

Abstract
The prefrontal cortex (PFC) underlies higher cognitive processes1 that are modulated by nicotinic acetylcholine receptor (nAChR) activation by cholinergic inputs2. PFC spontaneous default activity3 is altered in neuropsychiatric disorders4, including schizophrenia5—a disorder that can be accompanied by heavy smoking6. Recently, genome-wide association studies (GWAS) identified single-nucleotide polymorphisms (SNPs) in the human CHRNA5 gene, encoding the α5 nAChR subunit, that increase the risks for both smoking and schizophrenia7,8. Mice with altered nAChR gene function exhibit PFC-dependent behavioral deficits9–11, but it is unknown how the corresponding human polymorphisms alter the cellular and circuit mechanisms underlying behavior. Here we show that mice expressing a human α5SNP exhibit neurocognitive behavioral deficits in social interaction and sensorimotor gating tasks. Two-photon calcium imaging in awake mouse models showed that nicotine can differentially influence PFC pyramidal cell activity by nAChR modulation of layer II/III hierarchical inhibitory circuits. In α5-SNP-expressing and α5-knockout mice, lower activity of vasoactive intestinal polypeptide (VIP) interneurons resulted in an increased somatostatin (SOM) interneuron inhibitory drive over layer II/III pyramidal neurons. The decreased activity observed in α5-SNP-expressing mice resembles the hypofrontality observed in patients with psychiatric disorders, including schizophrenia and addiction5,12. Chronic nicotine administration reversed this hypofrontality, suggesting that administration of nicotine may represent a therapeutic strategy for the treatment of schizophrenia, and a physiological basis for the tendency of patients with schizophrenia to self-medicate by smoking13.
Impairment of cognitive processes that are critically reliant on the PFC is a signature of schizophrenia5 and is associated with low activity of the default mode network3. However, the cellular and circuit deficits underlying ‘hypofrontality’ remain unexplored. Because acetylcholine (ACh) release results in strong modulation of cortical activity via nAChRs14–16, it is not surprising that human nAChR gene variants are strongly linked to mental disorders with cognitive deficits17. Genetic variants within the CHRNA3–CHRNA5–CHRNB4 nAChR gene cluster contribute to the risk of habitual smoking7. Among the series of polymorphisms composing this human haplotype, the rs16969968 SNP leads to the substitution of aspartic acid at residue 398 by asparagine (D398N) in the human α5 subunit18. Both in vitro and ex vivo studies indicate that this SNP results in partial loss of function of α5-containing nAChRs18,19. In humans, this SNP is associated with reduced resting-state functional connectivity, and circuits involving the PFC are altered12.
Recently, the same genetic locus was associated with schizophrenia in a major GWAS8, but it is not yet clear whether this SNP is responsible for the hypofrontality observed in individuals with schizophrenia, which would directly implicate altered nicotinic signaling in this pathophysiology of schizophrenia. One obstacle preventing further mechanistic dissection of schizophrenia is the absence of animal models. Because genetic deletion of the α5 nicotinic subunit in mice leads to PFC-linked behavioral deficits, altered cholinergic excitation and aberrant neuronal morphology in the PFC20, we developed a mouse line expressing the human α5 variant to investigate the impact of this variant on behaviors associated with schizophrenia and to determine the neuronal and circuitry mechanisms underlying cognitive deficits.
Research on social cognition suggests that social interaction deficits, similar to emotional dysfunctions, represent characteristic markers of psychiatric disorders21. Because impaired social ability is a common feature of schizophrenia21, we used the three-chamber social test22 to determine whether α5-SNP-expressing (α5SNP) mice exhibit impairments in social interaction. The results indicated that α5SNP mice have deficits in sociability. Wild-type (WT) mice exhibited a clear preference for a novel social partner, spending 69.6 ± 2.9% of their time engaged in social interaction, while α5SNP littermates showed no preference between the social partner and an inanimate object (44.1 ± 5.3%) (Fig. 1a). The differences in social interaction cannot be attributed to the effects of differing genotypes on activity or general exploratory behavior because the WT and α5SNP mice did not differ in locomotor activity or combined time in the social and inanimate object interaction areas (Fig. 1a and Supplementary Discussion).
Figure 1.
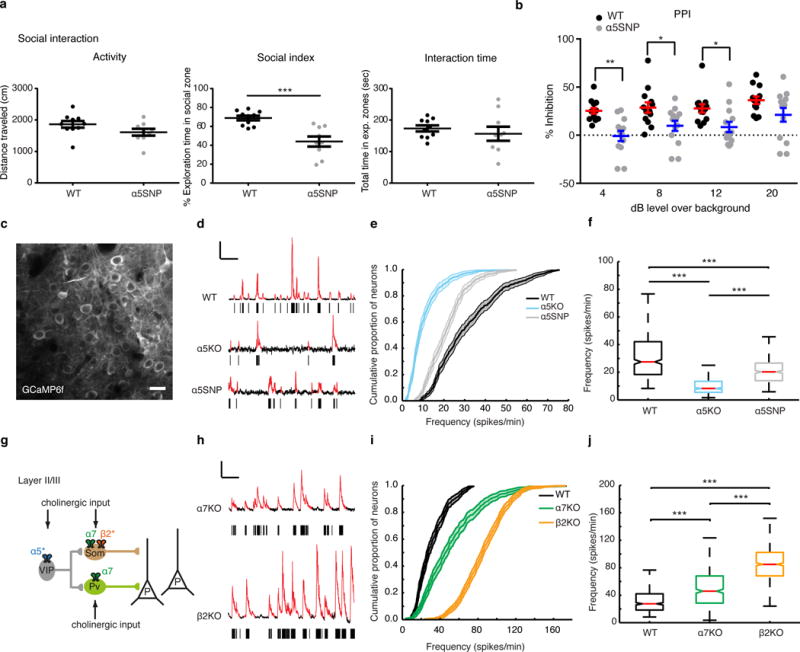
α5SNP mice exhibit reduced pyramidal cell activity, whereas α7KO and β2KO mice show increased activity, in layers II/III of the prelimbic cortex. (a) Quantifications of social and locomotor behavior in WT (n = 10) and α5SNP (n = 9) mice. Locomotor activity (left), social index (center) and total interaction time (right) are shown. ***P < 0.005, unpaired Student’s t test with Welch’s correction for unequal variance (t = 4.239, d.f. = 11.32); the mean ± s.e.m. for each group is shown. (b) Quantification of PPI behavior in WT and α5SNP mice (n = 13 mice per group) in response to prepulses of 4, 8, 12 and 20 dB. Two-way repeated-measures ANOVA showed the main effects of genotype (F1,24 = 10.83, P = 0.0031) and decibels (F3,72 = 8.96, P = 4.04. 10−5) but no significant interaction between the two (F3,72 = 1.01, P = 0.39). *P < 0.05, **P < 0.005, Sidak post hoc test used for genotype comparisons for a particular prepulse. The mean ± s.e.m. for each group is shown. (c) A representative in vivo two-photon image of GCaMP6f-expressing neurons in layers II/III of mouse PrLC. Scale bar in d,h, 20 μm. (d) Representative spontaneous Ca2+ transients recorded in WT, α5KO and α5SNP mice. The raster plots presented below each trace (black ticks) represent the estimated spikes for each of the automatically detected Ca2+ transients (highlighted in red in the trace above). Scale bars, 50% ΔF/F, 30 s. ΔF/F = (Fi – F0)/F0, where Fi is the fluorescence value for the ith frame of the measurement and F0 is the mean of the baseline fluorescence. (e) Population-averaged cumulative distribution functions (CDFs) of spontaneous firing rates (spikes/min) in WT (n = 11 mice, 2,205 cells), α5KO (n = 7 mice, 1,223 cells) and α5SNP (n = 11 mice, 1,417 cells) mice. The shaded regions in the CDFs reflect confidence intervals. (f) The median firing rates of WT, α5KO and α5SNP mice. For each box, the central mark shows the median, the edges of the box represent the 25th and 75th percentiles, and the whiskers extend to the most extreme data points that are not considered outliers. The ends of the whiskers represent the lowest data value still within 1.5 times the IQR (interquartile range) of the lower quartile and the highest data value still within 1.5 times the IQR of the upper quartile. ***P < 0.001, Kruskal–Wallis test. (g) Diagram of an inhibitory circuit. SOM interneurons express β2 and α7 nAChRs, whereas PV interneurons contain α7 nAChRs. The nicotinic excitation of VIP interneurons is mediated by α5-containing nicotinic receptors. Adapted from ref. 25 with permission of Nature Publishing Group. (h) Representative recordings of spontaneous Ca2+ transients in α7KO and β2KO mice and corresponding raster plots. (i) Population-averaged cumulative histograms of the distributions of spontaneous firing rates (spikes/min) in WT (n = 11 mice, 2,205 cells), α7KO (n = 4 mice, 1,171 cells) and β2KO (n = 7 mice, 3,199 cells) mice. (j) Median firing rates for WT, α7KO and β2KO mouse lines. ***P < 0.001, Kruskal-Wallis test.
Deficiencies in the prepulse inhibition (PPI) task are a measure of impaired sensorimotor gating and are observed in several human neuropsychiatric disorders23. The PPI task measures the ability of an animal to inhibit its startle in response to an acoustic tone (pulse) when the tone is preceded by a lower-intensity stimulus (prepulse). Examination of PPI has been particularly important in studying the neurobiology of schizophrenia because deficits in cognitive information gating are a clinically important feature of the disorder23. There is a high degree of similarity between measures of PPI in rodent models and humans, which is in accordance with the use of PPI as a cross-species measure of sensorimotor gating23. We found that α5SNP mice exhibited decreased PPI across a range of prepulse intensities (4, 8, 12 and 20 dB above background noise) relative to their WT littermates (Fig. 1b and Supplementary Discussion), suggesting that sensorimotor gating is disrupted by the α5 variant.
To determine the circuit mechanisms underlying the cognitive deficits attributed to dysfunction of α5-containing nAChRs and the human SNP, we assessed resting-state network activity using in vivo two-photon calcium imaging in the prelimbic cortex (PrLC) of the PFC in awake mice either lacking the α5 subunit (α5KO mice) or with knock-in of the α5 SNP (α5SNP mice) in comparison to WT mice. Neurons were transduced with an adeno-associated viral vector (AAV) expressing the genetically encoded calcium indicator GCaMP6f in order to monitor Ca2+ transients as a proxy for action potential firing (Fig. 1c and Supplementary Fig. 1). Because we estimate that 80% of the labeled neurons in layers II/III are pyramidal neurons (Supplementary Fig. 2), we consider that such experiments primarily indicate the activity of the pyramidal neuron population. Interestingly, we found that the median frequency and duration of Ca2+ transients were lower in both strains of transgenic mice than in WT mice (Supplementary Fig. 3). Spontaneous Ca2+ transients were deconvolved with a putative unitary (action potential–evoked) fluorescence response in order to estimate neuronal firing rates (Supplementary Figs. 4–6 and Supplementary Table 1). We found significantly lower layer II/III neuronal activity in the PrLC of α5KO (8.2 ± 0.5 spikes/min) and α5SNP (20.3 ± 0.6 spikes/min) mice when compared to WT mice (27.5 ± 0.9 spikes/min; Fig. 1d–f). Thus, loss of function of α5-containing nAChRs causes a decrease in layer II/III microcircuit activity, which parallels the decrease in hypofrontality that is a defining feature of schizophrenia in humans5.
The α5 subunit is present only in VIP interneurons in layers II/III24, which exert inhibitory control of SOM and parvalbumin (PV) interneurons in the PFC25. Since layer II/III pyramidal neurons do not express nAChRs, we hypothesized that loss of function of the α5 subunit would result in disinhibition of PV and SOM interneurons and, thus, reduced pyramidal cell activity (Fig. 1g). Because dendritic-targeting SOM interneurons express different nicotinic receptor subtypes, namely α7- and β2-containing nicotinic receptors15,26, from perisomatic-targeting PV cells15,27, which exclusively express α7-containing nAChRs, we compared PFC activity across transgenic mice with specific deletions of nAChR subunits differentially expressed in PV and SOM interneurons. We observed a strong increase in the median spike frequency of GCaMP6f-expressing pyramidal neurons in both KO lines, with the β2KO line exhibiting the highest frequencies (85 ± 1.1 spikes/min) in comparison to the α7KO line (45.9 ± 1.8 spikes/min) and WT mice (27.5 ± 0.9 spikes/min; Fig. 1h–j). Thus, the absence of either α7 or β2 subunits in layers II/III increases pyramidal neuron activity; this is most likely because of a decrease in the cholinergic drive of interneurons directly inhibiting pyramidal neurons.
Because α5 subunits are expressed in cortical layers that were outside of our imaging field in layers II/III15, we selectively re-expressed the WT α5 subunit in the PFC layer II/III interneurons of α5KO-GADCre mice by using a conditional lentiviral vector (Fig. 2a,b). We observed full restoration of pyramidal neuron activity to control levels after re-expression of the WT α5 subunit, whereas, when the α5 subunit encoding the human SNP was re-expressed in α5KO-GADCre mice, pyramidal cell activity was only partially restored, to levels between those of control mice and α5KO-GADCre mice (Fig. 2c). Since PV interneurons do not express the requisite β2 subunit to form a functional α5-containing nAChR, these results may be due to the expression of the α5 subunit in either VIP or SOM interneurons. Because the activity in pyramidal neurons is restored, re-expression of α5 in VIP interneurons likely represents the dominant effect on the circuit behavior. These data indicate that the hypofrontality in α5KO and α5SNP mice can be attributed exclusively to an effect on layer II/III GABAergic interneurons.
Figure 2.
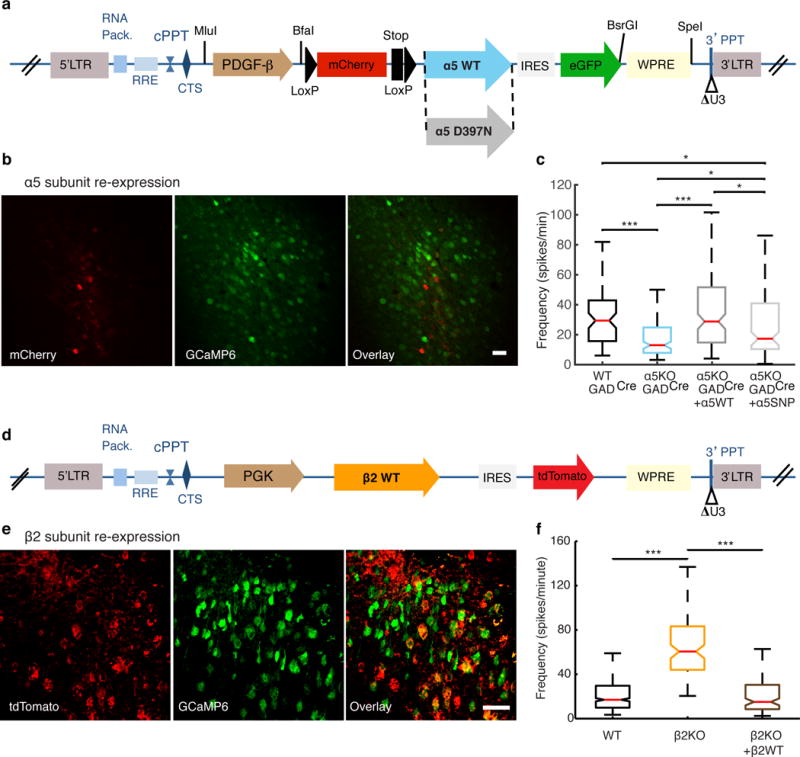
Targeted nicotinic acetylcholine receptor re-expression in layers II/III of the prelimbic cortex. (a) Schematic of conditional (‘floxed’) lentiviral vectors inducing the re-expression of WT (LV-α5WT) or SNP-containing (LV-α5SNP) α5 in interneurons in layers II/III of the PrLC. (b) Confocal images of coronal sections with mCherry (red) and GCaMP6f (green) expression in layers II/III of the PFC, and the merged image (right), after the LV-α5WT vector was specifically injected into layers II/III of the PFC of α5KO-GADCre mice. Scale bar, 50 μm. (c) Median frequency of spontaneous spikes from the pyramidal neurons of WT-GADCre (29.4 ± 2.7 spikes/min; n = 4 mice, 188 cells), α5KO-GADCre (14.3 ± 1.9 spikes/min; n = 3 mice, 258 cells), α5KO-GADCre + α5WT (28.8 ± 2.7 spikes/min; n = 3 mice, 243 cells) and α5KO-GADCre + α5SNP (19.3 ± 5.6 spikes/min; n = 3 mice, 134 cells) mice. *P < 0.05, ***P < 0.001, Kruskal–Wallis test. (d) Schematic of the lentiviral vector used for targeted rescue of functional β2 nAChRs specifically in layers II/III of the PrLC. (e) Confocal images of coronal sections with tdTomato (red) and GCaMP6f (green) expression in layers II/III of the PFC; the merged image is shown to the right. Scale bar, 50 μm. (f) The median frequency of spontaneous spikes found in WT (control injection of LV-PGK-mCherry-WPRE; 16.95 ± 0.92 spikes/min; n = 4 mice, 1,408 cells), β2KO (LV-PGK-mCherry-WPRE; 60.45 ± 3.55 spikes/min; n = 4 mice, 260 cells) and β2KO + β2WT (15.01 ± 2.23 spikes/min; n = 4 mice, 366 cells) mice. No significant difference was detected between WT and β2KO + β2WT mice (P = 0.87). ***P < 0.001, Kruskal-Wallis test.
In addition, because β2 subunits are also expressed in other cortical layers15, we monitored pyramidal cell activity in β2KO mice after re-expression of the β2 subunit. WT β2 subunit was re-expressed along with tdTomato as a reporter by targeted injection of a β2-IRES2-tdTomato bicistronic lentiviral vector into layers II/III of the PrLC of β2KO mice (Fig. 2d and Supplementary Fig. 7). In the PrLC, only the interneurons in layers II/III express nAChR α subunits, and therefore the lentivirus-mediated expression resulted in functional nAChRs containing β2 subunits exclusively in interneurons28. We found that the neuronal activity of β2KO mice was elevated when compared with that in WT mice, and the re-expression of WT β2 led to a normalization of activity (Fig. 2e,f and Supplementary Fig. 8). These results are consistent with layer II/III re-expression of β2 in SOM and VIP interneurons. When considered together, these manipulations demonstrate the critical role of layer II/III interneurons in controlling pyramidal neuron activity within the same layer.
Our results thus far suggest that the decrease in VIP activity in α5KO and α5SNP mice could result from a dishinhibition of pyramidal cells. To directly examine the influence of nicotinic signaling in each interneuron type, we examined the spiking activity of VIP, PV and SOM interneurons under conditions of α5 knockdown. We injected VIP-Cre, PV-Cre, or SOM-Cre with a Cre-dependent AAV vector to direct expression of GCaMP6f to each of these layer II/III interneuron populations. To specifically reduce expression of the α5 subunit in vivo, we used CRISPR-associated Cas9 endonuclease technology29. We injected the mice described above with a lentiviral vector expressing Cas9 and guide RNA (gRNA) targeting the Chrna5 gene (LV-CRISPR-α5) in layers II/III of the PFC (Fig. 3a,b). Choice of the gRNA sequence and analysis of potential off-target activity by next-generation sequencing (NGS) are described in the Supplementary Methods (Off target analysis using next generation sequencing). The rate of insertions/deletions was quantified to be 15.5% (Supplementary Fig. 9 and Supplementary Tables 2 and 3). Using the lentiviral vector in vivo, we found a strong reduction in the median spike frequency of VIP interneurons in VIP-Cre mice injected with LV-CRISPR-α5 lentivirus (3.49 ± 0.66 spikes/min) when compared to control mice injected with a control non-targeting CRISPR lentiviral vector (21.5 ± 2.17 spikes/min; Fig. 3c–f and Supplementary Fig. 10). We then examined how reduction of the α5 subunit in VIP interneurons could affect the spontaneous activity of PV and SOM interneurons, using PV-Cre or SOM-Cre mice together with AAV vectors for Cre-dependent expression of GCaMP6f. We did not observe any significant difference in PV-Cre mice injected with the LV-CRISPR-α5 lentivirus (6.07 ± 0.60 spikes/min) when compared to control mice (5.62 ± 0.45 spikes/min; Fig. 3g–j), which is consistent with weak innervation of PV interneurons by VIP interneurons25,30. In contrast, targeted injection of α5-targeting lentivirus into layers II/III resulted in an increase in SOM interneuron spontaneous activity (21.63 ± 0.67 spikes/min) when compared to control mice (5.57 ± 1.31 spikes/min; Fig. 3k–n), whereas pyramidal cell activity was reduced (Fig. 3o–r). Our data show that there is a central role for the α5 subunit in controlling VIP-mediated disinhibition via SOM interneuron inhibition of pyramidal neurons in layers II/III (Fig. 3s).
Figure 3.
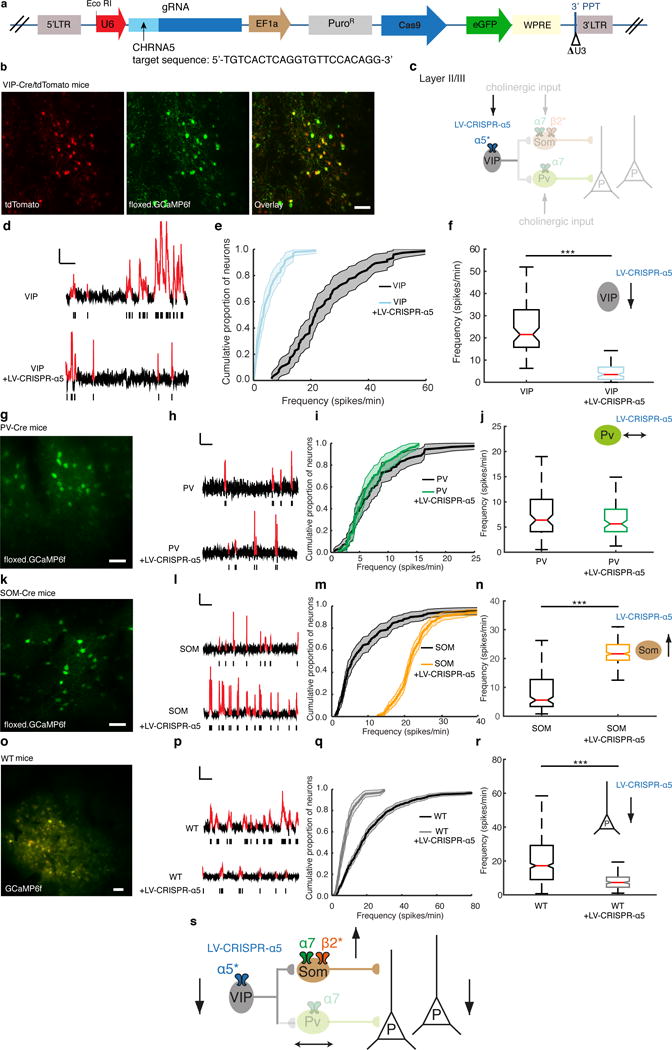
Nicotinic acetylcholine receptors modulate prefrontal cortex activity through inhibitory neurons. (a) Schematic of the α5-Cas9 lentiviral vector for delivery of Cas9 and gRNA targeting the Chrna5 gene (LV-CRISPR-α5) in layers II/III of the PFC. The arrow indicates the target sequence of the Chrna5 gene. (b) Representative in vivo two-photon images of GCaMP6f-expressing VIP interneurons in layers II/III of the PFC of VIP-Cre/tdTomato mice, with tdTomato shown in red and GCaMP6f shown in green. Scale bar, 50 μm. (c) Diagram of the inhibitory circuit showing that LV-CRISPR-α5 targets only the α5 nAChRs expressed by VIP interneurons in layers II/III of the PFC. Adapted from ref. 25 with permission of Nature Publishing Group. (d) Representative spontaneous Ca2+ transients (highlighted in red in the trace) of VIP interneurons (VIP-Cre/tdTomato control mice injected with a CRISPR nontargeting control lentiviral vector) and VIP + LV-CRISPR-α5 interneurons (VIP-Cre/tdTomato mice injected with the LV-CRISPR-α5 lentiviral vector) and corresponding raster plots (black ticks represent estimated spikes for each automatically detected Ca2+ transient). Scale bars, 50% ΔF/F, 30 s. (e) Population-averaged CDFs of spontaneous firing rates (spikes/min) in VIP (n = 4 mice, 177 cells) and VIP + LV-CRISPR- α5 (n = 4 mice, 268 cells) mice. The shaded regions in the CDFs reflect confidence intervals. (f) Median firing rates for VIP and VIP + LV-CRISPR-α5 mice. ***P < 0.001, Kruskal– Wallis test n = 4 mice for each group. (g) Representative in vivo two-photon image of GCaMP6f-expressing PV interneurons in layers II/III of the PFC of PV-Cre mice. Scale bar, 50 μm. (h) Representative spontaneous Ca2+ transients of PV interneurons and PV + LV-CRISPR-α5 interneurons and their corresponding raster plots. Scale bars, 50% ΔF/F, 30 s. (i) Population-averaged CDFs of spontaneous firing rates (spikes/min) in PV (n = 4 mice, 328 cells) and PV + LV-CRISPR-α5 (n = 4 mice, 280 cells) mice. (j) Median firing rates for PV and PV + LV-CRISPR-α5 mice. No significant difference was found between the groups (P = 0.55, Kruskal–Wallis test) n = 4 mice for each group. (k) Representative in vivo two-photon image of GCaMP6f-expressing SOM interneurons in layers II/III of the PFC of SOM-Cre mice. Scale bar, 50 μm. (l) Representative spontaneous Ca2+ transients of SOM interneurons and SOM + LV-CRISPR-α5 interneurons and their corresponding raster plots. Scale bars, 50% ΔF/F, 30 s. (m) Population-averaged CDFs of spontaneous firing rates (spikes/min) in SOM (n = 4 mice, 406 cells) and SOM + LV-CRISPR-α5 (n = 4 mice, 466 cells) mice. (n) Median firing rates for SOM and SOM + LV-CRISPR-α5 mice. ***P < 0.001, Kruskal– Wallis test n = 4 mice for each group. (o) Representative in vivo two-photon image of GCaMP6f-expressing pyramidal neurons in layers II/III of the PFC of WT mice. 100 nl of AAV1.CAG.tdTomato was co-injected to localize cells transduced by the CRISPR lentivirus (Online Methods). Scale bar, 50 μm. (p) Representative spontaneous Ca2+ transients of WT and WT + LV-CRISPR-α5 pyramidal neurons and corresponding raster plots. Scale bars, 50% ΔF/F, 20 s. (q) Population-averaged CDFs of spontaneous firing rates (spikes/min) in WT and WT + LV-CRISPR-α5 mice. (r) Median firing rates for WT (17.17 ± 11.18 spikes/min; n = 4 mice, 1,026 cells) and WT + LV-CRISPR-α5 (7.26 ± 0.62 spikes/min; n = 4 mice, 487 cells) mice. ***P < 0.001, Kruskal–Wallis test. (s) Diagram showing decrease in pyramidal neuronal activity due to decreased VIP interneuron inhibition of SOM interneurons in layers II/III. Adapted from ref. 25 with permission of Nature Publishing Group. All control mice were injected with a CRISPR nontargeting control lentiviral vector (Online Methods).
On the basis of evidence that the nicotine levels obtained from smoking can improve cognitive abilities31 and can mitigate certain schizophrenic symptoms6,32, we administered nicotine to WT and α5SNP mice by mini-pump infusion and examined alterations in layer II/III neuronal firing before and after infusion (Fig. 4a). The dose of nicotine was selected to maintain a plasma concentration analogous to that observed in smokers33. Pyramidal neuron activity significantly increased after 2 d of nicotine administration in WT mice and remained elevated after 2 weeks (Fig. 4b–e and Supplementary Fig. 11). Also, we found that nicotine treatment 1–2 weeks in duration increased pyramidal neuron activity in α5SNP mice to the levels of WT baseline activity (Fig. 4b–e). An explanation for these data is that chronic nicotine exposure desensitizes the β2-containing nAChRs expressed by interneurons. β2-dependent, but not α7-dependent, nAChR currents in the interneurons of PFC layers II/III were completely desensitized after exposure to the concentrations of nicotine obtained from smoking in slice preparations15. To test this in vivo, we administered the same dose of nicotine in SOM-Cre mice injected with AAV encoding Cre-dependent GCaMP6f expression and observed a significant reduction in SOM interneuron activity when compared to that in control mice (Fig. 4f–h). These results suggest that the normalization of neuronal firing in α5-deficient mice by nicotine administration is mediated by dominant nicotine-evoked desensitization of the β2-containing receptors on SOM interneurons. As a result, SOM inhibition of pyramidal cells is reduced. Together these data support the conclusion that layer II/III PFC activity is controlled by different nAChR subunits expressed specifically on different classes of interneurons. These experimental results argue strongly that nicotine, or nicotinic agonists, could normalize PFC activity and thus reduce some of the cognitive deficits found in humans with neuropsychiatric disorders.
Figure 4.
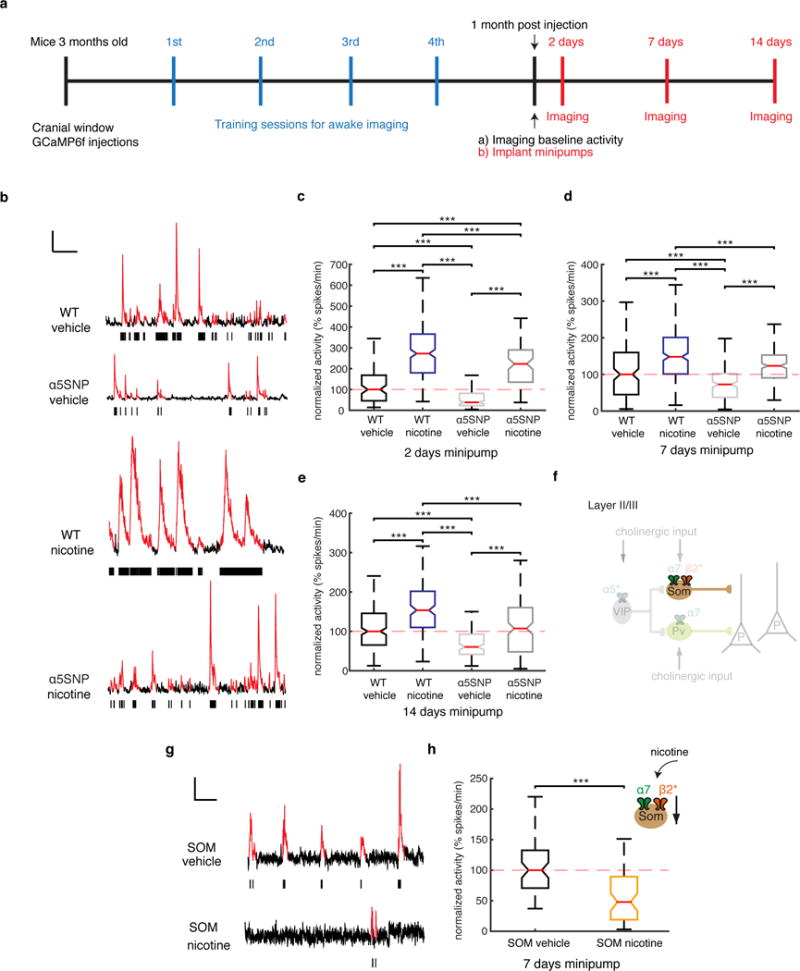
Effect of chronic nicotine on the prelimbic cortex activity of WT and α5SNP mice and desensitization of β2 nicotinic acetylcholine receptor subunits. (a) Timeline of the experiment. The arrows indicate the beginning of the imaging sessions. (b) Ca2+ transients (highlighted in red in the trace) of neurons in WT and α5SNP mice after vehicle (saline) and nicotine administration and corresponding raster plots (black ticks represent estimated spikes for each automatically detected Ca2+ transient). Scale bars, 50% ΔF/F, 30 s. (c) Comparison of the median activity (spikes/min; median ± s.e.m.) of WT and α5SNP mice with vehicle (n = 3 mice) or nicotine (n = 4 mice) infusion at 2 d after mini-pump implantation (WT vehicle, 203 cells; WT nicotine, 511 cells; α5SNP vehicle, 401 cells; α5SNP nicotine, 246 cells). Activities normalized with respect to values obtained in WT mice with vehicle (defined as 100%). (d) Comparison of the median activity of WT and α5SNP mice with either vehicle (n = 3 mice) or nicotine (n = 4 mice) infusion at 7 d after mini-pump implantation (WT vehicle, 300 cells; WT nicotine, 584 cells; α5SNP vehicle, 311 cells; α5SNP nicotine, 361 cells). Activities normalized as in c. (e) Comparison of the median activity of WT and α5SNP mice with vehicle (n = 3 mice) or nicotine (n = 4 mice) infusion at 14 d after mini-pump implantation (WT vehicle, 369 cells; WT nicotine, 440 cells; α5SNP vehicle, 289 cells; α5SNP nicotine, 182 cells). Activities were normalized as in c. ***P < 0.001, Kruskal–Wallis test. (f) Diagram of the inhibitory circuit showing that nicotine acts in SOM interneurons. Adapted from ref. 25 with permission of Nature Publishing Group. (g) Ca2+ transients (highlighted in red in the trace) of SOM interneurons after vehicle and nicotine administration and corresponding raster plots (black ticks represent estimated spikes for each automatically detected Ca2+ transient). Scale bars, 50% ΔF/F, 20 s. (h) Comparison of the median activity (spikes/min; median ± s.e.m.) of SOM interneurons after vehicle (n = 4 mice, 203 cells) or nicotine (n = 4 mice, 137 cells) infusion, 7 d after mini-pump implantation. Activities were normalized with respect to values obtained in SOM mice with vehicle (defined as 100%). In box-and-whisker plots in c–e,h, for each box, the central mark shows the median, the edges of the box represent the 25th and 75th percentiles, and the whiskers extend to the most extreme data points that are not considered outliers. The ends of the whiskers represent the lowest data value still within 1.5 times the IQR (interquartile range) of the lower quartile and the highest data value still within 1.5 times the IQR of the upper quartile. ***P < 0.001, Kruskal-Wallis test.
In summary, we show that endogenous ACh distinctly recruits specific interneuron types within an interneuron hierarchy that controls pyramidal neurons in layers II/III of the PrLC, through differential expression of nAChR subunits. We also provide the first experimental evidence, to our knowledge, that the α5 human polymorphism, which predisposes to nicotine addiction, provokes a decrease in neuronal activity in rodents that mirrors the hypofrontality observed in patients with psychiatric disorders, including schizophrenia and addiction5. Further, we demonstrate that chronic nicotine treatment of mice carrying the α5 SNP restores their neuronal activity deficits, and these findings together provide the basis for examining new pharmacotherapeutic strategies. Positive allosteric modulators of defined nAChR subunits, like the polymorphic α5 subunit, are strong candidates34. Finally, our results support the hypothesis that individuals with polymorphic CHRNA5 smoke to self-medicate in order to ameliorate at least some symptoms of cortical dysfunction.
METHODS
Subjects
Male α7−/− (α7KO)35, β2−/− (β2KO)36, α5−/− (α5KO)37 and WT C57BL/6J mice used in this study were maintained at Charles River Laboratories, and experiments were performed at 3 months of age. The experiments described in the present work were conducted in accordance with the guidelines on the ethical use of animals from the European Community Council Directive of 24 November 1986 (86/609/EEC) and in accordance with institutional animal welfare guidelines and were approved by Animalerie Centrale, Médecine du Travail and the Ethics Committee CETEA of Institut Pasteur, protocol numbers 2013-0056, 2013-0104 and 2015-0007.
Mice engineered to harbor the α5 D398N variant (α5SNP mice) were obtained via homologous recombination. They were generated by the Ingenious Targeting Laboratory using C57BL/6 embryonic stem cells and genomic constructs derived from a C57BL/6 BAC library. The targeting construct was mutated to convert an aspartic acid codon (GAT) to an asparagine codon (AAT) at amino acid position 397 of the mouse α5 subunit gene (Chrna5). The amino acid position 397 in mice is equivalent to position 398 in human CHRNA5. The neomycin selection cassette of the targeting construct was removed by breeding α5SNP mice to a C57BL/6J strain expressing a Zp3-cre transgene (C57BL/6-TgN(ZP3-Cre)93Knw, Jackson Laboratories). Removal of the neomycin cassette was verified by PCR.
GAD67-Cre mice were published previously38,39. To generate GAD67-Cre/tdTomato mice, GAD67-Cre mice were crossed with a ChAT-Cre/tdTomato line40. GAD67-Cre mice were bred onto a homozygous α5KO background to obtain α5KO-GADCre mice.
To generate VIP-Cre/tdTomato mice, homozygous VIP-IRES-Cre (VIPtm1(cre)Zjh, Jackson Laboratory) mice were crossed with homozygous Ai9 mice (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J, Jackson Laboratory) as previously described41. PV-Cre (008069, Jackson Laboratory) and SOM-Cre (013044, Jackson Laboratory) mice were bred onto a C57BL/6J background.
All experiments were performed using male mice.
Social interaction tests
Eight- to 12-week-old male WT and α5SNP littermates were tested for social behavior using a three-chamber social approach paradigm42. Experimental mice were habituated for 1 h in separate, clean holding cages and then were introduced into a three-chamber arena with only empty object-containment cages (circular metallic cages, Stoelting Neuroscience) for a 10-min acclimation phase in two 5-min sessions in a 3- to 4-h period (habituation periods I and II). The following day, the mice were placed in the center chamber (without access to the left and right social test areas) and were allowed to explore the center area for 5 min (open field period). After this exploration period, the barriers to the adjacent chambers were removed, allowing mice to explore the left and right arenas, which contained a social partner (i.e., unfamiliar C57BL/6J male mouse) in one chamber and an inanimate object (plastic toy) in the other chamber. Experimental mice were given 10 min to explore both chambers and were measured for approach behavior as interaction time (i.e., sniffing, approach) with targets in each chamber (within 2 cm, excluding non-nose contact or exploration). Sessions were video recorded, and object exploration time and total distance moved were analyzed using the Noldus tracking system. A social preference index was calculated as the percentage of time spent investigating the social target (unfamiliar C57BL/6J male mouse) out of the total exploration time for both objects. The analysis was conducted with investigators blinded to the genotypes of subjects. Arenas and their contents were thoroughly cleaned between testing sessions. Multiple mice from different home cages were used as social targets to prevent potential odorant confounds from target home cages.
Prepulse inhibition of the startle reflex
Male WT and α5SNP littermates ranging in age from 60–75 d were used for the study. The PPI apparatus (SR-Lab, San Diego Instruments) consists of a sound-attenuating chamber and a vented Plexiglas animal enclosure (4 cm in diameter) that rests on a 20 × 13 cm Plexiglas frame. Background (70 dB), prepulse (74, 78, 82 and 90 dB) and pulse (120 dB) bursts were generated through a computer using SR-Lab software (San Diego Instruments) and presented through a speaker mounted (13 cm) above the animal enclosure. The magnitude of the startle response was measured by a piezoelectric accelerometer mounted below the Plexiglas animal enclosure.
Each session was initiated with a 5-min acclimation period followed by initiation of the test session with five pulse-only trials (white noise, 120 dB/40 ms); each session also concluded with five identical pulse-only trials. Both the pre- and post-pulse trials were excluded from analysis. The remaining trials consisted of pseudorandomized presentation within ten trial blocks of a blank (no-stimulus) trial, a pulse-only trial and four different types of prepulse + pulse trials (20 ms at 74, 78, 82 or 90 dB followed by a pulse (120 dB) 100 ms after the prepulse). PPI was calculated using the means of ten trials for each type of stimulus: percent PPI = (pulse-only score − (prepulse + pulse score))/pulse-only score × 100.
Stereotaxic injections and chronic cranial windows
Mice were anesthetized with ketamine (10% in PBS; Imalgen 1000, Rhone Mérieux) and xylazine (2% in PBS; Rompun, Bayer), 10 ml/kg intraperitoneally (i.p.), and placed into a stereotaxic frame. Mice were kept on a thermal blanket, and their eyes were protected with artificial tear ointment. The scalp was washed with three alternating swabs of 70% ethanol and betadine. Surgical tools were sterilized using a hot glass-bead sterilizer. The skull was exposed, and 1% xylocaine was applied to the periosteum. A chronic cranial window was prepared as previously described43 (Supplementary Fig. 1). Briefly, the skull was gently thinned using a dental drill around the region of interest, and the thinned bone was removed using forceps, leaving the dura intact. For recordings of pyramidal neurons, 200 nl of AAV virus (2.2 × 1013 genome copies (GC)/ml; AAV1.syn.GCaMP6f.WPRE.SV40, University of Pennsylvania Vector Core) was injected bilaterally at the following coordinates—PrL: AP, +2.8 mm from the bregma; L, ±0.5 mm; DV, −0.3 to −0.1 mm from the skull—using a Nanoject II (Drummond Scientific) at the slow infusion setting. For recordings of interneurons in the Cre lines, 200 nl of AAV virus (2.1 × 1013 GC/ml; AAV1.Syn.Flex.GCaMP6f.WPRE.SV40, University of Pennsylvania Vector Core) was injected bilaterally at the same coordinates as described above. The glass pipette was left in situ for an additional 5 min, before being slowly withdrawn from the brain. The optical window was covered with a circular coverglass (5 mm in diameter) and was sealed to the skull with dental cement (Coffret SUPERBOND complet, Phymep). A sterile small stainless steel bar with a screw hole was embedded into the acrylic over the cerebellum to head-fix the mouse for imaging sessions, and all exposed skull was covered with dental cement.
Lentiviral expression vectors and stereotaxic procedure
The lentiviral expression vectors were derived from the pHR’ expression vectors first described by Naldini et al.44 with several subsequent modifications. In the lentivirus used in this study, expression of mCherry cDNA is under the control of the mouse phosphoglycerate kinase (PGK) promoter. Further details can be found in ref. 45. Briefly, to create the pLV-PGK-mCherry-WPRE vector, a two-step strategy was used. First, mCherry cDNA was amplified from the pRSET plasmid (a kind gift of the laboratory of S. Tajbakhsh, Stem Cells and Development lab, Institut Pasteur, Paris, France), using the following primers—forward, 5′-GTACTCGAGCCACCATGGTGAGCAAGGGC-3′; reverse, 5′-GCTGACGCGGCCGCTTACTTGTACAGCTCGTCCATG-3′—and the T/A cloning strategy was used to introduce mCherry cDNA into the pGEM-T Easy vector (Promega). Next, we took advantage of the XhoI site in the multiple-cloning region of the vector to replace the fragment encoding eGFP in the preexisting pTRIP-PGK-eGFP lentiviral vector45 with mCherry cDNA between the XhoI and BsrGI sites. This viral vector was used as a control injection for the β2 rescue experiments.
To create the pLV-dsmPGK-β2-IRES-tdTomato-WPRE vector, we used a previously described PTP-β2-IRES-GFP-WPRE vector45 and replaced the full IRES-GFP sequence with IRES-tdTomato available from Clotech (pLVX-IRES-tdTomato) by using the Gateway system. Lentivectors were diluted in PBS before stereotaxic injection to achieve a dose corresponding to 250 ng of p24 protein in 2 μl.
Production of the conditional lentivectors was performed as previously described34. Briefly, viral particles were generated by cotransfection of HEK-293T cells with the vector plasmid, a packaging plasmid and an envelope plasmid using the calcium phosphate protocol. At 2 d after transfection, viral particles were collected in the supernatant, treated with DNase I and MgCl2, passed through a 0.45-μm filter, concentrated by ultracentrifugation and resuspended in a small volume of PBS. Viral stocks were stored in small aliquots at −80 °C before use. Viral titers were estimated by quantification of the p24 capsid protein using the HIV-1 p24 antigen immunoassay (ZeptoMetrix) according to the manufacturer’s instructions. Lentivectors were diluted in PBS before stereotaxic injection to achieve a dose corresponding to 150 ng of p24 protein in 2 μl for re-expression of the α5 subunit. As controls, we used vectors carrying only eGFP under the same promoter and we injected the same volumes. The lentiviral vectors were injected bilaterally into the PrLC at the following coordinates—PrL: AP, +2.8 mm from the bregma; L, ±0.5 mm; DV, −0.25 to −0.1 mm from the skull—using a Nanoject II (Drummond Scientific) at the slow infusion setting.
CRISPR/Cas9 nuclease RNA-guided genome editing
We used CRISPR-associated Cas9 endonuclease technology for inactivation of the α5 subunit (Sigma-Aldrich). Three different gRNAs were designed by Sigma-Aldrich using an algorithm based on Bowtie sequence analysis software. gRNAs targeting a site in the first two-thirds of the coding sequence with at least 3 bp of mismatch with any other location in the host genome were proposed for testing by genomic cleavage detection assay (CEL-1 endonuclease). Assessment of the CRISPR/Cas9 on-target and off-target effects is described in the Supplementary Methods (Off target analysis using next generation sequencing) and Supplementary Tables 2 and 3. After screening of three target sites in the Chrna5 gene, the following target site was chosen: TGTCACTCAGGTGTTCCACAGG. The lentiviral vector uses a one-plasmid system consisting of an EF1-α-driven Cas9 gene and a U6-driven gRNA, with both puromycin and GFP coexpressed with Cas9. 2.5 μg of U6-gRNA was nucleofected with 5 μg of Cas9 plasmid. In vitro validation was performed in mouse N2a cells (Sigma-Aldrich), where the targeting efficiency was established to be 13.2% by enzymatic assay. N2a cells tested negative for mycoplasma. Insertions and deletions were detected at the gRNA target via mismatch assay (CEL-1 endonuclease) using the following primers: forward, 5′-GATTATTGATAACATTGAGTTTC-3′; reverse, 5′-CTCTAGTGGCTAACTCACAC-3′. As a control, we used a lentiviral plasmid vector that includes a gRNA sequence that does not target known human, mouse or rat genes (CRISPR12-1EA, Sigma-Aldrich). Viral particles were generated by cotransfection of HEK-293T cells with the vector plasmid, as described above. For the α5-Cas9 lentiviral vector, 49 ng of p24 protein in 1 μl was locally injected in layers II/III of the PFC, whereas the control virus was diluted in PBS before stereotaxic injection to achieve a dose corresponding to 50 ng of p24 protein in 1 μl. In addition, 100 nl of AAV1.CAG.tdTomato.WPRE.SV40 (1.52 × 1013 GC/ml; University of Pennsylvania Vector Core) was added to the solution to allow localization of the cells transduced with LV-CRISPR virus.
Mouse handling for awake imaging
Mice were habituated to the imaging environment by handling and training for 4 d, as described previously46,47. Briefly, the animals were habituated to the following conditions sequentially: (1) gentle handling, (2) free exploration of a 50-ml open-ended support tube (for the mouse to rest in during imaging), (3) incremental periods of mild head fixation in which the mouse’s head was briefly secured to the head post (5 s to 15 s) and (4) extended periods of head fixation on the mouse stage while the mouse rested in the support tube (5 min to 60 min).
Immunofluorescence
At the end of the final two-photon imaging session, immunofluorescence was performed in order to verify the injection site. Fixation was carried out by transcardiac perfusion of anesthetized mice using 4% paraformaldehyde (PFA), and the brains were removed and post-fixed by immersion in PFA for 2 d at 4 °C. The brains were then immersed in 30% sucrose in PBS overnight for cryoprotection at 4 °C. Serial 40-μm coronal sections were cut with a sliding microtome (Leica Microsystems), transferred to PBS, mounted on slides and coverslipped with ProLong Gold antifade reagent with DAPI (Invitrogen). Microscopy was carried out with a Zeiss epifluorescent microscope.
To determine expression patterns in β2KO-GADCre mice injected with β2-IRES2-tdTomato and in α5KO-GADCre mice injected with PDGF-floxed-mCherry-α5 WT/SNP-IRES2-eGFPP-WPRE, we anesthetized and perfused the mice as described above. Serial 40-μm coronal sections were cut with a sliding microtome, collected and washed in PBS treated with 0.2% Tween-20 (PBS-T), immersed in 10% normal horse serum blocking solution, and incubated overnight with primary antibodies against GFP (1:2,000; Abcam, ab38689) and RFP (1:2,000; Molecular Probes, R10367) followed by incubation with secondary antibodies conjugated to Alexa Fluor 488 (green; 1:500; Jackson ImmunoResearch Laboratories, 715-545-150) and Alexa Fluor 568 (red; 1:500; Abcam, ab175470). The sections were mounted on slides and coverslipped with ProLong Gold antifade reagent with DAPI. Microscopy was carried out with a Zeiss LSM-700 confocal microscope (Supplementary Fig. 7). For all antibodies, validation is provided at the manufacturer’s website.
Surgical implantation of mini-pumps
Mice were anesthetized with a xylazine and ketamine combination (15% xylazine/2.5% ketamine/82.5% PBS). Osmotic mini-pumps (model 2004, ALZET DURECT) were implanted subcutaneously at the nape of the neck as previously described48. Nicotine or saline was delivered at a constant rate (2.4 mg per kg body weight per day for nicotine).
In vivo two-photon imaging
In vivo imaging was performed with an Ultima IV two-photon laser-scanning microscope system (Bruker), using a 16x, 0.8 N.A. water immersion objective (Nikon) with the femtosecond laser (MaiTai DeepSee, Spectra Physics) tuned to 950 nm for imaging of cells expressing GCaMP6f and to 1020 nm for imaging of cells expressing tdTomato. Time-series movies of neuronal populations expressing GCaMP6f were acquired at the frame rate of 6.926 Hz (182 × 182 μm field of view; 0.71 μm/pixel). The duration of each focal plane movie was 216.564 s (1,500 frames) to track spontaneous neuronal activity. For interneuron recordings, time-series movies of interneuronal populations expressing GCaMP6f were acquired at the frame rate of 30.33 Hz (405 × 405 μm field of view; 0.79 μm/pixel). The duration of each focal plane movie was 164.897 s (5,000 frames).
Randomization and blinding
The mice from all experimental groups, both baseline and nicotine treated, were imaged in a random order. Experiments were performed with blinding for the β2 re-expressing mice and for recordings of the VIP-Cre and PV-Cre baseline and SOM-Cre nicotine-treated mice but not for the rest of the experiments. Analysis was performed with blinding for the social interaction test data, the activities of the α5KO, α5SNP and β2 re-expressing mice, and recordings of the baseline PV-Cre and SOM-Cre mice and SOM-Cre nicotine-treated mice, but not for the rest of the experiments.
Two-photon data analysis
Image analysis was performed offline with ImageJ software. The time series were corrected using Image Stabilizer (http://www.cs.cmu.edu/~kangli/code/Image_Stabilizer.html). Regions of interest (ROIs) were manually selected in FIJI, and detection of Ca2+ transients of individual neurons was performed automatically by using a custom-written toolbox in MATLAB (Mathworks). For details on the analysis method, see the Supplementary Methods (Two-photon data analysis).
Statistical analysis
Two-photon data
Kruskal–Wallis one-way analysis of variance combined with multiple-comparison testing was applied on the aggregated activity (transients/min and spikes/min) of the neurons in all mouse groups in order to study the statistical similarities between them. For each mouse category, we used interpolation in order to account for the different number of neurons recorded in the different animals. In this way, our statistical analysis assigned the same weight to each neuronal population recorded in the different mice of a given category. Levene’s test was used to compare the variances of the neural distributions that were similar between groups that were statistically compared. We used Welch’s F-test ANOVA as a complementary test for heteroscedasticity. This test gave similar results as the Kruskal–Wallis test with the same level of significance. We used the multiple-comparisons Tukey–Kramer test in order to determine which pairs of mean ranks were significantly different. Animals were not excluded from the analysis. The number of mice needed for our study was determined by preliminary experiments in anesthetized mice and pilot experiments in awake mice. Box plots, histograms and empirical cumulative distribution functions (Kaplan–Meier estimate) were used to compare the different distributions of neural activity between the mouse types. For each box, the central mark is the median, the edges of the box are the 25th and 75th percentiles, and the whiskers extend to the most extreme data points not considered outliers. The ends of the whiskers represent the lowest data value still within 1.5 times the IQR (interquartile range) of the lower quartile and the highest data value still within 1.5 times the IQR of the upper quartile. On the empirical cumulative distribution plots, 95% lower and upper confidence bounds were computed with Greenwood’s formula.
Behavioral data
Statistical analysis was performed using Prism (GraphPad) software. Social interaction task differences were assessed using unpaired Student’s t test. Because the variance between genotypes for the social index and interaction time measures were not equal, unpaired Student’s t test with Welch’s correction was used. Two-way repeated-measures ANOVA with Sidak post hoc test was used for the PPI data analysis. All data other than the aforementioned social interaction tests met the assumptions of the chosen statistical test: normality and equal variances.
Data availability
The custom-written toolbox in MATLAB (Mathworks, 2014b) is available upon request. All data and materials used in this study are available upon request.
METHODS
Methods and any associated references are available in the online version of the paper.
Supplementary Material
Acknowledgments
F.K. is a scholar of the Pasteur Paris University Doctoral Program (PPU) and received a stipend from the Stavros Niarchos Foundation. This work was supported by the CNRS UMR 3571, the Fondation de la recherche médicale (FRM grant DPA20140629803), the Agence nationale de la recherche (ANR), the Laboratoire d’Excellence BIO-PSY (including salary support to F.K., AAP Fin de thèse 2015), the programme PasteurInnov 2012, by the FP7 ERANET programme NICO-GENE, Grant Agreement n009 BLANC 20092009BLANC 20, by the European Commission FP7 RTD Project HEALTH-2009-Neurocyp.08-202088 Grant 242167, by the French National Cancer Institute Grant CANCEROPOLE IDF 2016-1-TABAC-01-IP-1 MASKOS (all to U.M.), and NIH CA089392 and DA015663 (to J.S.). The laboratories of U.M., B.S.G. and D.A.D. are part of the École des Neurosciences de Paris Ile-de-France RTRA network. U.M. and D.A.D. are members of the Laboratory of Excellence, LabEx BIO-PSY. As such this work was supported by French state funds managed by the ANR within the Investissements d’Avenir programme under reference ANR-11-IDEX-0004-02. B.S.G. is member of the Laboratory of Excellence, LabEx IEC. B.S.G. acknowledges support from the Russian Academic Excellence Project ‘5-100’. We would like to thank S. Pons for valuable support and discussions on lentiviral constructs, and M. Soudant for production of lentiviral vectors. We also acknowledge the Pasteur Institute Shared Neuroscience Department imaging facility funded by Ile de France Domaine d’Intérêt Majeur (DIM/NeRF). We acknowledge the GENIE Program and the Janelia Research Campus and specifically V. Jayaraman, Ph.D., D. S. Kim, Ph.D., L. L. Looger, Ph.D., K. Svoboda, Ph.D. from the GENIE Project, Janelia Research Campus, Howard Hughes Medical Institute for making the AAV.Syn.GCaMP6f and AAV.Syn.Flex.GCaMP6f available. Finally, we thank Drs G. Fond and M. Groszer from the LabEx BIO-PSY for comments on the manuscript.
Footnotes
The authors declare no competing financial interests.
Author Contributions F.K. and U.M. conceived and designed research. F.K. performed experiments. F.K, K.A.S. and D.A.D. established the imaging technique. F.K., M.R., D.T. and D.A.D. designed the analysis code. F.K. and M.R. analyzed data. H.O.N., J.L. and C.H. performed behavioral experiments. M.W. contributed the VIP-Cre/tdTomato mice. J.A.S. developed the α5 SNP mouse line. M.N., JP.C. and B.S.G. provided advice. F.K. wrote the original manuscript, it was reviewed and edited by D.A.D. and U.M., and all other authors edited it. U.M. supervised the research.
References and Notes
- 1.Dehaene S, Changeux JP. Experimental and theoretical approaches to conscious processing. Neuron. 2011;70:200–27. doi: 10.1016/j.neuron.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 2.Bloem B, Poorthuis RB, Mansvelder HD. Cholinergic modulation of the medial prefrontal cortex: the role of nicotinic receptors in attention and regulation of neuronal activity. Front Neural Circuits. 2014;8:17. doi: 10.3389/fncir.2014.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raichle ME. The Brain’s Default Mode Network. Annu Rev Neurosci. 2015;38:433–47. doi: 10.1146/annurev-neuro-071013-014030. [DOI] [PubMed] [Google Scholar]
- 4.Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 5.Barch DM, et al. Selective deficits in prefrontal cortex function in medication-naive patients with schizophrenia. Arch Gen Psychiatry. 2001;58:280–8. doi: 10.1001/archpsyc.58.3.280. [DOI] [PubMed] [Google Scholar]
- 6.de Leon J, Diaz FJ. A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophr Res. 2005;76:135–57. doi: 10.1016/j.schres.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42:441–7. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ripke S, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young JW, et al. Impaired attention is central to the cognitive deficits observed in alpha 7 deficient mice. Eur Neuropsychopharmacol. 2007;17:145–55. doi: 10.1016/j.euroneuro.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Guillem K, et al. Nicotinic acetylcholine receptor β2 subunits in the medial prefrontal cortex control attention. Science. 2011;333:888–91. doi: 10.1126/science.1207079. [DOI] [PubMed] [Google Scholar]
- 11.Bailey CDC, De Biasi M, Fletcher PJ, Lambe EK. The nicotinic acetylcholine receptor alpha5 subunit plays a key role in attention circuitry and accuracy. J Neurosci. 2010;30:9241–52. doi: 10.1523/JNEUROSCI.2258-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hong LE, et al. A genetically modulated, intrinsic cingulate circuit supports human nicotine addiction. Proc Natl Acad Sci. 2010;107:13509–13514. doi: 10.1073/pnas.1004745107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adler LE, et al. Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull. 1998;24:189–202. doi: 10.1093/oxfordjournals.schbul.a033320. [DOI] [PubMed] [Google Scholar]
- 14.Robbins TW, Roberts AC. Differential regulation of fronto-executive function by the monoamines and acetylcholine. Cereb Cortex. 2007;17(Suppl 1):i151–60. doi: 10.1093/cercor/bhm066. [DOI] [PubMed] [Google Scholar]
- 15.Poorthuis RB, et al. Layer-specific modulation of the prefrontal cortex by nicotinic acetylcholine receptors. Cereb Cortex. 2013;23:148–61. doi: 10.1093/cercor/bhr390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarter M, Parikh V, Howe WM. Phasic acetylcholine release and the volume transmission hypothesis: time to move on. Nat Rev Neurosci. 2009;10:383–90. doi: 10.1038/nm2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sinkus ML, et al. The human CHRNA7 and CHRFAM7A genes: A review of the genetics, regulation, and function. Neuropharmacology. 2015 doi: 10.1016/j.neuropharm.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bierut LJ, et al. Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry. 2008;165:1163–71. doi: 10.1176/appi.ajp.2008.07111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sciaccaluga M, et al. Crucial role of nicotinic 5 subunit variants for Ca2+ fluxes in ventral midbrain neurons. FASEB J. 2015;29:3389–3398. doi: 10.1096/fj.14-268102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Proulx E, Piva M, Tian MK, Bailey CDC, Lambe EK. Nicotinic acetylcholine receptors in attention circuitry: the role of layer VI neurons of prefrontal cortex. Cell Mol Life Sci. 2014;71:1225–44. doi: 10.1007/s00018-013-1481-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derntl B, Habel U. Deficits in social cognition: a marker for psychiatric disorders? Eur Arch Psychiatry Clin Neurosci. 2011;261:145–149. doi: 10.1007/s00406-011-0244-0. [DOI] [PubMed] [Google Scholar]
- 22.Nadler JJ, et al. Automated apparatus for quantitation of social approach behaviors in mice. Genes, Brain Behav. 2004;3:303–314. doi: 10.1111/j.1601-183X.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- 23.Braff DL, Geyer MA, Swerdlow NR. Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 2001;156:234–58. doi: 10.1007/s002130100810. [DOI] [PubMed] [Google Scholar]
- 24.Porter JT, et al. Selective excitation of subtypes of neocortical interneurons by nicotinic receptors. J Neurosci. 1999;19:5228–35. doi: 10.1523/JNEUROSCI.19-13-05228.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pi HJ, et al. Cortical interneurons that specialize in disinhibitory control. Nature. 2013;503:521–4. doi: 10.1038/nature12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poorthuis RB, Bloem B, Verhoog MB, Mansvelder HD. Layer-specific interference with cholinergic signaling in the prefrontal cortex by smoking concentrations of nicotine. J Neurosci. 2013;33:4843–53. doi: 10.1523/JNEUROSCI.5012-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex. 1997;7:476–86. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- 28.McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol. 1995;57:521–46. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- 29.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–55. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M. Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci. 2013;16:1068–76. doi: 10.1038/nn.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 2006;184:523–39. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- 32.Hambsch B, et al. Chronic nicotine improves short-term memory selectively in a G72 mouse model of schizophrenia. Br J Pharmacol. 2014;171:1758–71. doi: 10.1111/bph.12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matta SG, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- 34.Morel C, et al. Nicotine consumption is regulated by a human polymorphism in dopamine neurons. Mol Psychiatry. 2014;19:930–6. doi: 10.1038/mp.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Orr-Urtreger A, et al. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–71. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picciotto MR, et al. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–7. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- 37.Salas R, et al. The nicotinic acetylcholine receptor subunit alpha 5 mediates short-term effects of nicotine in vivo. Mol Pharmacol. 2003;63:1059–66. doi: 10.1124/mol.63.5.1059. [DOI] [PubMed] [Google Scholar]
- 38.Fuchs EC, et al. Genetically altered AMPA-type glutamate receptor kinetics in interneurons disrupt long-range synchrony of gamma oscillation. Proc Natl Acad Sci U S A. 2001;98:3571–6. doi: 10.1073/pnas.051631898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tolu S, et al. A versatile system for the neuronal subtype specific expression of lentiviral vectors. FASEB J. 2010;24:723–30. doi: 10.1096/fj.09-139790. [DOI] [PubMed] [Google Scholar]
- 40.Arroyo S, Bennett C, Aziz D, Brown SP, Hestrin S. Prolonged disynaptic inhibition in the cortex mediated by slow, non-α7 nicotinic excitation of a specific subset of cortical interneurons. J Neurosci. 2012;32:3859–64. doi: 10.1523/JNEUROSCI.0115-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prönneke A, et al. Characterizing VIP Neurons in the Barrel Cortex of VIPcre/tdTomato Mice Reveals Layer-Specific Differences. Cereb Cortex. 2015;25:4854–68. doi: 10.1093/cercor/bhv202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi GB, et al. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–9. doi: 10.1126/science.aad0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holtmaat A, et al. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4:1128–44. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naldini L, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–7. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 45.Maskos U, et al. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–7. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- 46.Lepousez G, Lledo PM. Odor discrimination requires proper olfactory fast oscillations in awake mice. Neuron. 2013;80:1010–24. doi: 10.1016/j.neuron.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 47.Guo ZV, et al. Procedures for behavioral experiments in head-fixed mice. PLoS One. 2014;9:e88678. doi: 10.1371/journal.pone.0088678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Besson M, et al. Long-term effects of chronic nicotine exposure on brain nicotinic receptors. Proc Natl Acad Sci U S A. 2007;104:8155–60. doi: 10.1073/pnas.0702698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The custom-written toolbox in MATLAB (Mathworks, 2014b) is available upon request. All data and materials used in this study are available upon request.