

Abstract
Objective
Syncope is sudden transient loss of consciousness and postural tone with spontaneous recovery; the most common form is vasovagal syncope(VVS). We previously demonstrated impaired post-synaptic adrenergic responsiveness in young VVS patientswas reversed by blocking nitric oxide synthase(NOS). We hypothesised that nitric oxide may account for reduced orthostatic tolerance in young recurrent VVS patients.
Methods
We recorded haemodynamics in supine VVS and healthy volunteers (aged 15–27 years), challenged with graded lower body negative pressure (LBNP) (−15, –30, −45 mm Hg each for 5 min, then −60 mm Hg for a maximum of 50 min) with and without NOS inhibitor NG-monomethyl-L-arginine acetate(L-NMMA). Saline plus phenylephrine (Saline+PE) was used as volume and pressor control for L-NMMA.
Results
Controls endured 25.9±4.0 min of LBNP during Saline+PE compared with 11.6±1.4 min for fainters (p<0.001). After L-NMMA, control subjects endured 24.8±3.2 min compared with 22.6±1.6 min for fainters. Mean arterial pressure decreased more in VVS patients during LBNP with Saline+PE (p<0.001) which was reversed by L-NMMA; cardiac output decreased similarly in controls and VVS patients and was unaffected by L-NMMA. Total peripheral resistance increased for controls but decreased for VVS during Saline+PE (p<0.001) but was similar following L-NMMA. Splanchnic vascular resistance increased during LBNP in controls, but decreased in VVS patients following Saline+PE which L-NMMA restored.
Conclusions
We conclude that arterial vasoconstriction is impaired in young VVS patients, which is corrected by NOS inhibition. The data suggest that both pre- and post-synaptic arterial vasoconstriction may be affected by nitric oxide.
INTRODUCTION
Syncope (fainting) is rapid transient loss of consciousness and postural tone due to cerebral hypoperfusion with spontaneous recovery.1 Postural faint, denoted ‘vasovagal syncope’ (VVS), is the most common form of syncope in the young.2 VVS is common and can be induced in otherwise healthy individuals at different thresholds of orthostatic stress.3
During orthostasis caudal translocation of blood reduces central blood volume.4 Venous return and cardiac output (CO) decrease. Baroreceptors detect a postural decrease in arterial and cardiopulmonary stretch, and maintain blood pressure (BP) by a compensatory increase in heart rate (HR), total peripheral resistance (TPR), elastic recoil of venous blood, and splanchnic venoconstriction.5 This is the normal reflex response to orthostasis.6
Most adults with VVS exhibit excessive pooling in the splanchnic circulation and lower extremities that contributes to increased central hypovolaemia, reduced CO and a sustained increase in TPR.7–9 Younger adults and children follow a different course: TPR first increases during orthostasis but then progressively falls, producing a fall in BP without necessarily requiring reduced CO.10–12 This is followed by hypotension-bradycardia. There is evidence for impaired arterial vasoconstriction in VVS in the young with particular impairment of the splanchnic regional vasculature.13–15
VVS may have evolved as a defence against haemorrhagic blood loss.16,17 Studies of haemorrhage in animals show reduced endogenous response to norepinephrine and reduced responsiveness to exogenous adrenergic vasoconstrictors which can be reversed by nitric oxide synthase (NOS) inhibition.18,19 We recently investigated the dose response to infusion of the α-1 adrenergic agonist phenylephrine before and after NOS inhibition with NG-monomethyl-L-arginine, monoacetate salt (L-NMMA).15 We found that splanchnic flow and resistance were poorly responsive to phenylephrine in VVS subjects compared with controls before L-NMMA, but were enhanced after L-NMMA.
We hypothesise that nitric oxide (NO) mediated effects may account for reduced orthostatic tolerance in young adult patients with recurrent VVS and can be reversed by NOS inhibition.
METHODS
Subjects
To test this hypothesis, we recruited 12 subjects with a history of recurrent fainting (eight female, 21.2±1.2 years) and 12 healthy non-fainting control subjects (eight female, 23.0±1.1 years), all in the age range of 15–27 years. The characteristics of the two groups are shown in table 1. There were no differences in age, weight and body mass index between the groups. Fainters were referred after experiencing at least three episodes of fainting within the last 12 months. The diagnosis of VVS was primarily based on clinical history. Diagnostic features encompassed predisposing situations, prodromal symptoms, physical signs and recovery symptoms.20 Control subjects were recruited from healthy volunteers free of clinical illness, syncope and orthostatic intolerance, using on-line advertisements on the New York Medical College network.
Table 1.
Control and vasovagal syncope (VVS) patient characteristics
| Control subjects | VVS subjects | |
|---|---|---|
| Number (female/male) | 12 (8/4) | 12 (8/4) |
| Age (years±SEM) | 23.0±1.1 | 21.2±1.2 |
| Age range (years) | 15–27 | 15–27 |
| Height (cm) | 168.6±2.6 | 170.1±4.2 |
| Weight (kg) | 68.1±3.1 | 65.4±6.1 |
| Heart rate (beats/min) | 63±2 | 62±2 |
| Systolic blood pressure (mm Hg) | 118±2 | 121±2 |
| Diastolic blood pressure (mm Hg) | 63±2 | 63±4 |
| Mean arterial pressure (mm Hg) | 81±2 | 81±2 |
This study was approved by the Institutional Review Board of New York Medical College. All subjects 18 years of age or older signed an informed consent; those younger than 18 assented to participate and their parent or legal guardian signed an informed consent.
Protocol
All patients and controls were instrumented on two separate days. On the first day they received intravenous administration of the NOS inhibitor L-NMMA dissolved in normal saline. On another day they received an equivolumic intravenous infusion of normal saline as a volume control. An amount of phenylephrine (PE), individualised for each subject, was added to the saline in order to produce BP and HR changes equivalent to the reflex changes caused by the infusion of the L-NMMA. This is referred to as Saline+PE. Subjects were blinded to infusions.
Due to constraints of performing experiments on human subjects, we undertook two studies in these subjects. The first of these studies has already been published.15 In this earlier study, we constructed dose–response curves for phenylephrine with and without L-NMMA and showed impaired post-synaptic α-1-adrenergic vasoconstriction in young adults with VVS which was corrected by NOS inhibition with L-NMMA. In the present study, we evaluated responses to increasing orthostatic tolerance imposed by lower body negative pressure. Thus, because baseline data and ‘stressors’ in each study were different, these two studies were, of course, different.
Instrumentation
Subjects arrived at our climate-controlled centre at 09:30 and were instructed about the tests and instrumentation. Following instrumentation, subjects remained supine for 30 min to acclimatise. Baseline data were acquired. Thereafter either L-NMMA or saline was given by infusion on different days. We placed paired electrodes using anatomic landmarks to estimate thoracic, splanchnic, pelvic and calf segmental blood volumes, blood flows and vascular resistance by impedance plethysmography.8 Forearm blood flow was also measured by venous occlusion plethysmography every 5 min.21
L-NMMA infusion
VVS patients and control subjects received the non-isoform specific NOS inhibitor L-NMMA delivered as a 500 μg/kg/min intravenous loading dose for 15 min, followed by a 50 μg/kg/min maintenance infusion. In all subjects, a steady state for HR, BP, TPR and CO was reached during the maintenance L-NMMA infusion within 40 min.
Saline + phenylephrine infusion (Saline+PE)
On another day, separated by at least 2 days to allow for the elimination of L-NMMA (half-life ~60 min), VVS patients and control subjects received saline delivered to simulate the fluid volume and timing of a loading dose of L-NMMA. During this maintenance phase a low dose of phenylephrine was slowly titrated until HR and BP were similar to HR and BP in the same subject after loading L-NMMA, as previously described.15 This dose of phenylephrine was maintained throughout the experiment. The phenylephrine dose never exceeded 0.2 μg/kg/min in any subject.
Lower body negative pressure (LBNP) protocol
After either L-NMMA or Saline+PE were infused and HR and BP stabilised, new post-L-NMMA (or post-Saline+PE) baseline haemodynamic data were acquired. Baseline haemodynamics were followed by sequential LBNP of −15, –30, and −45 mm Hg each for 5 min. Thereafter, LBNP was set to −60 mm Hg for a maximum of 50 min. At each level of LBNP we measured HR, BP, mean arterial pressure (MAP), ModelFlow CO, TPR, regional blood flows and blood volumes using beat-to-beat measurement modalities. Negative pressure applied to the chamber produces blood and fluid translocation from the chest and upper body to the lower body.22
Data analysis
During the experiments MAP was calculated as (systolic BP+2*diastolic BP)/3 and TPR was calculated as MAP/CO. The primary outcome variables were BP, HR, CO, TPR and time to stopping LBNP to measure the orthostatic intolerance threshold; secondary outcome variables were changes in splanchnic, pelvic and calf blood flows measured by impedance plethysmography. Thoracic (central) blood flow was measured as the CO. Paired t-tests compared the response to drug loading shown in table 1.
The LBNP data were analysed as two separate experiments (L-NMMA and Saline+PE), using repeated measures analysis of variance (RM-ANOVA). For each experiment, there was one between factor (GROUP, VVS patient vs control) and one within factor (PRESSURE, the applied LBNP). Within patients, outcomes were measured at baseline, −15, –30 and −45 mm Hg during Saline+PE, and at baseline, −15, –30, −45, and −60(1) mm Hg during L-NMMA, which constituted the repeated measures. The primary relationship of interest for both experiments was the GROUP by PRESSURE interaction, which describes how patients and controls differ in their response to the LBNP challenge under each experimental setting. For these analyses, we assumed a covariance structure of compound symmetry. Reported p values reflect the interaction term using the Greenhouse-Geisser correction. Further, we assumed that, if there were a response to the LBNP challenge for either group, the response would increase or decrease linearly. Thus, our interest was in the difference in slopes between the study groups, rather than the group difference at any particular negative pressure level. As such, no post-hoc pairwise comparisons of groups at pressure specific pressure levels were conducted. Values are presented as mean±SEM. Statistical significance was set at p≤0.05. NCSS 2007 (NCSS, LCC, and Kaysville) statistical software was used for statistical analyses.
Additional detailed information regarding subjects, Instrumentation, LBNP, L-NMMA and data analysis can be found in the online Supplementary file Materials and Methods section.
RESULTS
Baseline and post-drug data
These data are tabulated in table 2. There were no significant differences in systolic BP or diastolic BP between control and VVS groups, although control systolic pressures increased from baseline (p<0.05) after L-NMMA and Saline+PE. MAP was significantly increased for controls and VVS (p<0.01 for both). HR was not different for control and VVS at baseline and after loading, but was significantly reduced by L-NMMA (p<0.05) for both control and VVS compared with baseline. Neither respiratory rate nor end-tidal CO2 (ETCO2) (data not shown) were affected by either L-NMMA or Saline+PE in either group. CO was not reduced by L-NMMA for control and VVS compared with baseline, while TPR was increased by L-NMMA (p<0.05) compared with baseline for both control and VVS. There were no significant differences between L-NMMA and Saline+PE CO or TPR for either group. There was no effect of drugs on forearm blood flow. L-NMMA significantly reduced (p<0.05) splanchnic blood flow for both control and VVS.
Table 2.
Baseline and post-drug load characteristics (post-load = following L-NMMA or Saline+PE)
| Control baseline | Control post-load | VVS baseline | VVS post-load | |
|---|---|---|---|---|
| Systolic BP (mm Hg) | ||||
| LNMMA | 119±2 | 126±3* | 122±3 | 128±5 |
| Saline+PE | 117±2 | 126±3* | 120±2 | 127±4 |
| Diastolic BP (mm Hg) | ||||
| LNMMA | 63±2 | 73±4 | 63±4 | 77±3 |
| Saline+PE | 64±2 | 76±3 | 63±4 | 77±3 |
| MAP (mm Hg) | ||||
| LNMMA | 80±1 | 91±4* | 80±2 | 93±1* |
| Saline+PE | 83±2 | 93±3* | 82±1 | 94±2* |
| HR (beats/min) | ||||
| LNMMA | 63±2 | 56±4* | 62±4 | 58±5* |
| Saline+PE | 63±2 | 57±4* | 62±4 | 59±5* |
| CO (L/min) | ||||
| LNMMA | 4.9±0.4 | 5.1±0.3 | 4.9±0.4 | 4.8±0.4 |
| Saline+PE | 5.2±0.4 | 4.9±0.3 | 4.8±0.4 | 4.6±0.5 |
| TPR (mm Hg/L/min) | ||||
| LNMMA | 16.3±1.4 | 18.7±1.0* | 16.1±1.0 | 18.6±0.9* |
| Saline+PE | 16.0±1.0 | 19.0±1.1* | 15.5±1.2 | 21.1±1.0* |
| Splanchnic BF (L/min) | ||||
| LNMMA | 1.5±0.2 | 1.2±0.1* | 1.3±0.1 | 1.0±0.1* |
| Saline+PE | 1.2±0.2 | 1.2±0.2 | 1.2±0.1 | 1.2±0.1 |
| Forearm BF (mL%/min) | ||||
| LNMMA | 2.3±0.26 | 1.98±0.33 | 2.21±0.19 | 1.91±0.34 |
| Saline+PE | 2.11±0.30 | 2.01±0.27 | 2.22±0.25 | 2.18±0.12 |
p<0.05 compared with baseline.
BF, blood flow; BP, blood pressure; CO, cardiac output; HR, heart rate; LNMMA, NG-monomethyl-L-arginine; MAP, mean arterial pressure; PE, phenylephrine; TPR, total peripheral resistance.
Duration of LBNP
Control subjects withstood the effects of LBNP for 25.9±4.0 min during Saline+PE experiments, while fainters lasted only 11.6±1.4 min (p<0.001). After loading with L-NMMA, control subjects withstood the effects of LBNP for 24.8±3.2 min, while fainters lasted 22.6±1.6 min which was increased compared with VVS subjects receiving Saline+PE (p<0.001).
MAP and HR responses to LBNP
MAP (top panels) and HR (lower panels) during imposition of increasing negative pressures with LBNP are shown in figure 1. MAP decreased for all subjects during LBNP with Saline+PE (left panel) but decreased more in VVS patients compared with control subjects (p<0.001). In contrast, MAP decreased similarly during LBNP with L-NMMA (right panel).
Figure 1.
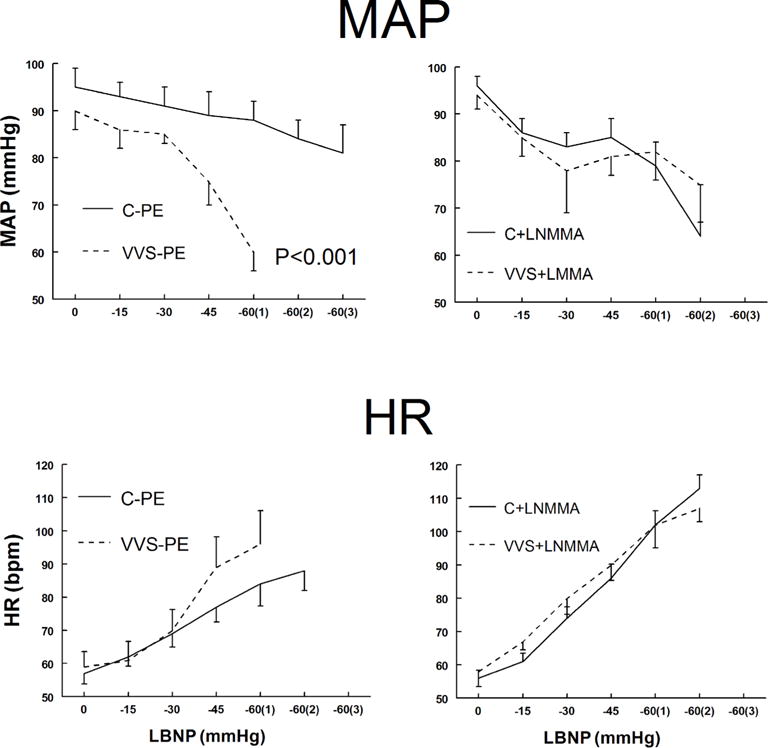
Mean arterial pressure (MAP) is shown in upper panels and heart rate (HR) is shown in lower panels during lower body negative pressure (LBNP) for control subjects (solid lines) and vasovagal syncope (VVS) subjects (dashed lines). Saline and low dose phenylephrine (PE) were infused to simulate the fluid volume and maintained (left panels) to assure that heart rate (HR) and mean arterial pressure (MAP) were similar to HR and MAP in each subject after loading NG-monomethyl-L-arginine (L-NMMA) (right panels). This is depicted as Saline+PE and is shown for control subjects who received saline and phenylephrine (C–PE), control subjects who received L-NMMA (C+LNMMA), patients with VVS who received saline and phenylephrine (VVS-PE) and patients with VVS who received L-NMMA (VVS+LNMMA). Following that, increasingly negative LBNP was applied. p<0.001 represents a significant difference in the group by pressure interaction effect.
HR increased during LBNP, and was similar for both groups during Saline+PE. There was a trend towards higher HR in VVS as pressure decreased. HR increased similarly for both groups after L-NMMA. Thus, MAP during LBNP was decreased in VVS after Saline+PE loading but was similar to control after L-NMMA loading. HR response was similar for both groups and decreased with L-NMMA.
CO and TPR responses to LBNP
CO and TPR during LBNP are shown in figure 2. CO decreased similarly with LBNP for control subjects and VVS subjects. The presence of L-NMMA decreased CO with LBNP similarly for both control and VVS.
Figure 2.
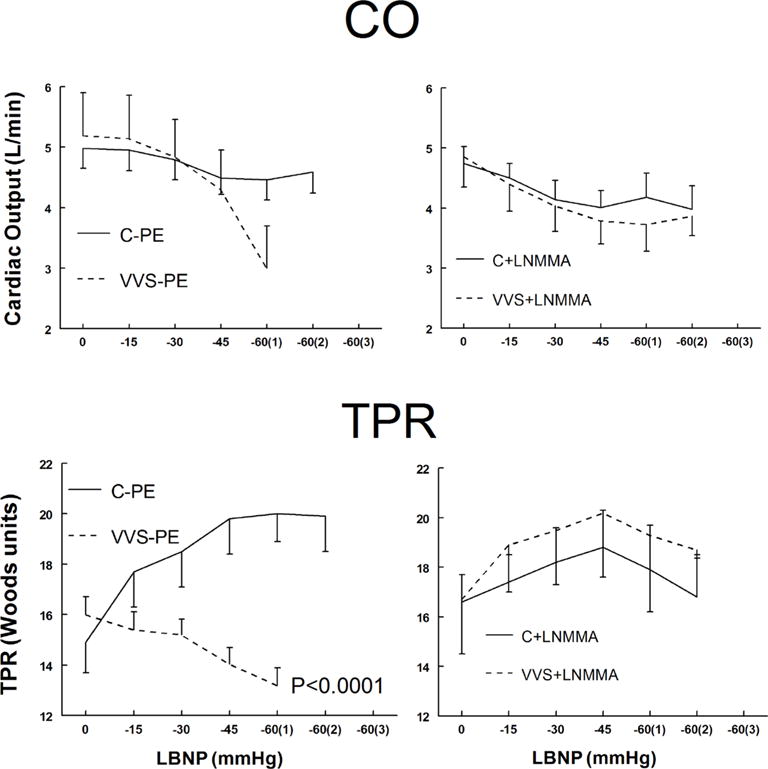
Cardiac output (CO), upper panels, and total peripheral resistance (TPR), lower panels, during lower body negative pressure (LBNP) for control subjects (solid lines) and vasovagal syncope (VVS) subjects (dashed lines). Saline and low dose phenylephrine (PE) were infused to simulate the fluid volume and maintained (left panels) to assure that heart rate (HR) and mean arterial pressure (MAP) were similar to HR and MAP in each subject after loading NG-monomethyl-L-arginine (L-NMMA) (right panels). This is depicted as Saline+PE and is shown for control subjects (C–PE), control subjects who received L-NMMA (C+LNMMA), patients with VVS who received saline and PE (VVS-PE) and patients with VVS who received L-NMMA (VVS+LNMMA). Following that, increasingly negative LBNP was applied. p<0.0001 represents a significant difference in the group by pressure interaction effect.
TPR increased significantly with LBNP for control but decreased for VVS (p<0.0001). There were similar changes in TPR with LBNP for both groups after L-NMMA. Thus, TPR was impaired in VVS after Saline+PE but was similar to control after L-NMMA loading.
Changes in regional blood volumes during LBNP
Figure 3 shows the percentage changes in central, splanchnic, pelvic and calf regional blood volumes during LBNP with Saline+PE and L-NMMA. There were no significant differences in regional blood volumes which also appeared little affected by L-NMMA. Pelvic and calf blood volumes increased for control and VVS as expected during LBNP. The decreases did not differ between groups suggesting that venous capacitance and micro-vascular filtration were similar in both cases.
Figure 3.
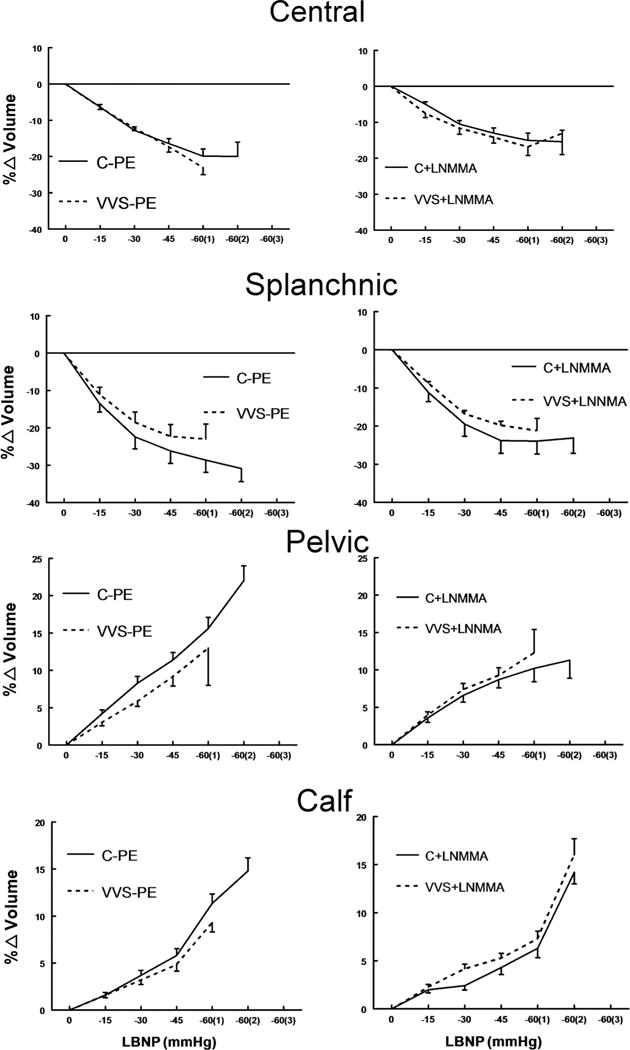
Change (%) in regional blood volumes. From top to bottom these are central (thoracic) blood volume, splanchnic blood volume, pelvic blood volume and calf blood volume. Control subjects (C) are shown as solid lines and vasovagal syncope (VVS) subjects are shown as dashed lines. Saline and low dose phenylephrine (PE) were infused to simulate the fluid volume and maintained (left panels) to assure that heart rate (HR) and mean arterial pressure (MAP) were similar to HR and MAP in each subject after loading NG-monomethyl-L-arginine (L-NMMA) (right panels). There were no statistically significant differences between control and VVS patients either with Saline+PE or L-NMMA.
Forearm vascular resistance, splanchnic blood flow and splanchnic vascular resistance responses to LBNP
Forearm vascular resistance (Rforearm), splanchnic blood flow (Fsplanchnic) and splanchnic arterial resistance (Rsplanchnic) during LBNP are shown in figure 4. Rforearm was not different for VVS and control during LBNP although it was increased compared with Saline+PE after L-NMMA.
Figure 4.
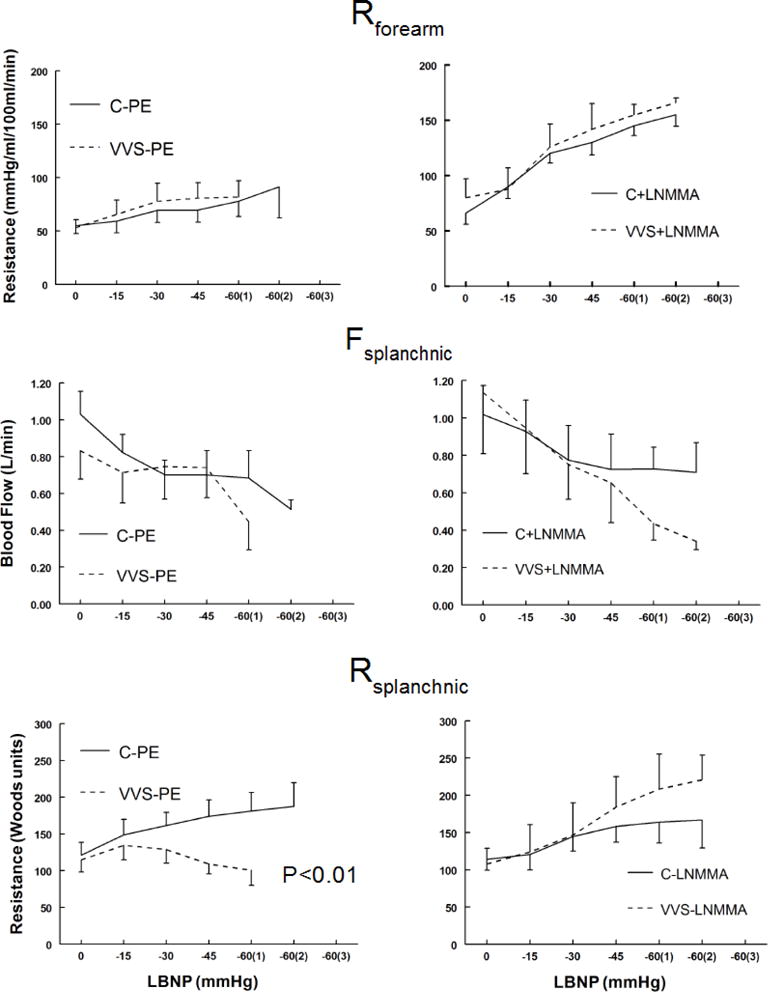
Forearm vascular resistance (Rforearm) shown in the upper panels increase similarly during the lower body negative pressure (LBNP) for control and vasovagal syncope (VVS) subjects during saline and phenylephrine (Saline+PE) and NG-monomethyl-L-arginine (L-NMMA). Control subjects (C) are shown as solid lines and VVS subjects are shown as dashed lines. Saline+PE are shown in left panels and L-NMMA response in right panels. Splanchnic blood flow (Fsplanchnic), shown in the middle panels, was also similar for VVS and control subjects during LBNP under Saline+PE and L-NMMA conditions. In contrast splanchnic arterial resistance (Rsplanchnic) decreased with LBNP during Saline+PE studies in VVS patients and was significantly different from results in controls in whom Rsplanchnic increased. p<0.01 represents a significant difference in the group by pressure interaction effect.
Fsplanchnic was similar for VVS and control subjects during LBNP under Saline+PE and L-NMMA conditions. Rsplanchnic in VVS patients decreased with LBNP during Saline+PE studies and was significantly different from results in controls in whom Rsplanchnic increased (p<0.01). Thus, splanchnic but not forearm vascular resistance was decreased compared with control in VVS during LBNP following Saline+PE; splanchnic vascular resistance was corrected (ie, not different from control) following L-NMMA.
DISCUSSION
The results of this study in young adults indicate that there is impaired arterial vasoconstriction in recurrent VVS patients compared with healthy volunteers resulting in reduced TPR and reduced BP during an imposed orthostatic challenge (LBNP). Tolerance for LBNP is greatly reduced in VVS patients compared with control subjects. Vasoconstrictive impairment and orthostatic tolerance is corrected following NOS inhibition using L-NMMA. Stated simply, there is impaired splanchnic vasoconstriction in VVS that is restored by NOS inhibition.
Arterial vasoconstriction is most impaired in the splanchnic vasculature and is corrected following L-NMMA. In contrast, forearm vasoconstriction is unimpaired in VVS patients and somewhat increased by L-NMMA.
We focused on whether LBNP had an effect on group differences; whether the response slopes differed as pressure was successively reduced. We were not concerned with differences at each pressure or in identifying the pressure at which the departure became significant.
Our observations receive support from findings of NO contributions to potentiated splanchnic hyporeactivity,23 impaired cardiovagal baroreflex24 and abnormal cerebral autoregulation25 which occur during the pre-syncopal phase of VVS.
In previous work15 we showed that post-synaptic adrenergic vasoconstriction was impaired in VVS. We wondered whether pre-synaptic adrenergic vasoconstriction was likewise compromised. This question could not be directly answered since sympathetic transduction at the synapse depends on both pre- and post-synaptic aspects and post-synaptic adrenergic vasoconstriction is compromised. Using a heuristic approach, and knowing TPR in VVS subjects with and without PE in the absence and presence of L-NMMA, our calculations suggest that VVS presynaptic adrenergic function is also impaired.
NO blunts adrenergic neurotransmission
NO is a gasotransmitter with pleiotropic effects and ubiquitous distribution. Three separate NOS isoforms are recognised: neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial NOS (eNOS). nNOS and eNOS are constitutively expressed and depend on calcium-calmodulin.26
So-called ‘nitrergic’ or ‘nitroxidergic’ nerves containing nNOS proliferate within the gastrointestinal tract, distribute with the parasympathetic nervous system and are often found close to adrenergic synapses of vascular smooth muscle.27 Nitrergic NO can act at pre- and post-synaptic sites to reduce sympathetic transduction at vascular smooth muscle.28 Effects are largest in the splanchnic vasculature29 and in the kidney30 where nitrergic nerve density is greatest. Effects are potentiated during sympathetic activation.
Splanchnic vasodilation occurs in real or simulated microgravity (bed rest in man, hindlimb suspension in rats) and associates with orthostatic intolerance, adrenergic hyporeactivity31,32 and enhanced production of NO via increased transcription of NOS isoforms.33,34 Increased NO has been reported in postural tachycardia syndrome and VVS,35–37 but results are controversial due to methodological issues.36
Nevertheless, nitrergic NO modulation of adrenergic vasoconstriction causes splanchnic vasodilation. Our data suggest that vasovagal syncope patients have increased production of NO within the splanchnic circulation resulting in a post-synaptic defect in adrenergic vasoconstriction, and likely an additional pre-synaptic defect. The defect has been shown to be present during LBNP, a form of orthostatic stress, leading to a blunted response to sympathetic stimulation and decreased orthostatic tolerance. We were unable to measure epinephrine release in this study for technical reasons. NO may have a role in epinephrine release in humans as it does in animals.38 While L-NMMA may have blunted epinephrine release, neurotransmitters were not measured in this study. Further information may be revealed in future studies that include measurements of these parameters.
Our heuristic approach suggests that pre-synaptic defects in addition to post-synaptic defects in adrenergic vasoconstriction are present. Pre-synaptic deficits could include reduction of central sympathetic activation and ganglionic transmission leading to reduced sympathetic nerve activity, reduced norepinephrine release, or impaired synthesis of norepinephrine. Post-synaptic effects could include decreased numbers of adrenergic receptors; however, this is unlikely given the fairly rapid normalising effects of L-NMMA. Post-synaptic effects could also include interference with adrenergic binding and with vascular smooth muscle transduction which are likely candidates for NO-induced downregulation. These potential mechanisms cannot be distinguished based on current data.
Our results, obtained in young adults, may differ from results in older adults. For example, in females the first peak of VVS occurs at 15–24 years, with another peak occurring after age 40.1 The mechanisms of VVS at various ages are unclear, but increased incidence with age has been attributed to the use of vasoactive medications, decreased vascular compliance, diminished cardiac function, physical deconditioning and poorer health. Therefore, our findings of impaired adrenergic vasoconstriction in VVS under conditions of orthostatic stress, which can be corrected by NOS inhibition, may be unique to young adults.
Limitations
Certain features of the study may be underpowered giving rise to type I errors in light of multiple outcomes in addition to multiple testing for each outcome. Thus, replication of these experiments is necessary to determine the robustness of our results and the use of a larger sample size would help verify the stability of our significant results. In addition, replication of these results with larger samples may also inform on additional significant differences currently deemed non-significant.
Use of LBNP is not precisely equivalent to upright posture but it is accepted as a satisfactory approximation for study.
The findings of this study may not be applicable across all age groups. Sex differences and menstrual phase were not distinguished. There were insufficient subjects for this purpose. However, no obvious difference in sympathetic nerve activity or vasoconstrictive ability by sex or menstrual phase has been found in prior investigations, although total blood volume and reduced BP occur in young women.39,40
Available evidence suggests that L-NMMA does not cross the blood-brain barrier in humans; thus, direct effects on the brain have been discounted in this study.41
Use of impedance methods carries limitations in measurement of vascular resistance but we have not found a better method at this point. Our past work8 validated impedance estimates of splanchnic blood flow against more invasive indocyanine green methods. Direct measurement of splanchnic pressures would remove remaining speculation, but this is challenging in humans as it requires invasive instrumentation which raises difficult ethical issues in volunteers and vasovagal subjects.
Conclusions
Our data demonstrate that impaired splanchnic adrenergic vasoconstriction in young patients with VVS, the most common form of syncope, accounts for orthostatic intolerance which can be reversed by NOS inhibition. We conclude that arterial vasoconstriction is impaired in young VVS patients, which is corrected by NOS inhibition. This suggests that both pre- and post-synaptic arterial vasoconstriction may be affected by NO.
Supplementary Material
Key messages.
What is already known about this subject?
Mechanisms of vasovagal syncope remain elusive.
What does this study add?
Our data demonstrate that impaired splanchnic adrenergic vasoconstriction in young patients with vasovagal syncope, the most common form of syncope, accounts for orthostatic intolerance which can be reversed by nitric oxide synthase inhibition.
How might this impact on clinical practice?
There could be a role for nitric oxide (NO) scavenging or reduced NO production in the treatment of vasovagal syncope.
Acknowledgments
Funding Funding for this project was provided by National Institutes of Health grant RO1 HL 112736 (JMS) and NIH R21NS 094644 (MSM).
Footnotes
Contributors JMS was responsible for the conception and design of the experiments, interpretation of data and drafting the article. RS was responsible for interpretation of the data and revising the manuscript. MLK was responsible for collection, assembly and interpretation of the data. AMG was responsible for collection, assembly and interpretation of the data. PV was responsible for biostatistical analysis and interpretation of the data and critically revising intellectual content of the article. MSM was responsible for design of the experiments, analysis and interpretation of the data and critically revising intellectual content of the article. All authors read and approved the final article.
Competing interests None declared.
Patient consent Consent for our research were signed by patient, and by guardian if under 18 years old. However, the data presented here are completely anonymized.
Ethics approval Committee for the Protection of Human Subjects of New York Medical College.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Moya A, Sutton R, Ammirati F, et al. Guidelines for the diagnosis and management of syncope (version 2009) Eur Heart J. 2009;30:2631–71. doi: 10.1093/eurheartj/ehp298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gowers WR. A lecture on vagal and vasovagal attacks. Lancet. 1907;173:716–24. [Google Scholar]
- 3.el Bedawi KM, Hainsworth R. Combined head-up tilt and lower body suction: a test of orthostatic tolerance. Clin Auton Res. 1994;4:41–7. doi: 10.1007/BF01828837. [DOI] [PubMed] [Google Scholar]
- 4.Rowell LB. Human cardiovascular control. New York, NY: Oxford University Press; 1993. [Google Scholar]
- 5.Donald DE, Rowlands DJ, Ferguson DA. Similarity of blood flow in the normal and the sympathectomized dog hind limb during graded exercise. Circ Res. 1970;26:185–99. doi: 10.1161/01.res.26.2.185. [DOI] [PubMed] [Google Scholar]
- 6.O’Leary DD, Kimmerly DS, Cechetto AD, et al. Differential effect of head-up tilt on cardiovagal and sympathetic baroreflex sensitivity in humans. Exp Physiol. 2003;88:769–74. doi: 10.1113/eph8802632. [DOI] [PubMed] [Google Scholar]
- 7.Mosqueda-Garcia R, Furlan R, Jt M, et al. The elusive pathophysiology of neurally mediated syncope [In process citation] Circulation. 2000;102:2898–906. doi: 10.1161/01.cir.102.23.2898. [DOI] [PubMed] [Google Scholar]
- 8.Stewart JM, Medow MS, Glover JL, et al. Persistent splanchnic hyperemia during upright tilt in postural tachycardia syndrome. Am J Physiol Heart Circ Physiol. 2006;290:H665–H673. doi: 10.1152/ajpheart.00784.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taneja I, Medow MS, Glover JL, et al. Increased vasoconstriction predisposes to hyperpnea and postural faint. Am J Physiol Heart Circ Physiol. 2008;295:H372–H381. doi: 10.1152/ajpheart.00101.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Jong-de Vos van Steenwijk CC, Wieling W, Johannes JM, et al. Incidence and hemodynamic characteristics of near-fainting in healthy 6- to 16-year old subjects. J Am Coll Cardiol. 1995;25:1615–21. doi: 10.1016/0735-1097(95)00056-a. [DOI] [PubMed] [Google Scholar]
- 11.Nowak JA, Ocon A, Taneja I, et al. Multiresolution wavelet analysis of time-dependent physiological responses in syncopal youths. Am J Physiol Heart Circ Physiol. 2009;296:H171–H179. doi: 10.1152/ajpheart.00963.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verheyden B, Liu J, van Dijk N, et al. Steep fall in cardiac output is main determinant of hypotension during drug-free and nitroglycerine-induced orthostatic vasovagal syncope. Heart Rhythm. 2008;5:1695–701. doi: 10.1016/j.hrthm.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Dietz NM, Halliwill JR, Spielmann JM, et al. Sympathetic withdrawal and forearm vasodilation during vasovagal syncope in humans. J Appl Physiol. 1997;82:1785–93. doi: 10.1152/jappl.1997.82.6.1785. [DOI] [PubMed] [Google Scholar]
- 14.Stewart JM, McLeod KJ, Sanyal S, et al. Relation of postural vasovagal syncope to splanchnic hypervolemia in adolescents. Circulation. 2004;110:2575–81. doi: 10.1161/01.CIR.0000145543.88293.21. [DOI] [PubMed] [Google Scholar]
- 15.Stewart JM, Suggs M, Merchant S, et al. Postsynaptic α1-adrenergic vasoconstriction is impaired in young patients with vasovagal syncope and is corrected by nitric oxide synthase inhibition. Circ Arrhythm Electrophysiol. 2016;9:e003828. doi: 10.1161/CIRCEP.115.003828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barcroft H, Mcmichael JE, Scarpey-Schafer EP. Posthaemorrhagic fainting. Study by cardiac output and forearm flow. Lancet. 1944;1:489–91. [Google Scholar]
- 17.Diehl RR. Vasovagal syncope and darwinian fitness. Clin Auton Res. 2005;15:126–9. doi: 10.1007/s10286-005-0244-0. [DOI] [PubMed] [Google Scholar]
- 18.Liu LM, Ward JA, Dubick MA. Hemorrhage-induced vascular hyporeactivity to norepinephrine in select vasculatures of rats and the roles of nitric oxide and endothelin. Shock. 2003;19:208–14. doi: 10.1097/00024382-200303000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Pieber D, Horina G, Sandner-Kiesling A, et al. Pressor and mesenteric arterial hyporesponsiveness to angiotensin II is an early event in haemorrhagic hypotension in anaesthetised rats. Cardiovasc Res. 1999;44:166–75. doi: 10.1016/s0008-6363(99)00194-7. [DOI] [PubMed] [Google Scholar]
- 20.Sheldon RS, Grubb BP, Olshansky B, et al. Heart Rhythm Society expert consensus statement on the diagnosis and treatment of postural tachycardia syndrome, inappropriate sinus tachycardia, and vasovagal syncope. Heart Rhythm. 2015;2015:e41–e63. doi: 10.1016/j.hrthm.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stewart JM. Transient orthostatic hypotension is common in adolescents. J Pediatr. 2002;140:418–24. doi: 10.1067/mpd.2002.122643. [DOI] [PubMed] [Google Scholar]
- 22.Hagan RD, Diaz FJ, Horvath SM. Plasma volume changes with movement to supine and standing positions. J Appl Physiol Respir Environ Exerc Physiol. 1978;45:414–7. doi: 10.1152/jappl.1978.45.3.414. [DOI] [PubMed] [Google Scholar]
- 23.Hamza SM, Kaufman S. Role of spleen in integrated control of splanchnic vascular tone: physiology and pathophysiology. Can J Physiol Pharmacol. 2009;87:1–7. doi: 10.1139/Y08-103. [DOI] [PubMed] [Google Scholar]
- 24.Liu JL, Murakami H, Zucker IH. Effects of NO on Baroreflex control of heart rate and renal nerve activity in conscious rabbits. Am J Physiol. 1996;270:R1361–R1370. doi: 10.1152/ajpregu.1996.270.6.R1361. [DOI] [PubMed] [Google Scholar]
- 25.Lavi S, Egbarya R, Lavi R, et al. Role of nitric oxide in the regulation of cerebral blood flow in humans: chemoregulation versus mechanoregulation. Circulation. 2003;107:1901–5. doi: 10.1161/01.CIR.0000057973.99140.5A. [DOI] [PubMed] [Google Scholar]
- 26.Busse R, Mülsch A. Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 1990;265:133–6. doi: 10.1016/0014-5793(90)80902-u. [DOI] [PubMed] [Google Scholar]
- 27.Toda N, Okamura T. The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol Rev. 2003;55:271–324. doi: 10.1124/pr.55.2.3. [DOI] [PubMed] [Google Scholar]
- 28.Storgaard T, Nedergaard OA. Prejunctional modulation by angiotensins of noradrenaline release from sympathetic neurons in isolated rabbit aorta. Naunyn Schmiedebergs Arch Pharmacol. 1997;356:706–11. doi: 10.1007/pl00005109. [DOI] [PubMed] [Google Scholar]
- 29.Kolo LL, Westfall TC, Macarthur H. Nitric oxide decreases the biological activity of norepinephrine resulting in altered vascular tone in the rat mesenteric arterial bed. Am J Physiol Heart Circ Physiol. 2004;286:296H–303. doi: 10.1152/ajpheart.00668.2003. [DOI] [PubMed] [Google Scholar]
- 30.Toda N, Okamura T. Modulation of renal blood flow and vascular tone by neuronal nitric oxide synthase-derived nitric oxide. J Vasc Res. 2011;48:1–10. doi: 10.1159/000317395. [DOI] [PubMed] [Google Scholar]
- 31.Arbeille PP, Besnard SS, Kerbeci PP, et al. Portal vein cross-sectional area and flow and orthostatic tolerance: a 90-day bed rest study. J Appl Physiol. 2005;99:1853–7. doi: 10.1152/japplphysiol.00331.2005. [DOI] [PubMed] [Google Scholar]
- 32.Overton JM, Tipton CM. Effect of hindlimb suspension on cardiovascular responses to sympathomimetics and lower body negative pressure. J Appl Physiol. 1990;68:355–62. doi: 10.1152/jappl.1990.68.1.355. [DOI] [PubMed] [Google Scholar]
- 33.Mueller PJ, Foley CM, Hasser EM. Hindlimb unloading alters nitric oxide and autonomic control of resting arterial pressure in conscious rats. Am J Physiol Regul Integr Comp Physiol. 2005;289:R140–R147. doi: 10.1152/ajpregu.00820.2004. [DOI] [PubMed] [Google Scholar]
- 34.Vaziri ND, Ding Y, Sangha DS, et al. Upregulation of NOS by simulated microgravity, potential cause of orthostatic intolerance. J Appl Physiol. 2000;89:338–44. doi: 10.1152/jappl.2000.89.1.338. [DOI] [PubMed] [Google Scholar]
- 35.Galetta F, Franzoni F, Plantinga Y, et al. Endothelial function in young subjects with vaso-vagal syncope. Biomed Pharmacother. 2006;60:448–52. doi: 10.1016/j.biopha.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 36.Ruiz GA, Sinigaglia S, Hermes R, et al. Role of nitric oxide in young patients with vasovagal syncope. Europace. 2010;12:987–90. doi: 10.1093/europace/euq148. [DOI] [PubMed] [Google Scholar]
- 37.Liao Y, Chen S, Liu X, et al. Flow-mediated vasodilation and endothelium function in children with postural orthostatic tachycardia syndrome. Am J Cardiol. 2010;106:378–82. doi: 10.1016/j.amjcard.2010.03.034. [DOI] [PubMed] [Google Scholar]
- 38.Figueroa XF, Poblete I, Fernández R, et al. NO production and eNOS phosphorylation induced by epinephrine through the activation of beta-adrenoceptors. Am J Physiol Heart Circ Physiol. 2009;297:H134–H143. doi: 10.1152/ajpheart.00023.2009. [DOI] [PubMed] [Google Scholar]
- 39.Fu Q, Witkowski S, Okazaki K, et al. Effects of gender and hypovolemia on sympathetic neural responses to orthostatic stress. Am J Physiol Regul Integr Comp Physiol. 2005;289:R109–R116. doi: 10.1152/ajpregu.00013.2005. [DOI] [PubMed] [Google Scholar]
- 40.Stickford AS, VanGundy TB, Levine BD, et al. Menstrual cycle phase does not affect sympathetic neural activity in women with postural orthostatic tachycardia syndrome. J Physiol. 2015;593:2131–43. doi: 10.1113/JP270088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang R, Witkowski S, Cui J, et al. Inhibition of nitric oxide synthase does not alter dynamic cerebral autoregulation in humans. Am J Physiol Heart Circ Physiol. 2004;286:H683–H689. doi: 10.1152/ajpheart.00373.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.