

Abstract
In drug-discovery, systematic variations of substituents on a common scaffold and bioisosteric replacements are often used to generate diversity and obtain molecules with better biological effects. However, this could saturate the small-molecule diversity pool resulting in drug-resistance. On the other hand, conventional drug-discovery relies on targeting known pockets on protein surfaces leading to drug resistance by mutations of critical pocket residues. Here, we present a two-pronged strategy of designing novel drugs that target unique pockets on a protein’s surface to overcome the above problems. Dihydrofolate reductase, DHFR, is a critical enzyme involved in thymidine and purine nucleotide biosynthesis. Several classes of compounds that are structural-analogues of the substrate dihydrofolate have been explored for their anti-folate activity. Here, we describe 10 novel small-molecule inhibitors of Escherichia coli DHFR, EcDHFR, belonging to the stilbenoid, deoxybenzoin and chalcone family of compounds discovered by a combination of pocket-based virtual ligand screening and systematic scaffold-hopping. These inhibitors show a unique uncompetitive or noncompetitive inhibition mechanism, distinct from those reported for all known inhibitors of DHFR, indicative of binding to a unique pocket distinct from either substrate or cofactor-binding pockets. Furthermore, we demonstrate that rescue mutants of EcDHFR, with reduced affinity to all known classes of DHFR inhibitors, are inhibited at the same concentration as the wild-type. These compounds also exhibit antibacterial activity against E. coli harboring the drug-resistant variant of DHFR. This discovery is the first report on a novel class of inhibitors targeting a unique pocket on EcDHFR.
Keywords: Mechanistic characterization, binding, inhibition, Escherichia coli dihydrofolate reductase, drug discovery, ononetin
Graphical Abstract
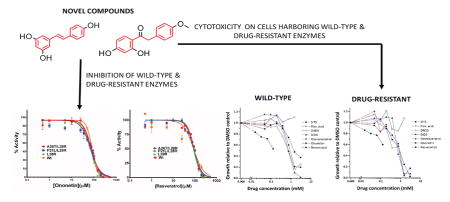
Introduction
Traditional drug-discovery initiatives have targeted pockets, predominantly active sites and in a few cases allosteric sites, on a protein’s surface1. The problem with this approach is the widespread occurrence of undesirable, and occasionally lethal, side effects shown by a majority of drugs due to off-target interactions apart from the rapid acquisition of drug resistance against these molecules due to mutations on the protein. It has been recently demonstrated that drug cross-reactivity may be a direct consequence of the remarkably small number of geometrically-distinct pockets2, 3. Incremental changes in the metabolites have failed to rescue this problem given the high-conservation of interaction profiles that govern ligand-protein interactions. This requires a new approach to designing drugs that can circumvent the limitations of targeting conventional protein pockets by using a series of ligands with incremental modifications.
Dihydrofolate reductase, DHFR, is an important enzyme in the de novo pathway of purine and thymidine synthesis4. Small-molecules targeting this enzyme have been shown to possess utility as potential antibiotics4 and anti-cancer agents5. However, this enzyme develops rapid resistance to available antifolates by acquiring mutations on residues critical for binding. It has been demonstrated that clinical-levels of resistance to known antifolates can be obtained after merely three rounds of directed-evolution efforts6. Further, attempts at understanding the evolutionary-paths for development of antibiotic resistance in DHFR led to the understanding that resistance evolves by sequential-fixation of mutations through ordered-pathways. The most prominent mutants conferring trimethoprim-resistance were either on the promoter (−9G>A; −35C>T) or on the DHFR protein (P21, A26, L28R, W30 and I94) and their combinations7. Furthermore, physical-chemical studies on these mutants have demonstrated that the decreased affinity to the drug comes at the cost of catalytic-efficiency and protein-stability8. To address the issue of rapid drug-resistance acquisition, several classes of compounds have been explored for their potential anti-folate activity.
Analogues of the cofactor NADPH have been reported as potential inhibitors of EcDHFR9. However, NADPH analogues would not be ideal candidates as clinically relevant inhibitors because of the extensive cross-reactivity that would arise due to possible targeting of NADPH binding sites in diverse enzymes and the extensive presence of nucleotide-binding pockets on several different proteins.
Most reported inhibitors for DHFRs are structural-analogues of its substrate dihydrofolate and act by competitively displacing the latter. Antifolates available as inhibitors of DHFRs contain the 2,4-diamino-1,3-diaza pharmacophore10. Predominant classes of dihydrofolate analogues employed as inhibitors include diaminoquinazoline11–16, diaminopyrimidine17–20, diaminopteridine21 and diaminotriazines22, 23. However, it should be noted that the development of rapid drug-resistance to anti-folate antibiotics belonging to any of the above-mentioned classes poses an immense challenge24.
This necessitates discovering novel scaffolds that can not only overcome the excessive reliance on folate-analogs but should also inhibit the enzyme by binding to unique sites on the enzyme. In a previous study employing our novel pocket-based VLS tool PoLi, we identified ononetin as a unique scaffold binding to EcDHFR25. The current study explores detailed inhibition-kinetics to demonstrate that ononetin is a novel inhibitor of EcDHFR showing a unique mechanism of inhibition. Further, using the principles of scaffold-hopping and structure-activity relationship (SAR), we discovered 10 novel small-molecule inhibitors of EcDHFR. These inhibitors have a novel-scaffold distinct from either the substrate or cofactor of EcDHFR and targets a unique pocket on the protein. Most importantly, this study demonstrates that these inhibitors are capable of inhibiting rescue-variants that show reduced affinity for most available antifolates and show cytotoxic effect against E. coli that harbor those DHFR variants.
Results and Discussions
Scaffold Hopping and SAR to assess the molecular features governing EcDHFR inhibition
The VLS algorithm, PoLi, predicted ononetin as potential binder of EcDHFR25. Consistent with VLS predictions, the small molecule showed binding to EcDHFR as assessed by differential scanning fluorimetry (DSF)25. To understand whether the demonstrated binding by ononetin translates into inhibition, we assessed its inhibitory effect on EcDHFR activity. Inhibition-studies indicate that ononetin inhibits the enzyme (Fig 2).
Fig 2.

Binding and Inhibition of EcDHFR by small-molecules (A) Single-concentration assessment of enzyme-inhibition by small-molecules. Each histogram represents the activity in the presence of 1mM inhibitor. (B) DSF curves for small-molecules showing inhibition. The inhibitors were kept fixed at 100μM. (C) IC50-determination for deoxybenzoin, stilbene and chalcones analogues for EcDHFR.
To validate the above observation and tease apart the molecular features that might be contributing to binding and inhibition, a systematic SAR and knowledge-based scaffold-hopping was employed. This led to the screening of twenty-two molecules belonging to the deoxybenzoins, isoflavones, flavones, stilbenoids, catecholamaine-derivatives and HPPD inhibitors (Fig 1 A). Fig 2 A summarizes the result from the inhibition assessment study. No binding to or inhibition of EcDHFR activity was detected for either isoflavones or flavones. Furthermore, if the number of degrees of freedom in the bonds connecting the two benzenes is less than 3, no significant-inhibition is observed. Lack of a benzene-ring or substitution of a cyclohexane instead of a benzene also led to abolishment of inhibition. Furthermore, substitutions on the benzene-ring that are not capable of either accepting or donating hydrogen bonds also abolished inhibition of the enzyme. These observations point out that the presence of two benzene rings, a linker with a minimum of three-degrees of freedom and presence of hydrogen bonding donors/acceptors on the benzene rings constitute the essential molecular features that determine whether or not a molecule will inhibit EcDHFR (Fig 1 B).
Fig 1.

Structures and molecular-features governing EcDHFR inhibition. (A) Structures of various small-molecules employed in this study classified based on parental scaffold (B) Minimal molecular-features that determine the inhibition potential of a small-molecule for EcDHFR. The number and positioning of hydrogen bonding donors/acceptors on the benzene-ring is representative and can vary depending on the potency of the inhibition (See Table 1).
Six stilbenoid compounds, viz., resveratrol, oxyresveratrol, SITS, DIDS, flavonic acid and DNDS, showed inhibition in the initial screen apart from that observed with ononetin. At 100 μM concentration, the inhibition by stilbenoids was more pronounced than that shown by ononetin (Fig 2 A). Subsequently, detailed binding (Fig 2 B) and concentration dependence of inhibition for the various small molecules was assessed (Table 1 and Fig 2 C).
Table 1.
Inhibition and binding parameters for the various small-molecules.
| S. No | Compound | IC50 (μM) | Kiapp (μM)2 | ΔTm(°C)3 |
|---|---|---|---|---|
| 1 | SITS | 32.3 ± 1.8 | 32.3 | NS |
| 2 | flavonic acid | 36.7 ± 3.1 | 36.7 | NS |
| 3 | DNDS1 | 45.3 ± 10.1 | 45.3 | 1.9 |
| 4 | DIDS | 51.0 ± 1.7 | 51.0 | NS |
| 5 | oxyresveratrol | 81.8 ± 1.8 | 81.8 | 1.9 |
| 6 | resveratrol | 95.7 ± 3.0 | 95.7 | 8.3 |
| 7 | ononetin | 83.4 ± 1.1 | 83.4 | 3.2 |
dinitro disulfonic acid;
calculated theoretically with the assumption that it is non-competitive inhibition, i.e. Ki=IC50 when S=Km or S≫Km or S ≪ Km;
all the thermal shifts were done at a single concentration of 0.5 mM small molecule
NS, no signal possibly due to interference with the extrinsic fluorophore dye.
As an additional step, we tested ten-compounds from the chalcones family, with an additional-linker bond that connects the two benzenes, for their potential in inhibiting EcDHFR. Scaffold-hopping (Fig S1 A) and preliminary SAR (Fig S1 B) was carried out for these chalcones. Three of the assessed compounds, with hydrogen bonding donors/acceptors on the benzene ring, showed unequivocal inhibition (Fig S1 B). This shows that any compound possessing 3–4 degrees of freedom connecting the two benzene moieties, with appropriate hydrogen bonding acceptors/donors, can inhibit EcDHFR.
To rule out non-specific protein-aggregation-induced inhibition, two hits belonging to the deoxybenzoin and stilbene families, respectively, were assessed for potentially causing aggregation of the protein. Experiments were performed with dynamic light scattering at 100 μM protein. Results indicate that there is no small-molecule-mediated aggregation of the protein sample, clearly showing that the inhibition is highly specific (Fig S2).
Detailed kinetic characterization of inhibition
Ononetin as uncompetitive inhibitor of H2F binding
To further understand the nature of the inhibition shown by the deoxybenzoin, ononetin, vis-à-vis H2F, we resorted to detailed inhibition-kinetics. Dihydrofolate was titrated at several fixed concentrations of ononetin and at saturating NADPH. The resulting curves from the experiment were globally fit to models for various types of inhibition. A sum-of-square F-test was performed to validate the non-linear fits. The experimental curves showed the best fit to model for uncompetitive-inhibition (R2-0.84) (Fig 3 A) giving an αKi, the equilibrium dissociation-constant for the uncompetitive-inhibitor, of 111.2 ± 12.37 μM (Table 2). For ease of visualization, the data were transformed and plotted as double-reciprocal Lineweaver-Burk plot, LB. Fig 3 B shows parallel lines on the LB-plot, which is further indicative of uncompetitive displacement of substrate dihydrofolate by ononetin. This suggests an ordered binding-event whereby H2F binding facilitates inhibitor binding. This is a very unique observation that, to the best of our knowledge, has not been seen with any other DHFR-inhibitors. As has been pointed out in the introduction, most known inhibitors of DHFR are analogues of H2F and competitively displace the latter. In contrast, the experimental results suggest that an additional binding site is created upon H2F binding that can accommodate the inhibitor ononetin.
Fig 3.

Typical inhibition-kinetics of EcDHFR by ononetin at low substrate/inhibitor concentrations (A) Competition-experiments of ononetin against H2F at saturating NADPH. (B) Lineweaver-Burk plot of the data from (A). (C) Competition-experiments of ononetin against NADPH at saturating concentration of H2F. (D) Lineweaver-Burk plot of the data from (C).
Table 2.
Inhibition parameters and mechanism of the small molecule ononetin for EcDHFR at different substrate and cofactor concentrations
| Inhibitor | Lower substrate* | Higher substrate | ||
|---|---|---|---|---|
| ononetin | Substrate | Inhibition Mechanism | αKi (μM) | Ki (μM)# |
| H2F | Uncompetitive | 111.2 ± 12.37 | 22.15 ± 10.94 | |
| NADPH | Uncompetitive | 113.2 ± 13.1 | 72.69 ± 22.18 | |
| resveratrol | H2F | Linear Mixed-type | 88.30 ± 6.02 | 31.48 ± 8.85 |
| SITS | H2F | Non-competitive | 39.63 ± 3.01 | 35.86 ± 11.23 |
| flavonic acid | H2F | Non-competitive | 76.25 ± 6.82 | 18.32 ± 9.68 |
The reported values are for the global non-linear fits.
values from fit to equation for substrate-inhibition at highest inhibitor concentration.
Note that the values reported here are not from global non-linear fits and differ from one inhibitor to another depending on the highest concentration employed for the respective inhibitor (Fig S5).
In uncompetitive-inhibition, the inhibitor usually binds reversibly to the ES complex yielding an inactive ESI-complex. Hence Vmax in the presence of the inhibitor will be lower than in its absence. However, the apparent Km of the substrate also decreases indicating an affinity increase. The increase results from the reaction ES + I giving ESI that uses up some ES causing the substrate-binding reaction E + S giving ES to proceed further to the right. However, this increased-affinity is misleading and essentially represents a locked enzyme that can no longer turnover the substrate and does not indicate inhibitor-mediated substrate-binding. In this case, the binding affinities for the inhibitor and the substrate are an order-of-magnitude different. While, for the substrate dihydrofolate it is in lower micromolar range, for the inhibitor, the Ki is almost 0.1 mM. Hence, though the effect of substrate-binding on inhibitor-binding is pronounced, the reverse is not true. Hence, it can be stated with reasonable confidence that substrate binding facilitates inhibitor binding.
Ononetin as uncompetitive inhibitors of NADPH binding
Inhibition of the cofactor NADPH binding by ononetin was also assessed. The experimental curves, once again, showed the best fit to uncompetitive inhibition model (Fig 3 C) yielding an αKi of 113.2 ± 13.1 μM. As above, the LB plots showed parallel lines further indicative of uncompetitive inhibition (Fig 3 D) and an ordered binding event whereby NADPH binding facilitates inhibitor binding. Further, this data is in agreement with reported mechanisms of DHFR inhibitors by folate-analogs16, 23. It has been demonstrated that the binding of and inhibition by folate-analogs is conditional to NADPH-binding16, 23.
Substrate inhibition like behavior at high substrate and ononetin concentrations
Careful inspection of the nonlinear plots in Fig 3 A and Fig 3 C shows pronounced nonlinearity at saturating substrate and high inhibitor concentrations. However, as has been demonstrated in previous studies16, 23 and inspection of curves at low inhibitor concentrations in Fig 3 A and Fig 3 C shows, no substrate-inhibition is seen in the absence of the inhibitor. This led us to hypothesize that the observed behavior might be indicative of a unique two-site binding inhibition mechanism displayed by ononetin.
To confirm this hypothesis, the experiment shown in Fig 3 A and 3 C, was extended to high substrate and cofactor concentrations. As seen in Fig S3 A and S3 C, the resulting curves show pronounced substrate-inhibition-like behavior at higher inhibitor concentration. Further, fitting the curves to equations of substrate-inhibition gave Ki values that kept decreasing (i.e. increasing potency of inhibition) with increasing substrate concentration indicative of substrate-inhibition like behavior by ononetin (Fig S3 A and S3 C). This pattern is clearly evident in the non-linear LB plot (Fig S3 B and S3 D). This is a unique kinetic behavior displayed by this inhibitor and is the first of this kind to be observed with any reported EcDHFR inhibitors. Taken together, these patterns are likely indicative of either substrate-inhibitor/cofactor-inhibitor/substrate-cofactor-inhibitor complex as true inhibitor or an alternate-site of binding for the inhibitor.
In summary, uncompetitive-inhibition by the inhibitor at low substrate and cofactor concentrations and substrate inhibition-like behavior at high substrate and cofactor concentrations indicates that the kinetic-model for inhibition may be more complex than preliminary analysis of the data suggests. Further, numerical-analysis of data by fitting it to global models with multiple unknown variables would likely lead to overfitting.
In order to understand whether either the substrate or cofactor molecule forms a complex with ononetin, wavelength scans on UV-Vis spectrophotometer were carried out with the individual metabolites and their combinations thereof. In summary, though no convincing evidence for complex formation between the substrate/cofactor and ononetin was seen, H2F and ononetin showed a spectrum that was markedly different than the spectrum of either H2F or ononetin (Fig S4). This was markedly different than the case of NADPH and ononetin where the composite spectrum was an additive equivalent of the two individual spectrums (Fig S4).
It has to be once again reiterated here that these compounds are the first to show inhibition that is not competitive either against the cofactor or the substrate.
Inhibition mechanism shown by stilbenoid inhibitors
We performed detailed inhibition-kinetics with a few representative stilbenoid compounds to understand whether they too show these unique-mechanisms of inhibition that is not competitive against H2F. Substrate dihydrofolate was titrated at several fixed concentrations of resveratrol, flavonic-acid and SITS and at saturating NADPH. Fig S5 shows the global experimental points and the non-linear fits of the experiments to the respective inhibition models for points that do not show substrate-inhibition like behavior. The plots show fits either to non-competitive or mixed-inhibition indicating, once again, to a distinct site of binding. Data for resveratrol fits well to the model for mixed-inhibition while data for SITS and flavonic acid fit the non-competitive inhibition model better than equivalent models (Fig S5 A–C). At high-substrate and inhibitor concentrations, the curve starts resembling substrate-inhibition like behavior as seen with ononetin, possibly indicative of two-site binding.
Novel class of inhibitors targeting drug-resistant EcDHFR variants
Often, protein targets develop resistance against a small-molecule drug by acquiring mutations that either function to weaken the binding or decrease the potency of inhibition of the small-molecule7. Such mutations come at an evolutionary cost vis-à-vis enzyme-stability and catalytic-turnover ability8. Previous work on the laboratory-evolution of resistance of E. coli against trimethoprim demonstrated that the resistance is acquired through exclusive mutations in the gene encoding DHFR. We wanted to test whether a subset of the newly-discovered small-molecule inhibitors could inhibit the drug-resistant variants of the EcDHFR. With this intention, drug-resistant single (L28R) and double mutants (A26T/L28R and P21L/L28R) were expressed and purified to homogeneity. Before assessing the new-molecules, classical and slow-onset tight binding inhibitors that are structural analogues of substrate dihydrofolate reported by us in our previous studies were assessed11, 15, 16, 23. The small-molecules belonged to the diaminotriazine, diaminopyrroloquinazoline and diaminopetridine class of molecules. The molecules were assessed at their respective IC50 values in duplicate across the wild-type, one single-mutant and the two double-mutants. As can be seen from Fig S6, though all the molecules inhibit the wild-type enzyme by ~ 50-fold as is expected, they fail to inhibit the mutant enzymes. However, molecules belonging to deoxybenzoin and stilbenoid family inhibited both the wild-type enzyme and the drug-resistant mutants (Fig 4 A). We would like to point out that these are the first set of molecules that inhibit these drug-resistant variants of EcDHFR and have the potential to be developed into next generation antibiotics against such rescue-variants of the opportunistic pathogen. Point estimation of inhibition could sometimes be misleading. Hence, for two promising hits, ononetin and resveratrol, IC50s were estimated for the wild-type and the three mutant-variants. For both ononetin (Fig 4 B) and resveratrol (Fig 4C), the IC50 curves are superimposable indicating that these new-class of molecules have equal potency in inhibiting both the drug-resistant mutant variants and the wild-type enzyme. It should be pointed out that the drug-resistance conferring mutations are localized either in or proximal to the substrate-binding pocket. These small-molecules inhibiting the mutants as potently as the wild-type indirectly vindicates our conclusions from the kinetic studies showing a distinct site of binding different from the substrate-binding site. Any overlap of binding-site with the mutation-harboring region would be expected to perturb the binding, and thus inhibition, either favorably or unfavorably as was seen with diaminotriazine, diaminopyrroloquinazoline and diaminopteridine group of compounds.
Fig 4.

Assessment of inhibition for EcDHFR wild-type and rescue-variants (A) by deoxybenzoin and stilbenoid class of small-molecules. The assessment was carried out in duplicate at a single-concentration of the inhibitor around its experimental IC50-values for the wild-type enzyme. (B) Estimation of IC50-values of ononetin for wild-type, A26T/L28R mutant, P21L/L28R mutant and L28R mutants. (C) Estimation of IC50-values of resveratrol for wild-type, A26T/L28R mutant, P21L/L28R mutant and L28R mutants.
Binding-site prediction and potential mechanism of inhibition
In addition to the large catalytic pocket comprised of the NADP and folate-binding sites, CAVITATOR detected three additional pockets (Table S2). These new pockets are much smaller than the catalytic-pockets both in terms of volume and number of amino-acids. The main catalytic-site has more than 40 amino-acids and a volume greater than 1000 Å3; whereas the new pockets have 12–15 amino-acids and volumes of approximately 150–300 Å3. Figure S7 shows the location of these predicted pockets with respect to the catalytic-site. Though being adjacent to the catalytic-site, these pockets do not share any amino-acids with the former. PKT2 is the largest of all three new pockets located below the catalytic-site as shown in Figure S7A. It is a shallow-pocket comprised mostly of loops that are wrapped around the beta-sheets (Figure S7 B) below the Met-20 loop. PKT3 is located close to the folate-binding-site (Figure S7 C) in a cleft between a helix and a beta-sheet behind the folate-binding-site. PKT4, on the other hand, is formed by the space between two short helices (Figure S7 D) located behind the binding-site of the adenine moiety of NADP.
Next, we compared these new-pockets with the available holo-pockets. Interestingly, PKT4 has significant similarity (statistical p-value of 0.89×10−4) with the bromo-resveratrol binding site of human Sirt326 (PDB_ID: 4C78). In human Sirt3, bromo-resveratrol is in contact with six amino-acids, among which five residues get perfectly aligned (two out of five are identical amino-acids) with amino-acids in PKT4 (Table S3 and Fig 5). According to the interactions between bromo-resveratrol and Sirt3 calculated by PoseView27 and reported on the corresponding PDB page, the carbonyl group of Met-311’s backbone is responsible for hydrogen-bonding with the bromo-resveratrol. In our pocket alignment, Met-311 of Sirt3 is aligned with Lys-106 of DHFR with a Cα - Cα distance of 0.171 Å. This perfect alignment of the hydrogen-bond acceptor carbonyl-groups, together with the alignment of the neighboring amino-acids (Fig 5), suggest that the pocket can accommodate resveratrol-like molecules. In addition to their pocket similarity, we would like to emphasize that both PKT4 in DHFR and bromo-resveratrol binding site in Sirt3 are located in a Rossmann-fold and there is weak global similarity (TM-Score = 0.42) between DHFR and Sirt3.
Fig. 5.
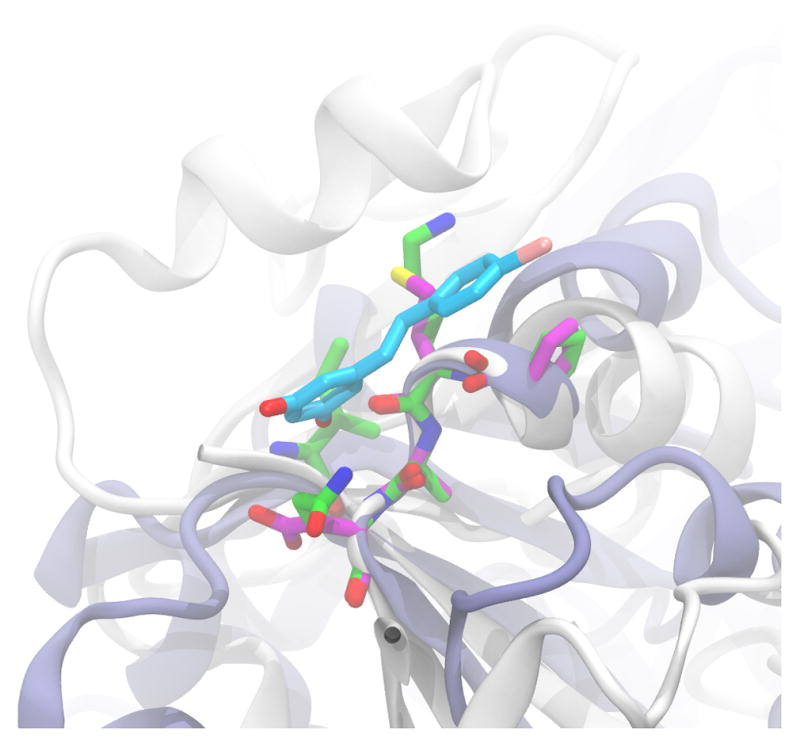
Superposition of EcDHFR (white) and its aligned pocket residues (green) onto the best template (ice blue), PDB_ID: 4C78, and its pocket residues (magenta) binding bromo-resveratrol (turquoise).
Figure S8 shows the structure of NADP-bound DHFR and bromo-resveratrol copied from Sirt3 using their pocket alignment. Amino-acids inside PKT4 are adjacent to the amino-acids (Leu-62, Val-78, Gln102) that are in contact with the adenine-moiety of NADP. One possible explanation for the mechanism of inhibition by resveratrol-like molecules could be the perturbations in the adenine-binding-site of NADP because of inhibitor binding at PKT4. However, this needs to be further investigated.
Inhibition of E. coli growth encoding wild-type vs mutant-variants of DHFR
Subsequent to obtaining the above mentioned results, we tested the activity of a subset of small-molecules targeting DHFR as bacterial growth inhibitors using E. coli cells. In addition, we also tested the E. coli mutant strain in which the chromosomal DHFR-coding gene folA was replaced by a drug-resistant variant (L28R) known to increase resistance to trimethoprim approximately 50-fold8, 28. The drug dose-response curves obtained for the wild-type and L28R mutant strains (Fig 6 A and Fig 6 B, respectively) clearly show that all compounds, except flavonic-acid and oxyresveratrol, inhibit growth of both strains within the range of concentrations tested. Moreover, in agreement with the enzymatic activity results obtained for the purified protein-variants, the calculated in-vivo IC50 values determined for drug-resistant strain are also comparable to those obtained for wild-type strain. Our results clearly point to an inhibition of DHFR activity targeted by these newly discovered small-molecules as the cause for the arrest of bacterial growth.
Fig 6.

Growth-inhibition of wild-type (A) and resistant (B) E. coli cells by deoxybenzoin and stilbenoid class of small-molecules. Growth measurements were performed in duplicate for each drug-concentration. (C) IC50-values determined from (A) and (B) by non-linear curve-fitting for each drug and for each E. coli strain.
It is important to note that resveratrol (the compound with highest inhibitory activity in-vivo) had been previously reported to have antibacterial activity29, 30, with an estimated IC50 of 0.25 mM, very similar to that determined in this work, although its mechanism of action has not been elucidated. To address if this molecule affects growth by specifically inhibiting DHFR, we tested the effect of resveratrol on E. coli cultures grown in media supplemented with a metabolic complement (folAmix) that is known to restore growth of cells treated with trimethoprim31, a potent-specific-inhibitor of DHFR. The presence of folAmix fully restores growth of cells inhibited by different resveratrol concentrations (Fig S9), indicating that this compound specifically blocks folate metabolism. This work provides the first evidence for the intracellular targeting of DHFR by this compound as one potential basis for the antibacterial activity demonstrated by this compound. Interestingly, introduction of an additional hydroxyl-group to resveratrol seems to render this molecule biologically inactive, possibly due to differences in cell permeation properties.
Discussion
Previous analysis of drug-protein interactions and the demonstration of the limited number of geometrically-distinct pockets on protein surfaces have implied that structurally-diverse and distinct small-molecules can bind to similar receptors2. However, all instances of diverse small-molecules binding to a given protein of interest have relied extensively on serendipitous findings and there are merely a handful of successful discoveries32. Moreover, the new small-molecules are always minor-alterations on the initial scaffolds thus expanding the novelty aspect incrementally33. This necessitates the development of tools that can search for novel-scaffolds in a more systematic manner. In the current work, we demonstrate that a virtual ligand-screening algorithm that relies extensively on pocket-similarity and ligand-pruning25, when combined with chemically intuitive scaffold hopping, can yield interesting leads in terms of completely novel-scaffolds that have the potential to bind and inhibit a medically-important receptor protein.
All the scaffolds described in the present work as inhibitors of DHFR, viz., stilbenoid, deoxybenzoin and chalcones, have no precedence whatsoever in either binding or inhibiting DHFR. Further, given that a substantial number of them like resveratrol, oxyresveratrol and ononetin are found in natural products like wine and legumes34, 35, future exploration of them as potential therapeutic agents makes cytotoxicity on human cells less of a concern, making them valuable lead-molecules. It has not escaped our attention that stilbenoids, like resveratrol, have been shown to possess anticancer activity with roles in modulating various pathways. However, none of these studies have ever indicated purine-metabolism as a potential target and DHFR as the receptor that also gets modulated34, 36. We, as an extension of our results, posit that, among other targets, resveratrol may bring about its anticancer-activity by targeting purine-metabolism through inhibiting human DHFR in cancer-cells. This is the subject of a potentially interesting future study.
This study is an important attempt in translational research. Starting from computational attempts at predicting small-molecular binders, it validated the predicted binding and assessed inhibition of the intended target. Furthermore, it demonstrated differential inhibition across the wild-type and drug-resistant variants of the target protein and concluded by showing cytotoxic-effects on E. coli harboring both wild-type and a drug-resistant rescue-variant harboring the mutated enzyme.
Future extensions of this work will include improvement of activity and cell-penetrability of the lead compounds to bring them into a viable therapeutic range as potential stand-alone drugs or components of natural-product formulations.
In conclusion, this is the first study that demonstrates the successful application of a pocket and ligand similarity based VLS algorithm to arrive at novel-scaffolds and to employ systematic scaffold-hopping to expand the small-molecules diversity space in inhibitor discovery. Ten molecules, with ability to inhibit the drug-resistant-variant of EcDHFR, have been discovered and characterized. These molecules inhibit the protein by displaying a unique-mechanism of inhibition and by binding to a novel-site on the enzyme.
Methods
Reagents
All reagents and chemicals were of high quality and were procured from Sigma-Aldrich Co., USA, Amresco, or Fisher-Scientific. Developmental Therapeutics Program (DTP) of the National Cancer Institute (NCI), provided a substantial fraction of the various small-molecules employed in this study while the rest were obtained from Sigma (Table S1).
Wild-type and mutant protein expression and purification
Wild-type and mutant proteins were expressed and purified as previously described8.
Binding studies using DSF
Binding of the molecules to EcDHFR was tested by DSF. The experiments were carried out following previously reported protocols from our lab11, 23. Briefly, the reactions were carried out in 96 well plates on the RealPlex quantitative-PCR instrument (Eppendorf). The reaction mixture contained 100mM HEPES pH 7.3 and 150mM NaCl with 5X concentration of Sypro orange. Various compounds were tested for binding at a final concentration of 500μM with 5μM of EcDHFR.
Dihydrofolate reductase assay
DHFR was assayed as previously reported16. Briefly, the conversion of NADPH was monitored as a decrease in absorbance at 340nm for 100 seconds. The amount of product formed was computed using a molar-extinction coefficient (ε) of 6.2 × 103 M−1cm−1 for β-NADPH at 340 nm37. The non-enzymatic hydrolysis of NADPH was normalized. Assays were initiated with enzyme and the initial velocities, where product formation was less than 5%, were measured for reaction mixtures containing 100mM HEPES pH 7.3 at ~ 22 ° C.
All the measurements were performed in duplicate, and the error-values indicated are standard-errors (S.E.). EcDHFR concentration was 16.7nM16. All the data were fit using the non-linear curve-fitting subroutines of GraphPad Prism, version 4.0 (GraphPad Software, Inc.).
Inhibition-kinetics
IC50 determination assays were carried out in 100mM HEPES pH 7.3, 60μM NADPH, 50μM H2F and variable concentration of each inhibitor. The enzyme concentration was as specified above. The curves were fit to equation (1),
| (1) |
where, I is the inhibitor concentration, and y is the percentage of activity.
Experimental Ki values were determined by titrating the substrates H2F and NADPH, around their respective Km values, at various fixed-concentrations of the inhibitors around their Kiapp values. The resulting [substrate] vs. velocity curves were fit to models of competitive-inhibition (equation 2), non-competitive-inhibition (equation 3), uncompetitive-inhibition (equation 4) and mixed-type-inhibition (equation 5) in order to discriminate between the different types of inhibition and to estimate the various inhibition constants (Ki).
Competitive:
| (2) |
Non-competitive:
| (3) |
Uncompetitive:
| (4) |
Linear Mixed-type:
| (5) |
where, v is the velocity of the reaction, Vmax is the maximum velocity, [S] is the substrate concentration, and [I] is the inhibitor concentration. Km is the Michaelis-Menten constant, and Ki is the inhibition constant. Visual-assessment of the type of inhibition was done with Lineweaver-Burk plots.
Plots showing substrate-inhibition behavior were fit using the following equation
| (6) |
The double-reciprocal plot for the substrate-inhibition was fit to the equation
| (7) |
In the above equation, [S] appearing in the denominator is the substrate for the enzyme, while [S] appearing in the numerator is the inhibitor and hence, the double reciprocal plot shows upward curvature at high substrate concentrations.
Computational approach to binding site prediction
We used nine experimentally-solved crystal structures38 of EcDHFR (PDB IDs: 1RX1, 1RX2, 1RX3, 1RX4, 1RX5, 1RX6, 1RX7, 1RX8, 1RX9) to detect geometrical-pockets (cavities) using the CAVITATOR39 pocket-detection algorithm. CAVITATOR uses a grid-based method to assign residues to a pocket while excluding the ledge associated residues. The consensus residue pockets were defined as those detected in more than 75% of the structures. Next, using APoc39, 40, we compared the predicted pockets of DHFR to the holo pockets of the HOLO-PDB template used in our previous study41. The library of holo-structures includes co-crystallized structures of protein-ligand complexes in which the ligand has at least six heavy-atoms. The binding-site is defined by considering protein residues with heavy-atom contacts with the ligand (distance less than 4.5Å). Finally, the results were sorted using the pocket-similarity score and their relevant statistical p-values reported by APoc.
Antibacterial activity assay
The E. coli strain in which chromosomal folA gene had been replaced by mutant L28R was a kind gift from Dr. Roy Kishony. Prior to growth-measurements, cells were grown overnight in minimal-M9-medium at 30°C. The following day, the cultures were used to inoculate a 384-well plate with M9-medium supplemented with different concentrations of drug (up to 3mM) or the corresponding volume of DMSO alone. Resveratrol and oxyresveratrol could not be tested above 0.25 mM due to insolubility. The starting optical-density was 0.005, and the growth measurements were performed at 30°C using a Tecan Infinite M200 Pro plate-reader. The ability of cells to grow at each drug-concentration was assessed by computing the area under the growth-curve (OD600nm) over a period of 15h. Since the solvent (DMSO) itself showed inhibitory effects on growth (down to approximately 30% at the highest DMSO concentration used), the extent of inhibition at each drug-concentration was normalized against reference growth-curves obtained in the presence of matching amounts of DMSO alone. Duplicate measurements were performed for each drug concentration. Rescue of growth by the mixture folAmix was tested by supplementing M9-media with 20 mg/L adenine, 80mg/mL inosine, 200mg/L thymine, 20mg/L methionine and 20mg/L glycine.
Supplementary Material
Acknowledgments
This project was funded by 1R35GM118039 of the Division of General Medical Sciences of the NIH. We would also like to thank the Developmental Therapeutics Program of the National Cancer Institute for providing the small-molecules used in this study.
Abbreviations
- EcDHFR
Escherichia coli dihydrofolate reductase
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced
- H2F
dihydrofolate
- H4F
tetrahydrofolate
- SAR
Structure-Activity Relationship
Footnotes
The Supporting Information is available free of charge on the ACS Publications website. The Supporting information contains the following: Methods and results for ononetin-substrate interaction and DLS. Fig S1. SAR-chalcones Fig S2. DLS Fig S3. Atypical inhibition-ononetin Fig S4. Wavelength-spectra to assess substrate-cofactor-inhibitor interactions. Fig S5. Inhibition-stilbenes Fig S6. Inhibition of wt and mutant EcDHFR by classical-inhibitors. Fig S7. EcDHFR and its alternate pockets. Fig S8. Predicted binding-site for ononetin-like molecules Fig S9. Growth-rescue of E. coli treated with resveratrol by metabolic-complementation. Table S1. Compound-list. Table S2. New-pockets. Table S3. Alignment of EcDHFR’s PKT4 and binding-site of bromo-resveratrol in hSirt3.
References
- 1.Panjkovich A, Daura X. Assessing the structural conservation of protein pockets to study functional and allosteric sites: implications for drug discovery. BMC Struct Biol. 2010;10:9. doi: 10.1186/1472-6807-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Skolnick J, Gao M. Interplay of physics and evolution in the likely origin of protein biochemical function. Proc Natl Acad Sci U S A. 2013;110:9344–9349. doi: 10.1073/pnas.1300011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skolnick J, Gao M, Roy A, Srinivasan B, Zhou H. Implications of the small number of distinct ligand binding pockets in proteins for drug discovery, evolution and biochemical function. Bioorg Med Chem Lett. 2015;25:1163–1170. doi: 10.1016/j.bmcl.2015.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schweitzer BI, Dicker AP, Bertino JR. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990;4:2441–2452. doi: 10.1096/fasebj.4.8.2185970. [DOI] [PubMed] [Google Scholar]
- 5.McGuire JJ. Anticancer antifolates: current status and future directions. Curr Pharm Des. 2003;9:2593–2613. doi: 10.2174/1381612033453712. [DOI] [PubMed] [Google Scholar]
- 6.Watson M, Liu JW, Ollis D. Directed evolution of trimethoprim resistance in Escherichia coli. FEBS J. 2007;274:2661–2671. doi: 10.1111/j.1742-4658.2007.05801.x. [DOI] [PubMed] [Google Scholar]
- 7.Toprak E, Veres A, Michel JB, Chait R, Hartl DL, Kishony R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet. 2011;44:101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rodrigues JV, Bershtein S, Li A, Lozovsky ER, Hartl DL, Shakhnovich EI. Biophysical principles predict fitness landscapes of drug resistance. Proc Natl Acad Sci U S A. 2016;113:E1470–1478. doi: 10.1073/pnas.1601441113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stone SRMA, Morrison JF. Interaction of analogues of nicotinamide adenine dinucleotide phosphate with dihydrofolate reductase from Escherichia coli. Biochemistry. 1984;23 [Google Scholar]
- 10.Kompis IM, Islam K, Then RL. DNA and RNA synthesis: antifolates. Chem Rev. 2005;105:593–620. doi: 10.1021/cr0301144. [DOI] [PubMed] [Google Scholar]
- 11.Srinivasan B, Zhou H, Kubanek J, Skolnick J. Experimental validation of FINDSITE(comb) virtual ligand screening results for eight proteins yields novel nanomolar and micromolar binders. J Cheminform. 2014;6:16. doi: 10.1186/1758-2946-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukunaga JY, Hansch C, Steller EE. Inhibition of dihydrofolate reductase. Structure-activity correlations of quinazolines. J Med Chem. 1976;19:605–611. doi: 10.1021/jm00227a006. [DOI] [PubMed] [Google Scholar]
- 13.Kuyper LF, Baccanari DP, Jones ML, Hunter RN, Tansik RL, Joyner SS, Boytos CM, Rudolph SK, Knick V, Wilson HR, Caddell JM, Friedman HS, Comley JC, Stables JN. High-affinity inhibitors of dihydrofolate reductase: antimicrobial and anticancer activities of 7,8-dialkyl-1,3-diaminopyrrolo[3,2-f]quinazolines with small molecular size. J Med Chem. 1996;39:892–903. doi: 10.1021/jm9505122. [DOI] [PubMed] [Google Scholar]
- 14.Rosowsky A, Hynes JB, Queener SF. Structure-activity and structure-selectivity studies on diaminoquinazolines and other inhibitors of Pneumocystis carinii and Toxoplasma gondii dihydrofolate reductase. Antimicrob Agents Chemother. 1995;39:79–86. doi: 10.1128/aac.39.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srinivasan B, Skolnick J, Zhou H. USPTO, editor. Molecules with potent DHFR binding affinity and antibacterial activity. Georgia Tech Research Corporation; United States: 2014. [Google Scholar]
- 16.Srinivasan B, Skolnick J. Insights into the slow-onset tight-binding inhibition of Escherichia coli dihydrofolate reductase: detailed mechanistic characterization of pyrrolo [3,2-f] quinazoline-1,3-diamine and its derivatives as novel tight-binding inhibitors. FEBS J. 2015;282:1922–1938. doi: 10.1111/febs.13244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baccanari DP, Daluge S, King RW. Inhibition of dihydrofolate reductase: effect of reduced nicotinamide adenine dinucleotide phosphate on the selectivity and affinity of diaminobenzylpyrimidines. Biochemistry. 1982;21:5068–5075. doi: 10.1021/bi00263a034. [DOI] [PubMed] [Google Scholar]
- 18.Chio LC, Queener SF. Identification of highly potent and selective inhibitors of Toxoplasma gondii dihydrofolate reductase. Antimicrob Agents Chemother. 1993;37:1914–1923. doi: 10.1128/aac.37.9.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li RL, Poe M. Quantitative structure-activity relationships for the inhibition of Escherichia coli dihydrofolate reductase by 5-(substituted benzyl)-2,4-diaminopyrimidines. J Med Chem. 1988;31:366–370. doi: 10.1021/jm00397a017. [DOI] [PubMed] [Google Scholar]
- 20.Hansch C, Li R, Blaney JM, Langridge R. Comparison of the inhibition of Escherichia coli and Lactobacillus casei dihydrofolate reductase by 2,4-diamino-5-(substituted-benzyl)pyrimidines: quantitative structure-activity relationships, X-ray crystallography, and computer graphics in structure-activity analysis. J Med Chem. 1982;25:777–784. doi: 10.1021/jm00349a003. [DOI] [PubMed] [Google Scholar]
- 21.Williams JW, Morrison JF, Duggleby RG. Methotrexate, a high-affinity pseudosubstrate of dihydrofolate reductase. Biochemistry. 1979;18:2567–2573. doi: 10.1021/bi00579a021. [DOI] [PubMed] [Google Scholar]
- 22.Blaney JM, Hansch C, Silipo C, Vittoria A. Structure-Activity Relationships of Dihydrofolate Reductase Inhibitors. Chem Rev. 1984;84:333–407. [Google Scholar]
- 23.Srinivasan B, Tonddast-Navaei S, Skolnick J. Ligand binding studies, preliminary structure-activity relationship and detailed mechanistic characterization of 1-phenyl-6,6-dimethyl-1,3,5-triazine-2,4-diamine derivatives as inhibitors of Escherichia coli dihydrofolate reductase. Eur J Med Chem. 2015;103:600–614. doi: 10.1016/j.ejmech.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lynch C, Pearce R, Pota H, Cox J, Abeku TA, Rwakimari J, Naidoo I, Tibenderana J, Roper C. Emergence of a dhfr mutation conferring high-level drug resistance in Plasmodium falciparum populations from southwest Uganda. J Infect Dis. 2008;197:1598–1604. doi: 10.1086/587845. [DOI] [PubMed] [Google Scholar]
- 25.Roy A, Srinivasan B, Skolnick J. PoLi: A Virtual Screening Pipeline Based on Template Pocket and Ligand Similarity. J Chem Inf Model. 2015;55:1757–1770. doi: 10.1021/acs.jcim.5b00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen GT, Gertz M, Steegborn C. Crystal structures of Sirt3 complexes with 4′-bromo-resveratrol reveal binding sites and inhibition mechanism. Chem Biol. 2013;20:1375–1385. doi: 10.1016/j.chembiol.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 27.Stierand K, Rarey M. Drawing the PDB: Protein-Ligand Complexes in Two Dimensions. ACS Med Chem Lett. 2010;1:540–545. doi: 10.1021/ml100164p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmer AC, Toprak E, Baym M, Kim S, Veres A, Bershtein S, Kishony R. Delayed commitment to evolutionary fate in antibiotic resistance fitness landscapes. Nat Commun. 2015;6:7385. doi: 10.1038/ncomms8385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang D, Lim YH. Resveratrol antibacterial activity against Escherichia coli is mediated by Z-ring formation inhibition via suppression of FtsZ expression. Sci Rep. 2015;5:10029. doi: 10.1038/srep10029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seukep JA, Sandjo LP, Ngadjui BT, Kuete V. Antibacterial and antibiotic-resistance modifying activity of the extracts and compounds from Nauclea pobeguinii against Gram-negative multi-drug resistant phenotypes. BMC Complement Altern Med. 2016;16:193. doi: 10.1186/s12906-016-1173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwon YK, Higgins MB, Rabinowitz JD. Antifolate-induced depletion of intracellular glycine and purines inhibits thymineless death in E. coli. ACS Chem Biol. 2010;5:787–795. doi: 10.1021/cb100096f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bohm HJ, Flohr A, Stahl M. Scaffold hopping. Drug Discov Today Technol. 2004;1:217–224. doi: 10.1016/j.ddtec.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 33.Sun H, Tawa G, Wallqvist A. Classification of scaffold-hopping approaches. Drug Discov Today. 2012;17:310–324. doi: 10.1016/j.drudis.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, Moon RC, Pezzuto JM. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 35.Straub I, Mohr F, Stab J, Konrad M, Philipp SE, Oberwinkler J, Schaefer M. Citrus fruit and fabacea secondary metabolites potently and selectively block TRPM3. Br J Pharmacol. 2013;168:1835–1850. doi: 10.1111/bph.12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Signorelli P, Ghidoni R. Resveratrol as an anticancer nutrient: molecular basis, open questions and promises. J Nutr Biochem. 2005;16:449–466. doi: 10.1016/j.jnutbio.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Horecker BL, Kornberg A. The extinction coefficients of the reduced band of pyridine nucleotides. J Biol Chem. 1948;175:385–390. [PubMed] [Google Scholar]
- 38.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 39.Gao M, Skolnick J. APoc: large-scale identification of similar protein pockets. Bioinformatics. 2013;29:597–604. doi: 10.1093/bioinformatics/btt024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao M, Skolnick J. A comprehensive survey of small-molecule binding pockets in proteins. PLoS Comput Biol. 2013;9:e1003302. doi: 10.1371/journal.pcbi.1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tonddast-Navaei S, Srinivasan B, Skolnick J. On the importance of composite protein multiple ligand interactions in protein pockets. J Comput Chem. 2017;38:1252–1259. doi: 10.1002/jcc.24523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.