

Abstract
Apert syndrome is an autosomal dominant craniosynostosis syndrome accompanied by limb anomalies. The fibroblast growth factor receptor 2 (FGFR2) gene is responsible for the disease and two different heterozygous mutations, p.Pro253Arg and p.Ser252Trp, have been defined as responsible in the majority of cases of Apert syndrome. In this case report, two patients with Apert syndrome with two different FGFR2 gene mutations are presented. Case-1, a 4-month-old boy with craniosynostosis and syndactyly was referred to pediatric genetic clinic. The molecular analysis revealed p.Pro253Arg mutation in the FGFR2 gene, which confirmed the diagnosis of Apert syndrome. Case-2, a 16-year-old girl with developmental delay, cleft palate, syndactyly, and craniosynostosis, was also diagnosed as having Apert syndrome. A molecular diagnosis identified a p.Ser252Trp heterozygous mutation in the FGFR2 gene. Case-1 underwent surgery for craniosynostosis at age 10 months and he was developmentally normal during the 2 year follow-up period. As a conclusion, early surgical intervention should be considered in cases of Apert syndrome to prevent intellectual disability.
Keywords: Apert syndrome, craniosynostosis, FGFR2
Introduction
Apert syndrome (AS) is a considerably rare syndrome characterized by craniosynostosis, mid-facial hypoplasia, and syndactyly in the hands and feet. Its prevalence has been reported as 1/65.000 and it constitutes approximately 4.5% of craniosynostosis syndromes (1).
Although AS is inherited in an autosomal dominant pattern, the great majority of cases occur as a result of spontaneous mutations. In 1995, Park et al. (2) showed that p.Ser252Trp (c.755C>G) and p.Pro253Arg (c.758C>G) mutations occurring in the fibroblast growth factor receptor 2 (FGFR2) gene located in chromosome 10q26 led to AS.
In this article, two patients with AS carrying p.Pro253Arg and p.Ser252Trp mutations as confirmed using molecular analysis are presented.
Case 1
A four-month-old male baby who was born by spontaneous vaginal delivery at the 39th gestational week with a birth weight of 3300 g from the first pregnancy of a 26-year-old healthy mother and a 25-year-old healthy father who had no consanguinity, was found to have a body weight of 7 kg (50th percentile), a height of 63 cm (25–50th percentile) and a head circumference of 42 cm (50–75th percentile) at presentation. The physical examination revealed a wide forehead, acrobrachycephaly, hypertelorism, proptosis, flattened nasal root, long philtrum, and low-set ears. Type III syndactyly involving all digits on both hands and feet was present (Picture 1). Other systems were found to be natural on examination. A diagnosis of AS was made with the clinical findings, and the diagnosis was confirmed with molecular analysis. The patient’s blood count and serum biochemistry tests were evaluated as normal. Abdominal ultrasonography revealed hydronephrosis, and transfontanelle ultrasonography revealed cavum septum pellucidum. A bone examination revealed bone syndactyly in the hands and feet. Brain three-dimensional tomography revealed synostosis in the coronal suture (Picture 2). Echocardiography, brain magnetic resonance, and a hearing test revealed no pathology. The patient was referred to the division of neurosurgery because of craniosynostosis and corrective surgery was performed at the age of 10 months. In the physical examination performed when the boy was aged two years, neuromotor development was evaluated as compatible with age.
Picture 1 a, b.

Image of case 1 showing dysmorphic facial signs (a), syndactyly in the hands and feet (b)
Picture 2 a, b.
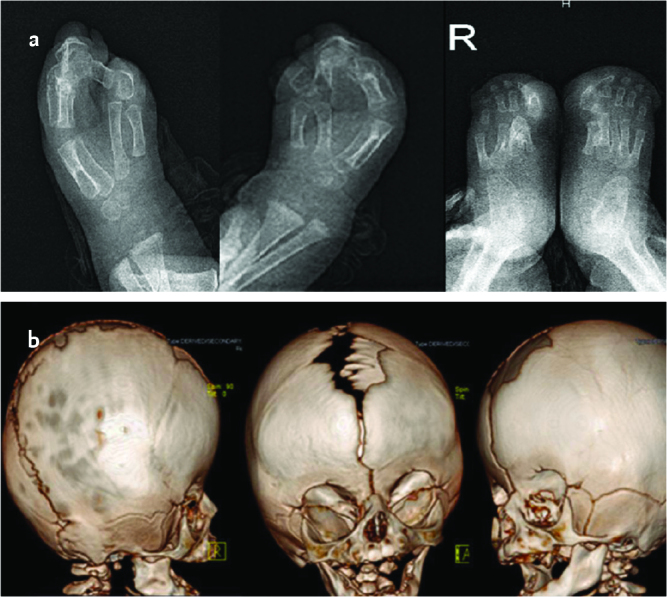
Images of direct X-ray showing syndactyly in the hands and feet in case 1 (a), three-dimensional computerized tomography images showing coronal suture synocytosis (b)
Case 2
A sixteen-year-old female patient was referred to our hospital because of craniosynostosis and syndactyly. She was born by spontaneous vaginal delivery at the 38th gestational week with a birth weight of 3400 g as the fourth living child from the sixth pregnancy of a 28-year-old healthy mother and a 28-year-old healthy father who had no consanguinity. It was learned that large fontanelle and syndactyly were found on physical examination, syndactyly corrective surgery was planned, but the family did not attend follow-up visits. At presentation, her body weight was found as 47 kg (25–50th percentile), her height was 143.8 cm (<3rd percentile), and her head circumference was 54.5 cm (50th percentile). On physical examination, short stature, wide forehead, acrobrachycephaly, hypertelorism, proptosis, down-slanting palpable fissures, flattened nasal root, long nose tip, long philtrum, cleft palate, malocclusion of the teeth, and low-set ears were found. Bilateral type I syndactyly involving the second and fifth digits in the hands and feet and wide distal phalanx in the thumb were present (Picture 3). Examination of the other systems were found as natural.
Picture 3 a, b.
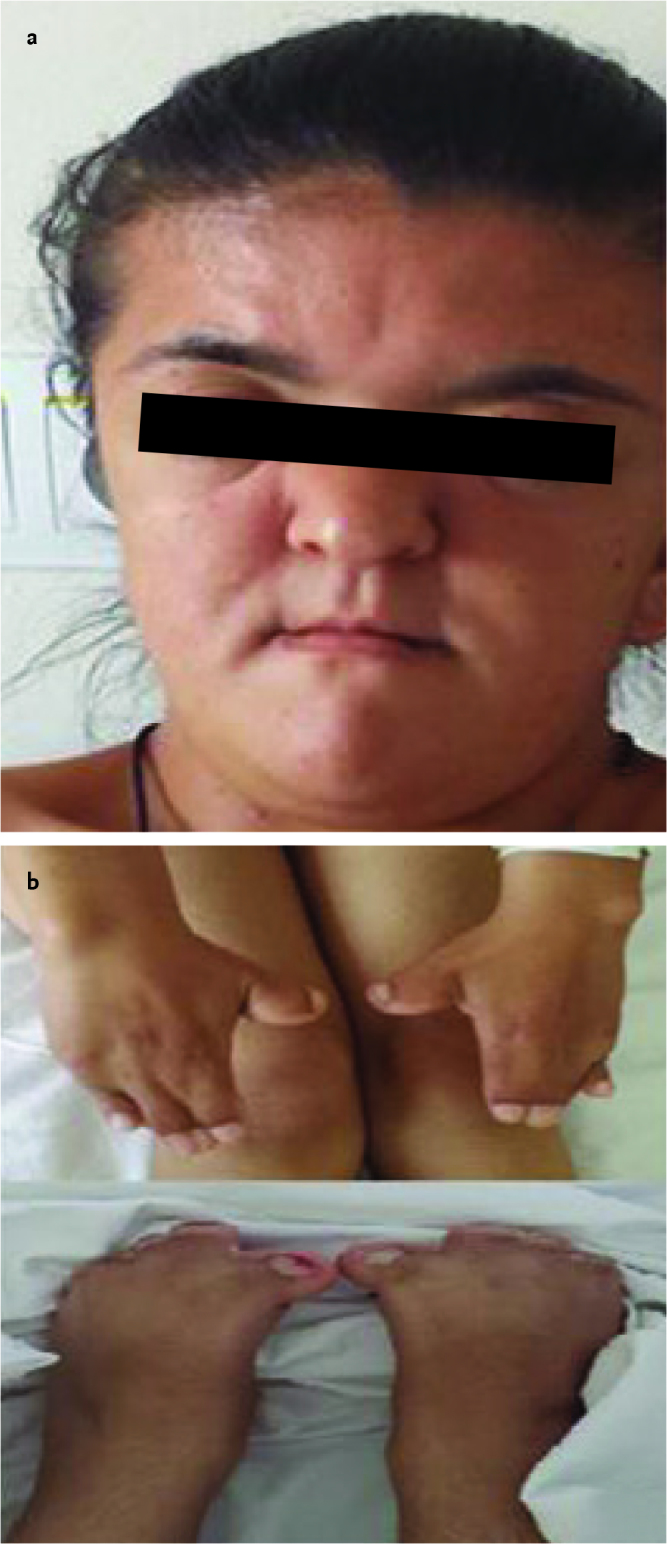
Image of case 2 showing dysmorphic facial signs (a), syndactyly involving the second and fifth digits in the hands and feet (b)
Complete blood count and serum biochemistry tests, abdominal ultrasonography, echocardiography, and hearing tests revealed no pathology. The patient’s intelligence quotient (IQ) was found as 58 in a cognitive evaluation performed using the Wechsler Intelligence Scale for Children-Revised (WISC-R) test. A whole bone examination revealed bilateral bone syndactyly involving the second and fifth digits in the hands and feet and fusion between the fourth and seventh cervical vertebrae. Brain three-dimensional computerized tomography imaging revealed synostosis in the coronal and sagittal sutures and maxillary and mandibular hypoplasia (Picture 4). Brain magnetic resonance imaging was evaluated as normal. A clinical diagnosis of AS was made with these findings.
Picture 4 a, b.
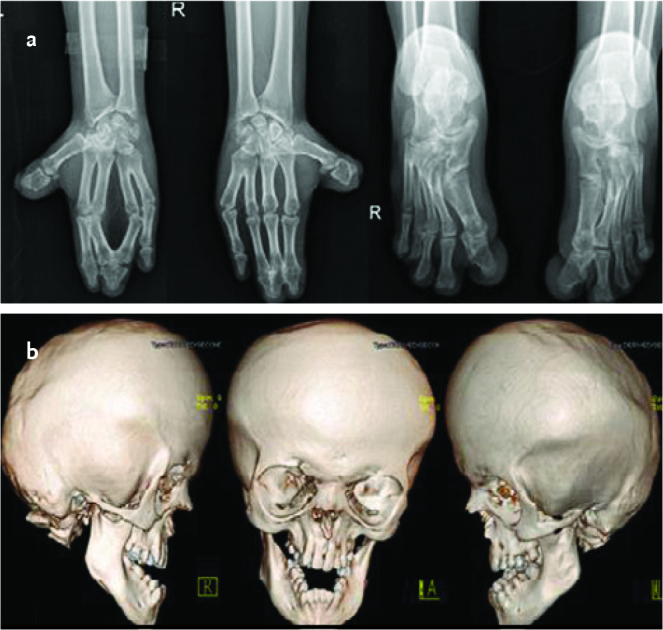
Images of X-ray showing syndactyly in the hands and feet in case 2 (a), three- dimensional computerized tomography images showing synocytosis in the coronal and sagittal sutures and maxillary and mandibular hypoplasia (b)
Informed consent was obtained from the families of both patients.
Molecular Analysis
After voluntary consent and written consent were obtained from both families, Sanger sequence analysis was performed using an ABI3130 automated sequence analyzer to determine FGFR2 gene mutations. The sequence obtained was compared with the reference sequences. The changes found were named according to the NM_001105 transcript. A heterozygous p.Pro253Arg mutation was found in case 1 and a heterozygous p.Ser252Trp mutation was found in case 2 (Picture 3). It was found that the parents of both patients did not carry mutations and the mutations in the patients occurred de novo.
Discussion
Apert syndrome occurs as a result of heterozygous missense mutations in the FGFR2 gene. It has been reported that the p.Ser252Trp mutation is responsible for 71% of cases and the p.Pro253Arg mutation accounts for 26% of cases (2). Similarly, the p.Ser252Trp mutation was found more frequently in Turkish patients in a study conducted by Nur et al. (3). Six new mutations leading to AS have been defined up to the present time in addition to these mutations, which are frequently observed in the FGFR2 gene (4).
The FGFR2 protein is a tyrosine kinase receptor composed of extracellular, transmembrane, and cytoplasmic regions (5). Craniosynostosis syndromes including Apert, Crouzon, Pfeiffer, Jackson-Weiss, and Beare-Stevenson syndromes, and FGFR2-related isolated coronal synostosis occur as a result of FGFR2 gene mutations localized in the 10q25.3-26 locus (2). It has been proposed that the fact that modifying genes and epigenetic factors are effective on phenotype in addition to FGFR2 mutations is a reason for the wide distribution of phenotypic changes occurring as a result of these mutations (6).
Two mutations that occur in the FGFR2 gene lead to some phenotypic differences. It has been reported that the malformation of cleft palate is observed more frequently in patients carrying the p.Ser252Trp mutation and syndactyly is more severe in patients carrying the p.Pro253Arg mutation (2). In addition, it has been found that urogenital anomalies including hydronephrosis and nephrolithiasis are observed more frequently with the p.Pro253Arg mutation (7). Similarly, it was observed that syndactyly was more severe and hydronephrosis accompanied the p.Pro253Arg mutation, and cleft palate malformation accompanied the p.Ser252Trp mutation in our cases. In AS, central nervous system anomalies, congenital heart diseases, genital anomalies, aplasia cutis, and excessive sweating may also be observed in addition to craniosynostosis and syndactyly (7, 8). In addition, atypical clinical findings including metopic suture synostosis, edema in the fundus oculi, epilepsy, and central apnea have also been described in patients carrying the p.Ser252Trp mutation (9). However, similar phenotypic findings were not found in our cases.
Varying degrees of mental retardation have been reported in approximately 50% of patients with AS and it has been shown that the timing of craniosynostosis corrective surgery is an important factor that affects mental development. It was found that the IQ value was 70 and above in 50% of patients who underwent surgery before the age of one year, but only 7.1% of patients who underwent surgery at older ages had a normal intelligence level (10). Similarly, the intellectual developmental steps were found as normal in our first patient who underwent surgery at the age of 10 months, and mental retardation was found in our second patient who did not present for follow-up visits until the age of 16 years.
In conclusion, genetic counseling should be provided according to the underlying mutation considering the phenotype-genotype relation in AS. In addition, early diagnosis and treatment of craniosynostosis at an appropriate time have paramount importance in terms of preventing mental retardation.
Footnotes
Informed Consent: Written informed consent was obtained from the patient’s parents.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - E.I., F.O. Design - E.I., T.A.; Supervision - H.O., F.O.; Materials - E.I., T.A.; Data Collection and/or Processing - E.I., T.A.; Analysis and/or Interpretation - E.I., H.O.; Literature Review - E.I., T.A.; Writing - E.I., T.A.; Critical Review - H.O., F.O.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support.
References
- 1.Cohen MM, Jr, Kreiborg S, Lammer EJ, et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. 1992;42:655–9. doi: 10.1002/ajmg.1320420505. https://doi.org/10.1002/ajmg.1320420505. [DOI] [PubMed] [Google Scholar]
- 2.Park WJ, Theda C, Maestri NE, et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995;57:321–8. [PMC free article] [PubMed] [Google Scholar]
- 3.Nur BG, Pehlivanoğlu S, Mıhçı E, et al. Clinicogenetic study of Turkish patients with syndromic craniosynostosis and literature review. Pediatr Neurol. 2014;50:482–90. doi: 10.1016/j.pediatrneurol.2014.01.023. https://doi.org/10.1016/j.pediatrneurol.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 4.Liu C, Cui Y, Luan J, Zhou X, Han J. The molecular and cellular basis of Apert syndrome. Intractable Rare Dis Res. 2013;2:115–22. doi: 10.5582/irdr.2013.v2.4.115. https://doi.org/10.5582/irdr.2013.v2.4.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carinci F, Pezzetti F, Locci P, et al. Apert and Crouzon syndromes: clinical findings, genes and extracellular matrix. J Craniofac Surg. 2005;16:361–8. doi: 10.1097/01.scs.0000157078.53871.11. https://doi.org/10.1097/01.SCS.0000157078.53871.11. [DOI] [PubMed] [Google Scholar]
- 6.Oldridge M, Lunt PW, Zackai EH, et al. Genotype-phenotype correlation for nucleotide substitutions in the IgII-IgIII linker of FGFR2. Hum Mol Genet. 1997;6:137–43. doi: 10.1093/hmg/6.1.137. https://doi.org/10.1093/hmg/6.1.137. [DOI] [PubMed] [Google Scholar]
- 7.Slaney SF, Oldridge M, Hurst JA, et al. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. 1996;58:923–32. [PMC free article] [PubMed] [Google Scholar]
- 8.Ibarra-Arce A, Ortiz de Zárate-Alarcón G, Flores-Pe-a LG, et al. Mutations in the FGFR2 gene in Mexican patients with Apert syndrome. Genet Mol Res. 2015;14:2341–6. doi: 10.4238/2015.March.27.19. https://doi.org/10.4238/2015.March.27.19. [DOI] [PubMed] [Google Scholar]
- 9.Spruijt B, Rijken BF, Joosten KF, et al. Atypical presentation of a newborn with Apert syndrome. Childs Nerv Syst. 2015;31:481–6. doi: 10.1007/s00381-014-2601-6. https://doi.org/10.1007/s00381-014-2601-6. [DOI] [PubMed] [Google Scholar]
- 10.Renier D, Arnaud E, Cinalli G, Sebag G, Zerah M, Marchac D. Prognosis for mental function in Apert’s syndrome. J Neurosurg. 1996;85:66–72. doi: 10.3171/jns.1996.85.1.0066. https://doi.org/10.3171/jns.1996.85.1.0066. [DOI] [PubMed] [Google Scholar]