (2) | 20 (3) | |
| 5 | 46 (7) | 45 (7) | |
| Unavailable | 36 (5) | 54 (8) | |
Confidence intervals (CIs) were calculated from the exact confidence interval, have not been adjusted for the multiple comparisons, and should not be used for definitive comparisons.
Complete response at the end of the randomized regimen is defined as the proportion of patients who had complete response20 at the end of treatment with either regimen (A+AVD or ABVD).
Overall response at the end of the randomized regimen is defined as the proportion of patients who had complete or partial response20 at the end of treatment with either regimen (A+AVD or ABVD).
Complete response at the end of frontline therapy is defined as the proportion of patients who had complete response after the completion of either the randomized regimen (A+AVD or ABVD) or alternate frontline therapy.
The Deauville score is a 5-point scale on which higher scores indicate greater uptake of 18F-fluorodeoxyglucose at involved sites on PET. A score of 1 indicates no uptake, a score of 2 uptake at an initial site that is less than or equal to the uptake at the mediastinum, a score of 3 uptake at an initial site that is greater than uptake at the mediastinum but less than or equal to uptake at the liver, a score of 4 uptake at an initial site that is moderately increased as compared with uptake at the liver, and a score of 5 markedly increased uptake at any site or uptake at a new site of disease. The absence of complete response at the end of primary chemotherapy was defined as a Deauville score of 3, 4, or 5.
According to investigator assessment, the 2-year modified progression-free survival rate was 81.0% (95% CI, 77.6 to 83.9) with the A+AVD regimen versus 74.4% (95% CI, 70.7 to 77.7) with the ABVD regimen, corresponding to a 27% lower overall risk of an event among patients treated with A+AVD than among those treated with ABVD (hazard ratio for progression, death, or modified progression, 0.73; 95% CI, 0.57 to 0.92; P = 0.007) (Fig. 1B). There was 91% concordance between independent review and investigator determination of a modified progression-free survival event.
Prespecified subgroup analyses of modified progression-free survival showed a hazard ratio of less than 1 for the A+AVD regimen versus the ABVD regimen in the majority of subgroups (Fig. 2). Certain subgroups of patients appeared to benefit more with A+AVD than with ABVD. These subgroups included patients from North America, patients with involvement of more than one extranodal site, patients with an IPS indicating a high risk of treatment failure (scores of 4 to 7), men, patients with stage IV disease, and patients younger than 60 years of age. The rates of negativity at PET2 (Deauville score, 1 to 3) were 89% with A+AVD versus 86% with ABVD.
Figure 2. Forest-Plot Analysis of Modified Progression-free Survival.
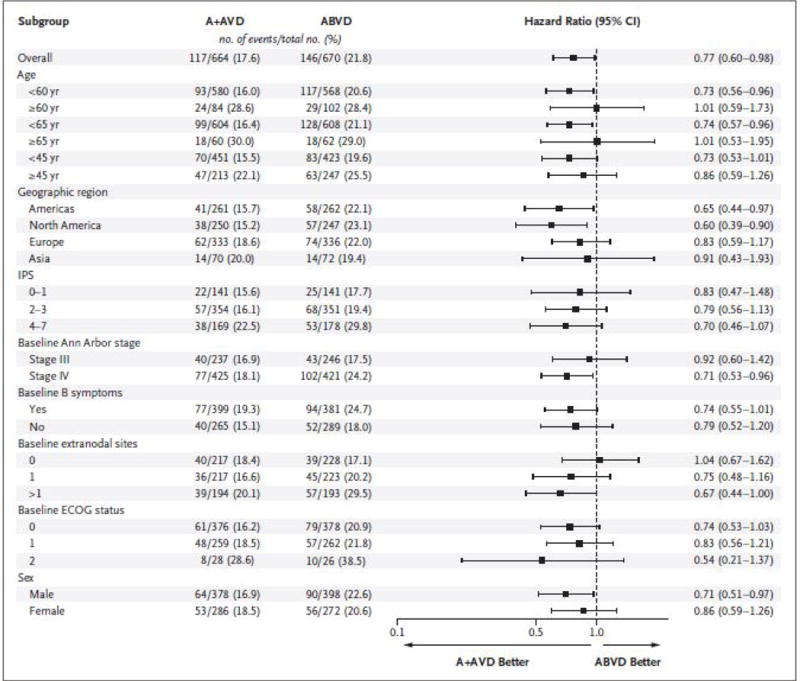
This forest plot shows modified progression-free survival according to the independent review committee in key prespecified subgroups. The hazard ratio for treatment with A+AVD versus ABVD and the 95% confidence intervals (CIs) were based on an unstratified Cox proportional-hazards regression model, with treatment as the explanatory variable. The intention-to-treat population included all the patients who underwent randomization. The International Prognostic Score (IPS) ranges from 0 to 7, with higher scores indicating increased risk of treatment failure: low risk, 0 or 1; intermediate risk, 2 or 3; and high risk, 4 to 7. The Ann Arbor staging system ranges from I to IV, with higher stages indicating more widespread disease. B symptoms consist of night sweats, unexplained fever (temperature >38°C), or loss of more than 10% of body weight. Values for the Eastern Cooperative Oncology Group (ECOG) performance status range from 0 to 5, with higher scores indicating greater disability.
There were 28 deaths in the A+AVD group (9 during treatment [within 30 days after the last dose of frontline therapy] and 19 during follow-up [31 days or more after the last dose of frontline therapy]) and 39 deaths in the ABVD group (13 during treatment and 26 during follow-up). The interim 2-year overall survival rate for the A+AVD group was 96.6% (95% CI, 94.8 to 97.7) and that for the ABVD group was 94.9% (95% CI, 92.9 to 96.4), which corresponded to a reduction in the risk of death of 28% in favor of the A+AVD regimen (hazard ratio, 0.72; 95% CI, 0.44 to 1.17; P = 0.19) (Fig. S2 in the Supplementary Appendix). Results for other secondary end points are shown in Table 3. Only 15 of 662 patients who received A+AVD and 9 of 659 patients who received ABVD switched to alternative chemotherapy during frontline therapy for reasons other than progressive disease (a Deauville score of 5 in 1 of 15 and 4 of 9 patients, respectively; adverse events in 12 of 15 and 1 of 9 patients, respectively; and other reasons in 2 of 15 and 4 of 9 patients, respectively) (Table S5 in the Supplementary Appendix).
Overall, fewer patients in the A+AVD group than in the ABVD group received subsequent anticancer therapies. Recipients of these therapies in the A+AVD group versus the ABVD group were as follows: radiation (in 52 patients in each group), chemotherapy (66 vs. 99), high-dose chemotherapy plus transplantation (36 vs. 54), immunotherapy (10 vs. 16), and chemotherapy plus radiation (2 vs. 3).
SAFETY
The median duration of treatment and the number of completed cycles were similar in the two groups (Table S6 in the Supplementary Appendix). The proportions of patients who received the regimens as intended, without dose modification such as delays, holds, or reductions, are shown in Table S6 in the Supplementary Appendix.
The safety profiles for both groups are summarized in Table 4, and in Table S7 in the Supplementary Appendix. Overall, neutropenia was reported in 58% of the patients receiving A+AVD and in 45% of the patients receiving ABVD, and febrile neutropenia was reported in 19% and 8%, respectively. In both groups, the incidence of febrile neutropenia was higher among patients 60 years of age or older than among those younger than 60 years of age (37% vs. 17% in the A+AVD group and 17% vs. 6% in the ABVD group). The incidence of febrile neutropenia was also higher in earlier rather than later cycles of therapy in both groups (9% in cycle 1 vs. 1 to 6% in cycles 2 through 6 in the A+AVD group and 4% in cycle 1 vs. ≤1% in cycles 2 through 6 in the ABVD group). The incidence of discontinuation of any trial drug due to neutropenia or febrile neutropenia was 1% or less in both groups.
Table 4.
Summary of Adverse Events in the Safety Population.*
| Events | A+AVD (N = 662) |
ABVD (N = 659) |
|---|---|---|
| no. (%) | ||
| Adverse events | ||
| Any adverse event | 653 (99) | 646 (98) |
| Grade ≥3 adverse event | 549 (83) | 434 (66) |
| Serious adverse event | 284 (43) | 178 (27) |
| Adverse event resulting in drug discontinuation | 88 (13) | 105 (16) |
| Death during treatment† | 9 (1) | 13 (2) |
| Death due to drug-related adverse events | 8 (1) | 7 (1) |
| Hospitalizations | 242 (37) | 186 (28) |
| Common adverse events‡ | ||
| Neutropenia | ||
| Any grade | 382 (58) | 295 (45) |
| Grade ≥3 | 357 (54) | 260 (39) |
| Constipation | ||
| Any grade | 279 (42) | 241 (37) |
| Grade ≥3 | 11 (2) | 4 (<1) |
| Vomiting | ||
| Any grade | 216 (33) | 183 (28) |
| Grade ≥3 | 23 (3) | 9 (1) |
| Fatigue | ||
| Any grade | 211 (32) | 211 (32) |
| Grade ≥3 | 19 (3) | 7 (1) |
| Peripheral sensory neuropathy | ||
| Any grade | 189 (29) | 111 (17) |
| Grade ≥3 | 31 (5) | 3 (<1) |
| Diarrhea | ||
| Any grade | 181 (27) | 121 (18) |
| Grade ≥3 | 19 (3) | 5 (<1) |
| Pyrexia | ||
| Any grade | 179 (27) | 147 (22) |
| Grade ≥3 | 19 (3) | 13 (2) |
| Peripheral neuropathy | ||
| Any grade | 174 (26) | 85 (13) |
| Grade ≥3 | 27 (4) | 6 (<1) |
| Abdominal pain | ||
| Any grade | 142 (21) | 65 (10) |
| Grade ≥3 | 21 (3) | 4 (<1) |
| Stomatitis | ||
| Any grade | 138 (21) | 104 (16) |
| Grade ≥3 | 10 (2) | 3 (<1) |
For a full summary of adverse events, including rates of drug-related adverse events and deaths, see Table S7 in the Supplementary Appendix.
Death during treatment is a death that occurred within 30 days after the last dose of frontline therapy.
The events listed include the most clinically important common adverse events. Adverse events (those of any grade that occurred in at least 20% of the patients in either group) excluded from the table are nausea, alopecia, weight loss, and anemia.
The rate of infections (determined in accordance with the MedDRA primary system organ-class term of “infections and infestations”) in the A+AVD group was 55% (361 of 662 patients) and the rate in the ABVD group was 50% (331 of 659 patients); rates of infection of grade 3 or higher were 18% (116 of 662 patients) and 10% (66 of 659 patients), respectively. Discussion with the independent data and safety monitoring committee (after 76% of enrollment was complete) led to the recommendation of primary prophylaxis with granulocyte colony-stimulating factor (G-CSF) for patients who were yet to be enrolled and who would receive the A+AVD regimen, owing to the higher incidence of febrile neutropenia in that group. In the A+AVD group, the incidence of febrile neutropenia was lower among the 83 patients who received primary prophylaxis with G-CSF (defined as use of G-CSF by day 5 of treatment) than among those who did not (11% [9 of 83] vs. 21% [119 of 579]) (Table 5). The occurrence of infections and infestations of grade 3 or higher was also lower among the patients who received G-CSF than among those who did not (11% [9 of 83 patients] vs. 18% [107 of 579 patients]).
Table 5.
Summary of Adverse Events in Patients Who Did and Those Who Did Not Receive Primary Prophylaxis with Granulocyte Colony-Stimulating Factor.
| Events | A+AVD (N = 662) |
ABVD (N = 659) |
||
|---|---|---|---|---|
| No (N = 579) |
Yes (N = 83) |
No (N = 616) |
Yes (N = 43) |
|
| number (percent) | ||||
| Febrile neutropenia during treatment | 119 (21) | 9 (11) | 49 (8) | 3 (7) |
| Any neutropenia* | 425 (73) | 29 (35) | 352 (57) | 9 (21) |
| Neutropenia grade ≥3* | 406 (70) | 24 (29) | 309 (50) | 8 (19) |
| Grade ≥3 adverse event | 502 (87) | 47 (57) | 414 (67) | 20 (47) |
| Infections and infestations (SOC) | 322 (56) | 39 (47) | 312 (51) | 19 (44) |
| Grade ≥3 infections and infestations (SOC) | 107 (18) | 9 (11) | 63 (10) | 3 (7) |
| Serious adverse event | 257 (44) | 27 (33) | 171 (28) | 7 (16) |
| Serious adverse events of febrile neutropenia, neutropenia, sepsis, neutropenic sepsis, pyrexia, or infections and infestations (SOC) | 190 (33) | 20 (24) | 107 (17) | 4 (9) |
| Deaths during treatment† | 8 (1) | 1 (1)‡ | 12 (2) | 1 (2) |
Neutropenia and neutropenia grade 3 or higher (neutrophil count <1000 per cubic millimeter) include the preferred terms of “neutropenia” and “neutrophil count decreased.” SOC denotes system organ class for the noted event.
Death during treatment is a death that occurred within 30 days after the last dose of frontline therapy.
The patient in the A+AVD group who had G-CSF primary prophylaxis received G-CSF for treatment of neutropenia, which occurred before day 5.
Peripheral neuropathy (determined on the basis of a standardized MedDRA query; see Table S8 in the Supplementary Appendix) occurred in 67% of the patients (442 of 662) receiving A+AVD and 43% of the patients (286 of 659) receiving ABVD. Grade 2 peripheral neuropathy occurred in 20% of the patients (130 of 662) in the A+AVD group versus 9% of the patients (57 of 659) in the ABVD group, and peripheral neuropathy of grade 3 or higher occurred in 11% of the patients (70 of 662) in the former group (with grade 4 occurring in 1 patient) versus 2% of the patients (11 of 659) in the latter. Among patients with peripheral neuropathy, a trial drug was discontinued in 10% in the A+AVD group (44 of 442) versus 4% in the ABVD group (11 of 286). Two thirds of the patients in the A+AVD group (295 of 442) who had peripheral neuropathy had resolution (43%, 191 of 442) or improvement by at least one grade (24%, 104 of 442) in terms of events related to peripheral neuropathy at the time of the last follow-up visit; at that time, 92% of ongoing events related to peripheral neuropathy were grade 1 (64%) or grade 2 (29%) in the A+AVD group. Pulmonary toxicity, defined as events related to interstitial lung disease (in accordance with a standardized MedDRA query), was reported in 2% of the patients (12 of 662) in the A+AVD group versus 7% (44 of 659) in the ABVD group; events of grade 3 or higher were reported in less than 1% of the patients (5 of 662) in the former group and 3% of the patients (21 of 659) in the latter.
During treatment, there were 9 deaths in the A+AVD group and 13 deaths in the ABVD group. In the A+AVD group, 7 deaths were associated with neutropenia (all occurred in patients who had not received primary prophylaxis with G-CSF before the onset of neutropenia, with the exception of 1 patient who entered the trial with preexisting neutropenia) and 2 deaths were due to myocardial infarction. In the ABVD group, 11 deaths were due to or associated with pulmonary-related toxicity and 1 death was due to cardiopulmonary failure. The cause of 1 death was unknown. Among the patients enrolled in the trial, 37% (242 of 662) in the A+AVD group and 28% (186 of 659) in the ABVD group were hospitalized during the trial.
Fertility was not formally assessed; however, similar numbers of pregnancies were reported in each treatment group, which suggests that there was no significant difference in the effect on fertility. At the time of this analysis, a total of 78 pregnancies were reported among trial participants and their partners (42 in the A+AVD group and 36 in the ABVD group).
DISCUSSION
This large, international, randomized phase 3 trial involving patients who had received a recent diagnosis of stage III or IV classic Hodgkin’s lymphoma showed that treatment with brentuximab vedotin plus AVD, as compared with standard treatment with ABVD, resulted in a statistically significant and clinically meaningful improvement in the rate of modified progression-free survival, with a difference at 2 years of 4.9 percentage points as assessed by an independent committee, whose members were unaware of group assignments and 6.6 percentage points as assessed by the trial investigators. These outcomes were associated with reductions in the overall risk of failure of the primary chemotherapy treatment of 23% as assessed by an independent review committee and 27% as assessed by the trial investigators.
The goal of frontline chemotherapy for Hodgkin’s lymphoma is to cure patients without the need for additional therapy. Because metabolically detectable residual disease is a reliable predictor of imminent progression, it is accepted practice to initiate subsequent chemotherapy or radiotherapy on the basis of a positive PET scan at the end of frontline treatment.21–23 In this context, the conventional end point of progression-free survival does not accurately assess the curative intent of frontline chemotherapy. Thus, in the ECHELON-1 trial, the primary end point was “modified” progression-free survival, which, in addition to disease progression or death, included modified progression, defined as evidence of non-complete response after the completion of front-line chemotherapy (based on independently assessed PET results) followed by subsequent anticancer therapy, as an event, thus accurately assessing the curative potential of the frontline chemotherapy.
The results of the interim overall survival analysis and all other secondary efficacy end points favored A+AVD, further supporting the conclusion that A+AVD is a more effective front-line treatment for advanced Hodgkin’s lymphoma than ABVD. Furthermore, the benefit of A+AVD was observed consistently in the majority of prespecified subgroups, including patients in whom there was involvement of more than one extranodal site, patients with an IPS indicating high risk for treatment failure (4 to 7), and patients with stage IV disease. The rate of positivity at PET2 was low, and a higher proportion of the patients treated with A+AVD than those treated with ABVD had negative results at PET2 (89% vs. 86%).
This trial shows that the addition of brentuximab vedotin and the elimination of bleomycin from frontline therapy in the A+AVD regimen lowers the incidence of pulmonary toxicity while improving efficacy as compared with the ABVD regimen. No new types of risk to patient safety were identified, although the incidence of febrile neutropenia was higher than expected and an increased incidence of infections was noted in the A+AVD group. The majority of the deaths during treatment in the A+AVD group were associated with febrile neutropenia; however, primary prophylaxis with G-CSF appeared to mitigate the increased risk of febrile neutropenia and its sequelae in the subgroup of 83 patients who received primary prophylaxis, resulting in reduced rates of neutropenia, febrile neutropenia, and serious infection. Peripheral neuropathy occurred more frequently in patients in the A+AVD group. The incidence of peripheral neuropathy of grade 3 or higher was increased by 9 percentage points in this group as compared with the ABVD group, and peripheral neuropathy was largely reversible, either resolving or abating in 67% of the patients in whom the condition had developed. Both the percentage of patients who received subsequent salvage chemotherapy and the percentage of patients who received high-dose chemotherapy followed by transplantation were approximately 33% lower among patients treated with A+AVD than among patients treated with ABVD; those treated with A+AVD were therefore less likely to be subject to the toxicities associated with aggressive salvage therapies.
The results of the ECHELON-1 trial are particularly important considering the opportunity A+AVD provides to administer a treatment to older patients that is at least equivalent in its effectiveness to ABVD, and to do so safely. Older patients with advanced Hodgkin’s lymphoma represent a special group, considering their incidence of disease (approximately 20% of all cases), lower rates of treatment efficacy, and typically higher rates of severe toxicity, particularly the pulmonary toxicity that is associated with bleomycin.6,24,25 When choosing frontline treatment, it is important to consider the lifetime burden of late and long-term adverse effects from salvage chemotherapy, radiotherapy, and transplantation (including infertility, pulmonary and cardiac toxicities, and secondary cancers).26,27 The A+AVD regimen is associated with more myelotoxicity (which can be ameliorated with prophylactic G-CSF) and neurotoxicity (which is largely reversible) than ABVD but substantially less pulmonary toxicity and appears to be more effective for the frontline treatment of advanced-stage classic Hodgkin’s lymphoma.
Supplementary Material
Acknowledgments
Supported by Millennium Pharmaceuticals, a wholly owned subsidiary of Takeda Pharmaceuticals, and Seattle Genetics.
We thank the patients who participated in this trial and their families, as well as the investigators and staff at all ECHELON-1 clinical sites; the members of the independent data and safety monitoring committee and the independent review committee; Jeanenne Chung, Vijay Maharaj, and Andy Chi of Millennium Pharmaceuticals and Eric Sievers and Naomi Hunder of Seattle Genetics for their contributions to the conception and execution of the ECHELON-1 trial; and Hannah Finnigan and Sarah Feeny of FireKite for writing support during the development of the manuscript.
APPENDIX
The authors’ full names and academic degrees are as follows: Joseph M. Connors, M.D., Wojciech Jurczak, M.D., Ph.D., David J. Straus, M.D., Stephen M. Ansell, M.D., Ph.D., Won S. Kim, M.D., Ph.D., Andrea Gallamini, M.D., Anas Younes, M.D., Sergey Alekseev, M.D., Árpád Illés, M.D., D.Sci., Marco Picardi, M.D., Ewa Lech-Maranda, M.D., Ph.D., Yasuhiro Oki, M.D., Tatyana Feldman, M.D., Piotr Smolewski, M.D., Ph.D., Kerry J. Savage, M.D., Nancy L. Bartlett, M.D., Jan Walewski, M.D., Robert Chen, M.D., Radhakrishnan Ramchandren, M.D., Pier L. Zinzani, M.D., Ph.D., David Cunningham, M.B. Ch.B., M.D., Andras Rosta, Ph.D., Neil C. Josephson, M.D., Eric Song, Ph.D., Jessica Sachs, M.D., Rachael Liu, Ph.D., Hina A. Jolin, Pharm.D., Dirk Huebner, M.D., and John Radford, M.D.
The authors’ affiliations are as follows: the University of British Columbia and the Department of Medical Oncology, British Columbia Cancer Agency Centre for Lymphoid Cancer, Vancouver, Canada (J.M.C., K.J.S.); the Department of Hematology, Jagiellonian University, Krakow (W.J.), the Department of Hematology, Institute of Hematology and Transfusion Medicine, Warsaw (E.L.-M.), the Department of Hematology and Transfusion Medicine, Center of Postgraduate Medical Education, Warsaw (E.L.-M.), the Department of Experimental Hematology, Medical University of Lodz, Lodz (P.S.), and the Department of Lymphoid Malignancy, the Maria Sklodowska-Curie Memorial Institute and Oncology Center, Warsaw (J.W.) — all in Poland; Lymphoma Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York (D.J.S., A.Y.); the Department of Internal Medicine, Division of Hematology, Mayo Clinic, Rochester, MN (S.M.A.); the Division of Hematology and Oncology, Sungkyunkwan University School of Medicine, Samsung Medical Center, Seoul, South Korea (W.S.K.); Research Innovation and Statistics, Antoine-Lacassagne Cancer Center, Nice, France (A.G.); Petrov Research Institute of Oncology, St. Petersburg, Russia (S.A.); the Department of Hematology, Faculty of Medicine, University of Debrecen, Debrecen (Á.I.), and the Department of Hematology, National Institute of Oncology, Budapest (A.R.) — both in Hungary; the Department of Advanced Biomedical Science, Federico II University Hospital, Naples (M.P.), and the Institute of Hematology Seràgnoli, University of Bologna, Bologna (P.L.Z.) — both in Italy; the Department of Lymphoma and Myeloma, M.D. Anderson Cancer Center, Houston (Y.O.); John Theurer Cancer Center, Hackensack University Medical Center, Hackensack, NJ (T.F.); the Department of Medicine, Division of Oncology, Washington University School of Medicine, St. Louis (N.L.B.); the Department of Hematology and Hematopoietic Cell Transplantation, City of Hope National Medical Center, Duarte, CA (R.C.); the Department of Hematology–Oncology, Barbara Ann Karmanos Cancer Center, Detroit (R.R.); Gastrointestinal and Lymphoma Unit, The Royal Marsden NHS Foundation Trust, Sutton (D.C.), and the Department of Medical Oncology, University of Manchester and the Christie NHS Foundation Trust, Manchester Academic Health Science Centre, Manchester (J.R.) — both in the United Kingdom; the Department of Clinical Development, Seattle Genetics, Bothell, WA (N.C.J., E.S.); and Oncology Clinical Research (J.S., H.A.J., D.H.) and Global Biostatistics (R.L.), Millennium Pharmaceuticals, Cambridge, MA.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Engert A. ABVD or BEACOPP for advanced Hodgkin lymphoma. J Clin Oncol. 2016;34:1167–9. doi: 10.1200/JCO.2015.64.8683. [DOI] [PubMed] [Google Scholar]
- 2.Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med. 1992;327:1478–84. doi: 10.1056/NEJM199211193272102. [DOI] [PubMed] [Google Scholar]
- 3.Carde P, Karrasch M, Fortpied C, et al. Eight cycles of ABVD versus four cycles of BEACOPPescalated plus four cycles of BEACOPPbaseline in stage III to IV, international prognostic score ≥ 3, high-risk Hodgkin lymphoma: first results of the phase III EORTC 20012 Intergroup Trial. J Clin Oncol. 2016;34:2028–36. doi: 10.1200/JCO.2015.64.5648. [DOI] [PubMed] [Google Scholar]
- 4.Gordon LI, Hong F, Fisher RI, et al. Randomized phase III trial of ABVD versus Stanford V with or without radiation therapy in locally extensive and advanced-stage Hodgkin lymphoma: an intergroup study coordinated by the Eastern Cooperative Oncology Group (E2496) J Clin Oncol. 2013;31:684–91. doi: 10.1200/JCO.2012.43.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canellos GP, Duggan D, Johnson J, Niedzwiecki D. How important is bleomycin in the adriamycin + bleomycin + vinblastine + dacarbazine regimen? J Clin Oncol. 2004;22:1532–3. doi: 10.1200/JCO.2004.99.010. [DOI] [PubMed] [Google Scholar]
- 6.Martin WG, Ristow KM, Habermann TM, Colgan JP, Witzig TE, Ansell SM. Bleomycin pulmonary toxicity has a negative impact on the outcome of patients with Hodgkin’s lymphoma. J Clin Oncol. 2005;23:7614–20. doi: 10.1200/JCO.2005.02.7243. [DOI] [PubMed] [Google Scholar]
- 7.Borchmann P, Goergen H, Kobe C, et al. Treatment reduction in patients with advanced-stage Hodgkin lymphoma and negative interim PET: final results of the international, randomized phase 3 trial HD18 by the German Hodgkin Study Group. Haematologica. 2017;102:S150. doi: 10.1016/S0140-6736(17)32134-7. abstract. [DOI] [PubMed] [Google Scholar]
- 8.Johnson P, Federico M, Kirkwood A, et al. Adapted treatment guided by interim PET-CT scan in advanced Hodgkin’s lymphoma. N Engl J Med. 2016;374:2419–29. doi: 10.1056/NEJMoa1510093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borchmann P, Eichenauer DA, Pluetschow A, et al. Targeted BEACOPP variants in patients with newly diagnosed advanced stage classical Hodgkin lymphoma: final analysis of a randomized phase II study. Blood. 2015;126:580. abstract. [Google Scholar]
- 10.Schwab U, Stein H, Gerdes J, et al. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature. 1982;299:65–7. doi: 10.1038/299065a0. [DOI] [PubMed] [Google Scholar]
- 11.Adcetris (brentuximab vedotin) Bothell, WA: Seattle Genetics; 2016. (package insert) ( http://www.seattlegenetics.com/application/files/2515/1059/6728/ADCETRIS_USPI_USP-BVP-2015-01625pdf.pdf) [Google Scholar]
- 12.Adcetris (brentuximab vedotin). EU summary of product characteristics. ( http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002455/WC500135055.pdf)
- 13.Younes A, Connors JM, Park SI, et al. Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncol. 2013;14:1348–56. doi: 10.1016/S1470-2045(13)70501-1. [DOI] [PubMed] [Google Scholar]
- 14.Connors JM, Ansell SM, Fanale M, Park SI, Younes A. Five-year follow-up of brentuximab vedotin combined with ABVD or AVD for advanced-stage classical Hodgkin lymphoma. Blood. 2017;130:1375–7. doi: 10.1182/blood-2017-05-784678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. N Engl J Med. 1998;339:1506–14. doi: 10.1056/NEJM199811193392104. [DOI] [PubMed] [Google Scholar]
- 16.Meignan M, Gallamini A, Haioun C, Polliack A. Report on the Second International Workshop on interim positron emission tomography in lymphoma held in Menton, France, 8-9 April 2010. Leuk Lymphoma. 2010;51:2171–80. doi: 10.3109/10428194.2010.529208. [DOI] [PubMed] [Google Scholar]
- 17.Lister TA, Crowther D, Sutcliffe SB, et al. Report of a committee convened to discuss the evaluation and staging of patients with Hodgkin’s disease: Cotswolds meeting. J Clin Oncol. 1989;7:1630–6. doi: 10.1200/JCO.1989.7.11.1630. [DOI] [PubMed] [Google Scholar]
- 18.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117:5019–32. doi: 10.1182/blood-2011-01-293050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649–55. [PubMed] [Google Scholar]
- 20.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–86. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 21.Barnes JA, LaCasce AS, Zukotynski K, et al. End-of-treatment but not interim PET scan predicts outcome in nonbulky limited-stage Hodgkin’s lymphoma. Ann Oncol. 2011;22:910–5. doi: 10.1093/annonc/mdq549. [DOI] [PubMed] [Google Scholar]
- 22.Engert A, Haverkamp H, Kobe C, et al. Reduced-intensity chemotherapy and PET-guided radiotherapy in patients with advanced stage Hodgkin’s lymphoma (HD15 trial): a randomised, open-label, phase 3 non-inferiority trial. Lancet. 2012;379:1791–9. doi: 10.1016/S0140-6736(11)61940-5. [DOI] [PubMed] [Google Scholar]
- 23.Spaepen K, Stroobants S, Dupont P, et al. Can positron emission tomography with [(18)F]-fluorodeoxyglucose after first-line treatment distinguish Hodgkin’s disease patients who need additional therapy from others in whom additional therapy would mean avoidable toxicity? Br J Haematol. 2001;115:272–8. doi: 10.1046/j.1365-2141.2001.03169.x. [DOI] [PubMed] [Google Scholar]
- 24.Engert A, Ballova V, Haverkamp H, et al. Hodgkin’s lymphoma in elderly patients: a comprehensive retrospective analysis from the German Hodgkin’s Study Group. J Clin Oncol. 2005;23:5052–60. doi: 10.1200/JCO.2005.11.080. [DOI] [PubMed] [Google Scholar]
- 25.Shenoy P, Maggioncalda A, Malik N, Flowers CR. Incidence patterns and outcomes for hodgkin lymphoma patients in the United States. Adv Hematol. 2011;2011:725219. doi: 10.1155/2011/725219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matasar MJ, Ford JS, Riedel ER, Salz T, Oeffinger KC, Straus DJ. Late morbidity and mortality in patients with Hodgkin’s lymphoma treated during adulthood. J Natl Cancer Inst. 2015;107(4):djv018. doi: 10.1093/jnci/djv018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng AK, van Leeuwen FE. Hodgkin lymphoma: late effects of treatment and guidelines for surveillance. Semin Hematol. 2016;53:209–15. doi: 10.1053/j.seminhematol.2016.05.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.