

Abstract
Adeno-associated virus (AAV) vectors have been used successfully in clinical trials for patients with hemophilia or blindness, but pre-existing neutralizing antibodies (Nab) are common in the general population and exclude many patients from clinical trials. Exploration of effective strategies to enhance AAV transduction and escape from Nab activity is still imperative. Previous studies have shown the compatibility of capsids from AAV serotypes and homology of recognition sites of AAV Nab located on different capsid subunits from one virion. In this study, we co-transfected AAV2 and AAV8 helper plasmids at different ratios (3:1, 1:1 and 1:3) to assemble haploid capsids and study both their transduction efficiency and Nab escape activity. After muscular injection, all of the haploid viruses induced higher transduction than their parental AAV vectors (2- to 9-fold over AAV2), with the highest of these being the haploid vector AAV2/8 3:1. After systemic administration, a 4-fold higher transduction in the liver was observed with haploid AAV2/8 1:3 than that with AAV8 alone. We then packaged the therapeutic factor IX cassette into haploid AAV2/8 1:3 capsids and injected them into FIX knockout mice via the tail vein. Higher FIX expression and improved phenotypic correction were achieved with the haploid AAV2/8 1:3 virus vector when compared to that of AAV8. Additionally, the haploid virus AAV2/8 1:3 was able to escape AAV2 neutralization and did not increase capsid antigen presentation capacity when compared to AAV8. To improve the Nab evasion ability of the haploid virus, we produced the triploid vector AAV2/8/9 by co-transfecting AAV2, AAV8 and AAV9 helper plasmids at a ratio of 1:1:1. After systemic administration, a 2-fold higher transduction in the liver was observed with the triploid vector AAV2/8/9 than that with AAV8. Nab analysis demonstrated that the triploid AAV2/8/9 vector was able to escape Nab activity from mouse sera immunized with parental serotypes. These results indicate that polyploid viruses might potentially acquire advantages from parental serotypes for enhancement of AAV transduction and evasion of Nab recognition without increasing capsid antigen presentation in target cells. Polyploid AAV vectors can be generated from any AAV serotype, whether natural, rational, library derived or a combination thereof, providing a novel strategy that should be explored in future clinical trials in patients with neutralizing antibodies.
Keywords: AAV, Gene therapy, Transduction, Polyploid, Neutralizing antibody
1. Introduction
Adeno-associated virus (AAV), a non-pathogenic parvovirus that requires a helper virus for efficient replication, is utilized as a viral vector for gene therapy because of its safety and simplicity. AAV has a broad host and cell-type tropism capable of transducing both dividing and non-dividing cells. To date, 12 AAV serotypes and > 100 variants have been identified [1,2]. Different serotype capsids have different infectivity rates in tissue and cultured cells, which depend on the primary receptor and co-receptors on the cell surface, or on the intracellular trafficking pathway itself. The primary receptors of some serotypes of AAV have been found, such as heparin sulfate proteoglycan (HSPG) for AAV2 and AAV3, and N-linked sialic acid for AAV5 [3,4]. The primary receptors of AAV7 and AAV8 have not been identified. Interestingly, AAV vector transduction efficiency in cultured cells may not always be translated into that of animals. For instance, AAV8 induces much higher transgene expression than other serotypes in the mouse liver [5], but not in cultured cell lines.
Of the 12 serotypes, several AAV serotypes and their variants have been used in clinical trials [6–8]. As the first characterized capsid, AAV2 has been most widely used in gene delivery, such as RPE 65 for Leber congenital amaurosis and Factor IX (FIX) for hemophilia B [8–10]. Although the application of AAV vectors has been proven safe and a therapeutic effect has been achieved in these clinical trials, one of the major challenges of the AAV vector is its low infectivity, and that it requires relatively large numbers of viral genomes [11,12]. AAV8 is another vector that has been used in several clinical trials for patients with hemophilia B. The results from AAV8/FIX liver-targeted delivery have demonstrated that there are distinct species-specific differences in transgene expression between mice, non-human primates, and humans [13–15]. While 1010 vector genome (vg) of AAV8 with FIX gene could reach supra-physiologic levels (> 100%) of FIX expression in FIX knock-out mice [15], only high doses (2 × 1012 vg/kg of body weight) could induce detectable FIX expression in humans [10,16]. Based on the results described above, the development of effective strategies to enhance AAV transduction is still necessary. The majority of people have been naturally exposed to AAVs, and as a result, a large portion of the population has neutralizing antibodies (Nabs) in their blood and other bodily fluids [17,18]. The presence of Nabs poses another major challenge for broader AAV applications in future clinical trials [18,19]. Many approaches have been explored to enhance AAV transduction or to evade Nab activity, especially the genetic modification of the AAV capsid based on rational design and directed evolution [20–24]. Although several AAV mutants have demonstrated high transduction, along with the capacity to escape Nab, in vitro or in animal models the modification of the capsid can potentially result in a different cell tropism than that of the parental AAVs [25].
Our original studies demonstrated the concept that the capsids from different AAV serotypes (AAV1 to AAV5) were compatible for assembly when contributed from separate AAV serotype capsids [26]. Most available AAV monoclonal antibodies have been characterized at the atomic level and recognize several sites located on different AAV subunits [27–31]. Additionally, recent studies utilizing chimeric AAV capsids have demonstrated that higher transduction can be achieved by swapping a structural domain for a primary receptor or for a tissue-specific motif from different serotypes by classic recombinogenic techniques. For example, the introduction of an AAV9 glycan receptor into an AAV2 capsid enhances AAV2 transduction [32], or substitution of a 100 aa domain from AAV6 into an AAV2 capsid increases muscle tropism [21]. While usually successful, these approaches are dependent on structural analysis knowledge and genetically engineered substrates, which may be time consuming and unpredictable in nature with respect to their final product. Based on these genetically altered AAV capsid genomes, we hypothesize that a polyploid AAV vector might induce a higher transduction efficiency without eliminating the tropism from the parental vectors. A polyploidy AAV vector is defined as a vector which is produced from the co-transfection of capsids from different serotypes parents, or mutant serotype parents that results in a wild-type AAV virion assembled from 60 intact capsomere subunits. Moreover, these polyploid capsids might have the ability to escape Nab since the majority of Nabs recognize conformational epitopes, and the polyploid virions would have subtle changes in their surface structure that might potentially alter such epitopes.
2. Materials and methods
2.1. Cell lines
HEK293 cells, Huh7 cells and C2C12 cells were maintained at 37 °C in 5% CO2 in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum and 1% penicillin–streptomycin.
2.2. Recombinant AAV virus production
Recombinant AAV was produced by a triple-plasmid transfection system [33]. A 15 cm dish of HEK293 cells was transfected with 9 μg of AAV transgene plasmid pTR/CBA-Luc, 12 μg of AAV helper plasmid containing AAV Rep and Cap genes, and 15 μg of Ad helper plasmid pXX6-80. To generate haploid AAV2/8 virions, AAV2 or AAV8 helper plasmids were co-transfected at three different ratios of 1:1, 1:3 and 3:1. To make triploid AAV2/8/9 vectors, the ratio of helper plasmid for each serotype was 1:1:1. Sixty hours post-transfection, HEK293 cells were collected and lysed. Supernatant was subjected to CsCl gradient ultra-centrifugation. Virus titer was determined by quantitative PCR using a pair of primers that were designed to bind to a homologous sequence on the ITR region. The qPCR assay was conducted using SYBR Green reagents and Light cycler 480 from Roche.
2.3. In vitro transduction assay
Huh7 and C2C12 cells were transduced by recombinant viruses with 1 × 104 vg/cell in a flat-bottom, 24-well plate. Forty-eight hours later, cells were harvested and evaluated by a luciferase assay system (Promega, Madison, WI).
2.4. Animal study
Animal experiments performed in this study were conducted with C57BL/6 mice and FIX−/− mice. The mice were maintained in accordance to NIH guidelines, as approved by the UNC Institutional Animal Care and Use Committee (IACUC). Six-week-old female C57BL/6 mice were injected with 3 × 1010 vg of recombinant viruses via retro-orbital injection. Luciferase expression was imaged 1 week post-injection using a Xenogen IVIS Lumina (Caliper Lifesciences, Waltham, MA) following intraperitoneal injection of D-luciferin substrate (Nanolight Pinetop, AZ). Bioluminescent images were analyzed using Living Image (PerkinElmer, Waltham, MA). For muscle transduction, 1 × 1010 particles of AAV/Luc were injected into the gastrocnemius of 6-week-old C57BL/6 females. Mice were imaged at the indicated time points.
FIX knockout male mice (FIX KO mice) received 1 × 1010 vg via tail vein injection. This delivery method has similar liver transduction and safety profile to retro-orbital injection (data not shown). At various time points after injection, blood was collected from the retro-orbital plexus. At week 6, mouse bleeding analysis was performed.
2.5. Quantitation of luciferase expression in the liver
Animals utilized for imaging studies were sacrificed at week 4 after recombinant virus injection and the livers were collected. Livers were minced and homogenized in passive lysis buffer. After the liver lysates were centrifuged, luciferase activity in supernatant was detected. Total protein concentration in tissue lysates were measured using the Bradford assay (BioRad, Hercules, CA).
2.6. Detection of AAV genome copy number in the liver
The minced livers were treated with Protease K and total genomic DNA was isolated by the PureLink Genomic DNA mini Kit (Invitrogen, Carlsbad, CA). The luciferase gene was detected by qPCR assay. The mouse lamin gene served as an internal control.
2.7. Human FIX expression, function and tail-bleeding time assays
To determine human FIX expression, one-stage hFIX activity and tail-bleeding time assays were performed as previously described [34,35].
2.8. Neutralization assay
Huh7 cells were seeded in a 48-well plate at a density of 105 cells for each well. Two-fold dilutions of the mouse antibody were incubated with AAV-Luc (1 × 108 vg) for 1 h at 37 °C. The mixture was added into cells and incubated for 48 h at 37 °C. Cells were lysed with passive lysis buffer (Promega, Madison, WI) and luciferase activity was measured. Nab titers were defined as the highest dilution for which luciferase activity was 50% lower than serum-free controls.
2.9. In vivo T cell proliferation
We have previously established a system in which the AAV2 or AAV8 virus capsid was engineered with a substitution of an ovalbumin immunodominant peptide sequence SIINFEKL (AAV2-OVA or AAV8-OVA) to study the capsid antigen presentation in a mouse model [36]. The haploid AAV2/8-OVA 1:3 vectors were produced in the same strategy as described above. As described previously [36], C57BL/6 mice were injected with AAV8-OVA or haploid AAV2/8-OVA 1:3 vector via retro-orbital administration. At day 3 after AAV injection, CFSE (Carboxyfluorescein succinimidyl ester)-labeled spleen cells from OT-1/Rag-1 mice, which express an H-2Kb-restricted T cell receptor (TCR) specific for the OVA-derived SIINFEKL peptide were transferred into injected mice. At day 10 after OT-1 cells transfer, spleen cells were harvested and the frequency of proliferating T cells was determined by the percentage of CFSE dilution.
2.10. Statistical analysis
The data were presented as mean ± SD. The Student t-test was used to compare the differences between two groups, while Tukey multiple comparison was used between three or more groups. p values of < 0.05 were considered a statistically significant difference.
3. Results
3.1. Characterization of the haploid viruses in vitro
To investigate the possibility of AAV serotype 2 and 8 capsid proteins to form a haploid virus and its transduction profile, we transfected the helper plasmids of AAV2 and AAV8 at ratios of 3:1, 1:1 and 1:3 to make haploid vectors. All of the haploid viruses were purified using a cesium gradient and tittered by qPCR. There was no significant difference in virus yield between the haploid viruses and their parental AAV2 or AAV8 vectors by one-way analysis of the variance (p = 0.762) (Supplementary Table 1). To determine whether the capsid proteins of the haploid viruses were expressed, Western blot analysis was performed on equivalent virus genomes from purified haploid viruses using monoclonal antibody B1, which recognizes the capsid proteins of AAV2 and AAV8. In all haploid viruses, the mixture of VP2 capsids from AAV2 and AAV8 was observed and was distinguishable based on slight changes in molecular weight. The intensity of the VP2 capsid from AAV2 or AAV8 in haploid viruses was also related to the ratio of the two helper plasmids (Fig. S1). These results suggested that the capsids from AAV2 and AAV8 were compatible and able to assemble into haploid AAV virions.
To determine whether the tropism of these haploid vectors was changed by mixing the capsid proteins, the transduction efficacy of the haploid viruses was analyzed by transducing human Huh7 and mouse C2C12 cell lines (Fig. 1). The transduction efficiency of AAV8 was much lower than that of AAV2 in both of these cell lines. The transduction from all haploid vectors was higher than that from AAV8 and the transduction efficiency was correlated with the increase of AAV2 structural subunits (Fig. 1). Although the haploid vector transduction was lower than AAV2 in Huh7 cells, haploid vector AAV2/8 3:1 induced a 3-fold higher transduction than that of AAV2 in C2C12 cells. This in vitro transduction data supports the premise that the virus preparation is composed of haploid vectors and not just the combination of individual serotype vectors. It further indicates that the haploid vector composites may enhance AAV transduction.
Fig. 1.
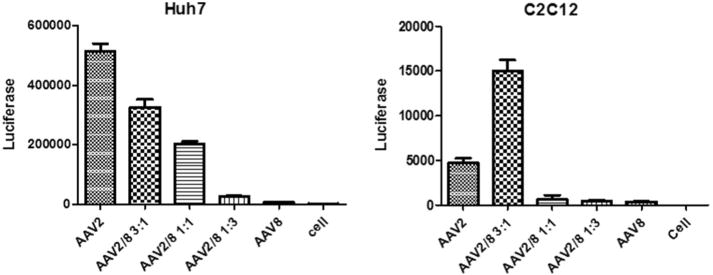
Transduction profiles of the haploid viruses in vitro. Haploid or parental viruses were added to Huh7 or C2C12 cells at 104 vg/cell. Cells were lysed for luciferase assay at 48 h post-transduction. The data represents an average of three separate infections, with the standard deviation indicated by an error bar.
3.2. Increased muscular transduction of haploid viruses
As described above, the transduction efficiency of the haploid virus AAV2/8 3:1 is higher than that of AAV2 and AAV8 in the muscle cell line C2C12. We then studied whether high transduction levels observed in vitro translated into mouse muscle tissues. Haploid AAV2/8 and their parental vectors were directly injected into the muscle of the hind legs in C57BL/6 mouse. As controls, the mixtures of AAV2 and AAV8 viruses at ratios of 3:1, 1:1, and 1:3 were also investigated. For a convenient comparison, one leg was injected with AAV2 and the opposite leg with haploid vector. A vector of 1 × 1010 vg for each virus was administered. Compared to AAV2, a similar muscular transduction was achieved for the parental AAV8 capsid (Fig. 2). Contrary to the result in C2C12 cells, an enhanced muscular transduction was observed from all of the haploid viruses (Fig. 2). The haploid vectors AAV2/8 1:1 and AAV2/8 1:3 achieved a 4- and a 2-fold higher transduction than AAV2, respectively. Notably, the muscular transduction of the haploid vector AAV2/8 3:1 was over 6-fold higher than that of AAV2. All of the controls (injections that were a result of physically mixing parental vectors), however, had similar transduction efficiencies as the AAV2 vector. Overall, these results suggest that the haploid composite virus is able to increase muscular transduction and further supports the premise that the viruses produced from co-transfection of two parental capsid plasmids are haploid in nature.
Fig. 2.
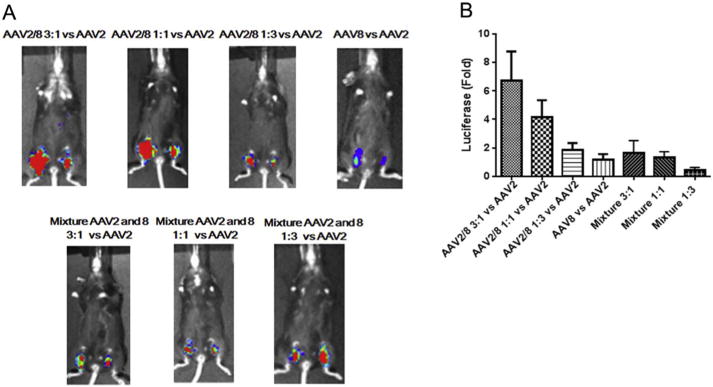
Transduction of the haploid viruses in mouse muscle. 1 × 1010 vg of the haploid viruses, parental viruses or viruses mixed with AAV2 and AAV8 were injected into C57 BL/6 mice via direct muscular injection. Each group included 4 mice. (A) After one week, luciferase gene expression was imaged by IVIS imaging system. (B) The photon signal was measured and calculated. The data represents an average of luciferase gene expression values for the 4 injected mice in each group, with the standard deviation indicated by an error bar. Face up: left leg-AAV8 or haploid or mixture viruses, right leg-AAV2.
3.3. Enhanced liver transduction of haploid viruses
Next, we evaluated the transduction efficiency of haploid viruses in the mouse liver. The mixtures of AAV2 and AAV8 viruses were also injected as controls. A dose of 3 × 1010 vg of AAV/luc vector was administered in C57BL mice via the retro-orbital vein and the imaging was carried out at day 3 post-AAV injection. The haploid virus AAV2/8 1:3 induced the highest transduction efficiency even over the other haploid combinations, the mixtures of parental viruses, and the parental AAV8 in mouse livers (Fig. 3A and B). The transduction efficiency of the haploid vector AAV2/8 1:3 was about 4-fold higher than that of AAV8 (Fig. 3B). The liver transduction from the other haploid viruses was lower than that from the parental vector AAV8, but higher than that of AAV2 (Fig. 3A and B). At day 7 post-injection, the mice were sacrificed, the livers were harvested, and the genomic DNA was isolated. The luciferase gene copy number in the liver was determined by qPCR. Different from the results for liver transduction efficiency, a similar AAV vector genome copy number was found in the liver regardless of virus composition (Fig. 3C). When transgene expression was normalized to gene copy number, the haploid vector AAV2/8 1:3 induced the highest relative transgene expression than any other haploid vector combination or parental serotypes (Fig. 3D). The transduction profile of haploid viruses in the liver was different from that in muscle transduction, in which all haploid viruses induced a higher transgene expression than that from parental serotypes, with the best preforming vector being from AAV2/8 3:1.
Fig. 3.
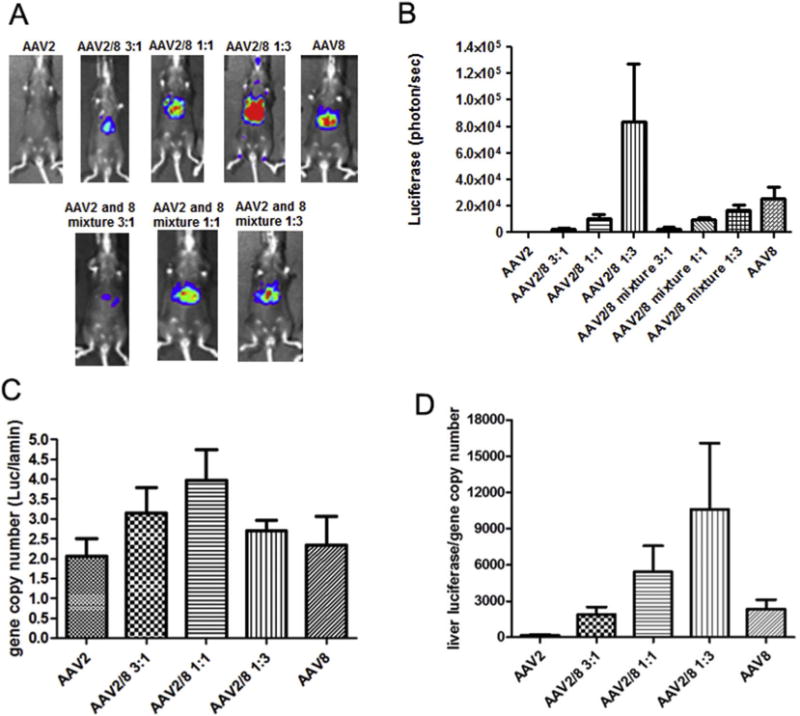
Transduction of the haploid viruses in mouse liver. 3 × 1010 vg of the haploid virus was administered via intravenous injection. At week 1 post-injection, luciferase gene expression was imaged by IVIS imaging system (A), and the photon signal was measured and calculated (B). At week 2 post-injection, mice were euthanized and their livers were harvested for DNA extraction AAV genome copy in the liver was measured by qPCR (C) and relatively luciferase expression per AAV genome copy number was calculated (D). The data represents the average and standard deviation from 4 mice.
3.4. Augmented therapeutic FIX expression and improved bleeding phenotypic correction with the haploid vector in a hemophilia B mouse model
Based on the above results, the haploid vector AAV2/8 1:3 induced much higher liver transduction than that of AAV8. We further tested if the haploid vector AAV2/8 1:3 would increase the therapeutic transgene expression in an animal disease model. We used human FIX (hFIX) as a therapeutic gene and injected the haploid vector AAV2/8 1:3/hFIX into FIX knockout (KO) mice via tail vein at a dose of 1 × 1010 vg/mouse. The haploid vector encodes the human-optimized FIX transgene and is driven by the liver-specific promoter, TTR. At week 1, 2, and 4 post-injection, ELISA and one-stage factor activity analyzed the hFIX expression and activity in circulation, respectively. At week 6, the blood loss for in vivo hFIX function was evaluated using a tail clipping assay. Consistent with the observation of high liver transduction with the haploid AAV vectors in wild-type C57BL/6 mice, the haploid vector AAV2/8 1:3 liver targeting produced much more hFIX than an AAV8 vector after 2 weeks post-injection (Fig. 4A). The higher hFIX protein expression of AAV2/8 1:3 correlated as predicted with high FIX activity (Fig. 4B). The blood loss for the mice with AAV2/8 1:3/hFIX injection was similar to that of wild-type C57BL/6 mice, and much less than that of KO mice (Fig. 4C). Although there was no significant difference of the blood loss between the mice with AAV8 and AAV2/8 1:3/hFIX injection in statistics, the AAV8-treated mice had a little more blood loss than that of AAV2/8 1:3-treated mice (Fig. 4C). This data shows that the haploid vector AAV2/8 1:3 increases the therapeutic transgene expression from the liver and improves disease phenotypic correction.
Fig. 4.
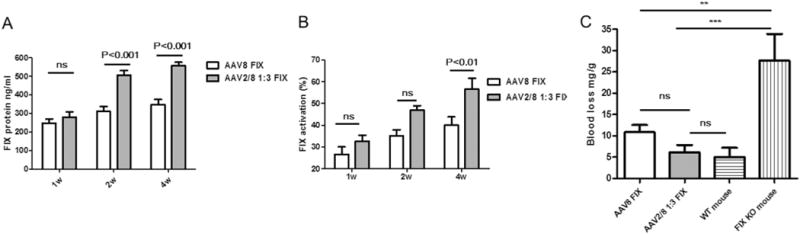
Therapeutic level of hFIX via haploid virus delivery. FIX knockout mice were injected with 1 × 1010 vg of each vector via tail vein. At 1, 2 and 4 weeks post-injection, blood samples were collected. (A) hFIX protein levels were tested by enzyme-linked immunosorbent assay. (B) hFIX function was tested by the hFIX-specific one-stage clotting assay. At week 6 post-injection, blood loss was determined by measuring the absorbance at A575 of hemoglobin content in the saline solution (C). The data represents the average and standard deviations from 5 mice (knock-out mice and normal mice, without AAV treatment, as controls) or 8 mice (AAV8 FIX or AAV2/8 1:3 FIX treated groups).
3.5. Ability of the haploid viruses AAV2/8 to escape Nab
Each individual haploid virus virion is composed of 60 subunits derived from different AAV serotype viral protein (VP) monomers. While the assembly of mixed capsid subunits from distinct serotypes generates infectious virions with enhanced transduction, they may also change the virion’s surface structure. It is well known that most AAV monoclonal antibodies recognize the conformational amino acid motif which contributed by different VP subunits on the surface of a single virion. Any surface structure change, therefore, potentially affects the Nab recognition. To study whether the haploid virus is able to escape Nabs generated in response to a parental vector, we firstly performed a Nab binding assay using monoclonal antibodies by an immune-blot assay. Three dilutions of virus-genome-containing particles were adsorbed to a nitrocellulose membrane and probed with Nab A20 or ADK8, which recognizes intact AAV2 or AAV8, respectively. All of the haploid viruses and the viruses with a mixture of AAV2 and AAV8 were recognized by the monoclonal antibodies A20 or ADK8 (Fig. S2). The reactivity of haploid viruses with A20 was increased by the incorporation of more AAV2 capsids into the haploid virus virion (Fig. S2 right panel). There was no obvious change for the recognition of anti-AAV8 Nab ADK8 among the haploid viruses, however, regardless of the capsid ratios (Fig. S2 left panel). Notably, the binding of the haploid AAV2/8 1:3 to A20 was much weaker than those of the parental AAV2 and of the virus with a mixture of AAV2 and 8 at a ratio of 1:3, which suggests that A20 binding sites are depleted on the haploid AAV2/8 1:3 virion surface.
We then analyzed the immunological profile of the haploid viruses against sera from AAV-immunized mice. Nab titers were used to evaluate the ability of the serum to inhibit vector transduction. Serum was collected from each mouse treated with parental viruses at week 4 post-injection. The neutralization profiles of the haploid viruses against A20 or ADK8 were similar to the data from a native immune-blot (Table 1, Fig. S2). There was no Nab cross-reactivity between AAV2 and AAV8. It is interesting to note that AAV8-immunized mouse serum had similar neutralizing activity against both the AAV8 virus and all of the haploid viruses, regardless of the amount of AAV8 capsid incorporation. The AAV8 serum also could not block the transduction from the viruses mixed with AAV2 and AAV8. It can be explained by the superior transduction of the haploid AAV2/AAV8 observed in a tested cell line. However, the haploid viruses partially escaped the neutralization from the AAV2 serum. The transduction of the haploid AAV2/8 1:1 resulted in a 16-fold decrease compared to that of the parental AAV2 after incubation of the virus and anti-AAV2 serum. The ability to escape the AAV2 serum Nab for haploid viruses was much higher than that for viruses mixed with AAV2 and AAV8. The haploid AAV2/8 1:3 almost completely escaped the AAV2 serum and A20 neutralization, which suggests that this haploid virus has the potential to be used for individuals who have anti-AAV2 Nabs (Table 1).
Table 1.
Neutralization antibody titer and cross-reactivity for the haploid virus AAV2/8.
| AAV2 | Vector
|
AAV8 | ||||||
|---|---|---|---|---|---|---|---|---|
| Haploid virus AAV2/8
|
Mixture virus AAV2 and 8
|
|||||||
| 3:1 | 1:1 | 1:3 | 3:1 | 1:1 | 1:3 | |||
| mAb | ||||||||
| A20 | 512 | 2048 | 32 | < 2 | ND | ND | ND | < 2 |
| ADK8 | < 2 | 512 | 512 | 1024 | ND | ND | ND | 1024 |
| Serum | ||||||||
| AAV2 | 4096 | 1024 | 256 | 8 | 4096 | 2048 | 1024 | < 2 |
| AAV8 | < 2 | 256 | 256 | 512 | < 2 | < 2 | < 2 | 512 |
3.6. The antigen presentation from the haploid AAV capsid is similar to that of AAV8 in vivo
We have previously demonstrated that the capsid antigen presentation from AAV2 and AAV8 is similar after systemic administration. To study the efficacy of the capsid antigen presentation, we produced a haploid AAV2/8 OVA 1:3 vector by the transfection of pXR2-OVA and pXR8-OVA at the ratio of 1:3. 1 × 1011 vg of AAV2/8-OVA and AAV8-OVA vectors were administered via retro-orbital injection into the C57BL/6 mice. Three days later, CFSE-labeled OT-1 mouse spleen cells were transferred into the C57BL/6 mice. At day 10 post-transferring OT-1 spleen cells, T cell proliferation was measured by flow cytometry. OT-1 T cell proliferation was significantly increased in mice receiving AAV2/8-OVA 1:3 or AAV8-OVA when compared to control mice without AAV vector administration (Fig. 5). There was no difference, however, for OT-1 cell proliferation between the AAV2/8-OVA 1:3 and AAV8-OVA groups. This result indicates that haploid vectors do not induce high capsid antigen presentation as a by-product of enhanced transduction after systemic administration.
Fig. 5.
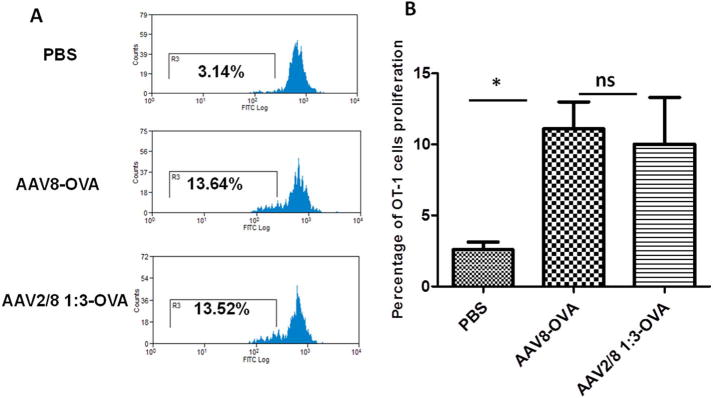
Capsid antigen presentation from the haploid vectors in vivo. The haploid vector AAV2/8-OVA 1:3 or the AAV8-OVA vectors were injected into C57BL/6 mice. Three days later, CFSE-labeled OT-1 T cells were transferred. At day 10 after transferring OT-1 T cells, the proliferation in the spleen was assessed via flow cytometry. (A) An example of histogram analysis. (B) Analysis of T cell proliferation. Each group contained five mice. Tukey multiple comparison test was used for statistical analysis. Asterisk indicates significant difference (p < 0.05), whereas ns indicates no significant difference (p > 0.05).
3.7. Improved Nab evasion ability with triploid vectors made from three serotypes
Our data described above demonstrated that haploid AAV2/8 viruses were not able to escape the AAV8 Nab activity efficiently, but had the capacity to evade the AAV2 Nab, which, as predicted, depended on the amount of parental virus capsid contributed from AAV8. To evaluate whether a haploid virus made from more serotypes VP subunits could improve the Nab escaping ability, we made the triploid virus AAV2/8/9 at a ratio of 1:1:1. After the injection of the triploid vector AAV2/8/9 into mice, the triploid virus AAV2/8/9 induced a 2-fold higher transduction in the liver when compared to AAV8. Interestingly, no difference in the level of liver transduction was observed among AAV8 and the haploid vectors AAV2/9 and AAV8/9, which were AAV helper plasmids made at a ratio of 1:1 (Fig. 6A and B). In this experiment, it was noted that the AAV9 systemic administration induced a higher liver transduction than AAV8. When a Nab assay was performed, the triploid AAV2/8/9 vector improved its Nab escape ability by about 20 fold, 32 fold and 8 fold, respectively when compared to AAV2, 8, and 9 (Table 2).
Fig. 6.
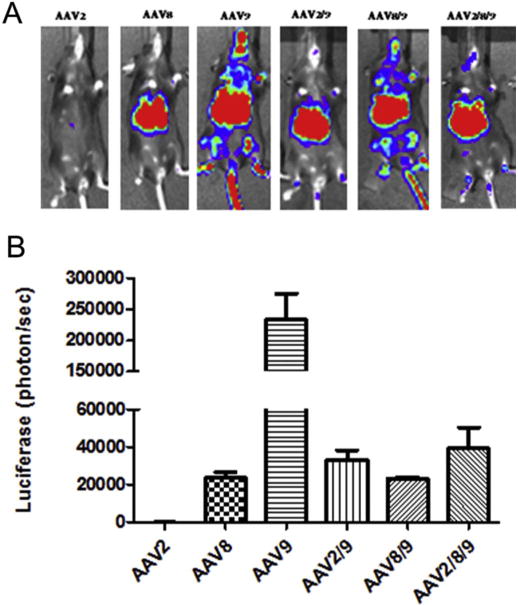
Liver transduction with the triploid virus AAV2/8/9. 3 × 1010 vg of the haploid viruses were injected via retro-orbital vein. At week 1 post-injection, luciferase gene expression was imaged by IVIS imaging system (A), and the photon signal was measured and calculated (B). The data represents the average and standard deviation from 5 mice.
Table 2.
Neutralization antibody titer and cross-reactivity for the triploid virus AAV2/8/9.
| AAV2 | AAV8 | AAV9 | AAV2/9 | AAV8/9 | AAV2/8/9 | |
|---|---|---|---|---|---|---|
| Serum AAV2 | > 2048 | < 2 | 512 | 128 | ||
| Serum AAV8 | < 2 | 128 | 32 | 4 | ||
| Serum AAV9 | < 2 | 16 | 2048 | 512 | 256 | |
| Serum AAV2/8/9 | 8 | 128 | 128 | 64 | 512 | 128 |
4. Discussion
In this study, haploid and triploid AAV virions were assembled from the capsid subunits derived from 2 or 3 serotypes. All of the haploid viruses achieved a higher transduction efficacy than the parental AAV2 vector in mouse muscle and liver, while the haploid virus AAV2/8 1:3 had a significant enhancement of liver transduction when compared to the parental AAV8 vector. Moreover, the haploid virus AAV2/8 1:3 which delivered human FIX induced much higher FIX expression and improved hemophilia phenotypic correction in FIX−/− mice than those of AAV8/hFIX. Importantly, the haploid virus AAV2/8 1:3 was able to escape the neutralization of anti-AAV2 serum and did not increase the capsid antigen presentation from transduced target cells. Integration of the AAV9 viral protein capsid subunits into haploid AAV2/8 virions further improved Nab escape capacity without comprising the enhanced transduction.
AAV2 and AAV8 are widely used for liver transduction. AAV8 induces a much higher level of transgene expression than AAV2 in mouse and non-human primates livers [5,14,15,37–39]. Recent clinical trials using AAV vectors, however, have raised concerns regarding the immunological responses to the AAV capsid and the limited levels of transgenic product [10,40,41]. Many groups have attempted to develop novel AAV vectors for the enhancement of transduction by engineering an AAV capsid. The most popular strategy for enhancing transduction efficacy is to modify the AAV capsid through rational design or directed evolution [8]. To rationally design novel AAV vectors for enhanced transduction in humans, we were the first to develop a chimeric virus AAV2.5 [37]. This capsid was used in AAV gene therapy clinical trials for DMD patients. Additional modifications have suggested that key residues can be genetically engineered on the backbone of a homologous AAV (e.g. AAV2G9) [32,42]. Both chimeric mutants, AAV2.5 and AAV2G9, induce a much higher transduction than AAV2 in mouse muscle and liver, respectively [32,42]. These observations indicate that these chimeric viruses may use properties from both AAV serotypes for enhanced transduction (for example, AAV2G9 uses two primary receptors-heparin and galactose for effective cell surface binding). This approach, however, more likely changes the tropism of novel AAV vectors, or even eliminates the cell tropism [25,43]. For example, we demonstrated that chimeric AAV2i8 (AAV2 with a heparan binding site swapped from AAV8) changes the parent AAV2 liver tropism to muscle tropic [43]. Other approaches to select AAV variants through error-prone PCR or a DNA shuffling library might be able to enhance transduction efficiency [23,44,45] or to escape pre-existing antibodies [21,46]. However, the potential limitations of these methods include long-term selection steps and unknown transduction efficiencies in vivo, especially in humans. The early studies have demonstrated that different serotypes of the AAV capsids are compatible for virion assembly [26,47]. More importantly each serotype of AAV uses distinct receptors or co-receptors for cell surface binding and different intracellular trafficking for effective infection [48]. All of these observations suggest that a higher transduction may be achieved by taking advantage of the infection pathways from different serotype AAV vectors if one single virion is made from different serotype VP capsid components. We tested this premise using capsids from two or three different AAV serotypes and found that all the haploid and triploid viruses had a higher transduction rate than at least one contributing parental vectors in vitro and in vivo. These results suggest an alternative approach of modifying the AAV capsids to enhance AAV transduction. Compared to rational design and directed evolution, the strategy of developing polyploid viral vectors does not require the detailed mechanism for the infection of individual parental AAV serotypes, is less labor intensive, and retain parental tropism.
Recent clinical trials demonstrated that a capsid specific CTL response was elicited, which was suggested to eliminate the AAV transduced hepatocytes [10]. One concern arises whether enhanced transduction from the haploid vectors would potentially present more capsid antigens on target cells for the recognition of capsid specific CTLs. When compared to that of AAV8 vector, the efficacy of the capsid antigen presentation from a haploid vector was not increased. This result has significant clinical relevance. Previous studies have demonstrated that the capsid antigen presentation is dose-dependent [36,49]. When less of the haploid vector is used to achieve the same therapeutic effect, low to no capsid antigen presentation was detected, strongly supporting the evaluation of such capsid in a clinical setting.
One of the most challenging aspects of efficient transduction in clinical trials is the broad prevalence of neutralizing antibodies to the AAV vectors [40,42]. Several studies have been explored to genetically modify AAV capsids for Nab evasion by rational mutation of Nab re-cognizing sites or by directed evolution approaches [30,46,31]. Although these mutated AAV vectors successfully escaped Nab against AAV, capsid mutations often change the AAV tropism and transduction efficiency. Additionally, the identification of Nab binding sites on AAV virions is far behind vector application in clinical trials, and it is impossible to figure out all the Nab binding sites from poly sera. Previous studies have demonstrated that the recognition sites of several AAV monoclonal antibodies are represented by three-dimensional protein motif from different subunits of one virion [28]. These studies suggest that the insertion of a capsid subunit from additional serotypes, as described here for haploid and triploid capsids, potentially changes the structure of the viron’s surface and eliminates Nab binding sites. It is true that when an AAV8 capsid is introduced into an AAV2 virion, the A20 binding ability and neutralizing activity from AAV2-immunized sera were dramatically decreased for the haploid viruses. It is interesting to point out that the assembly of AAV2 subunits into AAV8 virions did not reduce the capacity to bind the intact AAV8 monoclonal antibody ADK8 and did not escape the neutralizing activity of anti-AAV8 sera (Table 1). This observation suggests that all Nab recognition sites from polyclonal antiserum may be located on the same subunit of an AAV8 virion, similar to ADK8, which recognizes a linear epitope on the AAV8 variable region, VRVIII [50]. The result also suggests that the AAV8 capsids assembled into the AAV2 virions may play a major role in virus intracellular trafficking, although further experiments are still needed to warrant this hypothesis. When a triploid virus was made from the capsids of three serotypes, AAV2, 8, and 9, different from haploid vectors AAV2/8, the triploid AAV2/8/9 virus demonstrated an ability to escape the Nab activity of sera from AAV2, 8, or 9 immunized mice, which suggests that AAV8 and AAV9 share a similar transduction pathway.
One can argue that the virions made from the co-transfection of two or three AAV capsid plasmids are a mixture of individual parental serotype vectors and are not polyploid in nature. We cannot completely rule out the presence of some individual parental serotype vectors in this virus preparation. Several lines of evidence from this study support that the haploid and triploid virions do assemble from the transfections of two or three AAV helper plasmids at the specific input plasmid ratios, respectively. This includes the two VP2 bands of different sizes with western blot analysis, the different transduction efficiency and binding ability on target cells, as well as the Nab profile when compared to the parental vectors and the virus combinations with mixtures of the parental vectors.
5. Conclusion
This study demonstrated that capsids from AAV2, 8, and 9 were compatible for assembly into haploid and triploid virions. These haploid/triploid viruses enhanced the transduction efficiency in vitro and in vivo without increasing the vector capsid antigen load, and even escaped neutralization from the parental vector immunized sera. Application of the haploid virus to deliver a therapeutic transgene FIX was able to increase FIX expression and improve hemophilia phenotypic correction in mice with FIX deficiency. These results indicate that this unique class of AAV vectors easily exploits attributes of serotype specific variants by simply adding starting AAV helper plasmid constructs at specific ratios in the well established AAV production system. The system is not restricted to natural AAV serotypes but can utilize the chimeric or rational design of the AAV capsid if the starting material is available. All in all, the ability to increase transduction efficiency in target cells and evade Nabs, strongly suggest further testing and eventual analysis in future clinical trials.
Supplementary Material
Acknowledgments
We thank Violeta Zaric and Xiaojing Chen for their technical assistance. We thank Dr. Lauriel Earley for improving the manuscript. The authors acknowledge the UNC Biomedical Research Imaging Center (BRIC) Small Animal Imaging (SAI) facility for assistance of mouse imaging. This work was supported by National Institutes of Health Grants R01DK084033 and R01AI117408 (to C.L. and R.J.S.), R01HL125749 (To C.L.), P01HL112761, R01AI072176 (to R.J.S.), P30-CA016086-35-37, U54-CA151652-01-04 (to the BRIC SAI facility).
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2017.08.005.
Disclosure of conflict of interest
R. Jude Samulski is the founder, and a shareholder, at Asklepios BioPharmaceutical. He receives research support through the University of North Carolina from Asklepios BioPharmaceutical. He holds patents that have been licensed by UNC to Asklepios Biopharmaceutical, for which he receives royalties. He has consulted for Baxter Healthcare and has received payment for speaking.
References
- 1.Duan D. Systemic delivery of adeno-associated viral vectors. Curr Opin Virol. 2016;21:16–25. doi: 10.1016/j.coviro.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther. 2012;20:699–708. doi: 10.1038/mt.2011.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaludov N, Brown KE, Walters RW, Zabner J, Chiorini JA. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for Hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J Virol. 2001;75:6884–6893. doi: 10.1128/JVI.75.15.6884-6893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lisowski L, Tay SS, Alexander IE. Adeno-associated virus serotypes for gene therapeutics. Curr Opin Pharmacol. 2015;24:59–67. doi: 10.1016/j.coph.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 8.Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet. 2014;15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Mol Ther. 2013;21:509–519. doi: 10.1038/mt.2012.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nathwani AC, Tuddenham EGD, Rangarajan S, Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ, Harrington C, O’Beirne J, Smith K, Pasi J, Glader B, Rustagi P, Ng CYC, Kay MA, Zhou J, Spence Y, Morton CL, Allay J, Coleman J, Sleep S, Cunningham JM, Srivastava D, Basner-Tschakarjan E, Mingozzi F, High KA, Gray JT, Reiss UM, Nienhuis AW, Davidoff AM. Adenovirus-Associated Virus Vector–Mediated Gene Transfer in Hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward P, Walsh CE. Current and future prospects for hemophilia gene therapy. Expert Rev Hematol. 2016;9:649–659. doi: 10.1080/17474086.2016.1182859. [DOI] [PubMed] [Google Scholar]
- 12.Brantly ML, Spencer LT, Humphries M, Conlon TJ, Spencer CT, Poirier A, Garlington W, Baker D, Song S, Berns KI, Muzyczka N, Snyder RO, Byrne BJ, Flotte TR. Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 alphal-antitrypsin (AAT) vector in AAT-deficient adults. Hum Gene Ther. 2006;17:1177–1186. doi: 10.1089/hum.2006.17.1177. [DOI] [PubMed] [Google Scholar]
- 13.Nietupski JB, Hurlbut GD, Ziegler RJ, Chu Q, Hodges BL, Ashe KM, Bree M, Cheng SH, Gregory RJ, Marshall J, Scheule RK. Systemic administration of AAV8-α-galactosidase a induces humoral tolerance in nonhuman primates despite low hepatic expression. Mol Ther. 2011;19:1999–2011. doi: 10.1038/mt.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidoff AM, Gray JT, Ng CYC, Zhang Y, Zhou J, Spence Y, Bakar Y, Nathwani AC. Comparison of the ability of adeno-associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models. Mol Ther. 2005;11:875–888. doi: 10.1016/j.ymthe.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 15.Nathwani AC, Gray JT, Ng CYC, Zhou J, Spence Y, Waddington SN, Tuddenham EGD, Kemball-Cook G, McIntosh J, Boon-Spijker M, Mertens K, Davidoff AM. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107:2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nathwani AC, Reiss UM, Tuddenham EGD, Rosales C, Chowdary P, McIntosh J, Della Peruta M, Lheriteau E, Patel N, Raj D, Riddell A, Pie J, Rangarajan S, Bevan D, Recht M, Shen YM, Halka KG, Basner-Tschakarjan E, Mingozzi F, High KA, Allay J, Kay MA, Ng CYC, Zhou J, Cancio M, Morton CL, Gray JT, Srivastava D, Nienhuis AW, Davidoff AM. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N Engl J Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 19.Kotterman MA, Yin L, Strazzeri JM, Flannery JG, Merigan WH, Schaffer DV. Antibody neutralization poses a barrier to intravitreal adeno-associated viral vector gene delivery to non-human primates. Gene Ther. 2015;22:116–126. doi: 10.1038/gt.2014.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li C, Diprimio N, Bowles DE, Hirsch ML, Monahan PE, Asokan A, Rabinowitz J, Agbandje-McKenna M, Samulski RJ. Single amino acid modification of adeno-associated virus capsid changes transduction and humoral immune profiles. J Virol. 2012;86:7752–7759. doi: 10.1128/JVI.00675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Wu S, Albright B, Hirsch M, Li W, Tseng Y-S, Agbandje-McKenna M, McPhee S, Asokan A, Samulski RJ. Development of patient-specific AAV vectors after neutralizing antibody selection for enhanced muscle gene transfer. Mol Ther. 2016;24:53–65. doi: 10.1038/mt.2015.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, Govindaswamy L, Agbandje-McKenna M, Leichtle S, Redmond DE, Jr, McCown TJ, Petermann KB, Sharpless NE, Samulski RJ. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther. 2008;16:1252–1260. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 23.Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, Wilson EM, Nygaard S, Grompe M, Alexander IE, Kay MA. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, Herzog RW, Zolotukhin I, Warrington KH, Weigel-Van Aken KA, Hobbs JA, Zolotukhin S, Muzyczka N, Srivastava A. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci U S A. 2008;105:7827–7832. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu P, Xiao W, Conlon T, Hughes J, Agbandje-McKenna M, Ferkol T, Flotte T, Muzyczka N. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and Construction of AAV2 vectors with altered tropism. J Virol. 2000;74:8635–8647. doi: 10.1128/jvi.74.18.8635-8647.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, Samulski RJ. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gurda BL, DiMattia MA, Miller EB, Bennett A, McKenna R, Weichert WS, Nelson CD, Chen W-j, Muzyczka N, Olson NH, Sinkovits RS, Chiorini JA, Zolotutkhin S, Kozyreva OG, Samulski RJ, Baker TS, Parrish CR, Agbandje-McKenna M. Capsid antibodies to different adeno-associated virus serotypes bind common regions. J Virol. 2013;87:9111–9124. doi: 10.1128/JVI.00622-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCraw DM, O’Donnell JK, Taylor KA, Stagg SM, Chapman MS. structure of adeno-associated virus-2 in complex with neutralizing monoclonal antibody A20. Virology. 2012;431:40–49. doi: 10.1016/j.virol.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wobus CE, Hügle-Dörr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2–cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74:9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lochrie MA, Tatsuno GP, Christie B, McDonnell JW, Zhou S, Surosky R, Pierce GF, Colosi P. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J Virol. 2006;80:821–834. doi: 10.1128/JVI.80.2.821-834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huttner NA, Girod A, Perabo L, Edbauer D, Kleinschmidt JA, Buning H, Hallek M. Genetic modifications of the adeno-associated virus type 2 capsid reduce the affinity and the neutralizing effects of human serum antibodies. Gene Ther. 2003;10:2139–2147. doi: 10.1038/sj.gt.3302123. [DOI] [PubMed] [Google Scholar]
- 32.Shen S, Horowitz ED, Troupes AN, Brown SM, Pulicherla N, Samulski RJ, Agbandje-McKenna M, Asokan A. Engraftment of a galactose receptor footprint onto adeno-associated viral capsids improves transduction efficiency. J Biol Chem. 2013;288:28814–28823. doi: 10.1074/jbc.M113.482380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1:1412–1428. doi: 10.1038/nprot.2006.207. [DOI] [PubMed] [Google Scholar]
- 34.Sun J, Hakobyan N, Valentino LA, Feldman BL, Samulski RJ, Monahan PE. Intraarticular factor IX protein or gene replacement protects against development of hemophilic synovitis in the absence of circulating factor IX. Blood. 2008;112:4532–4541. doi: 10.1182/blood-2008-01-131417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schuettrumpf J, Herzog RW, Schlachterman A, Kaufhold A, Stafford DW, Arruda VR. Factor IX variants improve gene therapy efficacy for hemophilia B. Blood. 2005;105:2316–2323. doi: 10.1182/blood-2004-08-2990. [DOI] [PubMed] [Google Scholar]
- 36.He Y, Weinberg MS, Hirsch M, Johnson MC, Tisch R, Samulski RJ, Li C. Kinetics of adeno-associated virus serotype 2 (AAV2) and AAV8 capsid antigen presentation in vivo are identical. Hum Gene Ther. 2013;24:545–553. doi: 10.1089/hum.2013.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarkar R, Tetreault R, Gao G, Wang L, Bell P, Chandler R, Wilson JM, Kazazian HH. Total correction of hemophilia A mice with canine FVIII using an AAV 8 serotype. Blood. 2004;103:1253–1260. doi: 10.1182/blood-2003-08-2954. [DOI] [PubMed] [Google Scholar]
- 38.Nathwani AC, Gray JT, McIntosh J, Ng CYC, Zhou J, Spence Y, Cochrane M, Gray E, Tuddenham EGD, Davidoff AM. Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood. 2007;109:1414–1421. doi: 10.1182/blood-2006-03-010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. Clades of adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manno CS, Arruda VR, Pierce GF, Glader B, Ragni M, Rasko J, Ozelo MC, Hoots K, Blatt P, Konkle B, Dake M, Kaye R, Razavi M, Zajko A, Zehnder J, Nakai H, Chew A, Leonard D, Wright JF, Lessard RR, Sommer JM, Tigges M, Sabatino D, Luk A, Jiang H, Mingozzi F, Couto L, Ertl HC, High KA, Kay MA. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 41.Jiang H, Couto LB, Patarroyo-White S, Liu T, Nagy D, Vargas JA, Zhou S, Scallan CD, Sommer J, Vijay S, Mingozzi F, High KA, Pierce GF. Effects of transient immunosuppression on adenoassociated, virus-mediated, liver-directed gene transfer in rhesus macaques and implications for human gene therapy. Blood. 2006;108:3321–3328. doi: 10.1182/blood-2006-04-017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, Li J, Wang B, Monahan PE, Rabinowitz JE, Grieger JC, Govindasamy L, Agbandje-McKenna M, Xiao X, Samulski RJ. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther. 2012;20:443–455. doi: 10.1038/mt.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asokan A, Conway JC, Phillips JL, Li C, Hegge J, Sinnott R, Yadav S, DiPrimio N, Nam HJ, Agbandje-McKenna M, McPhee S, Wolff J, Samulski RJ. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol. 2010;28:79–82. doi: 10.1038/nbt.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marsic D, Govindasamy L, Currlin S, Markusic DM, Tseng Y-S, Herzog RW, Agbandje-McKenna M, Zolotukhin S. Vector design tour de force: integrating combinatorial and rational approaches to derive novel adeno-associated virus variants. Mol Ther. 2014;22:1900–1909. doi: 10.1038/mt.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang L, Jiang J, Drouin LM, Agbandje-Mckenna M, Chen C, Qiao C, Pu D, Hu X, Wang DZ, Li J, Xiao X. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci U S A. 2009;106:3946–3951. doi: 10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno-associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24:198–204. doi: 10.1038/nbt1182. [DOI] [PubMed] [Google Scholar]
- 47.Hauck B, Chen L, Xiao W. Generation and characterization of chimeric recombinant AAV vectors. Mol ther. 2003;7:419–425. doi: 10.1016/s1525-0016(03)00012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rabinowitz JE, Bowles DE, Faust SM, Ledford JG, Cunningham SE, Samulski RJ. Cross-dressing the virion: the transcapsidation of adeno-associated virus serotypes functionally defines subgroups. J Virol. 2004;78:4421–4432. doi: 10.1128/JVI.78.9.4421-4432.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hui DJ, Basner-Tschakarjan E, Chen Y, Davidson RJ, Buchlis G, Yazicioglu M, Pien GC, Finn JD, Haurigot V, Tai A, Scott DW, Cousens LP, Zhou S, De Groot AS, Mingozzi F. Modulation of CD8(+) T cell responses to AAV vectors with IgG-derived MHC class II epitopes. Mol Ther. 2013;21:1727–1737. doi: 10.1038/mt.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gurda BL, Raupp C, Popa-Wagner R, Naumer M, Olson NH, Ng R, McKenna R, Baker TS, Kleinschmidt JA, Agbandje-McKenna M. Mapping a neutralizing epitope onto the capsid of adeno-associated virus serotype 8. J Virol. 2012;86:7739–7751. doi: 10.1128/JVI.00218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.