

Abstract
Chronic kidney disease in children is an irreversible process that may lead to end-stage renal disease. The mortality rate in children with end-stage renal disease who receive dialysis increased dramatically in the last decade, and it is significantly higher compared with the general pediatric population. Furthermore, dialysis and transplant patients, who have developed end-stage renal disease during childhood, live respectively far less as compared with age/race-matched populations.
Different reports show that cardiovascular disease is the leading cause of death in children with end-stage renal disease and in adults with childhood-onset chronic kidney disease, and that children with chronic kidney disease are in the highest risk group for the development of cardiovascular disease.
Urea, which is generated in the liver during catabolism of amino acids and other nitrogenous metabolites, is normally excreted into the urine by the kidneys as rapidly as it is produced. When renal function is impaired, increasing concentrations of blood urea will steadily accumulate. For a long time, urea has been considered to have negligible toxicity. However, the finding that plasma urea is the only significant predictor of aortic plaque area fraction in an animal model of chronic renal failure -accelerated atherosclerosis, suggests that the high levels of urea found in chronic dialysis patients might play an important role in accelerated atherosclerosis in this group of patients. The aim of this review was to provide novel insights into the role played by urea in the pathogenesis of accelerated cardiovascular disease in renal failure.
Keywords: Atherogenesis, atherosclerosis, cardiovascular disease, chronic renal failure, urea, vascular toxicity
Introduction
Chronic kidney disease (CKD) in children is an irreversible process that may lead to end-stage renal disease (ESRD). A significant percentage of children with CKD will develop kidney failure by 20 years of age (1). It is a consolidated evidence that despite a prevalence ranging from 15–74.7 cases per million children reported in literature, CKD carries a significant impact in pediatric age groups, reducing life expectancy significantly (2).
The mortality rate in children with ESRD who receive dialysis increased dramatically in the last decade, and it is estimated to be at least 30 times higher than that in the general pediatric population (3). Furthermore, dialysis and transplant patients, who developed ESRD during childhood, lived respectively 40–50 years less, and 20–25 years less, compared with an age- and- race-matched population in the United States of America (3).
Reports from different international registries show that cardiovascular disease (CVD) is the leading cause of death in both children with ESRD and in adults with childhood-onset CKD (4). It is in fact well known that CKD increases up to 30-fold the risk of CVD in adult patients compared with the general population, and CVD risk remains 10 to 20 times higher after stratification of rage, sex, and presence of diabetes (5). The American Heart Association classified children with CKD in the highest risk group for the development of CVD, alongside individuals with homozygous familial hypercholesterolemia, diabetes mellitus type 1, heart transplantation or coronary aneurisms due to Kawasaki disease (6).
As with adult patients, children with CKD show an accelerated atherosclerosis, and early markers of atherosclerosis, such as increased carotid artery intima-media thickness (IMT) and carotid arterial wall stiffness, are frequently found in this patient population (4).
Clinic
Cardiovascular disease in patients with CKD results from a combination of traditional (e.g. hypertension, dyslipidemia, abnormal glucose metabolism and obesity) and CKD-related cardiovascular risk factors (CVRFs) (e.g. increased calcium-phosphorus product, hyperparathyroidism and anemia) (5). Although children with CRF have higher levels of general risk factors for CVD, these traditional CVRFs do not fully account for the high risk of atherosclerosis, CVD, and total mortality in these patients (4). Growing evidence supports a major role for nontraditional CVRFs in the pathogenesis of accelerated atherogenesis in this population (7). Uremia, in which various small molecules accumulate in the blood because of decreased renal excretion (8), has been considered a non-traditional CVRFs involved in the pathogenesis of the vascular changes seen in patients with chronic renal failure (CRF).
Traditionally, the term ‘uremia’ indicated increased serum concentrations of urea in patients with CRF. Urea, a small water soluble molecule, is generated in the liver during the catabolism of amino acids and other nitrogenous metabolites, and is normally excreted into the urine by the kidneys as rapidly as it is produced. When renal function is impaired, increasing concentrations of blood urea steadily accumulate. Therefore, because of its abundance, urea has been routinely used as a surrogate marker of renal function, protein intake, and dialysis adequacy (9).
For a long time, urea has been considered to have negligible toxicity, in spite of its high absolute concentrations reported in the blood of patients with CRF, because short-term increases in urea concentration have been shown not to affect either acute organ function or acute clinical outcomes (9). In 1972, Johnson et al. (10) found that in patients with far-advanced renal failure, blood urea concentrations of less than 50 mM [blood urea nitrogen (BUN) of 140 mg/dL] were well tolerated when blood urea concentration was maintained by adding urea to the dialysate solution and 30 years later, the Hemodialysis (HEMO) study showed that increasing the urea reduction rate from 66% to 75% did not alter survival in patients with an increased dialysis dose (11). However, the view that urea is simply an innocent bystander was recently challenged by several new observations.
Research effects
The finding that plasma urea is the only significant predictor of aortic plaque area fraction in an animal model of CRF-accelerated atherosclerosis (12), suggests that the high levels of urea found in patients under chronic dialysis might play an important role in accelerated atherosclerosis in this group of patients. The observation that oral administration of urea accelerates atherogenesis in non-uremic ApoE/mice fed with a high-fat diet in the absence of other factors and toxins that accumulate in CRF supported this hypothesis (13). Moreover, recent data demonstrated that the survival of patients on daily hemodialysis, which improves the removal of uremic toxins including urea, was 2- to 3-fold greater than that of patients dialyzed less frequently, and several recent in vitro and in vivo studies indicated a possible vascular toxicity of urea (14).
The aim of this review was to provide novel insights into the role played by urea in the pathogenesis of accelerated CVD in renal failure
Effects of urea on endothelial functions
Endothelial injury caused by various CVRFs is an initial step in the development of atherosclerosis (15). In uremic patients, evidence of endothelial dysfunction has been identified at early stages of the disease (7), and endothelial dysfunction has been shown to be the best predictor of subsequent cardiovascular events (15). Current data suggest that endothelial dysfunction appears early in children with renal failure, on chronic dialysis, and after renal transplantation, and is followed by arterial medial calcification (16). This calcification causes arterial wall stiffening and subsequently left ventricular hypertrophy (4, 16). Moreover, endothelial dysfunction appears to be associated with high serum levels of inflammatory markers, such as C-reactive protein, interleukin (IL)-6, IL-1β, tumor necrosis factor (TNF)-α, and fibrinogen in patients with CKD (17). Under these conditions, levels of cell adhesion molecules, such as monocyte chemotactic protein (MCP)-1 and vascular cell adhesion molecule (VCAM)-1 are up-regulated in order to promote monocyte infiltration into the activated endothelium (18).
D’Apolito et al. (19) showed that urea, at the concentration seen at the early stage of CRF, was able to reduce the activity of endothelial cell anti-atherosclerotic enzyme prostaglandin I2 (PGI2) synthase (20) in cultured primary human aortic endothelial cells (HAEC). The reduction in PGI2 synthase activity was associated with a significant increase in mRNA expression of the major inflammatory mediator nuclear factor kappa B (NFκB) subunit p65 (21), and with NFκB activation, as shown by the elevation of mRNA expression of NFκB specific target genes, VCAM1, endoglin, and vascular endothelial growth inhibitor (VEGI) (19). Moreover, urea increase in the HAEC the protein level of MCP-1 and VCAM1, two NFκB-dependent pro-atherogenic endothelial cell surface molecules expressed early in the development of atherosclerosis (19). Therefore, urea appears to be one of the factor that causes endothelial activation, which starts the atherosclerotic process in CRF.
Urea toxicity and endothelial precursor cells
Damaged endothelial cells can be replaced by endothelial progenitor cells (EPCs), a bone-marrow-derived mononuclear cell population, which play a key role in the preservation of vascular integrity (22) by promoting new endothelial growth and endothelial repair mechanisms (23). Therefore, a decreased EPCs number and function can contribute to impaired angiogenesis and atherosclerosis progression (24).
In non-uremic patients at risk for coronary artery disease, the number of circulating EPCs is reduced and the EPCs function is impaired (25). Uremia and compromised renal function are associated with a greater reduction in EPCs availability and function (26), which can contribute to the increased morbidity and mortality of these patients. In fact, in patients with CRF, there was a direct correlation between the percentage of senescent EPCs and the risk of developing CVD. Children on hemodialysis also show a reduction in EPCs number (27). Recently, we reported that urea, in addition to inducing endothelial damages, also had a toxic effect on EPCs, causing EPC dysfunction (28). Urea at the concentration seen in the very early stage of renal failure, impaired the ability of EPCs isolated from a healthy donor to form endothelial cell colony forming units (EC-CFU), and to differentiate into CD31 and vascular endothelial growth factor receptor 2–positive cells (29). A large number of studies used the reduced capacity of EPCs to form EC-CFU as an indicator of EPC dysfunction associated with cardiovascular disorders (29). Moreover, urea causes senescence in EPCs with a senescence-associated secretory phenotype (SASP) involving the production of factors that reinforce senescence arrest, alter the microenvironment, and trigger inflammation (30). An accelerated EPC senescence together with a reduction of EPC numbers has been shown in patients with CRF (31). Then, the increased concentration of urea can accelerate the atherosclerosis process in CRF by reducing endothelial repair mechanisms.
Urea toxicity and vascular smooth muscle cells
Vascular smooth muscle cells (VSMC) are the predominant cellular elements of the vascular media, which is responsible for vasoconstriction and dilation in response to normal or pharmacologic stimuli.
Recent studies have clarified the role played by VSMCs in the pathogenesis of atherosclerotic lesions. Under pathologic conditions, cells in mature vessels can undergo a reverse phenotypic shift from the normal contractile state to either synthetic, proliferative cells or to apoptotic and senescent cells (32). Although VSMC proliferation is beneficial throughout atherogenesis by having a reparative effect, VSMC cell death and senescence promote both atherogenesis and multiple features of plaque instability.
Moderate uremia has been shown to modulate the phenotype of aortic smooth muscle cells (33). Recently, Trecherel et al. (34) demonstrated that urea was able to induce the expression of a pro-apoptotic member of the BCL2 family, the Bcl-xL/Bcl-2-associated death promoter (BAD) protein, in VSMC. Urea-induced BAD overexpression may account for the increased apoptosis observed in the arterial wall of patients with uremia. In fact, BAD can sensitize the cells to the pro-apoptotic effect of oxidative stress, exerted for instance, by oxidized cholesterol (35).
Urea effect on insulin sensitivity and secretion
It is well known that insulin resistance is associated with endothelial dysfunction (36). Insulin induces vasodilatory effects by promoting the release of endothelial nitric oxide (37). Insulin-resistant states reduce nitric oxide production and exaggerated release of endothelin, in endothelial cells (37). In non-uremic people with normal glucose tolerance, insulin resistance markedly increases cardiovascular disease risk, even after adjustment for known risk factors such as low-density lipoprotein (LDL), triglycerides, high-density lipoprotein (HDL), and systolic blood pressure (38).
Insulin resistance is a well-documented feature of ESRD, and the rate of death among patients undergoing hemodialysis is greater than in those with more severe insulin resistance (39). The insulin resistance present in ESRD appears to start much earlier in the course of CKD when renal failure is subclinical (40). Although the cause of insulin resistance in CKD is unknown, several abnormalities associated with ESRD might interfere with insulin signaling (41). It has been shown that urea at the concentration seen in CRF can cause insulin resistance. In fact, urea infusion in normal animals is sufficient to induce the same degree of insulin resistance and elevated insulin resistance–associated adipokines, retinol binding protein (RBP4), and resistin, seen in uremic mice (41).
Treatment of 3T3-L1 adipocytes with urea at disease-relevant concentrations increases modification of insulin-signaling molecules by O-Linked β-N-acetylglucosamine (O-GlcNAc), and reduces insulin-stimulated insulin receptor substrate (IRS) and Akt phosphorylation and glucose transport. These negative signaling changes directly correlate with decreased insulin sensitivity and elevated levels of insulin resistance-associated adipokines. (42).
Several studies have described that the increased insulin resistance present in uremia was frequently associated with defective insulin secretion (43). Koppe et al. (44) demonstrated that urea could also directly impair pancreatic β-cell function. Exposure of mouse and human islets to urea concentrations seen in the very early stage of renal failure increased islet protein O-GlcNAcylation, causing inhibition of glycolysis and, consequently, impaired insulin secretion (44).
Similarly, insulin secretion was impaired in normal mice treated orally with urea for 3 weeks and it was restored by inhibition of O-GlcNAcylation (44). Therefore, increased concentrations of urea, as seen in CRF, can also accelerate the atherosclerotic process by inducing both insulin resistance and impaired insulin secretion
Urea Toxicity: mechanism of action
Urea exerts its toxic action through reactive oxygen species (ROS) production. Elevated concentrations of urea have been described to increase intracellular ROS generation in various cells, such as vascular endothelial cells, VSMCs, renal tubular cells, adipocytes, and beta cells (19, 28, 34, 42, 44). Normalization of intracellular ROS levels prevented each of the described urea effects such endothelial cell and EPC dysfunctions, EPC senescence, adipocyte insulin resistance, and impaired insulin of beta cells (18, 28, 42, 44). Furthermore, in normal mice either infused or treated orally with urea, antioxidant molecules can both restore insulin sensibility and normalize insulin secretion (42, 44). Oxidative stress is common in patients with CKD, and reduction of oxidative stress has a number of beneficial effects (45). Considering that the normal range of urea nitrogen in human blood or serum (BUN) is 5 to 20 mg/dL, or 1.8 to 7.1 mmol/L urea, we have shown that a small increase of urea concentration to 10 nmol/L in EPCs was already able to significantly increase ROS production. Ten nanomoles per liter is a urea concentration seen in the very early stage of CRF (28).
Cellular ROS can be produced by both mitochondrial and cytosolic mechanisms (46). In endothelial cells and in EPCs, it has been found that urea increased ROS production through the activation of both mitochondrial and cytosolic ROS generating mechanisms. Inhibition of either resulted in the complete normalization of urea-induced ROS (19, 28). These observations are consistent with the emerging concept of crosstalk between mitochondria and NADPH oxidases, the major cytosolic sources of superoxide in endothelial cells (47). An increase of mitochondrial ROS in some cell types may activate NADPH oxidases, and activation of NADPH oxidases may increase production of mitochondrial ROS. In pathologic conditions such as CRF, a threshold may be reached in which sufficient ROS are produced to activate an ongoing feed-forward cycle.
The mechanisms by which urea activates intracellular ROS production remain to be explored. It is known that urea at the concentration seen in patients with CRF can increase carbamylation of intracellular proteins (9). This post-translational modification may alter the function of cytosolic, nuclear, and mitochondrial proteins involved in the regulation of ROS production. Carbamylation occurs in the covalent binding of isocyanic acid to a-amino groups of free amino acids or N-terminus of proteins, and to e-amino groups of lysine residues (9). Isocyanic acid derived from the rapid conversion of its unreactive form cyanate, which originates from urea dissociation in aqueous solution. Furthermore, it has been shown that the increased level of urea in the serum of patients with CRF can carbamylate lipoprotein, which can also promote the atherosclerotic process. Carbamylated low-density lipoproteins (cLDL) can induce endothelial cell dysfunction, and carbamylated high-density lipoproteins (cHDL) lose their cardiovascular protective capacity (9, 48, 49). Several studies have shown a direct correlation between protein carbamylation levels and atherosclerosis and mortality in patients with CRF (50).
Conclusion
In summary, urea, long considered to have negligible toxicity in patients with CRF, no longer appears to be an innocent consequence of reduced renal function. Several studies in the last ten years have shown that urea can participate in the progression of the atherosclerotic process seen in patients with CRF. Urea, at disease-relevant concentrations, can induce endothelial dysfunction, EPC senescence, and increased susceptibility of apoptosis of VSMCs; each of which is involved in atherosclerosis progression. Moreover, elevated levels of urea can induce insulin resistance and impair pancreatic insulin secretion, which could contribute to the high cardiovascular morbidity observed in patients with CRF (Figure 1). Finally, elevated concentrations of urea may induce carbamylation of lipoproteins, which can also promote the atherosclerotic process. A reduction in the high morbidity and mortality caused by CRF may be achieved by novel therapeutics that directly target urea-induced ROS because urea’s pro-atherosclerotic effects are caused by its capacity to induce ROS production.
Figure 1.
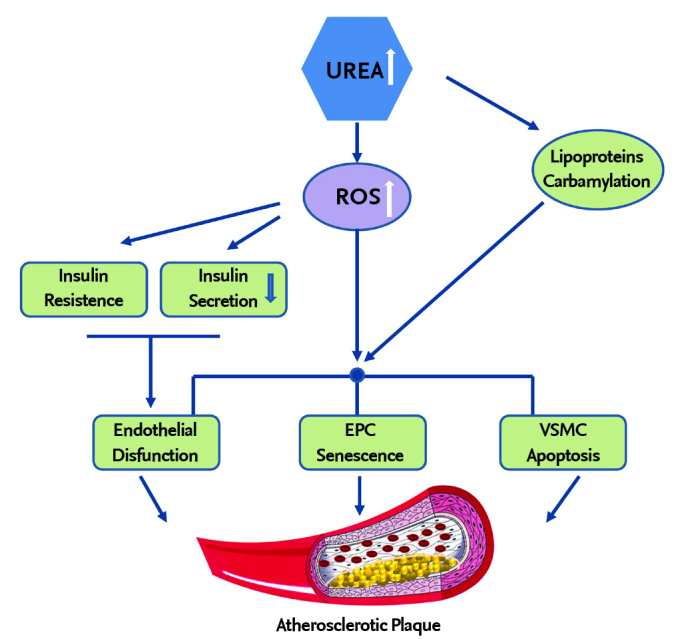
The mechanism which shows that urea accelerates atherosclerosis in patients with chronic renal failure VSMC: vascular smooth muscle cells; EPC: Endothelial precursor cells; ROS: reactive oxygen species
Footnotes
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.; Design - I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.; Supervision - I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.; Funding - I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.; Materials - I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.; Data Collection and/or Processing - I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.; Analysis and/or Interpretation I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.; Literature Review - I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.; Writing - I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.; Critical Review - I.G., M.D’A., M.B., A.B.M., A.L.C, M.S., P.F., M.P.M.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.Wong CJ, Moxey-Mims M, Jerry-Fluker J, Warady BA, Furth SL. CKiD (CKD in Children) Prospective Cohort Study: a review of current findings. Am J Kidney Dis. 2012;60:1002–11. doi: 10.1053/j.ajkd.2012.07.018. https://doi.org/10.1053/j.ajkd.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warady BA, Chadha V. Chronic kidney disease in children: the global perspective. Pediatr Nephrol. 2007;22:1999–2009. doi: 10.1007/s00467-006-0410-1. https://doi.org/10.1007/s00467-006-0410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.US Renal Data System. USRDS 2011 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2011. [Google Scholar]
- 4.Mitsnefes MM. Cardiovascular disease in children with chronic kidney disease. J Am Soc Nephrol. 2012;23:578–85. doi: 10.1681/ASN.2011111115. https://doi.org/10.1681/ASN.2011111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schiffri EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation. 2007;116:85–97. doi: 10.1161/CIRCULATIONAHA.106.678342. https://doi.org/10.1161/CIRCULATIONAHA.106.678342. [DOI] [PubMed] [Google Scholar]
- 6.Kavey RE, Allada V, Daniels SR, et al. Cardiovascular risk reduction in high-risk pediatric patients: a scientific statement from the American Heart Association Expert Panel on Population and Prevention Science; the Councils on Cardiovascular Disease in the Young, Epidemiology and Prevention, Nutrition, Physical Activity and Metabolism, High Blood Pressure Research, Cardiovascular Nursing, and the Kidney in Heart Disease; and the Interdisciplinary Working Group on Quality of Care and Outcomes Research, endorsed by the American Academy of Pediatrics. Circulation. 2006;114:2710–38. doi: 10.1161/CIRCULATIONAHA.106.179568. https://doi.org/10.1161/CIRCULATIONAHA.106.179568. [DOI] [PubMed] [Google Scholar]
- 7.Chade AR, Lerman A, Lerman LO. Kidney in early atherosclerosis. Hypertension. 2005;45:1042–9. doi: 10.1161/01.HYP.0000167121.14254.a0. https://doi.org/10.1161/01.HYP.0000167121.14254.a0. [DOI] [PubMed] [Google Scholar]
- 8.Vanholder R, Baurmeister U, Brunet P, et al. A bench to bedside view of uremic toxins. J Am Soc Nephrol. 2008;19:863–70. doi: 10.1681/ASN.2007121377. https://doi.org/10.1681/ASN.2007121377. [DOI] [PubMed] [Google Scholar]
- 9.Massy ZA, Pietrement C, Touré F. Reconsidering the lack of urea toxicity in dialysis patients. Semin Dial. 2016;29:333–7. doi: 10.1111/sdi.12515. https://doi.org/10.1111/sdi.12515. [DOI] [PubMed] [Google Scholar]
- 10.Johnson WJ, Hagge WW, Wagoner RD, Dinapoli RP, Rosevear JW. Effects of urea loading in patients with far-advanced renal failure. Mayo Clin Proc. 1972;47:21–9. [PubMed] [Google Scholar]
- 11.Eknoyan G, Beck GJ, Cheung AK, et al. Effect of dialysis dose and membrane flux in maintenance hemodialysis. N Engl J Med. 2002;347:2010–9. doi: 10.1056/NEJMoa021583. https://doi.org/10.1056/NEJMoa021583. [DOI] [PubMed] [Google Scholar]
- 12.Ivanovski O, Szumilak D, Nguyen-Khoa T, et al. The antioxidant N-acetylcysteine prevents accelerated atherosclerosis in uremic apolipoprotein E knockout mice. Kidney Int. 2005;67:2288–94. doi: 10.1111/j.1523-1755.2005.00332.x. https://doi.org/10.1111/j.1523-1755.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 13.Apostolov OE, Ray D, Savenka AV, Shah SV, Basnakian AG. Chronic uremia stimulates LDL carbamylation and atherosclerosis. J Am Soc Nephrol. 2010;21:1852–7. doi: 10.1681/ASN.2010040365. https://doi.org/10.1681/ASN.2010040365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanholder R, Massy Z, Argiles A, et al. Chronic kidney disease as a cause of cardiovascular morbidity and mortality. Nephrol Dial Transpl. 2005;20:1048–56. doi: 10.1093/ndt/gfh813. https://doi.org/10.1093/ndt/gfh813. [DOI] [PubMed] [Google Scholar]
- 15.Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:27–32. doi: 10.1161/01.CIR.0000131515.03336.f8. https://doi.org/10.1161/01.CIR.0000131515.03336.f8. [DOI] [PubMed] [Google Scholar]
- 16.Lilien MR, Groothoff JW. Cardiovascular disease in children with CKD or ESRD. Nat Rev Nephrol. 2009;5:229–35. doi: 10.1038/nrneph.2009.10. https://doi.org/10.1038/nrneph.2009.10. [DOI] [PubMed] [Google Scholar]
- 17.Jofre R, Rodriguez-Benitez P, Lopez-Gomez JM, Perez-Garcia R. Inflammatory syndrome in patients on hemodialysis. J Am Soc Nephrol. 2006;17:274–80. doi: 10.1681/ASN.2006080926. https://doi.org/10.1681/ASN.2006080926. [DOI] [PubMed] [Google Scholar]
- 18.Ross R. Atherosclerosis. An inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. https://doi.org/10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 19.D’Apolito M, Du X, Pisanelli D, et al. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis. 2015;239:393–400. doi: 10.1016/j.atherosclerosis.2015.01.034. https://doi.org/10.1016/j.atherosclerosis.2015.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zou MH, Shi C, Cohen RA. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with Thromboxane/Prostaglandin H2 Receptor–Mediated Apoptosis and Adhesion Molecule Expression in Cultured Human Aortic Endothelial Cells. Diabetes. 2002;51:198–203. doi: 10.2337/diabetes.51.1.198. https://doi.org/10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- 21.Marchetti V, Menghini R, Rizza S, et al. Benfotiamine counteracts glucose toxicity effects on endothelial progenitor cell differentiation via Akt/FoxO signaling. Diabetes. 2006;55:2231–7. doi: 10.2337/db06-0369. https://doi.org/10.2337/db06-0369. [DOI] [PubMed] [Google Scholar]
- 22.Asahara T, Takahashi T, Masuda H, et al. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999;18:3964–72. doi: 10.1093/emboj/18.14.3964. https://doi.org/10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yuen DA, Connelly KA, Advani A, et al. Culture modified bone marrow cells attenuate cardiac and renal injury in a chronic kidney disease rat model via a novel antifibrotic mechanism. PLoS One. 2010;5:e9543. doi: 10.1371/journal.pone.0009543. https://doi.org/10.1371/journal.pone.0009543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tepper OM, Galiano RD, Capla JM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–6. doi: 10.1161/01.cir.0000039526.42991.93. https://doi.org/10.1161/01.CIR.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 25.Hill JM, Zalos G, Halcox JP, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. https://doi.org/10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 26.de Groot K, Bahlmann FK, Sowa J, et al. Uremia causes endothelial progenitor cell deficiency. Kidney Int. 2004;66:641–6. doi: 10.1111/j.1523-1755.2004.00784.x. https://doi.org/10.1111/j.1523-1755.2004.00784.x. [DOI] [PubMed] [Google Scholar]
- 27.Jie KE, Lilien MR, Goossens MH, Westerweel PE, Klein MK, Verhaar MC. Reduced endothelial progenitor cells in children with hemodialysis but not predialysis chronic kidney disease. Pediatrics. 2010;126:e990–3. doi: 10.1542/peds.2009-3346. https://doi.org/10.1542/peds.2009-3346. [DOI] [PubMed] [Google Scholar]
- 28.D’Apolito M, Colia AL, Lasalvia M, et al. Urea-induced ROS accelerate senescence in endothelial progenitor cells. Atherosclerosis. 2017;263:127–36. doi: 10.1016/j.atherosclerosis.2017.06.028. https://doi.org/10.1016/j.atherosclerosis.2017.06.028. [DOI] [PubMed] [Google Scholar]
- 29.Padfield GJ, Short A, Mills NL, et al. The constituents and mechanisms of generation of ‘endothelial cell-colony forming units’. Cardiovasc Res. 2013;100:288–96. doi: 10.1093/cvr/cvt182. https://doi.org/10.1093/cvr/cvt182. [DOI] [PubMed] [Google Scholar]
- 30.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. https://doi.org/10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 31.Goligorsky MS, Yasuda K, Ratliff B. Dysfunctional endothelial progenitor cells in chronic kidney disease. Am Soc Nephrol. 2010;21:911–9. doi: 10.1681/ASN.2009111119. https://doi.org/10.1681/ASN.2009111119. [DOI] [PubMed] [Google Scholar]
- 32.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. https://doi.org/10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madsen M, Aarup A, Albinsson S, et al. Uremia modulates the phenotype of aortic smooth muscle cells. Atherosclerosis. 2017;257:64–70. doi: 10.1016/j.atherosclerosis.2016.12.022. https://doi.org/10.1016/j.atherosclerosis.2016.12.022. [DOI] [PubMed] [Google Scholar]
- 34.Trecherel E, Godin C, Louandre C, et al. Upregulation of BAD, a pro-apoptotic protein of the BCL2family, in vascular smooth muscle cells exposed to uremic conditions. Biochem Biophys Res Commun. 2012;417:479–83. doi: 10.1016/j.bbrc.2011.11.144. https://doi.org/10.1016/j.bbrc.2011.11.144. [DOI] [PubMed] [Google Scholar]
- 35.Shroff RC, McNair R, Figg N, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118:1748–57. doi: 10.1161/CIRCULATIONAHA.108.783738. https://doi.org/10.1161/CIRCULATIONAHA.108.783738. [DOI] [PubMed] [Google Scholar]
- 36.Mather KJ, Steinberg HO, Baron AD. Insulin resistance in the vasculature. J Clin Invest. 2013;123:1003–4. doi: 10.1172/JCI67166. https://doi.org/10.1172/JCI67166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lteif A, Vaishnava P, Baron AD, Mather KJ. Endothelin limits insulin action in obese/insulin-resistant humans. Diabetes. 2007;56:728–34. doi: 10.2337/db06-1406. https://doi.org/10.2337/db06-1406. [DOI] [PubMed] [Google Scholar]
- 38.Hanley AJ, Williams K, Stern MP, Haffner SM. Homeostasis model assessment of IR in relation to the incidence of cardiovascular disease: The San Antonio Heart Study. Diabetes Care. 2002;25:1177–84. doi: 10.2337/diacare.25.7.1177. https://doi.org/10.2337/diacare.25.7.1177. [DOI] [PubMed] [Google Scholar]
- 39.Bodlaj G, Ber J, Pichler R, Biesenbach G. Prevalence, severity and predictors of HOMA-estimated insulin resistance in diabetic and nondiabetic patients with end-stage renal disease. J Nephrol. 2006;19:607–12. [PubMed] [Google Scholar]
- 40.Lorenzo C, Nath SD, Hanley AJ, Abboud HE, Haffner SM. Relation of low glomerula filtrationrate to metabolic disorders in individuals without diabetes and with normoal albiminuria. Clin J Am Soc Nephrol. 2008;3:783–9. doi: 10.2215/CJN.02730707. https://doi.org/10.2215/CJN.02730707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takenaka T, Kanno Y, Ohno Y, Suzuki H. Key role of insulin resistance in vascular injury among hemodialysis patients. Metabolism. 2007;56:153–9. doi: 10.1016/j.metabol.2006.08.010. https://doi.org/10.1016/j.metabol.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 42.D’Apolito M, Du X, Zong H, et al. Urea induced ROS generation causes insulin resistance in mice with chronic renal failure. J Clin Investig. 2010;120:203–13. doi: 10.1172/JCI37672. https://doi.org/10.1172/JCI37672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fadda GZ, Hajjar SM, Perna AF, Zhou XJ, Lipson LG, Massry SG. On the mechanism of impaired insulin secretion in chronic renal failure. J Clin Invest. 1991;87:255–61. doi: 10.1172/JCI114979. https://doi.org/10.1172/JCI114979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koppe L, Nyam E, Vivot K, et al. Urea impairs β cell glycolysis and insulin secretion in chronic kidney disease. J Clin Invest. 2016;126:3598–612. doi: 10.1172/JCI86181. https://doi.org/10.1172/JCI86181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dounousi E, Papavasiliou E, Areti Makedou M, et al. Oxidative stress is progressively enhanced with advancing stages of CKD. Am J Kidney Dis. 2006;48:752–60. doi: 10.1053/j.ajkd.2006.08.015. https://doi.org/10.1053/j.ajkd.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 46.Menini S, Amadio L, Oddi G, et al. Deletion of p66Shc longevity gene protects against experimental diabetic glomerulopathy by preventing diabetes-induced oxidative stress. Diabetes. 2006;55:1642–50. doi: 10.2337/db05-1477. https://doi.org/10.2337/db05-1477. [DOI] [PubMed] [Google Scholar]
- 47.Dikalov S. Crosstalk between mitochondria and NADPH oxidases. Free Radic Biol Med. 2011;51:1289–301. doi: 10.1016/j.freeradbiomed.2011.06.033. https://doi.org/10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ok E, Basnakian AG, Apostolov EO, Barri YM, Shah SV. Carbamylated low-density lipoprotein induces death of endothelial cells: a link to atherosclerosis in patients with kidney disease. Kidney Int. 2005;68:173–8. doi: 10.1111/j.1523-1755.2005.00391.x. https://doi.org/10.1111/j.1523-1755.2005.00391.x. [DOI] [PubMed] [Google Scholar]
- 49.Holzer M, Gauster M, Pfeifer T, et al. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid Redox Signal. 2011;14:2337–46. doi: 10.1089/ars.2010.3640. https://doi.org/10.1089/ars.2010.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berg AH, Drechsler C, Wenger J, et al. Carbamylation of serum albumin as a risk factor for mortality in patients with kidney failure. Sci Transl Med. 2013;5:175ra29. doi: 10.1126/scitranslmed.3005218. https://doi.org/10.1126/scitranslmed.3005218. [DOI] [PMC free article] [PubMed] [Google Scholar]