

Abstract
Donohue syndrome (Leprechaunism) is characterized by severe insulin resistance, hyperinsulinemia, postprandial hyperglycemia, preprandial hypoglycemia, intrauterine and postnatal growth retardation, dysmorphic findings, and clinical and laboratory findings of hyperandrogenemia due to homozygous or compound heterozygous inactivating mutations in the insulin receptor gene. A female newborn presented with lack of subcutaneous fat tissue, bilateral simian creases, hypertrichosis, especially on her face, gingival hypertrophy, cliteromegaly, and prominent nipples. Her laboratory tests revealed hyperandrogenism, postprandial hyperglycemia and preprandial hypoglycemia, and very high concurrent insulin levels. She was diagnosed as having Donohue syndrome. Metformin and continuous nasogastric feeding were administrated. During follow-up, relatively good glycemic control was obtained. However, severe hypertrophic obstructive cardiomyopathy and severe malnutrition developed. She died aged 75 days of severe heart failure and pneumonia. Her insulin receptors gene analysis revealed a compound heterozygous mutation. One of these mutations was a p.R813 (c.2437C>T) mutation, which was defined previously and shown also in her father, the other mutation was a novel p.777-790delVAAFPNTSSTSVPT mutation, also shown in her mother. The parents were heterozygous for these mutations.
Keywords: Donohue syndrome, hyperandrogenism, hyperinsulinism, insulin receptor, insulin resistance, newborn
Introduction
Insulin is an anabolic hormone that is involved in cellular uptake of glucose, the inhibition of hepatic gluconeogenesis, reduction of lipolysis, increase in lipogenesis, and prevention of protein destruction (1).
Donohue syndrome is a condition in which binding of insulin to insulin receptors (INSR) and signalization of insulin is disrupted. This condition is caused by autosomal recessive, homozygous or compound heterozygous loss-of-function mutations in the INSR gene (19th chromosome). More than 150 mutations have been reported in the insulin receptor gene. Severe insulin resistance and hyperinsulinism develop in these patients. Patients are lost in early infancy because of intervening infections and heart failure. Rabson-Mendenhall syndrome is another phenotype of this condition. In this syndrome, patients have been reported to have the same clinical findings as seen in Donohue syndrome with a slightly lower degree of severity and can live up to the second decade (2–4).
In this article, a patient with Donohue syndrome who was followed up from the neonatal period because of problems caused by severe insulin resistance and compensatory hyperinsulinism and was found to have compound heterozygous mutation in the INSR gene is presented.
Case
Our patient was a female baby who was born at the 37th gestational week by normal vaginal spontaneous delivery from the first pregnancy of a 29-year-old mother who developed polyhydramnios and hypertension during her pregnancy. There was no consanguinity between the mother and father. The physical examination revealed a birth height of 43 cm (<3 p) and a birth weight of 1630 g (<3rd percentile). She had a cachectic and old appearance, coarse face, extraverted nostrils, dry and loose skin, decreased subcutaneous adipose tissue, bilateral simian creases, diffuse hypertrichosis, which was especially prominent on the face, gingival hypertrophy, cliteromegaly, and prominent nipples (Picture 1–2). Laboratory tests revealed hyperandrogenism (Table 1). Cliteromegaly and the presence of hyperandrogenism suggested the diagnosis of congenital adrenal hyperplasia (CAH). Her karyotype was found to be 46 XX. Hypoglycemia attacks were observed in the first postnatal days. Hypoglycemia was thought to occur in relation with low birth weight or CAH. Adrenal insufficiency and CAH were not found on normal-dose adrenocorticotropic hormone (ACTH) stimulation test. Hyperinsulinism was considered when postprandial hyperglycemia (270 mg/dL) and preprandial hypoglycemia attacks were found on the postnatal 21st day. In this period, the blood glucose levels were found as 178 mg/dL and 22 mg/dL, respectively, whereas concurrent insulin levels were found to be >1000 mIU/mL and 516 mIU/mL, and C-peptide (CPE) levels were found as 58 ng/mL and 12 ng/mL. A diagnosis of Donohue syndrome was made with these clinical and laboratory findings. Metformin at a dose of 50 mg/kg was initiated to correct hyperinsulinism and the dose was increased to 100 mg/kg subsequently. Continuous nasogastric feeding was initiated. The blood glucose levels were kept between 60 and 120 mg/dL with these approaches (Graphic 1). On the 42nd day, a small atrial septal defect and minimal septal hypertrophy were found on echocardiography and proponolol treatment was initiated. In the follow-up echocardiogram performed on the 49th day, it was found that severe hypertrophic cardiomyopathy developed and a diuretic drug was added to the treatment. In the follow-up, direct hyperbilirubinemia and nephrocalcinosis developed. Pelvic ultrasonography revealed marked enlargement in the ovaries (right ovary 29 x 15 mm, left ovary 24 x 14 mm). The patient did not gain weight despite sufficient nutritional supplement. Severe weight loss and subcutaneous adipose tissue loss developed. She was internalized in the intensive care unit because of severe pneumonia and heart failure. Mechanical ventilation and supportive treatment (antibiotic, digitalis, diuretic) were initiated. The patient was lost at the age of 75 days when she had a body weight of 1800 g. Hepatitis and severe pneumonia were found on postmortem biopsy.
Table 1.
Sex hormone levels of our patient on the postnatal 14th day
| FSH (mIU/mL) | 0.27 |
| LH (mIU/mL) | 0.1 |
| Estradiol (pg/mL) | 84.67 |
| DHEA-S (μg/mL) | 224 |
| 17 OH Progesterone (ng/mL) | 50.2 |
| Total testosterone (ng/dL) | 196 |
| Androstenedione (ng/mL) | 10 |
DHEAS: Dihydroepiandrostenedione sulphate; FSH: Follicle-stimulating hormone; LH: Luteinizing hormone; 17 OH Progesterone: 17 hydroxyprogesterone
Graphic 1.
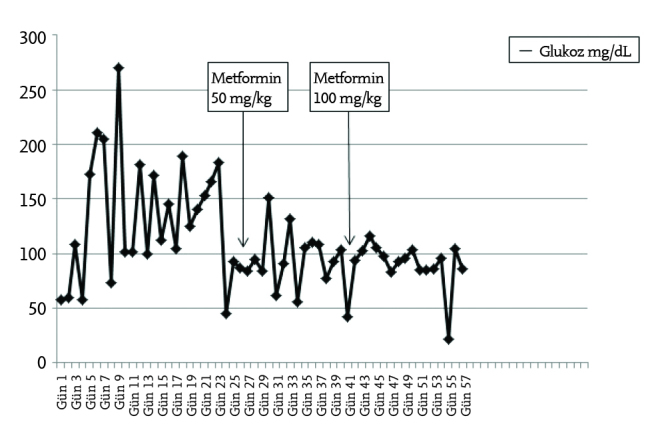
Blood glucose level monitoring of our patient
Obesity, acanthosis nigricans, hyperinsulinism, and disrupted glucose tolerance were found in the patient’s parents. The father’s glycated hemoglobin (HbA1c) value was within the normal limits, but the mother’s was found as 6.45%.
New-generation sequence analysis (Intergen Laboratory) revealed a compound heterozygous mutation in exon 12 region of the INSR gene in our patient. One of these mutations was a 14- amino acid deletion mutation (p.777-790delVAAFPNTSSTSVPT), which has not been previously reported, and was found in our patient’s mother as a heterozygous mutation. The other mutation was a known nonsense p.R813(c.2437C>T) mutation, which caused the occurrence of an early stop codon in the 813th amino acid position, and was found as a heterozygous mutation in the father.
Verbal consent was obtained from the patient’s family.
Discussion
Severe insulin resistance develops in Donohue syndrome because of INSR dysfunction. The metabolic and mitogenic actions of insulin cannot be fulfilled. Thus, metabolic disorders including severe compensatory hyperinsulinism, preprandial hypoglycemia, postprandial hyperglycemia attacks, and hyperandrogenism are observed in these patients. In addition, growth retardation develops from the intrauterine period because of the loss and lack of development of muscle and adipose tissue (2–5). The presence of severe hyperinsulinism concurrently with hyper-hypoglycemia attacks suggested severe insulin resistance in our patient. A diagnosis of Donohue syndrome was made with the presence of typical clinical and laboratory findings, the fact that these clinical findings had a severe course and the fact that we lost our patient at an early age.
Insulin and proinsulin show structural similarity with insulin-like growth factors (IGF) IGF-1 and IGF-2. In addition, INSR and type I IGF receptor also show structural similarity and use common postreceptor signal pathways. Insulin-like growth factors-1 and IGF-2 execute their biologic actions by way of both the insulin receptor and type I IGF receptor. Insulin like growth factors have the capacity to execute the effects of insulin involved in glucose and protein metabolism (1, 6).
In patients with Donohue syndrome, intracellular uptake of glucose is disrupted, insulin-dependent adipose-muscle tissues are lost and not replaced, and atrophy develops. In addition, it has been proposed that INSR-related IGF-1 and growth hormone signalization irregularities in some tissues might be involved in the growth retardation that develops in these patients (2, 4). Severe marasmus developed in our patient who did not gain any weight in the follow-up.
In absence of insulin receptors or IGF receptors, the present receptor has been shown to execute the other receptor’s function (1, 6). Therefore, hyperinsulinism causes pseudoacromegalic growth in many soft tissues, although severe growth retardation is found in patients with Donohue syndrome. This condition is especially prominent in androgen-dependent tissues (2). Pseudoacromegalic findings include elfin faces; large and low-set ears; wide nostrils; thick lips; gingival hypertrophy; large mouth, hands and feet; hypertrichosis; dysplastic nails; and acanthosis nigricans (2, 3–5). Similar clinical findings were observed in our patient. It has been reported that organ disorders including abdominal swelling, renal and hepatic enlargement, cholestasis, and hepatic fibrosis may also develop in these patients (2, 7, 8). Insulin receptor expression is found in all segments of the nephron in the kidneys. It has been proposed that insulin might be involved in calcium reabsorption in both the proximal and distal tubules (8). It has been determined that hypercalciuria and nephrocalcinosis are found in patients with insulin receptor gene mutations (2, 8). Direct hyperbilirubinemia and nephrocalcinosis developed in our patient during the follow-up.
Insulin increases the effects of gonadotropins synergistically (2, 9). With this effect, excessive growth occurs in sex hormone-dependent tissues in addition to the above-mentioned pseudoacromegalic findings in conditions of hyperinsulinism. Findings including breast hyperplasia, prominent nipples, ovarian cysts, juvenile granulosa cell ovarian tumor, Leydig cell hyperplasia, enlarged penis and cliteromegaly develop (2). Our patient had prominent nipples and cliteromegaly. Pelvic ultrasonographic examinations performed with an interval of one month revealed markedly enlarged ovaries with adolescence size and an increased number of follicles.
Insulin increases androgen synthesis in ovarian theca cells with its luteinizing hormone-like action. In addition, it increases free testosterone levels by decreasing the synthesis of sex-hormone binding protein in the liver. In patients with insulin resistance/hyperinsulinism, ovarian hyperandrogenism and virilization develop. On the other hand, it is known that testosterone increases insulin resistance by way of many mechanisms (9). Increased androgen levels and cliteromegaly were found in our patient. These findings prompted the initial investigation for CAH in our patient.
It has been reported that hypertrophic cardiomyopathy develops in babies with congenital hyperinsulinism including infants of diabetic mothers and in other conditions of insulin resistance (2, 4, 7). Hypertrophic obstructive cardiomyopathy also developed in our patient. Although the blood glucose levels had a stable course, our patient was lost because of severe heart failure secondary to severe hypertrophic obstructive cardiomyopathy and lung infection.
There is no treatment for this condition. It is somewhat difficult to control hypo-hyperglycemia attacks. Metformin and/or rosiglitazone is given to increase insulin sensitivity in treatment. However, the efficiency of these drugs is limited (4, 5). Metformin treatment was initiated in our patient. In addition, continuous nasogastric feeding was initiated to avoid abnormal insulin synthesis, which would be stimulated by oral intake. With this approach, the blood glucose levels had a course within relatively normal limits. It has been reported that insulin at a very high dose was used for hyperglycemia in some patients with Donohue syndrome (4, 5). It has been reported that an insulin pump was primarily implanted in a newborn with Donohue syndrome, but subsequent blood glucose levels could only be controlled with very high doses of insulin glargine. In addition, it was reported that the giant ovarian cysts found in this patient recovered with insulin glargine treatment (5). Our patient had no requirement for insulin treatment.
Recombinant IGF-1 (rIGF-1) decreases blood glucose and insulin levels in minutes by way of IGF-1 receptors (4). Use of rIGF-1 is recommended in patients with Donohue syndrome. However, experience in use of rIGF-1 is limited. Moreover, it has also been reported that it does not provide sufficient efficacy, causes renal hypertrophy and proliferative retinopathy, and increased development of cardiac hypertrophy (3–5, 7). Recombinant IGF-1 could not be given to our patient because it could not be provided during her life time.
Donohue syndrome is caused by homozygous and compound heterozygous loss-of-function mutations in the INSR gene (2, 3). There is an ineffective allele in cases of heterozygous INSR gene mutation and the clinical picture is silent. Insulin resistance is present in these patients who carry a risk for the development of diabetes in the presence of obesity (2, 3). The parents of our patient who had a heterozygous INSR gene mutation had clinical findings of insulin resistance as mentioned above including obesity, acanthosis nigricans, hyperinsulinism, and disrupted glucose tolerance.
It has been reported that families with a risk of having a child with Donohue syndrome could have healthy children with preimplantation genetic analysis (10). Performing genetic analysis and providing genetic counselling will prevent repeat experiences of this heavy process by patients, physicians and families. Genetic counseling was provided to our patient’s family in which both parents were carriers for this condition, thereby carrying a risk for having another child with Donohue syndrome.
In this article, our experiences related with the problems and monitoring of a patient with Donohue syndrome who died of hepatitis and pneumonia have been presented.
Picture 1–2.
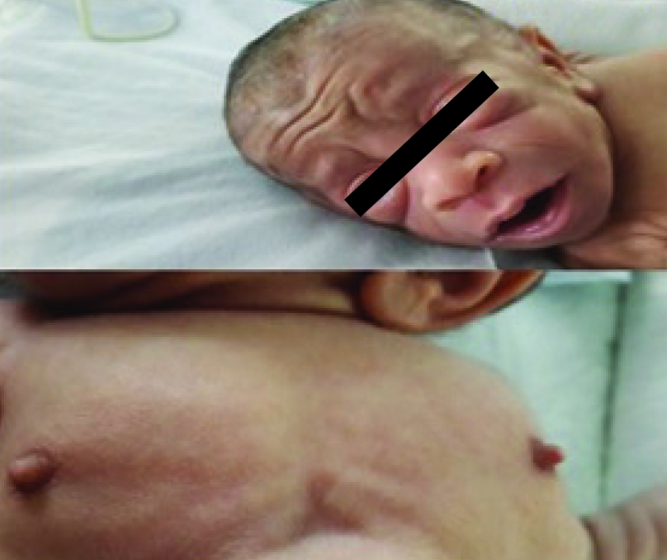
Appearance of our patient’s face and nipples
Footnotes
Informed Consent: Verbal informed consent was obtained from patients’ parents who participated in this study.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - B.K., N.T.; Design - B.K.; Supervision - B.K.; Funding - N.T., E.A.; Data Collection and/or Processing - E.A., P.K., S.D.A., Ö.B., B.K.; Analysis and/or Interpretation - B.K.; Literature Review - B.K. ;Writing - B.K.; Critical Review - B.K.; Other - N.T.
Conflict of Interest: No conflict of interest was declared by the authors
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.McDonald A, Williams RM, Regan FM, Semple RK, Dunger DB. IGF-I treatment of insulin resistance. Eur J Endocrinol. 2007;157:51–6. doi: 10.1530/EJE-07-0271. https://doi.org/10.1530/EJE-07-0271. [DOI] [PubMed] [Google Scholar]
- 2.Semple RK, Savage DB, Halsall DJ, O’Rahilly S. Syndromes of severe insulin resistance and/or lipodystrophy. In: Weiss RE, Refetoff S, editors. Genetic diagnosis of endocrine disorders. USA: Academic Press; 2010. pp. 39–52.https://doi.org/10.1016/B978-0-12-374430-2.00004-3 [Google Scholar]
- 3.Nobile S, Semple RK, Carnielli RP. A novel mutation of the insulin receptor gene in a preterm infant with Donohue syndrome and heart failure. J Ped End Met. 2012;25:363–6. doi: 10.1515/jpem-2011-0448. https://doi.org/10.1515/jpem-2011-0448. [DOI] [PubMed] [Google Scholar]
- 4.Semple RK, Williams RM, Dunger DB. What is the best management strategy for patients with severe insulin resistance? Clin Endocrinol. 2010;73:286–90. doi: 10.1111/j.1365-2265.2010.03810.x. https://doi.org/10.1111/j.1365-2265.2010.03810.x. [DOI] [PubMed] [Google Scholar]
- 5.Çelik AY, Odabaş D, Pirgon O. Leprechaunismli bir infantta bozulmuş glukoz regülasyonu ve dev over kistlerinin yüksek poz insülin glarjin ile tedavisi-olgu sunumu. Güncel Ped. 2010;8:119–22. [Google Scholar]
- 6.Lamothe B, Baudry A, Christoffersen CT, et al. Insulin receptor-deficient cells as a new tool for dissecting complex interplay in insulin and insulin-like growth factors. FEBS Lett. 1998;426:381–5. doi: 10.1016/s0014-5793(98)00377-9. https://doi.org/10.1016/S0014-5793(98)00377-9. [DOI] [PubMed] [Google Scholar]
- 7.Tinka H, Bratanic N, Podkrajsek KT, et al. Severe progressive obstructive cardiomyopathy and renal tubular dysfunction in Donohue syndrome with decreased insulin receptor autophosphorylation due to a novel INSR mutation. Eur J Ped. 2013;172:1125–9. doi: 10.1007/s00431-012-1901-7. https://doi.org/10.1007/s00431-012-1901-7. [DOI] [PubMed] [Google Scholar]
- 8.Simpkin A, Cochran E, Cameron F, et al. Insulin receptor and the kidney: nephrocalcinosis in patients with recessive INSR mutations. Nephron Physiol. 2014;128:55–61. doi: 10.1159/000366225. https://doi.org/10.1159/000366225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hisrchberg AL. Polycystic ovary syndrome, obesity and reproductive implications: hyperandrogenism and insulin resistance. CME released 24.8.2014; valid for credit through 24.8.2010. Available from: https://www.medscape.org/viewarticle/707476.
- 10.Dokuzeylül N, Kahraman S. Preimplantation genetic diagnosis for Donohue syndrome (Leprechaunism) J Turk-German Gyn Assoc. 2009;10:122–3. [Google Scholar]