

Summary paragraph
Recent advances in enzyme engineering and design have expanded nature’s catalytic repertoire to functions that are new to biology1–3. Yet only a subset of these engineered enzymes can function in living systems4–7. Finding enzymatic pathways that forge chemical bonds not found in biology is particularly difficult in the cellular environment, as this hinges on the discovery not only of new enzyme activities but also reagents that are simultaneously sufficiently reactive for the desired transformation and stable in vivo. Here we report the discovery, evolution, and generalisation of a fully genetically-encoded platform for producing chiral organoboranes in bacteria. Escherichia coli harbouring wild-type cytochrome c from Rhodothermus marinus8 (Rma cyt c) were found to form carbon–boron bonds in the presence of borane-Lewis base complexes, through carbene insertion into B–H bonds. Directed evolution of Rma cyt c in the bacterial catalyst provided access to 16 novel chiral organoboranes. The catalyst is suitable for gram scale biosynthesis, offering up to 15300 turnovers, 6100 h–1 turnover frequency, 99:1 enantiomeric ratio (e.r.), and 100% chemoselectivity. The enantio-preference of the biocatalyst could also be switched to provide either enantiomer of the organoborane products. Evolved in the context of whole-cell catalysts, the proteins were more active in the whole-cell system than in purified forms. This study establishes a DNA-encoded and readily engineered bacterial platform for borylation; engineering can be accomplished at a pace which rivals the development of chemical synthetic methods, with the ability to achieve turnovers that are two orders of magnitude (over 400-fold) greater than that of known chiral catalysts for the same class of transformation9–11. This tunable method for manipulating boron in cells opens a whole new world of boron chemistry in living systems.
Boron-containing natural products are synthesised in the soil by the myxobacterium Sorangium cellulosum as antibiotics against Gram-positive bacteria12. In the sea, these molecules give the Jurassic red alga Solenopora jurassica its distinct pink colouration13; they are also produced by the bioluminescent bacterium Vibrio harveyi for cell-cell communications14 (Extended Data Fig. 1). To prepare boron-containing biomolecules, living organisms produce small molecules that spontaneously react with boric acid available in the environment15,16. While this non-enzymatic method for capturing boron is sufficient for an organism’s survival, it is limited to a substrate’s inherent affinity towards boric acid, and lacks tunability and generality for synthetic biology applications. Moreover, organisms that produce organoboranes (compounds that contain carbon–boron bonds) are unknown.
We envisioned that enzyme-catalyzed borylation could provide living organisms the ability to produce boron-containing products tailored to our needs. Such an enzyme is not known in nature, but we hypothesised that existing natural proteins might be repurposed and engineered to perform this task. In the past, we and others have exploited the promiscuity of natural and engineered haem proteins for various non-natural reactions4,6,7,17. The resulting enzymes are fully genetically-encoded and carry out their synthetic functions in their bacterial expression hosts. Here, we focused on introducing boron motifs to organic molecules enantioselectively, as this would generate boron-containing carbon-stereocentres, which are important structural features in functional organoboranes such as the FDA-approved chemotherapeutics Velcade® and Ninlaro®18. They are also versatile precursors for chemical derivatisation through stereospecific carbon–boron to carbon–carbon/carbon–heteroatom bond conversion19–21.
Though boron reagents applicable for carbon–boron bond formation in water are known22,23, their biocompatibility, cell permeability, stability, and reactivity in living systems, where biomolecules, nucleic acids, and metal ions abound, are uncertain. Nevertheless, since boron reagents designed for in vivo chemical biology applications are precedented24–26, we reasoned that reagents suitable for biological borylation could be found. We identified borane-Lewis base complexes as potential candidates due to their aqueous stability and reactivity towards carbenoid B–H insertion9–11,27 (Extended Data Fig. 2), a mechanistic pathway we believed might be adapted for use in the biological environment due to its orthogonality to living systems’ existing biochemistry.
We first set out to assess whether biological organoborane production might be feasible in a bacterial cell. When E. coli BL21(DE3) cells harbouring wild-type cytochrome c from Rhodothermus marinus, a Gram-negative, thermohalophilic bacterium from submarine hot springs in Iceland8 (Rma cyt c), were incubated with N-heterocyclic carbene borane28,29 (NHC-borane) 1 and ethyl 2-diazopropanoate (Me-EDA) 2 in neutral buffer (M9-N minimal medium, pH 7.4) at room temperature, in vivo production of organoborane 3 was observed, with 120 turnovers (calculated with respect to the concentration of Rma cyt c expressed in E. coli; Fig. 1a, b) and an e.r. of 85:15 (R/S isomer = 6; Fig. 1c). Since the pET22b/pEC86 expression system translocates Rma cyt c to the E. coli periplasm for post-translational maturation (during which the haem cofactor is covalently ligated to the cyt c apoprotein)30, we assumed that borylation takes places in the periplasmic compartment. In the absence of Rma cyt c, E. coli yielded only a trace amount of borylation product with very low stereoselectivity (Extended Data Table 1). Both substrates and the organoborane product were stable under these conditions. The haem cofactor alone could also promote the borylation reaction, although with no stereoselectivity. Other cytochrome c proteins, cytochromes P450, and globins also demonstrated carbon–boron bond forming ability, but their selectivities were unsatisfactory (Extended Data Table 1).
Figure 1. Discovery, evolution and characterisation of a bacterial catalyst for borylation.

a, Reaction scheme shows a representative in vivo borylation reaction between NHC-borane 1 and diazo ester 2 to yield organoborane 3. Standard substrate loading is 10 mM for both 1 and 2. The absolute configuration of biosynthesised 3 was assigned to be R by X-ray crystallography. b, c Sequential site-saturation mutagenesis of Rma cyt c targeting active-site amino acid residues M100, V75 and M103 improved the turnover and enantioselectivity of bacterial production of organoborane 3. Whole-cell Rma cyt c variants were compared using E. coli cells at concentration OD600 = 15. Total turnover numbers (TTNs) were calculated with respect to the concentration of Rma cyt c expressed in E. coli and represent the total number of turnovers obtained from the catalyst under the stated reaction conditions. d, X-ray crystal structure of wild-type Rma cyt c (PDB: 3CP5). e, Turnover frequencies (TOFs) of BORWT and BORR1 as whole-cell catalysts, cell lysates, or purified proteins for the production of organoborane 3. f, Purified and whole-cell BORR1 were preincubated with NHC-borane 1, Me-EDA 2, or organoborane 3 before they were used as borylation catalysts to determine the inactivation effects of 1–3. The numbers shown represent the %TTN retained after preincubation, and are relative to a control (no incubation) of the same type of catalyst (purified protein or whole cell). Bars and numbers above bars represent mean values averaged over four biological replicates. Individual data points are shown as overlays. NHC, N-heterocyclic. BSA, bovin serum albumin. Single-letter abbreviations for the amino acid residues are as follows: D, Asp; M, Met; R, Arg; T, Thr; and V, Val.
To improve the performance of this whole-cell catalyst, we subjected the wild-type Rma cyt c (which we refer to as BORWT hereafter) to site-saturation mutagenesis, sequentially targeting active-site amino acid residues M100, V75 and M103, which are closest to the haem iron in BORWT (within 7Å, Fig. 1d). Each single-site site-saturation mutagenesis library was cloned using the 22c-trick method31, screened as whole-cell catalysts in 96-well plates for improved borylation enantioselectivity, and the best variant was used to parent the next round of mutation and screening. With a single mutation M100D replacing the distal axial ligand, the first-generation biocatalyst exhibited 16-fold improvement in turnover over the wild-type (1850 TTN, Fig. 1b), with 88:12 e.r. (R/S isomer = 7; Fig. 1c). The M100D mutation also substantially improved carbene transfer reactivity for Si–H insertion catalyzed by Rma cyt c6. This improvement in catalytic performance is likely due to removal of the axial ligand from the haem iron, which opens a site primed for iron carbenoid formation and subsequent product formation32. Two subsequent rounds of mutagenesis and screening led to variant BORR1 (V75R M100D M103T), which exhibited a turnover of 2490 and an e.r. of 97.5:2.5 (R/S isomer = 39). This genetically programmed biological function is readily scalable from analytic to mmol scale – with 0.5 mmol substrates, BORR1 produced organoborane 3 in 97.5:2.5 e.r. and 75% isolated yield (3000 TTN). The absolute configuration of product 3 was unambiguously assigned to be R by X-ray crystallography.
With an excellent borylating bacterium in hand, the properties and potential of the system were assessed. We characterised the initial rates of in vivo borylation and found that screening for improved enantioselectivity also led to an overall rate enhancement: whole-cell BORR1 is 15 times faster than BORWT, with a turnover frequency of 6100 h–1. Interestingly, as purified protein or in cell lysate, both BORR1 and BORWT are orders of magnitude slower (Fig. 1e). When isolated BOR R1 protein and whole-cell BORR1 were preincubated with Me-EDA 2 before the borylation reaction, the isolated protein retained only ~50% of its activity, whereas whole-cell BORR1 retained >90% activity (Fig. 1f). NHC-borane 1 and organoborane product 3 did not inactivate the enzyme. Me-EDA likely inactivates BORR1 through carbene transfer to the haem cofactor and/or nucleophilic side chains of the protein, a mechanism we previously studied in detail for a cytochrome P450-based carbene transferase33. The intact periplasm apparently protects BORR1 from inactivation by Me-EDA, and carbene transfer to yield the organoborane product is generally faster than protein inactivation pathway(s) under those conditions. Similar observations have been reported for other protein-based carbene transfer reaction systems.7,34 Analysis of colony-forming units shows that in vivo organoborane production does not dramatically reduce the viability of the E. coli (Extended Data Fig. 3).
We next explored the scope of boron reagents that could function in the cellular environment. Ten boron reagents were tested under turnover-optimised conditions: though the size, solubility and lipophilicity of these reagents varied, all were found to permeate the cell membrane and give the desired products in excellent selectivities and turnovers (Fig. 2a). Various substitutions on the NHC nitrogen are tolerated (3 to 10). The reaction is chemoselective in the presence of terminal olefins (5), which could function as a reaction handle suitable for downstream biological or bio-orthogonal derivatisation. Sterically more demanding tetra- and penta-substituted NHCs are also accepted (7 to 10). Beside imidazole-based boron reagents, triazolylidene borane and picoline borane could also be used for in vivo borylation, yielding products 11 and 12 in 1070 TTN and 2440 TTN, respectively, with uniformly high selectivities (96:4 e.r.). On gram scale, in vivo borylation produced 740 mg of picoline organoborane 12 with 2910 TTN, 96:4 e.r. and 42% isolated yield (64% based on recovered starting material, Fig. 2b). The absolute configuration of 12 was assigned to be R by X-ray crystallography. When substrates were added portion-wise at regular time intervals to E. coli expressing BORR1 (we tested the sequential addition of up to eight equivalents of substrates over a period of 12 hours, Fig. 2c; Extended Data Table 2), organoborane 3 was produced with 10400 turnovers (50% yield, 96:4 e.r.), whereas organoborane 9 was obtained with 15300 turnovers (73% yield, 96:4 e.r.). No significant loss in activity or enantioselectivity was observed, demonstrating the potential of this bacterial catalyst for biosynthesis and incorporation into natural or engineered metabolic pathways.
Figure 2. Scope of chiral organoborane production in E. coli.

a, d, Scope of boron reagent and diazo ester for borylation catalysed by E. coli harbouring BORR1. Standard substrate loading is 10 mM for both substrates. Reactions conducted in duplicate. b, Gram scale synthesis (8.4 mmol) of organoborane 12 catalysed by whole-cell BORR1 (OD600 = 30). The small scale preparation of 12 (2440 TTN, 96:4 e.r.) was also reported for comparison. The absolute configuration of biosynthesised 12 was assigned to be R by X-ray crystallography. c, Biosynthesis of organoboranes 9 (blue) and 3 (orange) catalysed by whole-cell BORR1 (OD600 = 15). One substrate equivalent (8 mM final concentration of boron reagent and diazo ester) was added to the reaction every 75 min. Reactions conducted in biological duplicate. Bn, benzyl; e.r., enantiomeric ratio.
Systematic modification of the diazo ester substituents from Et to Me, i-Pr or Bn revealed that the borylation ability of BORR1 is not limited to Me-EDA (3, 13 to 15, Fig. 2d). The protein’s relative insensitivity to steric bulk of the ester might indicate that in the putative iron carbenoid intermediate this moiety is solvent-exposed rather than embedded within the active site. By re-randomising the 103 position in BORR1, a residue we believe might modulate loop dynamics for improved binding of this substrate, the borylation turnover of 15 improved (from 2560 to 4200 TTN) using V75R M100D M103D (BORR2, Fig. 3a). From the same site-saturation library, a borylation catalyst for trifluoromethyl-substituted diazo ester (CF3-EDA) was also discovered (V75R M100D M103F, BORR3). Acceptor/acceptor diazo reagents such as CF3-EDA are less reactive towards carbenoid formation due to their electron-deficient nature and have not been employed before this for enzymatic carbene-transfer reactions. The present system tolerates this class of substrates and yielded product 16 with 95:5 e.r. and 1560 TTN.
Figure 3. Expanding the generality and utility of biological borylation.

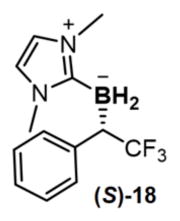
a, The generality of in vivo borylation was expanded through directed evolution to accommodate bulky substrates (15, 17) and less reactive acceptor/acceptor diazo reagents (16), to move beyond diazo ester-based substrates ((R)-18, (S)-18), and to provide either enantiomer of the organoborane products ((R)-18, (S)-18). Reactions conducted in biological quadruplicate. Solid arrows represent site-saturation mutagenesis studies. BORP* was discovered in the M100D V75X site-saturation mutagenesis library for Me-EDA borylation. Amino acid residues targeted during directed evolution are depicted in the X-ray crystal structure of wild-type Rma cyt c (PDB: 3CP5). The absolute configuration of biosynthesised (R)-18 was assigned by X-ray crystallography. b, Derivatisation of biocatalytic product. Organoborane (R)-18 was biosynthesised with E. coli harbouring BORP2 (OD600 = 30) on 1.3 mmol scale in 40% isolated yield (70% based on recovered starting material) for derivatisation studies. Conversion to pinacol borane 19 was achieved with retention of the stereogenic carbon centre (stereoselectivity determined after derivatisation to alcohol 20). The yield reported for 19 was determined by 19F NMR. We demonstrated the stereospecific transformation of 19 to alcohol 20 and Mattheson homologation-oxidation product 21. Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; F, Phe; G, Gly; L, Leu; M, Met; P, Pro; R, Arg; T, Thr; V, Val; and Y, Tyr.
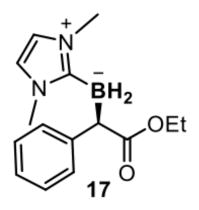
To further broaden the generality of this borylation platform, we re-examined the evolutionary landscape from BORWT to BORR1 to search for promiscuous mutants that might unlock new reactivities. Double mutant V75P M100D (BORP*) stood out as highly productive but poorly selective (69:31 e.r.) for Me-EDA borylation in the M100D V75X site-saturation library. As proline-mediated helix kinks are known to induce structural and dynamic changes to proteins, we asked whether the V75P mutation might provide access to a unique reaction space. Ethyl 2-diazophenylacetate (Ph-EDA) is a bulky donor/acceptor diazo reagent inactive towards BORWT, but when added to E. coli harbouring BORP* with NHC-borane 1, Ph-EDA was transformed to organoborane 17 in 100 TTN and 75:25 e.r. (Fig. 3a). By accumulating three additional loop mutations though directed evolution (M99Y, T101A and M103F; Extended Data Table 3), BORP* evolved into a synthetically useful catalyst (BORP1) for the borylation of Ph-EDA, supporting 340 turnovers with an e.r. of 94:6.
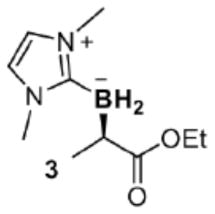
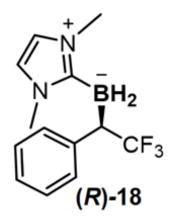
BORP* also allows us to move beyond diazo ester-based substrates and apply bacterial production to a different class of chiral organoboranes: though inactive towards BORWT, CF3-substituted (diazomethyl)benzene (CF3-DMB) reacted with NHC-borane 1 in the presence of BORP* to yield organoborane (R)-18 in vivo with 74 turnovers and modest selectivity (79:21 e.r.). We enhanced this through three cysteine mutations at Y71, M89 and M99 (BORP2; Extended Data Table 3) to produce organoborane (R)-18 in 96:4 e.r. and 1010 TTN. Through X-ray crystallography, the absolute configuration of (R)-18 was unambiguously assigned as R.
Finally, we asked whether the stereochemical preference of biological borylation could be switched. Towards this end, examination of the M100D V75X site-saturation library for CF3-DMB borylation led us to identify a variant (V75G M100D; BORG*) having an inverted stereochemical preference to BORP* in the carbon–boron bond-forming step (31:69 e.r. for R/S isomer; 340 TTN). The selectivity of BORG* was further tuned through mutations M89F, T98V, M99L, T101L and M103F (BORG1; Extended Data Table 3) to yield organoborane (S)-18 with 90:10 e.r. and 1120 TTN.
Chiral α-trifluoromethylated organoboranes are useful synthetic building blocks that combine the unique properties of fluorinated motifs with the versatile synthetic applications of organoboranes35; however, methods for their asymmetric preparation are rare11,36. Our ability to biosynthesise both enantiomers of these molecules may have applications in pharmaceutical and agrochemical synthesis. For example, product (R)-18 was converted to pinacol boronate 19 with retention of the stereogenic carbon centre (Fig. 3b). Through well-established stereospecific transformations19–21, pinacol boronates can be diversified into a broad array of chiral compounds. We demonstrated the transformation of 19 to alcohol 20, a motif found in compounds useful for the treatment of cancer37 and neurodegenerative diseases38, and the Mattheson homologation-oxidation product 21, both of which were obtained with good stereocontrol.
In conclusion, we present a platform for biological borylation, which can be tuned and configured through DNA manipulation. Microorganisms are powerful alternatives to chemical methods for producing pharmaceuticals, agrochemicals, materials, and fuels. They are available by fermentation at large scale and low cost, and their genetically-encoded synthetic prowess can be systematically modified and optimised. Borylation chemistry can now be added to biology’s vast synthetic repertoire.
Methods
Detailed experimental methods are available in the Supplementary Information.
Materials
Plasmid pET22b(+) was used as a cloning vector, and cloning was performed using Gibson assembly39. The cytochrome c maturation plasmid pEC8630 was used as part of a two-plasmid system to express prokaryotic cytochrome c proteins. Cells were grown using Luria-Bertani medium or HyperBroth (AthenaES) with 100 μg/mL ampicillin and 20 μg/mL chloramphenicol (LBamp/chlor or HBamp/chlor). Cells without the pEC86 plasmid were grown with 100 μg/mL ampicillin (LBamp or HBamp). Electrocompetent Escherichia coli cells were prepared following the protocol of Sambrook et al.40. T5 exonuclease, Phusion polymerase, and Taq ligase were purchased from New England Biolabs (NEB, Ipswich, MA). M9-N minimal medium (abbreviated as M9-N buffer; pH 7.4) was used as a buffering system for whole cells, lysates, and purified proteins, unless otherwise specified. M9-N buffer was used without a carbon source; it contains 47.7 mM Na2HPO4, 22.0 mM KH2PO4, 8.6 mM NaCl, 2.0 mM MgSO4, and 0.1 mM CaCl2.
Plasmid construction
All variants described in this paper were cloned and expressed using the pET22b(+) vector (Novagen). The gene encoding Rma cyt c (UNIPROT ID B3FQS5) was obtained as a single gBlock (IDT), codon-optimized for E. coli, and cloned using Gibson assembly39 into pET22b(+) (Novagen) between restriction sites NdeI and XhoI in frame with an N-terminal pelB leader sequence (to ensure periplasmic localization and proper maturation; MKYLLPTAAAGLLLLAAQPAMA) and a C-terminal 6xHis-tag. This plasmid was co-transformed with the cytochrome c maturation plasmid pEC86 into E. cloni® EXPRESS BL21(DE3) cells (Lucigen).
Cytochrome c expression and purification
Purified cytochrome c proteins were prepared as follows. One litre HBamp/chlor in a 4 L flask was inoculated with an overnight culture (20 mL, LBamp/chlor) of recombinant E. cloni® EXPRESS BL21(DE3) cells containing a pET22b(+) plasmid encoding the cytochrome c variant, and the pEC86 plasmid. The culture was shaken at 37 °C and 200 rpm (no humidity control) until the OD600 was 0.7 (approximately 3 hours). The culture was placed on ice for 30 minutes, and isopropyl β-D-1-thiogalactopyranoside (IPTG) and 5-aminolevulinic acid (ALA) were added to final concentrations of 20 μM and 200 μM, respectively. The incubator temperature was reduced to 20 °C, and the culture was allowed to shake for 22 hours at 200 rpm. Cells were harvested by centrifugation (4 °C, 15 min, 4,000xg), and the cell pellet was stored at −20 °C until further use (at least 24 hours). The cell pellet was resuspended in buffer containing 100 mM NaCl, 20 mM imidazole, and 20 mM Tris-HCl buffer (pH 7.5 at 25 °C) and cells were lysed by sonication (2 minutes, 2 seconds on, 2 seconds off, 40% duty cycle; Qsonica Q500 sonicator). Cell debris was removed by centrifugation for 20 min (5000xg, 4 °C). Supernatant was sterile filtered through a 0.45 μm cellulose acetate filter and purified using a 1 mL Ni-NTA column (HisTrap HP, GE Healthcare, Piscataway, NJ) using an AKTA purifier FPLC system (GE healthcare). The cytochrome c protein was eluted from the column by running a gradient from 20 to 500 mM imidazole over 10 column volumes. The purity of the collected cytochrome c fractions was analysed using sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Pure fractions were pooled and concentrated using a 3 kDa molecular weight cut-off centrifugal filter and dialyzed overnight into 0.05 M phosphate buffer (pH = 7.5) using 3 kDa molecular weight cut-off dialysis tubing. The dialyzed protein was concentrated again, flash-frozen on dry ice, and stored at −20 °C. The concentration of cytochrome c was determined in triplicate using the hemochrome assay described below.
Cytochrome P450 and globin expression and purification
Purified P450s and globins were prepared differently from the cytochrome c proteins, and described as follows. One litre HBamp in a 4 L flask was inoculated with an overnight culture (20 mL, LBamp) of recombinant E. cloni® EXPRESS BL21(DE3) cells containing a pET22b(+) plasmid encoding the P450 or globin variant. The culture was shaken at 37 °C and 200 rpm (no humidity control) until the OD600 was 0.7 (approximately 3 hours). The culture was placed on ice for 30 minutes, and IPTG and ALA were added to final concentrations of 0.5 mM and 1 mM, respectively. The incubator temperature was reduced to 20 °C, and the culture was allowed to shake for 20 hours at 200 rpm. Cells were harvested by centrifugation (4 °C, 15 min, 4,000xg), and the cell pellet was stored at −20 °C until further use (at least 24 hours). The cell pellet was resuspended in buffer containing 100 mM NaCl, 20 mM imidazole, and 20 mM Tris-HCl buffer (pH 7.5 at 25 °C). Hemin (30 mg/mL, 0.1 M NaOH; Frontier Scientific) was added to the resuspended cells such that 1 mg of hemin was added for every 1 gram of cell pellet. Cells were lysed by sonication (2 minutes, 1 seconds on, 2 seconds off, 40% duty cycle; Qsonica Q500 sonicator). Cell debris was removed by centrifugation for 20 min (27,000xg, 4 °C). Supernatant was sterile filtered through a 0.45 μm cellulose acetate filter, and purified using a 1 mL Ni-NTA column (HisTrap HP, GE Healthcare, Piscataway, NJ) using an AKTA purifier FPLC system (GE healthcare). The P450 and globin proteins were eluted from the column by running a gradient from 20 to 500 mM imidazole over 10 column volumes. The purity of the collected protein fractions was analysed using SDS-PAGE. Pure fractions were pooled and concentrated using a 10 kDa molecular weight cut-off centrifugal filter and buffer-exchanged with 0.1 M phosphate buffer (pH = 8.0). The purified protein was flash-frozen on dry ice and stored at −20 °C. P450 and globin concentrations were determined in triplicate using published extinction coefficients and the hemochrome assay described below.
Hemochrome assay
A solution of sodium dithionite (10 mg/mL) was prepared in M9-N buffer. Separately, a solution of 1 M NaOH (0.4 mL) was mixed with pyridine (1 mL), followed by centrifugation (10,000xg, 30 seconds) to separate the excess aqueous layer gave a pyridine-NaOH solution. To a cuvette containing 700 μL protein solution (purified protein or heat-treated lysate) in M9-N buffer, 50 μL of dithionite solution and 250 μL pyridine-NaOH solution were added. The cuvette was sealed with Parafilm, and the UV-Vis spectrum was recorded immediately. Cytochrome c concentration was determined using ε550–535 = 22.1 mM−1cm−1.41 Protein concentrations determined by the hemochrome assay were in agreement with that determined by the bicinchoninic acid (BCA) assay (Thermo Fisher) using bovine serum albumin (BSA) for standard curve preparation.
Mutagenesis library construction
Cytochrome c site-saturation mutagenesis libraries were generated using a modified version of the 22-codon site-saturation method31. For each site-saturation library, oligonucleotides were ordered such that the coding strand contained the degenerate codon NDT, VHG or TGG. The reverse complements of these primers were also ordered. The three forward primers were mixed together in a 12:9:1 ratio, (NDT:VHG:TGG) and the three reverse primers were mixed similarly. Two PCRs were performed, pairing the mixture of forward primers with a pET22b(+) internal reverse primer, and the mixture of reverse primers with a pET22b(+) internal forward primer. The two PCR products were gel purified, ligated together using Gibson assembly39, and transformed into E. cloni® EXPRESS BL21(DE3) cells.
Mutagenesis library screening in whole cells
Single colonies were picked with toothpicks off of LBamp/chlor agar plates, and grown in deep-well (2 mL) 96-well plates containing LBamp/chlor (400 μL) at 37 °C, 250 rpm shaking, and 80% relative humidity overnight. After 16 hours, 30 μL aliquots of these overnight cultures were transferred to deep-well 96-well plates containing HBamp/chlor (1 mL) using a 12-channel EDP3-Plus 5–50 μL pipette (Rainin). Glycerol stocks of the libraries were prepared by mixing cells in LBamp/chlor (100 μL) with 50% v/v glycerol (100 μL). Glycerol stocks were stored at −78 °C in 96-well microplates. Growth plates were allowed to shake for 3 hours at 37 °C, 250 rpm shaking, and 80% relative humidity. The plates were then placed on ice for 30 min. Cultures were induced by adding 10 μL of a solution, prepared in sterile deionized water, containing 2 mM IPTG and 20 mM ALA. The incubator temperature was reduced to 20 °C, and the induced cultures were allowed to shake for 20 hours (250 rpm, no humidity control). Cells were pelleted (4,000xg, 5 min, 4 °C), resuspended in 380 μL M9-N buffer, and the plates containing the cell suspensions were transferred to an anaerobic chamber. To deep-well plates of cell suspensions were added NHC-borane substrate (10 μL per well, 400 mM in MeCN) and diazo reagent (10 μL per well, 400 mM in MeCN). The plates were sealed with aluminium sealing tape, removed from the anaerobic chamber, and shaken at 500 rpm for 6 h (24 h for reactions with Ph-EDA or CF3-DMB due to their lower aqueous solubility). After quenching with hexanes/ethyl acetate (4:6 v/v, 0.6 mL), internal standard was added (20 μL of 20 mM 1,2,3-trimethoxybenzene in toluene). The plates were then sealed with sealing mats and shaken vigorously to thoroughly mix the organic and aqueous layers. The plates were centrifuged (4,000xg, 5 min) and the organic layer (200 μL) was transferred to autosampler vials with vial inserts for gas chromatography-mass spectrometry (GC-MS) or chiral high performance liquid chromatography (HPLC)/supercritical fluid chromatography (SFC) analysis. Hits from library screening were confirmed by small-scale biocatalytic reactions.
Cell lysate preparation
Cell lysates were prepared as follow: E. coli cells expressing Rma cyt c variant were pelleted (4,000xg, 5 min, 4 °C), resuspended in M9-N buffer and adjusted to the appropriate OD600. Cells were lysed by sonication (2 minutes, 1 seconds on, 2 seconds off, 40% duty cycle; Qsonica Q500 sonicator), aliquoted into 2 mL microcentrifuge tubes, and the cell debris was removed by centrifugation for 10 min (14,000xg, 4 °C). The supernatant was sterile filtered through a 0.45 μm cellulose acetate filter, and the concentration of cytochrome c protein lysate was determined using the hemochrome assay. Using this protocol, the protein concentrations we typically observed for OD600 = 15 lysates are in the 8 – 15 μM range for wild-type Rma cyt c and 1 – 10 μM for other Rma cyt c variants.
Small-scale whole-cell bioconversion
In an anaerobic chamber, NHC-borane (10 μL, 400 mM in MeCN) and diazo reagent (10 μL, 400 mM in MeCN) were added to E. coli harbouring Rma cyt c variant (380 μL, adjusted to the appropriate OD600) in a 2 mL crimp vial. The vial was crimp-sealed, removed from the anaerobic chamber, and shaken at 500 rpm at room temperature for 6 h (24 h for reactions with Ph-EDA or CF3-DMB). At the end of the reaction, the crimp vial was opened and the reaction was quenched with hexanes/ethyl acetate (4:6 v/v, 0.6 mL), followed by the addition of internal standard (20 μL of 20 mM 1,2,3-trimethoxybenzene in toluene). The reaction mixture was transferred to a microcentrifuge tube, vortexed (10 seconds, 3 times), then centrifuged (14,000xg, 5 min) to completely separate the organic and aqueous layers (the vortex-centrifugation step was repeated if complete phase separation was not achieved). The organic layer (200 μL) was removed for GC-MS and chiral SFC/HPLC analysis. All biocatalytic reactions reported were performed in replicates (duplicates to quadruplicates) from at least two biological replicates. The total turnover numbers (TTNs) reported are calculated with respect to Rma cyt c expressed in E. coli and represent the total number of turnovers obtained from the catalyst under the stated reaction conditions. For reactions using OD600 = 15 E. coli cells, the catalyst loadings are 0.0001 – 0.0015 mol% of enzymes with respect to the limiting reagent in the reaction. The gborylation product/gdry cell weight ratios ranged from ~0.05 (wild-type) to ~2 (engineered variant).
Cell viability assay
The colony forming units (cfu) of whole-cell reactions (+ borylation) and controls without borylation reagents (− borylation) were determined with biological replicates according to the following procedures. Six 2 mL screw cap vials containing 380 μL suspension of E. coli harbouring BORR1 (OD600 = 15) were transferred to an anaerobic chamber. To three of these vials were added NHC-borane 1 (10 μL, 400 mM in MeCN) and Me-EDA 2 (10 μL, 400 mM in MeCN). These vials were capped and shaken at 500 rpm in the anaerobic chamber (+ borylation). The remaining three vials were capped and shaken in the absence of reagents 1 and 2 (− borylation). After 2.5 hours, all six vials were removed from the anaerobic chamber. Aliquots of cell suspension were removed the vials and subjected to serial dilution to obtain stock solutions of 106, 107, and 108-fold dilution. 50 μL of each stock solution was plated on LBamp/chlor agar plates and incubate at 37 °C overnight. The cfu of the cell suspensions were calculated based on the colony counts of 107-dilution plate. The cfu for each vial are shown in Extended Data Fig. 2.
Biosynthesis of organoboranes 9 and 3 via serial substrate addition
Twelve 2 mL screw cap vials containing 400 μL suspension of cells harbouring Rma cyt c BORR1 (OD600 = 15) and 100 μL of glucose (250 mM) were transferred to an anaerobic chamber. The twelve vials were grouped into four group sets to determine the yield, TTN, and e.r. for reactions involving the stepwise addition of 2, 4, 6 or 8 equivalents of reagents. Each equivalent is 2.5 μL solution of NHC-BH3 substrate in MeCN (2 M) and 2.5 μL Me-EDA solution in MeCN (2 M). The time interval between each equivalent was 75 minutes. All four group sets were shaken at 480 rpm in the anaerobic chamber until the completion of the addition and reaction for the last group set. The vials were then removed from the anaerobic chamber and quenched with 1 mL of 4:6 hexanes/ethyl acetate and 100 μL internal standard (1,2,3-trimethoxybenzene, 20 mM in toluene). The reaction mixture was transferred to a microcentrifuge tube, vortexed (10 seconds, 3 times), then centrifuged (14,000xg, 5 min) to completely separate the organic and aqueous layers (the vortex-centrifugation step was repeated if complete phase separation was not achieved). The organic layer was removed. Another 1 mL of 4:6 hexanes/ethyl acetate and 100 μL internal standard were added for a second round of extraction and the organic solutions of two rounds of extraction were combined. 300 μL of the extract was taken for GC-MS and chiral HPLC analysis to determine the yield, TTN, and e.r..
Data Availability
Data supporting the findings of this study are available within the paper and its Supplementary Information, or are available from the corresponding author upon reasonable request. Crystallographic coordinates and structure factors have been deposited with the Cambridge Crystallographic Data Centre (CCDC) under accession codes 1572198 for organoborane 3, 1572200 for organoborane 18, and 1572201 for organoborane 12.
Extended Data
Extended Data Figure 1.

Examples of boron-containing natural products
Extended Data Figure 2. Summary of known catalytic systems for metal-carbenoid insertion reactions of boranes.

a, Rh2(esp)2-catalysed borylation of diazo esters with NHC-boranes.27 b, Cu(MeCN)4PF6-catalysed borylation of diazo esters with phosphine-borane.9 c, [Rh(C2H4)2Cl]2-catalysed borylation of diazo esters with amine-borane adducts.10 d, Cu(MeCN)4PF6-catalysed borylation of CF3-substituted (diazomethyl)benzene with phosphine-borane.11 e, Rh2(R-BTPCP)4-catalysed borylation using alkynes as carbene precursors.42 f, I2-catalysed borylation of diazo esters with NHC-boranes.43
Extended Data Figure 3. Effect of biological borylation on E. coli cell viability.
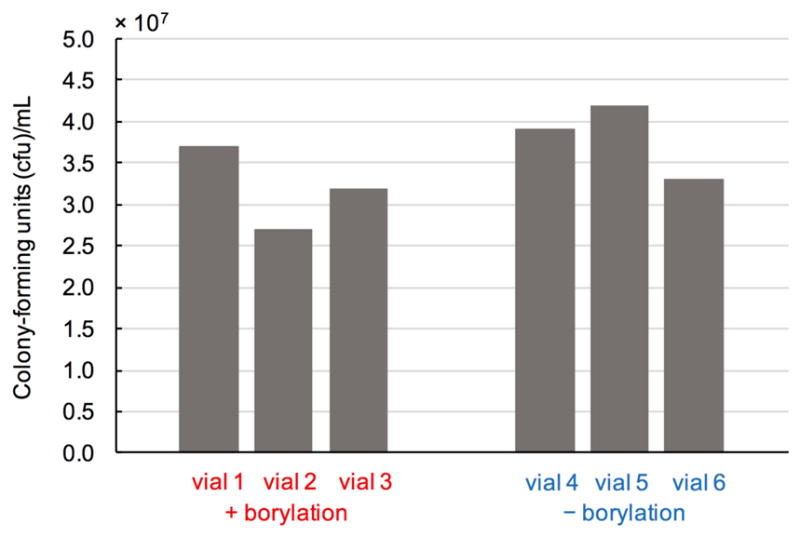
Cell viability assay was performed in biological triplicate, see Methods section for experimental protocol.
Extended Data Table 1.
Preliminary borylation experiments with haem and haem proteins using NHC-borane (1) and Me-EDA (2) as substrates
| Catalyst | TTN | e.r. |
|---|---|---|
| None | 0 | N/A |
| Haemin | 80 ± 5 | 0 |
| Haemin + BSA | 170 ± 10 | 54:46 |
| E. coli cell background | trace | 55:45 |
| R. marinus cyt c | 120 ± 20 | 85:15 |
| H. thermophilus cyt c | 140 ± 10 | 55:45 |
| P. ferrireducens protoglobin Y60V | NR | - |
| P411 CIS | trace | n.d. |
| BM3 P450 wild-type | NR | - |
| BM3 Hstar | trace | n.d. |
N/A – not applicable; NR – no product was detected; n.d. – not determined; Experiments with cytochromes c, globin, or cytochromes P450 were performed using E. coli harbouring the corresponding protein (OD600 = 15). Reactions were performed in biological triplicate. TTNs reported represent mean values averaged over three biological replicates, and the error bars indicate one standard deviation. Within instrument detection limit, variability in e.r. was not observed. Unreacted starting materials were observed at the end of all reactions and no attempt was made to optimize these reactions. Experiments with hemin were performed using 100 μM hemin, 10 mM NHC-borane 1, 10 mM Me-EDA 2, 10 mM Na2S2O4. Experiments with hemin and BSA were performed using 100 μM hemin in the presence of BSA (0.75 mg/mL) instead. Experiments to determine E. coli cell background reaction was performed with E. cloni® EXPRESS BL21(DE3) cells containing a pET22b(+) plasmid encoding halohydrin dehalogenase (HHDH) from Agrobacterium tumefaciens (UNIPROT ID Q93D82) instead of Rma cyt c. A. tumefaciens HHDH is inactive towards NHC-borane 1 and Me-EDA 2. P411 CIS4 and BM3 Hstar44 are previously reported engineered BM3 P450 variants.
Extended Data Table 2.
Biosynthesis of organoboranes 3 and 9 via serial substrate addition
| ||||||
|---|---|---|---|---|---|---|
| total equiv. of reagents | biological replicate 1 | biological replicate 2 | ||||
|
| ||||||
| yield% | TTN | e.r. | yield% | TTN | e.r. | |
| 2 | 42 | 2270 | 97.5 : 2.5 | 40 | 2130 | 97.5 : 2.5 |
| 4 | 43 | 4560 | 97:3 | 37 | 3840 | 97 : 3 |
| 6 | 57 | 9000 | 96.5 : 3.5 | 43 | 6800 | 96.5 : 3.5 |
| 8 | 50 | 10500 | 96.5 : 3.5 | 48 | 10300 | 96.5 : 3.5 |

| ||||||
|---|---|---|---|---|---|---|
| total equiv. of reagents | biological replicate 1 | biological replicate 2 | ||||
|
| ||||||
| yield% | TTN | e.r. | yield% | TTN | e.r. | |
| 2 | 63 | 3300 | 97.5 : 2.5 | 59 | 3100 | 97.5 : 2.5 |
| 4 | 67 | 7000 | 97 : 3 | 55 | 5800 | 97 : 3 |
| 6 | 71 | 11100 | 96.5 : 3.5 | 69 | 10900 | 96.5 : 3.5 |
| 8 | 75 | 15800 | 96 : 4 | 72 | 14800 | 96 : 4 |
Experiments performed in biological duplicate. See Methods section for experimental protocol.
Extended Data Table 3.
Directed evolution of whole cell Rma cyt c for improved enantioselectivity in the biosynthesis of organoboranes 17, (R)-18 and (S)-18
| |
|---|---|
| mutations | e.r. of 17 |
| M100D V75P | 75 : 25 |
| M100D V75P M99Y | 81 : 19 |
| M100D V75P M99Y T101A | 89 : 11 |
| M100D V75P M99Y T101A M103F | 94 : 6 |
| |
|---|---|
| mutations | e.r. of (R)-18 |
| M100D V75P | 76 : 24 |
| M100D V75P M89C | 90 : 10 |
| M100D V75P M89C Y71C | 94 : 6 |
| M100D V75P M89C Y71C M99C | 96 :4 |
| |
|---|---|
| mutations | e.r. of (S)-18 |
| M100D V75G | 73 : 27 |
| M100D V75G M89F M103F | 78 : 22 |
| M100D V75G M89F M103F T101L | 86: 14 |
| M100D V75G M89F M103F T101L M99L | 88 : 12 |
| M100D V75G M89F M103F T101L M99L T98V | 90 : 10 |
Supplementary Material
Acknowledgments
This work was supported in part by the National Science Foundation, Office of Chemical, Bioengineering, Environmental and Transport Systems SusChEM Initiative (grant CBET-1403077), and the Gordon and Betty Moore Foundation through Grant GBMF2809 to the Caltech Programmable Molecular Technology Initiative. X.H. is supported by a Ruth L. Kirschstein NIH Postdoctoral Fellowship (F32GM125231). We thank O. F. Brandenberg, S. Brinkmann-Chen, T. Hashimoto, R. D. Lewis, and D. K. Romney for discussions and/or comments on the manuscript, and N. W. Goldberg and A. Zutshi for experimental assistance. We are grateful to S. Virgil (Caltech Center for Catalysis and Chemical Synthesis), N. Torian (Caltech Mass Spectrometry Laboratory), M. K. Takase and L. Henling (Caltech X-ray Crystallography Facility) for analytical support; and H. Gray for providing the pEC86 plasmid.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions S.B.J.K. and X.H. designed the research with guidance from F.H.A. S.B.J.K., X.H., Y.G., K.C. performed the experiments and analysed the data. S.B.J.K., X.H. and F.H.A. wrote the manuscript with input from all authors.
The authors declare no competing financial interests.
Readers are welcome to comment on the online version of the paper.
A provisional patent application has been filed through the California Institute of Technology based on the results presented here.
References
- 1.Renata H, Wang ZJ, Arnold FH. Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew Chem Int Ed. 2015;54:3351–3367. doi: 10.1002/anie.201409470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyster TK, Ward TR. Genetic optimization of metalloenzymes: Enhancing enzymes for non-natural reactions. Angew Chem Int Ed. 2016;55:7344–7357. doi: 10.1002/anie.201508816. [DOI] [PubMed] [Google Scholar]
- 3.Hammer SC, Knight AM, Arnold FH. Design and evolution of enzymes for non-natural chemistry. Curr Opin Green Sustainable Chem. 2017 https://doi.org/10.1016/j.cogsc.2017.06.002.
- 4.Coelho PS, et al. A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat Chem Biol. 2013;9:485–487. doi: 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeschek M, et al. Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature. 2016;537:661–665. doi: 10.1038/nature19114. [DOI] [PubMed] [Google Scholar]
- 6.Kan SBJ, Lewis RD, Chen K, Arnold FH. Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life. Science. 2016;354:1048–1051. doi: 10.1126/science.aah6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tinoco A, Steck V, Tyagi V, Fasan R. Highly diastereo- and enantioselective synthesis of trifluoromethyl-substituted cyclopropanes via myoglobin-catalyzed transfer of trifluoromethylcarbene. J Am Chem Soc. 2017;139:5293–5296. doi: 10.1021/jacs.7b00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stelter M, et al. A novel type of monoheme cytochrome c: biochemical and structural characterization at 1.23 Å resolution of Rhodothermus marinus cytochrome c. Biochemistry. 2008;47:11953–11963. doi: 10.1021/bi800999g. [DOI] [PubMed] [Google Scholar]
- 9.Cheng QQ, Zhu SF, Zhang YZ, Xie XL, Zhou QL. Copper-catalyzed B–H bond insertion reaction: a highly efficient and enantioselective C–B bond-forming reaction with amine-borane and phosphine–borane adducts. J Am Chem Soc. 2013;135:14094–14097. doi: 10.1021/ja408306a. [DOI] [PubMed] [Google Scholar]
- 10.Chen D, Zhang X, Qi WY, Xu B, Xu MH. Rhodium(I)-catalyzed asymmetric carbene insertion into B–H bonds: highly enantioselective access to functionalized organoboranes. J Am Chem Soc. 2015;137:5268–5271. doi: 10.1021/jacs.5b00892. [DOI] [PubMed] [Google Scholar]
- 11.Hyde S, et al. Copper-catalyzed insertion into heteroatom–hydrogen bonds with trifluorodiazoalkanes. Angew Chem Int Ed. 2016;55:3785–3789. doi: 10.1002/anie.201511954. [DOI] [PubMed] [Google Scholar]
- 12.Irschik H, Schummer D, Gerth K, Hofle G, Reichenbach H. The tartrolons, new boron-containing antibiotics from a myxobacterium, Sorangium cellulosum. J Antibiot. 1995;48:26–30. doi: 10.7164/antibiotics.48.26. [DOI] [PubMed] [Google Scholar]
- 13.Wolkenstein K, Sun H, Falk H, Griesinger C. Structure and absolute configuration of Jurassic polyketide-derived spiroborate pigments obtained from microgram quantities. J Am Chem Soc. 2015;137:13460–13463. doi: 10.1021/jacs.5b08191. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, et al. Structural identification of a bacterial quorum-sensing signal containing boron. Nature. 2002;415:545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]
- 15.Elshahawi SI, et al. Boronated tartrolon antibiotic produced by symbiotic cellulose-degrading bacteria in shipworm gills. Proc Natl Acad Sci USA. 2013;110:E295–E304. doi: 10.1073/pnas.1213892110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dembitsky VM, Al Aziz Al Quntar A, Srebnik M. Natural and synthetic small boron-containing molecules as potential inhibitors of bacterial and fungal quorum sensing. Chem Rev. 2011;111:209–237. doi: 10.1021/cr100093b. [DOI] [PubMed] [Google Scholar]
- 17.Prier CK, Zhang RK, Buller AR, Brinkmann-Chen S, Arnold FH. Enantioselective, intermolecular benzylic C–H amination catalysed by an engineered iron-haem enzyme. Nat Chem. 2017;9:629–634. doi: 10.1038/nchem.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das BC, et al. Boron chemicals in diagnosis and therapeutics. Future Med Chem. 2013;5:653–676. doi: 10.4155/fmc.13.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 20.Leonori D, Aggarwal VK. Lithiation-borylation methodology and its application in synthesis. Acc Chem Res. 2014;47:3174–3183. doi: 10.1021/ar5002473. [DOI] [PubMed] [Google Scholar]
- 21.Leonori D, Aggarwal VK. Stereospecific couplings of secondary and tertiary boronic esters. Angew Chem Int Ed. 2015;54:1082–1096. doi: 10.1002/anie.201407701. [DOI] [PubMed] [Google Scholar]
- 22.Tehfe MA, et al. A water-compatible NHC-borane: Photopolymerizations in water and rate constants for elementary radical reactions. ACS Macro Lett. 2012;1:92–95. doi: 10.1021/mz200087g. [DOI] [PubMed] [Google Scholar]
- 23.Handa S, Wang Y, Gallou F, Lipshutz BH. Sustainable Fe-ppm Pd nanoparticle catalysis of Suzuki-Miyaura cross-couplings in water. Science. 2015;349:1087–1091. doi: 10.1126/science.aac6936. [DOI] [PubMed] [Google Scholar]
- 24.Chang MCY, Pralle A, Isacoff EY, Chang CJ. A selective, cell-permeable optical probe for hydrogen peroxide in living cells. J Am Chem Soc. 2004;126:15392–15393. doi: 10.1021/ja0441716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halo TL, Appelbaum J, Hobert EM, Balkin DM, Schepartz A. Selective recognition of protein tetraserine motifs with a cell-permeable, pro-fluorescent bis-boronic acid. J Am Chem Soc. 2009;131:438–439. doi: 10.1021/ja807872s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J, Bertozzi CR. A Bioorthogonal reaction of N-oxide and boron reagents. Angew Chem Int Ed. 2015;54:15777–15781. doi: 10.1002/anie.201508861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Curran DP. Insertion of reactive rhodium carbenes into boron–hydrogen bonds of stable N-heterocyclic carbene boranes. J Am Chem Soc. 2013;135:12076–12081. doi: 10.1021/ja4056245. [DOI] [PubMed] [Google Scholar]
- 28.Curran DP, et al. Synthesis and reactions of N-heterocyclic carbene boranes. Angew Chem Int Ed. 2011;50:10294–10317. doi: 10.1002/anie.201102717. [DOI] [PubMed] [Google Scholar]
- 29.Würtemberger-Pietsch S, Radius U, Marder TB. 25 years of N-heterocyclic carbenes: activation of both main-group element-element bonds and NHCs themselves. Dalton Trans. 2016;45:5880–5895. doi: 10.1039/c5dt04106f. [DOI] [PubMed] [Google Scholar]
- 30.Arslan E, Schulz H, Zufferey R, Künzler P, Thöny-Meyer L. Overproduction of the Bradyrhizobium japonicum c-type cytochrome subunits of the cbb3 oxidase in Escherichia coli. Biochem Biophys Res Commun. 1998;251:744–747. doi: 10.1006/bbrc.1998.9549. [DOI] [PubMed] [Google Scholar]
- 31.Kille S, et al. Reducing codon redundancy and screening effort of combinatorial protein libraries created by saturation mutagenesis. ACS Synth Biol. 2013;2:83–92. doi: 10.1021/sb300037w. [DOI] [PubMed] [Google Scholar]
- 32.Mara MW, et al. Metalloprotein entatic control of ligand-metal bonds quantified by ultrafast X-ray spectroscopy. Science. 2017;356:1276–1280. doi: 10.1126/science.aam6203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Renata H, et al. Identification of mechanism-based inactivation in P450-catalyzed cyclopropanation facilitates engineering of improved enzymes. J Am Chem Soc. 2016;138:12527–12533. doi: 10.1021/jacs.6b06823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernandez KE, et al. Highly stereoselective biocatalytic synthesis of key cyclopropane intermediate to ticagrelor. ACS Catal. 2016;6:7810–7813. doi: 10.1021/acscatal.6b02550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Argintaru OA, Ryu D, Aron I, Molander GA. Synthesis and applications of α-trifluoromethylated akylboron compounds. Angew Chem Int Ed. 2013;52:13656–13660. doi: 10.1002/anie.201308036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang Q, Guo T, Yu Z. Copper-catalyzed asymmetric borylation: Construction of a stereogenic carbon center bearing both CF3 and organoboron functional groups. J Org Chem. 2017;82:1951–1960. doi: 10.1021/acs.joc.6b02772. [DOI] [PubMed] [Google Scholar]
- 37.Kanouni T, Stafford JA, Veal JM, Wallace MB. WO 2014/151106 A1. Histone demethylase inhibitors. filed 12 Mar. 2014, and issued 25 Sep. 2014.
- 38.Scopes D. US 2012/0289523 A1. Pyrrolo [3,2-E] [1,2,4] triazolo [1,5-A] pyrimidines derivatives as inhibitors of microglia activation. filed 7 Oct. 2010, and issued 15 Nov. 2012.
- 39.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 40.Sambrook J, Frisch E, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; New York: 1989. [Google Scholar]
- 41.Berry EA, Trumpower BL. Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal Biochem. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 42.Yang JM, Li ZQ, Li ML, He Q, Zhu SF, Zhou QL. Catalytic B-H bond insertion reactions using alkynes as carbene precursors. J Am Chem Soc. 2017;139:3784–3789. doi: 10.1021/jacs.6b13168. [DOI] [PubMed] [Google Scholar]
- 43.Allen TH, Kawamoto T, Gardner S, Geib SJ, Curran DP. N-Heterocyclic carbene boryl iodides catalyze insertion reactions of N-heterocyclic carbene boranes and diazoesters. Org Lett. 2017;19:3680–3683. doi: 10.1021/acs.orglett.7b01777. [DOI] [PubMed] [Google Scholar]
- 44.Wang ZJ, Renata H, Peck NE, Farwell CC, Coelho PS, Arnold FH. Improved cyclopropanation activity of histidine-ligated cytochrome P450 enables the enantioselective formal synthesis of levomilnacipran. Angew Chem Int Ed. 2014;53:6810–6813. doi: 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this study are available within the paper and its Supplementary Information, or are available from the corresponding author upon reasonable request. Crystallographic coordinates and structure factors have been deposited with the Cambridge Crystallographic Data Centre (CCDC) under accession codes 1572198 for organoborane 3, 1572200 for organoborane 18, and 1572201 for organoborane 12.