

Abstract
Background
Bats are natural reservoirs for several highly pathogenic and novel viruses including coronaviruses (CoVs) (mainly Alphacoronavirus and Betacoronavirus). Lyle’s flying fox (Pteropus lylei)‘s roosts and foraging sites are usually in the proximity to humans and animals. Knowledge about age-specific pattern of CoV infection in P. lylei, prevalence, and viral shedding at roosts and foraging sites may have an impact on infection-age-structure model to control CoV outbreak.
Methods
P. lylei bats were captured monthly during January–December 2012 for detection of CoV at three areas in Chonburi province; two human dwellings, S1 and S2, where few fruit trees were located with an open pig farm, 0.6 km and 5.5 km away from the bat roost, S3. Nested RT-PCR of RNA-dependent RNA polymerase (RdRp) gene from rectal swabs was used for CoV detection. The strain of CoV was confirmed by sequencing and phylogenetic analysis.
Results
CoV infection was found in both juveniles and adult bats between May and October (January, in adults only and April, in juveniles only). Of total rectal swab positives (68/367, 18.5%), ratio was higher in bats captured at S1 (11/44, 25.0%) and S2 (35/99, 35.4%) foraging sites than at roost (S3) (22/224, 9.8%). Juveniles (forearm length ≤ 136 mm) were found with more CoV infection than adults at all three sites; S1 (9/24, 37.5% vs 2/20, 10%), S2 (22/49, 44.9% vs 13/50, 26.0%), and S3 (10/30, 33.3% vs 12/194, 6.2%). The average BCI of CoV infected bats was significantly lower than uninfected bats. No gender difference related to infection was found at the sites. Phylogenetic analysis of conserved RdRp gene revealed that the detected CoVs belonged to group D betacoronavirus (n = 64) and alphacoronavirus (n = 4).
Conclusions
The fact that CoV infection and shedding was found in more juvenile than adult bats may suggest transmission from mother during peripartum period. Whether viral reactivation during parturition period or stress is responsible in maintaining transmission in the bat colony needs to be explored.
Keywords: Coronavirus, Chiroptera, Pteropus, Thailand
Background
Coronaviruses (CoVs) are classified into four genera: Alphacoronavirus (αCoV), Betacoronavirus (βCoV), Gammacoronavirus, and Deltacoronavirus [1]. CoVs infect wide variety of mammals and birds, causing upper and lower respiratory, hepatic, enteric and neurological illnesses with varying severity. Bat CoVs (BtCoVs) are likely the gene source of αCoV and βCoV, while avian CoVs are sources of Gammacoronavirus, and Deltacoronavirus [2]. Although there is single lineage in αCoV, βCoVs are further separated into four lineages (A – D) [3]. Lineage A βCoV, including bovine CoVs, human CoV (HCoV)-OC43 and related viruses, have been detected in various mammals such as cows, horses, deer, antelopes, camels, giraffes, waterbucks, dogs, and humans worldwide, but not in bats. Lineages B-D βCoVs have been detected in bats worldwide [4].
Currently, six CoV strains are known to cause human infection; four CoVs cause mild respiratory illness, including two αCoVs: HCoV-NL63 and HCoV-229E, and two βCoVs: HCoV-HKU1and HCoV-OC43 [5]. The other two βCoVs cause severe respiratory tract infection with high-fatality rates, such as severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), belonging to lineages B and C, respectively. Bat related MERS-CoVs phylogenetically-related to humans have been previously discovered in Tylonycteris pachypus (BtCoV-HKU4) and Pipistrellus abramus (BtCoV-HKU5) in Hong Kong, in 2006 [6], Neoromicia capensis (NeoCoV) from South Africa, in 2011 [7], and Pipistrellus cf. hesperidus (PREDICT/PDF-2180 CoV) from Uganda, in 2013 [8]. SARS-like BtCoV was initially identified from the genus Rhinolophus in 2005, after the SARS outbreak in humans in 2002–2003, and identification of virus in palm civets (Paguma larvata) from live animal market in Guangdong, China in 2003 [9, 10].
BtCoVs have been identified in many insectivorous and frugivorous (family Pteropodidae) bats on many continents: America, Europe, Africa, and Asia [4]. Different species of Pteropodidae have been identified as a major source of lineage D βCoV (HKU9) in Africa (Rousettus aegyptiacus, Kenya [11], Pteropus rufus and Eidolon dupreanum, Madagascar [12]), and Asia (R. leschenaulti, China [13], Cynopterus brachyotis, Philippines [14], Ptenochirus jagori, Philippines [15], Pteropus giganteus, Bangladesh [16], Cynopterus sphinx, Thailand [17].
Thailand is home to 146 bat species (125 insectivorous and 21 frugivorous) [18]. The prevalence and diversity of BtCoVs in Thailand has been studied in the last decade [17, 19, 20]. CoVs were found in 11 insectivorous bat species and in 2 frugivorous bat species. However, data from Pteropus bats have been lacking despite Pteropus being the biggest colony of Pteropodidae in Thailand. Three species (P. lylei, P. vampyrus and P. hypomelanus) are reservoirs of Nipah virus (NiV) in Thailand [21]. The prevalence of NiV RNA in urine of P. lylei has been seasonally detected during the months of May and June [22].
P. lylei (Lyle’s flying fox (LFF)) ranges from Yunnan in China, and extends to Cambodia, Thailand, and Vietnam [23]. Up to 20 colonies have been identified in Thailand [24] and the largest known colony comprises of about 10,000 individuals [22]. It shares foraging areas with other frugivorous bats in fruit trees, from which the fruits are also shared by humans. Moreover, trees in populated temple grounds and cultivated land are common roosting sites for LFF. Thus, consumption of partially eaten fruit, uncooked meat, or contact with saliva, urine or faeces, which can be contaminated with bat viruses, poses a risk of viral transmission from LFF to humans or domestic animals.
The potential for emergence of zoonotic viruses into the human population depends on the prevalence of the virus in its host species, host range mutations within viral quasispecies, and the degree to which the reservoir host interacts with humans [25]. To better understand the prevalence, persistence, phylogeny, and potential for interaction with humans, here we describe a comprehensive longitudinal study to detect CoV in LFF, and factors influencing infectivity. Bat rectal swabs were collected monthly from their roosting area and from two human dwellings (foraging sites) nearby. Individual bats were weighed and forearm (FA) lengths were measured for further characterization on its body composition index (BCI). Our results demonstrated for the first time that α- and β-CoVs are endemically circulating in LFFs in Thailand, and that age and BCI are significantly different between infected and uninfected bats.
Methods
Study sites
LFFs were captured from the largest colony in Thailand (total population of around 10,000 bats) [22] at Chonburi province (Luang temple, 13,830,018.9”N, 101809054.9″E, 6 m asl) in Central Thailand. Bats were sampled from three different sites: two human dwellings (bat foraging areas) situated at a mean distance of 0.6 km (S1) and 5.5 km (S2 with a small open-system pig farm, 40 pigs) from a bat roost, and the bat roost (S3). Sampling was carried out under protocols approved and permitted by the Department of National Parks, Wildlife and Plant Conservation, Thailand (No. 0909.204/2686) and the Animal Use Protocol No.1473001 approved by Chulalongkorn University Animal Care and Use Committee.
Bat capture and sample collection
LFFs were captured monthly during January – December 2012 from the three sites, S1–3 (Table 1). At S1 and S2, bats were captured 2 nights per month, where the nets were set in the late evening, and captured animals were removed immediately. At S3, 10–20 bats were captured using mist-nets on the same nights as S1 and S2. Bats were not euthanized, and they were released after measurements were taken and samples were collected. Bats were identified morphometrically and species, sex, reproductive status, FA length and body mass were determined. Rectal swab was collected from each individual bat and immediately put into Lysis buffer (bioMérieux, SA, France). The samples were transported to laboratory on ice within 48 h and stored at -80o C until further analysis.
Table 1.
CoV positive bats, categorized by possible influential factors - collection site, age, sex and BCI (367 bats)
| Possible influential factor | CoV PCR results: No. Positive / no. of tested (%) | ||||
|---|---|---|---|---|---|
| S1 (house) | S2 (pig farm) | S3 (roost) | ALL SITES | ||
| Agea | Juvenile (J) | 9/24 (37.5) | 22/49 (44.9) | 10/30 (33.3) | 41/103 (39.8) |
| Adult (A) | 2/20 (10.0) | 13/50 (26.0) | 12/194 (6.2) | 27/264 (10.2) | |
| Total | 11/44 (25.0) | 35/99 (35.4) | 22/224 (9.8) | 68/367 (18.5) | |
| Ratio (J:A) | 24:20 = 1.20 | 49:50 = 0.98 | 30:194 = 0.15 | 103:264 = 0.39 | |
| Sex | Male | 6/27 (22.2) | 16/51 (31.4) | 14/142 (9.9) | 36/220 (16.4) |
| Female | 5/17 (29.4) | 19/48 (39.6) | 8/82 (9.8) | 32/147 (21.8) | |
| Total | 11/44 (25.0) | 35/99 (35.4) | 22/224 (9.8) | 68/367 (18.5) | |
| Mean BCIb (352 bats) |
All bats | 1.83 | 1.94 | 2.90 | 2.50 |
| Positive | 1.63 | 1.76 | 2.15 | 1.86 | |
| Negative | 1.89 | 2.04 | 2.99 | 2.65 | |
aJ = juvenile bat (forearm length ≤ 136 mm) A = adult bat (forearm length > 136 mm)
bMean BCI is calculated from 352 bats those FA length and body mass were measured
Nucleic acid extraction and CoV RNA detection
Total nucleic acid was extracted from 1 ml of suspended rectal swab using easyMAG® platform (bioMérieux, SA, France). Elution volume was 50 μl. Hemi-nested Reverse Transcription PCR (RT-PCR) was performed using broadly reactive consensus PCR primers for CoV, targeting the RNA-dependent RNA polymerase (RdRp) gene. A total of 5 μl of extracted nucleic acid was added to 50 μl of reaction mixture of OneStep RT-PCR kit (QIAGEN, Hilden, Germany), per manufacturer’s instructions, and reacted with each forward primer and reverse primer [14]. Hemi-nested PCR amplifications were performed using 2 μl of first amplification product and 48 μl of reaction mixture containing 1.0 unit of Platinum Taq DNA polymerase in 2.5 mM MgCl2, 400 μM dNTPs, 0.6 μM of second forward primer and 0.6 μM of the same reverse primer as the first round of RT-PCR. Amplification product of 434 bp was visualized using 2% agarose gel electrophoresis. All positive PCR products were further sequenced for confirmation and strain characterization.
Sequencing and phylogenetic analysis
The RdRp PCR products were gel purified using the NucleoSpin® Gel and PCR Clean-up kit (MACHEREY-NAGEL GmbH & Co. KG), and sequenced directly using an automated ABI PRISM 377 DNA sequencer. When multi peaks were shown in chromatogram at same position from direct sequencing, PCR products were cloned using the pGEM®-T Easy Vector System and the LigaFast™ Rapid DNA Ligation System (Promega) before sequencing. Five colonies were picked up for sequencing. Sequences were cleaned using Bio-edit program and aligned with reference sequences collected from GenBank. Alignments were performed using Multiple Alignment using Fast Fourier Transform (MAFFT) [26]. Phylogenetic trees were created based on 357 and 299 bp RdRp gene sequence using the maximum likelihood method. Bootstrap values were determined using 1000 replicates via RaxmlGUI 1.3 with outgroup (Bulbul CoV/HKU11–934/Pycnonotus jocosus/CHN/2007/FJ376619) using the GTRI substitution model [27]. The phylogenetic tree was visualised using the FigTree program, version 1.4.2 [28].
Statistical analysis
We considered the relative level of CoV infection in variables of bat. We used Chi-square and Fisher’s exact tests to determine the prevalence pattern of CoV by examining whether cues recorded in each kind of variables (location, sex, age and season) differed from expected. All statistical tests were completed in R statistic computing (version 3.2.2) with p < 0.05 interpreted as being statistically significant. The body condition index (BCI) was defined as body mass divided by FA length. To assess differences in BCI between CoV infected and uninfected bats, ANOVA with Tukey’s test for pair-wise comparisons was used for analysis.
Results
Sample collection
Only the P. lylei species (LFF) was included in this study. A total of 367 bats (220 male and 147 female) were captured and sampled. Total number of captured bats from sites S1, S2 and S3 were 44, 99, and 224 respectively (Table 1). FA length (≤ 136 mm) was used to distinguish between juvenile and adult [29]. Body mass and FA lengths were determined for 352 bats (95.9%). FA lengths of juveniles (n = 96) ranged from 79.23–136.0 mm, and in adults (n = 256) ranged from 136.47–170.0 mm. Body mass of juveniles ranged from 124.0–307.0 g, while adults ranged from 212.0–658.0 g. The BCI in juveniles and adults ranged from 1.08–2.32 and 1.43–4.27 respectively. The ratio of juvenile and pup per adult bats captured from sites S1 (24:20, 1.2) and S2 (49:50, 0.9) were similar, but a lower ratio was found at site S3 (30:194, 0.15) (Table 1). Number of bats trapped/captured each month varied between 14 and 46 bats; minimum of 10 was captured each month at their roost (S3) as control. Juvenile bats were not captured in January through to March for testing, as the ratio of juvenile and adult bats in natural population is low due to the LFF’s breeding cycle, which is once a year from November to February [22]. Thus new-borns are delivered in February/March, and weaning juvenile bats are mostly observed in May. Of the 147 females captured, 18 were at active breeding age evident by either lactating (n = 10, which the specimens were available for testing from 9 pups), being pregnant (n = 4) or having enlarged nipples indicative of previous lactation (n = 4).
Virus detection-prevalence
Sixty eight of 367 (18.5%) rectal swabs from LFF were positive for CoV by family wide CoV PCR [14]. As shown in Table 1, CoV RNA positive bats were found in 16.4% (36/220) male bats, and 21.8% (32/147) in female bats. There was no significant difference in the rate of infectivity when comparing sex (p > 0.05).
The number of CoV positive juvenile and adult bats from all three sites (n = 367 bats) were 39.8% (41/103) and 10.2% (27/264), respectively (Table 2). CoV positive juvenile bats from sites S1-S3 were 37.5% (9/24), 44.9% (22/49), and 33.3% (10/30), respectively. CoV positive adult bats from sites S1-S3 were 10% (2/20), 26.0% (13/50), and 6.2% (12/194), respectively (Table 1). Statistical analysis showed that CoV infectivity between juvenile and adult bats significantly differed (p < 0.01), indicating that CoV infection favoured juvenile LFFs.
Table 2.
Number of bats PCR-positive for coronavirus by month and agea from SI, S2 and S3
| Month | Juvenile | Adult | Total | |||
|---|---|---|---|---|---|---|
| Tested | Positive (%) | Tested | Positive (%) | Tested | Positive (%) | |
| January | 0 | 0 (0) | 14 | 4 (28.6) | 14 | 4 (28.6) |
| February | 0 | 0 (0) | 14 | 0 (0) | 14 | 0 (0) |
| March | 0 | 0 (0) | 20 | 0 (0) | 20 | 0 (0) |
| April | 12 | 2 (16.7) | 34 | 0 (0) | 46 | 2 (4.3) |
| May | 21 | 13 (61.9) | 18 | 3 (16.7) | 39 | 16 (41.0) |
| June | 20 | 11 (55. 0) | 9 | 3 (33.3) | 29 | 14 (48.3) |
| July | 21 | 7 (33.3) | 25 | 7 (28.0) | 46 | 14 (30.4) |
| August | 9 | 4 (44.4) | 26 | 5 (19.2) | 35 | 9 (25.7) |
| September | 12 | 3 (25.0) | 23 | 3 (13.0) | 35 | 6 (17.1) |
| October | 4 | 1 (25.0) | 23 | 2 (8.7) | 27 | 3 (11.1) |
| November | 3 | 0 (0) | 33 | 0 (0) | 36 | 0 (0) |
| December | 1 | 0 (0) | 25 | 0 (0) | 26 | 0 (0) |
| Total | 103 | 41 (39.8) | 264 | 27 (10.2) | 367 | 68 (18.5) |
aJuvenile bat: forearm length ≤ 136 mm
None of the rectal swabs from the 18 pregnant or lactating adult female bats tested positive for CoV. Interestingly, three attached pubs, but not their mothers, from a total of 9 pairs were found positive for CoV RNA. BCI of the two pups with available data were 1.69 (211 g body mass/125 mm FA) and 1.42 (170 g body mass/120 mm FA), which were lower than the uninfected mean for juvenile bats (1.72) (Table 3).
Table 3.
Range and mean of forearm (FA), body mass, and body condition index (BCI) of bats in this study (352 bats) classified by age and/or CoV infection status
| Bat characteristics | FA (mm) Range (mean) |
Body Mass (g) Range (mean) |
BCI Range (mean) |
|---|---|---|---|
| CoV positive bats | 117–160.28 (135.14) | 141–538 (255.19) | 1.11–3.36 (1.86) |
| CoV negative bats | 79.23–170.0 (147.55) | 124–658 (396.47) | 1.08–4.27 (2.65) |
| CoV positive juvenile bats | 117–136 (128.25) | 141–295 (206.75) | 1.11–2.26 (1.61) |
| CoV negative juvenile bats | 79.23–135.82 (128.82) | 124–307 (221.79) | 1.08–2.32 (1.72) |
| CoV positive adult bats | 136.67–160.28 (145.34) | 212–538 (326.96) | 1.52–3.36 (2.23) |
| CoV negative adult bats | 136.47–170 (151.51) | 215–658 (439.19) | 1.43–4.27 (2.88) |
| Total Juvenile bats | 79.23–136.0 (128.58) | 124–307 (215.52) | 1.08–2.32 (1.67) |
| Total Adult bats | 136.47–170 (150.86) | 212–658 (427.35) | 1.43–4.27 (2.81) |
The prevalence of CoV infection in bats from sites S1-S3 were 25.0% (11/44), 35.4% (35/99) and 9.8% (22/224), respectively (Table 1). Statistical analysis showed that the number of CoV infected bats at the different sites differed significantly (p < 0.01). Frequency of viral detection was higher at these two foraging sites than the roost (χ2 = 36.31, p < 0.001). However, the prevalence of CoV infection in juvenile bats from the 3 sites was similar, 37.5%, 44.9%, and 33.3%, respectively (Table 1). Age and conditions of bats which may reflect their physical health and fitness may influence the selection of foraging site and their vulnerability to infection. During the same year of study, tracked bats from this colony mostly foraged in farmland, plantations, and gardens with the maximum linear distances from 2.2–23.6 km between day roosts and foraging areas [30].
Temporal dynamics of viral shedding
Combining data from all sites, the CoV positive bats were found in 8 of 12 months, except February, March, November and December. High prevalent seasons were from May to August, with highest in June (14/29, 48.3%). Highest prevalence in juveniles were found in May (13/21, 61.9%), and in adults in June (3/9, 33.3%). There was higher prevalence of CoV infection among juvenile than adult bats during April–October (Table 2). In January, 4 CoV positive adult bats were found at S3. Individual BCI of the one female and 3 male bats were 2.83, 1.77, 1.84, 2.75, respectively, which were lower than the mean BCI in uninfected adult bats (2.88) (Table 3). We analysed the monthly prevalence of CoV infectivity in juvenile and adult bats, and combined (Table 2). There was significant difference in the seasonal prevalence of CoV infection and shedding in adults (p < 0.05), but not in juvenile bats or combined.
BCI – Infected bats
There was significant difference between FA and body mass of CoV positive bats compared to uninfected bats (p value < 0.01). BCI analysis was performed on bats with complete data of FA and body mass (352 bats). The mean BCI of captured bats in the study varied each month (Fig. 1). BCI of total tested bats (Fig. 1) and uninfected bats (Fig. 2) show similar seasonality. The lowest mean BCI of both total number of tested bats and CoV-positive bats were found in June (mean 2.05 and 1.62 respectively) (Figs. 1 and 2) when CoV infection (48.28%) was most prevalent (Table 2). In uninfected bats, the lowest mean BCI was found in July (2.12) rather than June (2.44) (Fig. 2). From this study, the CoV infected bats had significantly lower mean BCI than uninfected bats, 1.86 and 2.65 respectively (p value < 0.01) (Table 3). The BCI of CoV infected bats varied between 1.11 and 3.36 (mean 1.86) whilst the BCI of uninfected bats was between 1.08 and 4.27 (mean 2.65). The BCI of uninfected juvenile bats was between 1.08 and 2.32 (mean 1.72), whilst the BCI of uninfected adult bats varied between 1.43 and 4.27 (mean 2.88). The BCI of CoV infected juvenile bats was between 1.11 and 2.26 (mean 1.61), whilst the BCI of CoV infected adult bats varied between 1.52 and 3.36 (mean 2.23). The mean BCI of infected juvenile and adult bats were significantly lower than uninfected juvenile and adult bats respectively, (p value < 0.01, p value < 0.01 respectively) (Fig. 3). There was statistically significant difference in the mean body mass (p value < 0.01) and mean FA length (p value < 0.01) between CoV infected and uninfected bats (Table 3).
Fig. 1.
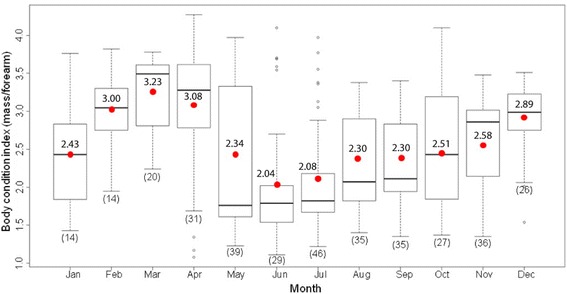
Body condition indices (BCI) of 352 bats captured in the study from January to December 2012. Bats were captured monthly at three sites (S1-S3). Numbers in brackets indicate sample size from 3 sites. Boxes depict the 25th and 75th percentiles, lines within boxes mark the median, red spot and number represent mean, whiskers represents minimum and maximum values, and circles indicates outliers
Fig. 2.
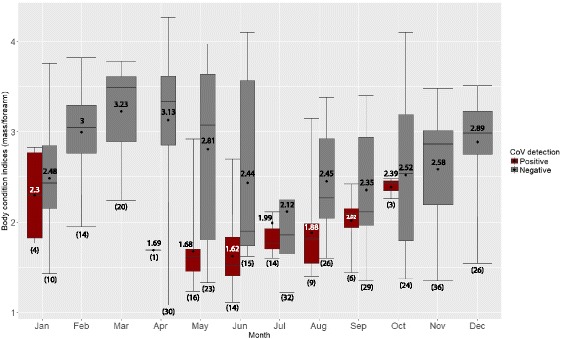
Body condition indices (BCI) of bats tested negative (gray) and positive (brown) in the study. Bats were captured monthly from January to December 2012 at three sites. Rectal swabs from 352 bats were tested for CoV by PCR. Numbers in brackets indicate sample size from 3 sites. Boxes depict the 25th and 75th percentiles, lines within boxes mark the median, spot and number represent mean, whiskers represents minimum and maximum values, and circles indicates outliers
Fig. 3.

Body condition indices (BCI) of bats tested negative (gray) and positive (brown) in the study. Rectal swabs from 352 bats were tested for CoV by PCR. Forearm length ≤ 136 mm was used to classify bats as juvenile. Numbers in brackets indicate sample size. Boxes depict the 25th and 75th percentiles, lines within boxes marks the median, whiskers represents minimum and maximum values, and circles indicates outliers
Phylogenetic analyses
Sixty-eight CoV sequences were deposited in GenBank with accession MG256395-MG256474 and MG333996-MG333999. Phylogenetic analysis of 357 bp of RdRp gene using raxmlGUI program revealed that 64 of 68 detected CoVs belonged to βCoV genus, roosting with Hong Kong strain, BtCoV HKU9 (R. lechenaulti, EF065513) and Kenya (R. aegyptiacus, GU065422), while the other 4 belonged to group 1A αCoV (Fig. 4a). The βCoVs from this study clustered in the same clade and shared 95.5–100% nucleotide identity with each other (98.3–100% identity of 118 amino acids). Two individual bats (BRT55709 and BRT55734) were found to be co-infected with multiple strains of the same βCoV species (difference of 1–2 amino acids). These viruses had amino acids differing from the HKU9 BtCoV, group D βCoV reference strain by 11.7–14.2%. They formed a different clade to other CoVs from the same bat genus (Pteropus) from Madagascar’s P. rufus (Fig. 4b). However, they were in the same clade with CoVs from different bat species captured at the same site with this study; Cynopterus sphinx, Scotophilus heathii, and Scotophilus kuhlii (Genbank accession numbers KJ868722, KJ020607, KJ020608, respectively) [17].
Fig. 4.

Maximum likelihood phylogenetic trees of coronavirus (CoV) generated using 357 (a-Bat CoVs) and 299 (b-Lineage D βCoVs) nucleotides of the RdRp gene sequences of CoVs from Thailand; in LFF (this study-blue), other bat species from previous study in Thailand [15] (green), from Madagascar’s Pteropus rufus [17] (b, pink) and reference strains of CoVs group (black). Only the representative sequences of LFF βCoVs were used for analysis. The raxmlGUI 1.3 and the GTRI substitution model with 1000 bootstrap were used for generating both phylogenetic trees. Trees were visualized using FigTree 1.4.2. BuCoV/HKU11–934/Pyc_joc/CHN/2007/FJ376619 was used as an outgroup for tree A
Discussion
This is the first longitudinal study of CoV infection in wild bats in Thailand, where 367 LFF bats were captured monthly for one year at one roosting site and two foraging sites close to the bat roost. One fourth of bats were juvenile, and 59.9% were male.
The ratios between captured juvenile and adult bats were different at the bat roost and foraging sites. Only 13% of juvenile (30/224) bats were captured at the bat roost in the year of the study, whereas half of the juvenile bats were captured from both foraging sites (24/44, 49/99, from S1 and S2 respectively). The maximum linear distances between roosts and foraging areas of LFF at this site varied from 2.2–22.3 km [30]. Foraging sites near roost, even with limited food sources, may be practical for young or unhealthy bats that are unable to fly far.
CoV RNA was detected in approximately 18% of all bats sampled, which is in the same range as the study in China (16%, [31]; 15.8%, [32]), and Germany (9.8%, [33]). The prevalence of CoV infection in Pteropus bats (P. rufus) from Madagascar was similar to this study (17.1%, 13/76) [12]. On the other hand, the prevalence in this study was higher than the two previous studies in Thailand by Wacharapluesadee et al. (6.7%, 47/626) [17] and Gouilh et al. (10.5%, 28/265) [20]. This may be the result of a bias from the cross sectional study of these two previous studies or an indication of difference in prevalence rate in different bat species.
Ratios of captured bat genders in this study were roughly similar at foraging sites. At the roost, male bats were predominantly captured. CoV infection was not correlated with sex of bat, neither at the roost nor at the foraging sites. This finding is similar to the studies from Germany [33] and Colorado, USA [25].
In our study, CoV infection was found to be associated with younger ages; 39.8% of juvenile bats versus 10.2% adult bats were positive for CoV RNA. Similar findings have been reported from the study in insectivorous bats from USA (19% juvenile versus 9% adult bats positive for CoV) [25] and Vespertilionid bats in Germany (23.7% juvenile versus 15.9% sub-adult versus 8.5% adult bats positive for CoV) [33]. These findings support the hypothesis that young bats may be more susceptible to CoV infection, and serves to propagate and play an important role in maintaining the virus within bat colonies. The divergence in rate of CoV infection from different study sites (Table 1) was likely to be influenced by the age and body condition of bats.
Three of 9 unweaned pups were CoV RNA positive, while their mothers and all lactating female carrying pups were negative for CoV. It may be possible there was a placental transmission, after which the virus was then cleared from adult female bats. Another possibility is that the unweaned bats acquired infection from contaminated secretion of other bats hanging from the same tree. However, the study by Gloza-Rausch et al. 2008 [33], where 54 of 178 (30%) of studied female bats were lactating, found higher rate of CoV infection in lactating bats (22.4%) than in non-lactating bats (9.7%) which supports the first scenario. It is to be noted that limited number of lactating bats were included in our study (9 of 147, 6.1%). Targeting mother-pup pairs in future studies would be required to confirm the vertical (placental) transmission of CoV in LFF.
Seasonal prevalence was mostly related to the number of juvenile bats captured for testing in each month (Table 2), except in January when all four CoV positive bats were adult. Notably, these positive adult bats had lower BCI (2.83, 1.77, 1.84, 2.75) than the mean uninfected adult bats (2.88). Three of the 4 infected adult bats had lower body mass (444, 429, 258, 276 g) than mean uninfected adult bats (439 g). The mean body mass of infected bats was significantly lower than in uninfected bats (Table 3). This is similar to the study where Hipposideros pomona bats in Hong Kong with HKU10 CoV infection had lower body mass than uninfected bats, even though they appeared to be healthy [34]. These bats seemed to be in poor condition, serving as the other group in addition to juvenile bats that further maintained the virus within the population.
Sixty eight CoVs were detected from this study, forming 2 genetically distinct strains. Sixty four belonged to βCoV (SARS-related group) with relatively close homology to the reference virus, BtCoV-HKU9 [6]. Four belonged to αCoV, and their sequences related to CoVs previously detected in insectivorous bats in Thailand such as H. lekaguli, H. armiger and Taphozous melanopogon [17]. This supports the possibility of interspecies transmission, rather than virus-host specific sharing, between bats of different suborder (Pteropus in Pteropodidae, Hipposideridae and Emballonuridae) that do not share food, foraging sites, or roosts, similar to the earlier HKU10 CoV study between R. leschenaulti and H. pomona bats [15]. The evolution of CoVs in different host species-order should be further studied in order to understand the route of spillover and transmission.
Bats from different species-genus that share foraging sites may also share infections and particular CoV strains, for example βCoV from LFF (this study), C. sphinx, S. Heathii, and S. kuhlii [17] (Fig. 4a-b). βCoV from same bat genus in different geographic region displayed distinct clusters (Fig. 4b), for example P. rufus from Madagascar (cluster 2–3) [12] and LFF from this study (cluster 4). This demonstrated the βCoV inclines interspecies sharing rather than virus-host specific sharing.
Given the mobility of LFF in Thailand, where the maximum linear distance between day roosts and foraging areas for LFF is 23.6 km [29], and its tendency for sharing habitat with other colonies, the detected strains of CoVs from this study may be found in LFFs all over the region. The high prevalence of CoV in this study suggests circulation of infection within the bat colony. Study of CoV diversity from other LFF colony in Thailand and region is required to improve our understanding of the evolution and spillover patterns of CoV.
Conclusions
Our study found that CoV transmission in LFF occurred throughout the year at a baseline level, and the months surrounding the birthing season (May–August) represented times of increased infection among juveniles. The CoV prevalence in LFF related mostly to the age of bat rather than location, sex or season. The interspecies transmission of CoV among different bat genus or family demonstrated the possibility of spillover and the potential for emergence of zoonotic viruses into the human population. This data provides the first long-term monitoring of CoV circulation in nature and identifies ecological drivers. The relationship between animal age and infectivity to other bat species should be further investigated to confirm this phenomenon. Additional studies on CoV diversity among Pteropus bat species in Thailand and neighbouring countries, as well as aspects of the virus-host interaction are needed to understand the origins, evolution, maintenance patterns, dispersal and zoonotic potential of CoV across the region.
Acknowledgements
We gratefully acknowledge the local support from the Thai Red Cross Society, Chulalongkorn University, Kasetsart University and the Department of National Parks Wildlife and Plant Conservation. We would also like to thank Siriporn Ghai for her intensive editing of the manuscript.
Funding
This study was supported by a research grant from the Thailand Research Fund (RDG5420089); the Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University (RES560530148-HR); Ratchadaphiseksomphot Fund of Faculty of Medicine, Chulalongkorn University; Health and Biomedical Science Research Program by National Research Council of Thailand and Health System Research Institute (P-13-50154); the Research Chair Grant (P-13-01091); Cluster and Program Management Office (CPMO) (P-15-50535); the National Science and Technology Development Agency (NSTDA),Thailand; and the USAID Emerging Pandemic Threats PREDICT 2 project (07–306–7119-52272).
Availability of data and materials
The GenBank accession numbers for coronavirus sequences reported in this paper are: MG256395-MG256474 (βCoV) and MG333996-MG333999 (αCoV) for 357 bp partial RdRp gene from Pteropus lylei.
Abbreviations
- ANOVA
Analysis of variance
- BCI
Body composition index
- bp
Base pairs
- BtCoV
Bat coronavirus
- CoV
Coronavirus
- DNA
Deoxyribonucleic acid
- FA
Forearm
- LFF
Lyle’s flying fox
- MERS
Middle East respiratory syndrome
- NiV
Nipah virus
- nt
nucleotides
- PCR
Polymerase Chain Reaction
- RdRp
RNA-dependent RNA polymerase
- RNA
ribonucleic acid
- RT-PCR
Reverse Transcription - Polymerase Chain Reaction
- S1
Study site 1 – human dwelling (0.6 km from bat roost)
- S2
Study site 2 – human dwelling, small open-pig farm (40 pigs) (5.5 km from bat roost)
- S3
Study site 3 – Bat roost
- SARS
Severe acute respiratory syndrome
- αCoV
Alphacoronavirus
- βCoV
Betacoronavirus
Authors’ contributions
SW and TH participated in the design of the study and drafted the manuscript. PD, TK, SY, SN, PP, and PM conducted sampling of bat specimens. TK carried out the molecular genetic studies. AC and AR participated in the sequence alignment and data analysis. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Bat specimens were collected with permission from the Department of National Parks, Wildlife and Plant Conservation (No. 0909.204/2686) and the Animal Use Protocol No.1473001 approved by Chulalongkorn University Animal Care and Use Committee.
Consent for publication
Not Applicable.
Competing interests
The authors claim no conflict of interest in the publication of this information.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Supaporn Wacharapluesadee, Phone: (+66) 2256-4000, Email: spwa@hotmail.com.
Prateep Duengkae, Email: prateepd@hotmail.com.
Aingorn Chaiyes, Email: chaiyes.stou@gmail.com.
Thongchai Kaewpom, Email: thongchai_ae@hotmail.com.
Apaporn Rodpan, Email: wava_38@hotmail.com.
Sangchai Yingsakmongkon, Email: sangchai59@hotmail.com.
Sininat Petcharat, Email: cakeinarak@hotmail.com.
Patcharakiti Phengsakul, Email: chubby_f8@hotmail.com.
Pattarapol Maneeorn, Email: lotterwildlifevet@gmail.com.
Thiravat Hemachudha, Email: fmedthm@gmail.com.
References
- 1.Adams MJ, Carstens EB. Ratification vote on taxonomic proposals to the international committee on taxonomy of viruses. Arch Virol. 2012;157:1411–1422. doi: 10.1007/s00705-012-1299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woo PC, Sk L, Lam CS, Lau CC, Tsang AK, Lau JH, et al. Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J Virol. 2012;86:3995–4008. doi: 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Groot RJ, Baker SC, Baric R, Enjuanes L, Gorbalenya AE, Holmes KV, et al. Family Coronaviridae. In: King AMQ, Lefkowitz E, Adams MJ, Carstens EB, et al., editors. Virus taxonomy: ninth report of the international committee on taxonomy of viruses. Amsterdam: Elsevier; 2012. pp. 806–820. [Google Scholar]
- 4.Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir Res. 2014;101:45–56. doi: 10.1016/j.antiviral.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan JF, Li KS, To KK. Cheng VC, Chen H, Yuen KY. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoVEMC) the beginning of another SARS-like pandemic? J Inf Secur. 2012;65:477–489. doi: 10.1016/j.jinf.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo PC, Wang M, Lau SK, Xu H, Poon RW, Guo R, et al. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J Virol. 2007;81:1574–1585. doi: 10.1128/JVI.02182-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ithete NL, Stoffberg S, Corman VM, Cottontail VM, Richards LR, Schoeman MC, et al. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg Infect Dis. 2013;19:1697–1699. doi: 10.3201/eid1910.130946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anthony SJ, Gilardi K, Menachery VD, Goldstein T, Ssebide B, Mbabazi R, et al. Further evidence for bats as the evolutionary source of Middle East respiratory syndrome coronavirus. MBio. 2017;8(2):e00373-17. doi: 10.1128/mBio.00373-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 11.Tong S, Conrardy C, Ruone S, Kuzmin IV, Guo X, Tao Y, et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg Infect Dis. 2009;15:482–485. doi: 10.3201/eid1503.081013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Razanajatovo NH, Nomenjanahary LA, Wilkinson DA, Razafimanahaka JH, Goodman SM, Jenkins RK, et al. Detection of new genetic variants of Betacoronaviruses in endemic frugivorous bats of Madagascar. Virol J. 2015;12:42. doi: 10.1186/s12985-015-0271-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lau SK, Poon RW, Wong BH, Wang M, Huang Y, Xu H, et al. Coexistence of different genotypes in the same bat and serological characterization of Rousettus bat coronavirus HKU9 belonging to a novel Betacoronavirus subgroup. J Virol. 2010;84:11385–11394. doi: 10.1128/JVI.01121-10. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Watanabe S, Masangkay JS, Nagata N, Morikawa S, Mizutani T, Fukushi S, et al. Bat coronaviruses and experimental infection of bats, the Philippines. Emerg Infect Dis. 2010;16:1217–1223. doi: 10.3201/eid1608.100208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsuda S, Watanabe S, Masangkay JS, Mizutani T, Alviola P, Ueda N, et al. Genomic and serological detection of bat coronavirus from bats in the Philippines. Arch Virol. 2012;157:2349–2355. doi: 10.1007/s00705-012-1410-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anthony SJ, Epstein JH, Murray KA, Navarrete-Macias I, Zambrana-Torrelio CM, Solovyov A, et al. A strategy to estimate unknown viral diversity in mammals. MBio. 2013;4:e00598-13. doi: 10.1128/mBio.00598-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wacharapluesadee S, Duengkae P, Rodpan A, Kaewpom T, Maneeorn P, Kanchanasaka B, et al. Diversity of coronavirus in bats from eastern Thailand. Virol J. 2015;12:57. doi: 10.1186/s12985-015-0289-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karnpun S, Wongwai A, Soisook P. Cave-dwelling bats of Thailand. Bangkok: Wildlife Research Section, Department of National Parks Wildlife and Plant Conservation; 2016. [Google Scholar]
- 19.Wacharapluesadee S, Sintunawa C, Kaewpom T, Khongnomnan K, Olival KJ, Epstein JH, et al. Group C betacoronavirus in bat guano fertilizer, Thailand. Emerg Infect Dis. 2013;19:1349–1351. doi: 10.3201/eid1908.130119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gouilh MA, Puechmaille SJ, Gonzalez JP, Teeling E, Kittayapong P, Manuguerra JC. SARS-coronavirus ancestor's foot-prints in south-east Asian bat colonies and the refuge theory. Infect Genet Evol. 2011;11:1690–1702. [DOI] [PMC free article] [PubMed]
- 21.Wacharapluesadee S, Lumlertdacha B, Boongird K, Wanghongsa S, Chanhome L, Rollin P, et al. Bat Nipah virus, Thailand. Emerg Infect Dis. 2005;11:1949–1951. doi: 10.3201/eid1112.050613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wacharapluesadee S, Boongird K, Wanghongsa S, Ratanasetyuth N, Supavonwong P, Saengsen D, et al. A longitudinal study of the prevalence of Nipah virus in Pteropus Lylei bats in Thailand: evidence for seasonal preference in disease transmission. Vector Borne Zoonotic Dis. 2010;10:183–190. doi: 10.1089/vbz.2008.0105. [DOI] [PubMed] [Google Scholar]
- 23.Bumrungsri S, Suyanto A, Francis C. Pteropus lylei. In: The IUCN Red List of Threatened Species. 2008. 10.2305/IUCN.UK.2008.RLTS.T18734A8513517.en. Accessed 12 Nov 2017.
- 24.Wacharapluesadee S, Olival KJ, Kanchanasaka B, Duengkae P, Kaewchot S, Srongmongkol P, et al. Surveillance for Ebola virus in wildlife, Thailand. Emerg Infect Dis. 2015;21:2271–2273. doi: 10.3201/eid2112.150860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osborne C, Cryan PM, O'Shea TJ, Oko LM, Ndaluka C, Calisher CH, et al. Alphacoronaviruses in new world bats: prevalence, persistence, phylogeny, and potential for interaction with humans. PLoS One. 2011;6:e19156. doi: 10.1371/journal.pone.0019156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silvestro D, Michalak I. raxmlGUI: a graphical front-end for RAxML. Org Divers Evol. 2012;12:335–337. doi: 10.1007/s13127-011-0056-0. [DOI] [Google Scholar]
- 28.Rambaut A. FigTree Tree Figure Drawing Tool, Version 1.4.2, (2014). Available from: http://tree.bio.ed.ac.uk/software/figtree/. Accessed 12 Nov 2017.
- 29.Wanghongsa S, Boongird K. On the population number and distribution of Lyle's flying fox (Pteropus lylei) in central plain. In: Wildlife research section annual report. Department of National Parks Wildlife and Plant Conservation. Bangkok; 2003. p. 89–100.
- 30.Weber N, Duengkae P, Fahr J, Dechmann DK, Phengsakul P, Khumbucha W, et al. High-resolution GPS tracking of Lyle's flying fox between temples and orchards in central Thailand. J Wildl Manag. 2015;79:957–968. doi: 10.1002/jwmg.904. [DOI] [Google Scholar]
- 31.Lau SK, Woo PC, Li KS, Huang Y, Wang M, Lam CS, et al. Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology. 2007;367:428–439. doi: 10.1016/j.virol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu DK, Poon LL, Chan KH, Chen H, Guan Y, Yuen KY, Peiris JS. Coronaviruses in bent-winged bats (Miniopterus spp.) J Gen Virol. 2006;87:2461–2466. doi: 10.1099/vir.0.82203-0. [DOI] [PubMed] [Google Scholar]
- 33.Gloza-Rausch F, Ipsen A, Seebens A, Göttsche M, Panning M, Drexler JF, et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg Infect Dis. 2008;14:626–631. doi: 10.3201/eid1404.071439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lau SK, Li KS, Tsang AK, Shek CT, Wang M, Choi GK, et al. Recent transmission of a novel alphacoronavirus, bat coronavirus HKU10, from Leschenault's rousettes to pomona leaf-nosed bats: first evidence of interspecies transmission of coronavirus between bats of different suborders. J Virol. 2012;86:11906–11918. doi: 10.1128/JVI.01305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The GenBank accession numbers for coronavirus sequences reported in this paper are: MG256395-MG256474 (βCoV) and MG333996-MG333999 (αCoV) for 357 bp partial RdRp gene from Pteropus lylei.