

Abstract
Apoptosis is a cellular suicide program that plays a critical role in development and human diseases, including cancer. Cancer cells evade apoptosis, thereby enabling excessive proliferation, survival under hypoxic conditions, and acquired resistance to therapeutic agents. Among various mechanisms that contribute to the evasion of apoptosis in cancer, metabolism is emerging as one of the key factors. Cellular metabolites can regulate functions of pro- and antiapoptotic proteins. In turn, p53, a regulator of apoptosis, also controls metabolism by limiting glycolysis and facilitating mitochondrial respiration. Consequently, with dysregulated metabolism and p53 inactivation, cancer cells are well-equipped to disable the apoptotic machinery. In this article, we review how cellular apoptosis is regulated and how metabolism can influence the signaling pathways leading to apoptosis, especially focusing on how glucose and lipid metabolism are altered in cancer cells and how these alterations can impact the apoptotic pathways.
1. INTRODUCTION
Apoptosis is a form of programmed cell death. Dysregulation of apoptosis is often associated with human diseases. Excessive apoptosis can cause degenerative disorders, whereas insufficient apoptosis can result in neoplastic diseases. In this regard, cancer cells avoid apoptosis by disabling the cell death machinery through various mechanisms, thereby gaining an excessive survival advantage (for review, see Fernald and Kurokawa, 2013). Cancer cells often evade apoptosis by overexpressing or stabilizing antiapoptotic BCL-2 family proteins while transcriptionally or posttranscriptionally inhibiting proapoptotic BCL-2 family members. Additionally, another common mechanism that contributes to cancer cell survival is the inactivation of transcription factors that trigger apoptosis, such as the tumor suppressor p53.
In addition to the marked resistance to apoptosis, cancer cells are also characterized by altered metabolic profiles (Dang, 2012a; DeBerardinis et al., 2008; Pavlova and Thompson, 2016; vander Heiden et al., 2009). Accumulating evidence strongly suggests that this metabolic shift plays a significant role in cancer cell survival, invasion, and metastasis (Buchakjian and Kornbluth, 2010; Fernald and Kurokawa, 2013; Hannun and Obeid, 2008; Semenza, 2007). However, it remains to be fully elucidated how cellular metabolism is reprogrammed and how these modifications benefit cancer cells.
Metabolism can directly or indirectly regulate the apoptotic machinery, and cancer cells harness altered metabolism to evade apoptosis. In this review, we will discuss how cancer metabolism affects the apoptosis pathway. We will first look at the general overview of how apoptosis takes place. Then, we will focus on how the glycolytic metabolism pathway, which is often upregulated in cancer cells, can influence cell death signaling. Importantly, p53 suppresses the glycolytic pathway. Thus, the loss of p53 function, which is seen in many cancers, might promote glycolytic flux. Finally, we will illustrate the role of lipid metabolism in the inhibition of apoptosis in cancer cells.
2. OVERVIEW OF APOPTOSIS PATHWAYS
Apoptosis is a form of programmed cell death, which is morphologically characterized by membrane-blebbing, chromatin condensation, and DNA fragmentation. Cysteine proteases, called caspases (Cysteine ASPartate proteASEs), mediate this process by cleaving substrate proteins at specific aspartate residues. The targets of caspases include hundreds of vital proteins, such as cytoskeletal proteins, kinases, and transcription factors (Fischer et al., 2002; Lüthi and Martin, 2007). The apoptotic pathway is the intermediary process that connects various stress signals with execution of caspase-mediated cell death. The mechanism of how caspases are activated differs depending on cell death stimuli and cell type.
2.1 Caspases
Caspases are categorized into two groups: initiator caspases and effector caspases (Fig. 1). Both initiator and effector caspases contain a catalytic site comprised of a small and a large subunit. Initiator apoptotic caspases (e.g., caspases-2, -8, and -9) have a large N-terminal prodomain that interacts with a specific adaptor protein required for dimerization-induced activation (see later). The dimerization of the initiator caspases takes place within a specific protein complex (e.g., PIDDosome for caspase-2, DISC for caspase-8, and apoptosome for caspase-9; see later). Once activated, the initiator caspases cleave effector caspases. Without a large prodomain, effector caspases (e.g., caspases-3, -6, and -7) do not need an adaptor protein for dimerization-induced activation. Rather, effector caspases spontaneously dimerize but are only activated upon cleavage between the small and large subunits by an active initiator caspase. Activated effector caspases then cleave a number of protein substrates to initiate apoptosis, leading to subsequent dismantling of cellular components (Fischer et al., 2002; Kurokawa and Kornbluth, 2009; Lüthi and Martin, 2007; Taylor et al., 2008).
Figure 1.

Structure of caspases. All caspases have a large subunit and a small subunit, which are indispensible for protease activity. Caspases can be divided into two types: initiators and effectors. While the effector caspases (i.e., caspases-3, -6, and -7) have a short prodomain, the initiators have a long prodomain which plays a crucial role for proximity-induced activation mediated by the interaction with adaptor proteins. Caspases-1, -4, -5, and -12 are implicated in inflammation, whereas caspases-8/-10 and caspases-2/-9 initiate the extrinsic and intrinsic apoptosis pathways, respectively. CARD; caspase recruitment domain, DED; death effector domain.
2.2 Intrinsic and Extrinsic Pathways
Depending on cellular stress, apoptosis can be initiated via two pathways, intrinsic or extrinsic (Fig. 2). The intrinsic apoptosis pathway is activated in response to intracellular stress, such as DNA damage and metabolic stress. Extrinsic apoptosis pathway is initiated by binding of death ligands (e.g., TNF and TRAIL) to their cognate death receptors (e.g., TNF-R1 and TRAIL-R1/R2) on the plasma membrane. Notably, while the intrinsic and extrinsic pathways are induced in response to different cellular stimuli, both pathways eventually culminate in the activation of effector caspases, especially caspase-3.
Figure 2.
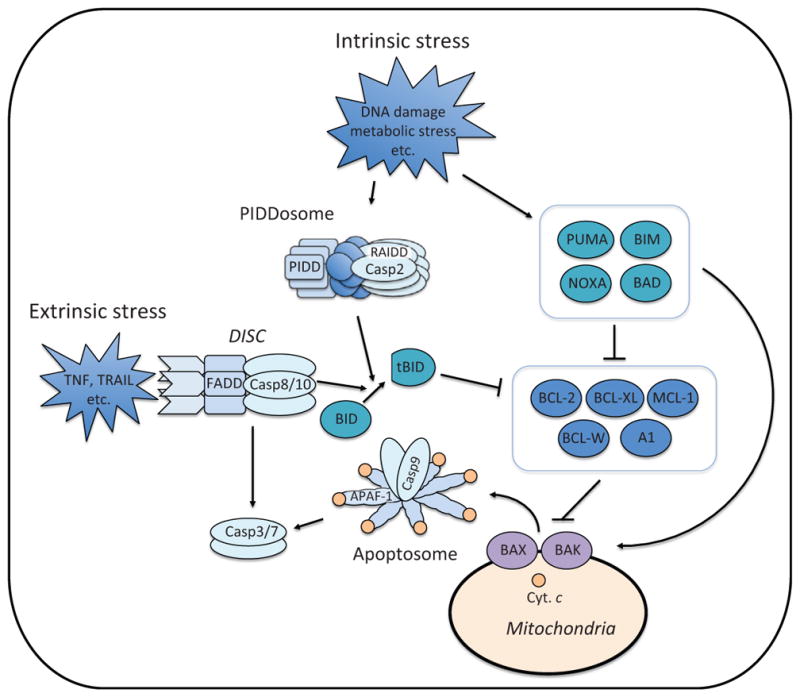
Apoptosis pathways. Intrinsic stress (e.g., DNA damage and metabolic stress) induces the expression and/or activation of proapoptotic BH3-only proteins, which either suppress antiapoptotic BCL-2 family proteins or directly activate BAX and BAK (Fig. 3). BAX and BAK induce mitochondrial outer membrane permeabilization (MOMP). Upon MOMP, cytochrome c (Cyt. c) is released from the mitochondrial intermembrane space. The release of cytochrome c triggers the formation of apoptosome in the cytoplasm, which recruits and activates caspase-9. Active caspase-9 cleaves and activates effector caspases, caspases-3 and -7. Intrinsic stress also engages the formation of PIDDosome which is composed of PIDD, RAIDD, and caspase-2. Through proximity-induced activation, casapse-2 becomes active and cleaves BID. Once cleaved, BID (tBID: truncated BID) becomes an active BH3-only protein. Extrinsic stress (e.g., TNF and TRAIL) is mediated through the death receptor, which forms the DISC (Death Inducing Signaling Complex) with FADD and caspase-8. Active caspase-8 directly cleaves and activates effector caspases or triggers the intrinsic apoptosis pathway through BID cleavage. TNF; tumor necrosis factor, TRAIL; TNF-related apoptosis-inducing ligand.
2.3 Activation of Initiator Caspases
The intrinsic apoptosis pathway involves mitochondria outer membrane permeabilization (MOMP; see later the section for MOMP). Upon MOMP, proapoptotic proteins are released from the mitochondrial intermembrane space into the cytoplasm. Among the key proteins in the intermembrane space of mitochondria is cytochrome c, a component of the electron transport chain (ETC). Once cytoplasmic, cytochrome c initiates the intrinsic pathway of apoptosis by binding to the adaptor protein APAF-1 as it triggers its oligomerization (Li et al., 1997). APAF-1 heptomer, together with cytochrome c and the cofactor dATP, forms the caspase-activating apoptosome platform (Figure 2). The apoptosome then activates caspase-9 by facilitating the dimerization of this initiator caspase (Acehan et al., 2002; Shi, 2002; Shiozaki et al., 2002).
The extrinsic pathway is initiated by the activation of caspase-8 or -10. Caspases-8 and -10 are activated through another caspase-activating platform: DISC (Death Inducing Signaling Complex) consisting of the death receptor, the adaptor protein FADD, and caspase-8 (or -10) (Fig. 2). Upon ligand binding, oligomerization of the death receptor is induced, leading to recruitment of FADD which, in turn, recruits and activates caspase-8 (or -10). Although the activation of caspase-8 (or -10) takes place near the plasma membrane, self-cleavage between the prodomain and the small subunit allows active caspases-8 and 10 (composed of the small and large subunits) to be freely released from the DISC.
The mechanism of caspase-2 activation is somewhat complicated and sometimes controversial. Canonically, caspase-2 is oligomerized and activated in the PIDDosome, which consists of the two adaptor proteins RAIDD (also known as CRADD) and PIDD (Park et al., 2007; Tinel et al., 2007; Tinel and Tschopp, 2004) (Fig. 2). Although the structure and assembly of the PIDDosome have been resolved (Park et al., 2007), it remains unclear how PIDDosome formation is regulated as opposed to cytochrome c- and a death ligand-induced apoptosome and DISC formation, respectively. Interestingly, PIDD can be induced by the tumor suppressor p53 (see later). However, caspase-2 activation can take place in the absence of wild type p53 (Castedo et al., 2004; Sidi et al., 2008). Moreover, recent studies show that activation of caspase-2 does not always require RAIDD and PIDD (Manzl et al., 2009, 2012; Ribe et al., 2012). It has also been shown that caspase-2 can be activated through the DISC consisting of a CD95, FADD, and caspase-8 (Lavrik et al., 2006; Olsson et al., 2009). Therefore, activation of caspase-2 may be regulated by multiple pathways and, most likely, in a context-dependent manner.
2.4 Mitochondria Outer Membrane Permeabilization (MOMP)
MOMP is regulated by a delicate balance between pro- and antiapoptotic B cell lymphoma 2 (BCL-2) family proteins and is often considered as a point of no return in the apoptosis pathway (Fig. 2). BCL-2 family proteins are characterized by the presence of BCL-2 homology domains (BH1-4) (Youle and Strasser, 2008) (Fig. 3A). Interacting through the BH domains, proapoptotic BCL-2 proteins promote MOMP, while antiapoptotic BCL-2 proteins antagonize this effect by direct interaction with their proapoptotic family members (Chen et al., 2005; Czabotar et al., 2014; Letai et al., 2002; Tait and Green, 2010). Among the BCL-2 family proteins, BAX and BAK play the most critical and direct role for MOMP induction as it is these two proteins that oligomerize to create “pores” on the mitochondrial outer membrane in response to apoptotic stimuli (Große et al., 2016; Salvador-Gallego et al., 2016). Other members of BCL-2 family proteins are required to directly or indirectly regulate BAX/BAK activation. Mice lacking both Bax and Bak are prenatally lethal because of defects in neuronal development owing to the absence of apoptosis (Lindsten et al., 2000). Moreover, mouse embryonic fibroblasts (MEFs) derived from the Bax/Bak double knockout mice are extremely resistant to various apoptotic stimuli (Lindsten et al., 2000; Wei et al., 2001).
Figure 3.

Regulation of BCL-2 family proteins. BCL-2 family proteins are divided into three groups: antiapoptotic BCL-2 family proteins (BCL-2, BCL-W, BCL-xL, MCL-1, and A1), proapoptotic multidomain proteins (BAX, BAK, and BOK), and proapoptotic BH3-only proteins (BIM, PUMA, BID, BAD, and NOXA).
Other proapoptotic BCL-2 family proteins include BID, BIM, BAD, NOXA, and PUMA (Fig. 3). These pro-apoptotic proteins contain only one BH domain, namely BH3 domain, and are called BH3-only proteins (Fig. 3). There are two models proposed to explain how activation of BAX and BAK is regulated by BCL-2 family proteins (Chen et al., 2005; Czabotar et al., 2014; Letai et al., 2002; Tait and Green, 2010): the indirect activator model and the direct activator-derepressor model (Fig. 4). In the indirect activator model, antiapoptotic BCL-2 family proteins constitutively bind and suppress BAX and BAK to inhibit MOMP, while competitive interaction between pro- and antiapoptotic BCL-2 proteins promotes BAX/BAK activation by disrupting the binding between BAX/BAK and the antiapoptotic BCL-2 proteins (Fig. 4). In the direct activator-derepressor model, proapoptotic BCL-2 family proteins could be divided to direct activators and derepressors (sensitizers). Direct activators, such as BIM, release BAX/BAK from anti-apoptotic BCL-2 family proteins for activation, while derepressors, such as BAD, suppress the function of antiapoptotic BCL-2 family proteins, circuitously contributing to BAX/BAK activation (Fig. 4). Notably, there is certain binding specificity between pro- and antiapoptotic BCL-2 family proteins, which add layers of complexity to the regulation of BAX/BAK activation (Fig. 5).
Figure 4.
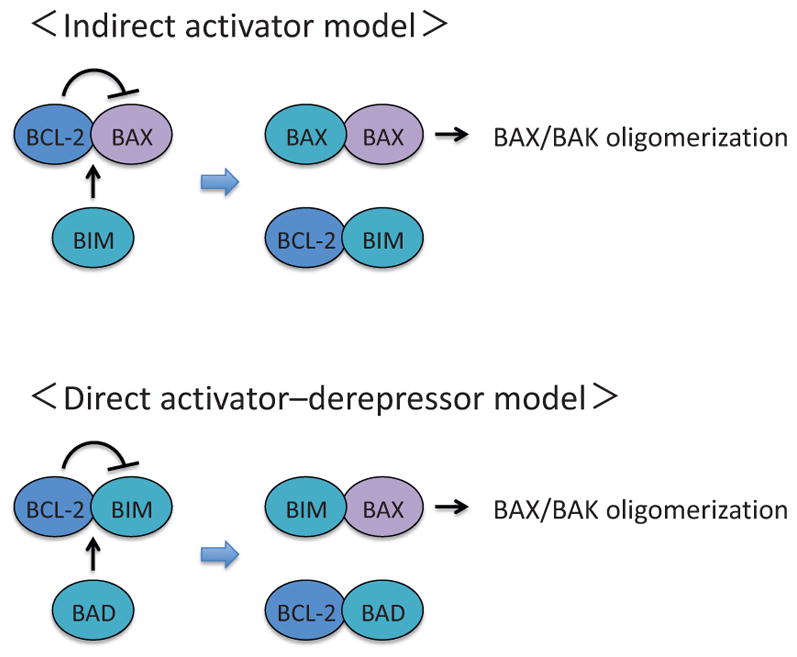
The indirect activator model and the direct activator-derepressor model. In the indirect activator model, activation of BAX and BAK can be directly suppressed by any of the antiapoptotic BCL-2 family proteins. Upon apoptotic stimuli, BH3-only proteins are induced and occupy the antiapoptotic BCL-2 family proteins, which will release BAX and BAK to form active pore-forming oligomers. In the direct activator-derepressor model, the activation of BAX/BAK is prevented by the suppression of BH3-only proteins by the antiapoptotic BCL-2 family proteins. Upon induction of BH3-only proteins, derepressors (e.g., BAD and NOXA) competitively bind to antiapoptotic BCL-2 family proteins (e.g., BID, BIM, PUMA), which will release direct activators and allow for subsequent oligomerization of BAX and BAK.
Figure 5.
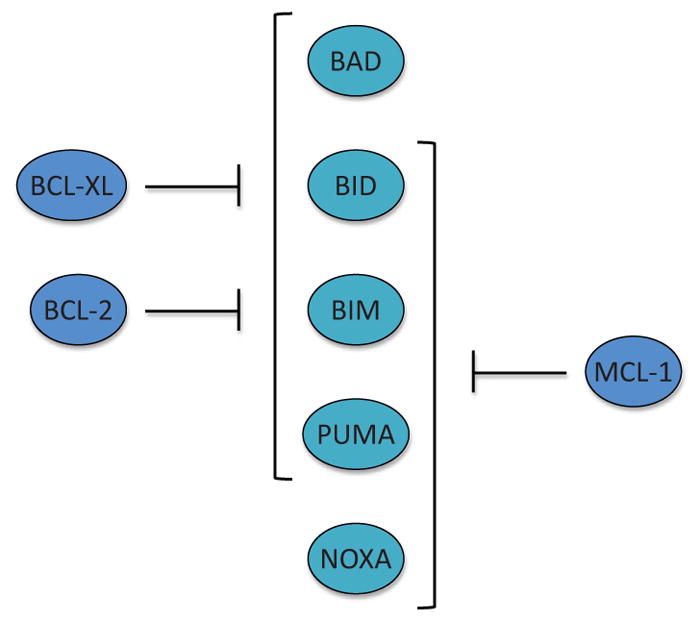
The interaction between antiapoptotic BCL-2 family proteins and proapoptotic BCL-2 family members. BCL-2 and BCL-xL interact with all of the BH3-only proteins except for NOXA, while MCL-1 can bind to all of the BH3-only protein except for BAD. Of note, BIM, PUMA, and BID are capable of interacting with all of the antiapoptotic BCL-2 family proteins. Furthermore, BIM, PUMA, and BID can directly activate BAX and BAK in the direct activator–derepressor model.
It should be noted that the proapoptotic BID is unique in that it is cleaved into its active tBID form only after cleavage by caspases-2 and -8/10, but not caspase-9. BID is considered to be the only BCL-2 family protein that can mediate the cell death signaling from PIDDosome and DISC to MOMP (Fig. 2).
3. REGULATION OF APOPTOSIS THROUGH GLYCOLYTIC PATHWAY
Enhanced glucose uptake is one of the hallmarks of cancer. Since Otto Warburg initially described that cancer cells exhibit increased conversion of glucose to lactate (Warburg, 1956), a number of studies have shown that enhanced anaerobic glycolysis is a unique characteristic of cancer cells. Furthermore, it is widely accepted that the “Warburg effect” is part of the fundamental metabolic changes cancer cells undergo in order to develop the propensity to grow and proliferate uncontrollably as well as to inhibit apoptosis.
Glucose uptake is regulated by the growth factor signaling pathway that stimulates expression of the glucose transporter (GLUT). Once transported into a cell, glucose is phosphorylated by hexokinases and is converted to glucose-6-phosphate (G6P) (Fig. 6). G6P then enters either the glycolysis pathway or the pentose phosphate pathway. The glycolysis pathway ultimately produces a net of two molecules of pyruvate and ATP per glucose molecule. In mitochondria, pyruvate is converted to acetyl-CoA and can then enter into the Krebs cycle. NADH generated through the Krebs cycle and glycolysis is used in the mitochondrial ETC, producing 32 ATPs per glucose molecule (oxidative phosphorylation) (Fig. 6).
Figure 6.

Metabolism pathways. Extracellular glucose is transported into a cell through the glucose transporter (GLUT), phosphorylated by hexokinase (HK), and converted to glucose-6-phosphate (G6P). Thereafter, G6P is metabolized through either the glycolysis pathway or the pentose phosphate pathway. In the pentose phosphate pathway, G6P is converted to ribose-5-phosphate (R5P), a precursor of nucleotide synthesis, while NADPH is produced as a by-product. In the glycolysis pathway, the final product, pyruvate, is converted to acetyl-CoA, and enters the Krebs cycle in the mitochondria. NADH is generated in the Krebs cycle and used for oxidative phosphorylation where ATPs are produced. On the other hand, citrate generated in the Krebs cycle is released into the cytoplasm where it is converted to acetyl-CoA and then malonyl-CoA. Acetyl-CoA and malonyl-CoA are used for fatty acid production by fatty acid synthase (FASN). Fatty acids are further metabolized to fatty acyl-CoA and used for membrane synthesis, formation of lipid droplets, and signaling lipid production. G6PD; G6P dehydrogenase, 6PG; 6-phosphogluconolactone, F6P; fructose-6-phosphate, F1,6BP; fructose-1,6-bisphosphate, PFK1; phosphofructokinase 1, TIGAR; TP53-induced glycolysis and apoptosis regulator, LDHA; lactate dehydrogenase A, PDH; pyruvate dehydrogenase, PDK; pyruvate dehydrogenase kinase, AIF; apoptosis-inducing factor, SCO2; synthesis of cytochrome c oxidase 2, ACC; acetyl-CoA carboxylase.
The pentose phosphate pathway produces NADPH and fatty acids. NADPH is a source of the ROS scavenger glutathione (GSH), whereas fatty acids are critical for generation of lipid membranes. The pentose phosphate pathway also generates ribose-5-phosphate, a precursor of nucleotides. Therefore, this pathway is crucial for biomass production. It is considered that highly proliferative cells, such as cancer cells, require large amounts of lipid membranes, nucleotides, in addition to amino acids and, therefore, upregulate the pentose phosphate pathway to compensate such demands.
3.1 Hypoxia-Inducible Factor-1
Altered glucose metabolism in cancer is, at least in part, linked to cancer microenvironment. As a solid tumor develops, cell density increases, which creates an environment with low oxygen levels (hypoxia) in the tumor (Dang, 2012b; Semenza, 2002). In a hypoxic condition, tumor cells switch their energy-producing metabolism from mitochondrial oxidative phosphorylation to the glycolytic pathway involving the pentose phosphate pathway (Fig. 6). Hypoxia-Inducible Factor-1 (HIF-1) is a key transcription factor that regulates gene expression under hypoxic conditions (Semenza, 2012, 2010a). HIF-1 consists of two subunits, HIF-1α and HIF-1β. While HIF-1β protein is constitutively expressed and present in excess, HIF-1α protein has a short half-life (Huang et al., 1998; Salceda and Caro, 1997). Therefore, the regulation of HIF-1 activity is considered to occur at the level of HIF-1α protein availability. In the presence of oxygen, HIF-1α is hydroxylated at a specific proline residue by prolyl hydroxylase PHD, using O2 as a substrate. This modification creates a binding site for von Hippel-Lindau (VHL) protein, and VHL recruits components of a ubiquitin E3 ligase complex for proteasomal degradation of HIF-1α (Epstein et al., 2001; Iwai et al., 1999; Kamura et al., 2000; Lisztwan et al., 1999). Therefore, in a hypoxic condition, VHL-mediated degradation of HIF-1α is prevented and HIF-1α accumulates, which leads to upregulation of HIF-1 target genes (Lu et al., 2002; Mole et al., 2009) (Fig. 7). Glucose transporters GLUT1 and GLUT3, and hexokinases HK1 and HK2 are the direct transcriptional targets of HIF-1 (Iyer et al., 1998). Other target genes of HIF-1 include PDK1 [pyruvate dehydrogenase (PDH) kinase] that inhibits conversion of pyruvate to acetyl-CoA (Kim et al., 2006; Papandreou et al., 2006), LDHA (lactate dehydrogenase A) that promotes conversion of pyruvate to lactate (Semenza et al., 1996), in addition to BNIP3 and BNIP3L that mediate clearance of damaged mitochondria (Bellot et al., 2009; Zhang et al., 2008). Many of these HIF-1 target gene products function to suppress mitochondrial biogenesis but promote glycolysis (Semenza, 2012).
Figure 7.

Target genes of HIF-1. HIF-1 promotes glycolysis by transcriptionally upregulating GLUT1, GLUT3, HK1, and HK2. HIF-1 also suppresses oxidative phosphorylation by the upregulation of gene expression of BNIP3, BNIP3L, LDHA, and PDK1. In addition, HIF-1 can inhibit apoptosis by suppressing the expression of BID. BNIP3; BCL-2/E1B-19 kDa interacting protein 3, BNIP3L; BNIP3-like.
In a hypoxic environment, the tumor suppressor p53 is induced, which could lead to cell cycle arrest and apoptosis (Chandel et al., 2000; Kilic et al., 2007). Nevertheless, solid tumors often exhibit marked resistance to various therapeutic treatments. Accumulating evidence indicates that HIF-1 plays a critical role in apoptosis resistance under hypoxia by upregulation of glucose uptake and glycolysis (Dong and Wang, 2004; Fulda and Debatin, 2007; Kilic et al., 2007; Malhotra and Brosius, 1999). In addition, HIF-1 directly suppresses expression of BID through promoter binding (Erler et al., 2004).
Overexpression of HIF-1 is observed in many different types of cancers (Semenza, 2010b). Using xenograft mouse models, it has been shown that HIF-1 activity is correlated with tumor formation (Ryan et al., 1998, 2000; Unruh et al., 2003). Interestingly, HIF-1 expression is also upregulated by the PI3K/AKT pathway (Elstrom et al., 2004; Zhong et al., 2000). Moreover, some oncogenes augment HIF-1-mediated upregulation of glycolysis. Ras oncoprotein promotes glycolysis through upregulation of MYC and HIF-1 activity (Lim et al., 2004; Sears et al., 1999). In turn, MYC corporately works with HIF-1 to induce expression of PDK and hexokinases (Kim et al., 2007). Tumors derived from H-RAS-transformed HIF-1α-deficient MEFs lead to reduction of tumor growth in vivo (Ryan et al., 2000, 1998). Importantly, such HIF-1α-deficient tumors exhibit better response to chemotherapeutics because of enhanced apoptosis, which underscores the antiapoptotic function of HIF-1α in cancer cells (Unruh et al., 2003). Several anticancer agents have been shown to inhibit HIF-1 activity by decreasing HIF-1α at mRNA or protein level (Semenza, 2013).
Although HIF-1 is critical for hypoxia-induced apoptosis resistance, it should be noted that HIF-1-independent mechanisms also exist. For instance, HIF-1-independent upregulation of antiapoptotic BCL-2, BCL-xL, and cIAP-2 as well as HIF-1-independent downregulation of proapoptotic BAX and BAD have been reported under hypoxic conditions (Dong et al., 2001; Erler et al., 2004; Kilic et al., 2007). These changes take place even after RNAi knockdown of HIF-1α (Erler et al., 2004; Kilic et al., 2007) or in HIF-1α-deficient MEFs (Dong et al., 2001). The HIF-1-independent mechanism of hypoxia-induced apoptosis resistance remains elusive.
3.2 AKT
The serine/threonine kinase AKT promotes cell survival and is often activated in cancer cells (Altomare and Testa, 2005; Bellacosa et al., 2005). Positively regulated by growth factors through phosphoinositide 3-kinase (PI3K), AKT increases glycolysis and lactate production (Edinger and Thompson, 2002; Elstrom et al., 2004; Rathmell et al., 2003; Wieman et al., 2007). Thus, AKT plays an important role in cellular survival mediated by growth factor signaling (Rathmell et al., 2003; Yamaguchi and Wang, 2001) (Fig. 8). The role of AKT in the regulation of antiapoptotic machinery by growth factors has been extensively studied in interleukin-3 (IL3)-dependent murine lymphocyte lines. In these cells, IL3 activates glycolysis (vander Heiden et al., 2001) and upregulates glucose transporter expression in an AKT-dependent manner (Plas et al., 2001; Rathmell et al., 2003; Wieman et al., 2007). Intriguingly, AKT promotes the localization of GLUT1 to the plasma membrane and increase cell survival (Wieman et al., 2007). In addition, AKT activates glycolysis through mitochondrial localization of hexokinase (Gottlob et al., 2001; Majewski et al., 2004a). In the presence of glucose, AKT increases the association of hexokinase with the outer mitochondrial membrane and prevents mitochondrial cytochrome c release (Majewski et al., 2004a,b).
Figure 8.
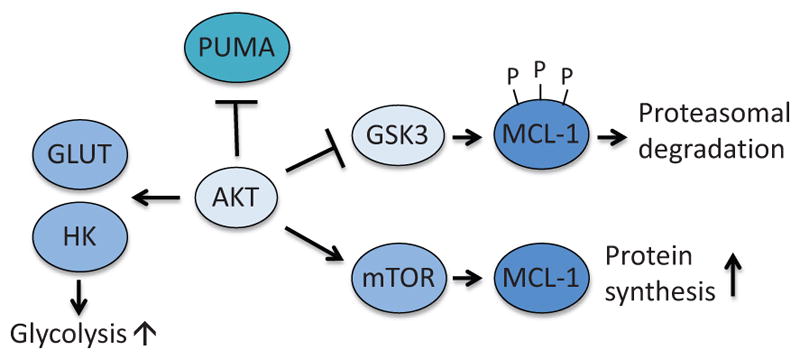
Anti-apoptotic function of AKT. AKT prevents apoptosis by suppressing induction of PUMA and by inhibiting GSK3-dependent degradation of MCL-1. In addition, AKT promotes translocation of GLUT to cellular membrane and activation of HK. GSK3; glycogen synthase kinase 3.
AKT also mediates metabolic protection of apoptosis through regulation of BCL-2 family proteins, especially MCL-1 and PUMA (Fig. 8). MCL-1 is characterized by rapid protein turnover. Among the mechanisms that regulate the stability of MCL-1 protein is phosphorylation by the serine/threonine kinase GSK3. GSK3 phosphorylates MCL-1 at multiple sites, which triggers ubiquitination and proteasomal degradation (Maurer et al., 2006). AKT downregulates this process by phosphorylating and inactivating GSK3. Upon growth factor stimulation, AKT becomes active, which results in suppression of GSK3-mediated degradation of MCL-1 (Zhao et al., 2007). In the presence of glucose, AKT also promotes MCL-1 protein synthesis by stimulating mTOR-mediated protein translation (Coloff et al., 2011a).
PUMA is a BH3-only protein that can be induced by the tumor suppressor p53 in response to various cellular stresses (Jeffers et al., 2003). Interestingly, glucose metabolism also suppresses p53-mediated PUMA induction and apoptosis (Zhao et al., 2008). Active AKT suppresses PUMA induction in the presence of glucose and is critical for AKT-mediated cell survival (Coloff et al., 2011b). Since PUMA is one of the most critical mediators of p53-dependent apoptosis (Jeffers et al., 2003), these studies suggest that AKT could metabolically suppress the tumor suppressor function of p53 in cancer cells.
3.3 BAD
The BH3-only protein BAD inhibits antiapoptotic BCL-2 and BCL-xL, and facilitates BAX/BAK activation in response to apoptotic stimuli (Chen et al., 2005; Youle and Strasser, 2008) (Figs. 3 and 5). BAD is a substrate of various kinases, such as AKT, protein kinase A (PKA), and c-Jun NH2-terminal kinase (JNK), which are regulated by the growth factor signaling pathway and the glycolysis pathway (Datta et al., 1997; del Peso et al., 1997) (Fig. 9). On the other hand, BAD is dephosphorylated by PP2A in response to growth factor withdrawal (Chiang et al., 2003). The primary phosphorylation sites of BAD are at Ser112, Ser136, and Ser155. Phosphorylation of these sites is inhibitory for BAD’s proapoptotic function, as it creates a docking site for the adaptor protein 14-3-3. Once bound, 14-3-3 sequesters BAD from its interaction with BCL-xL (Zha et al., 1996). The critical role of BAD phosphorylation was shown by a knock-in mouse model in which the three serine residues were mutated to nonphosphorylatable alanine residues (S112A, S136A, and S155A). The knock-in mice and cells isolated from the mice exhibited enhanced sensitivity to various apoptotic stimuli, including DNA damage (Datta et al., 2002). Interestingly, 14-3-3 itself can also be directly phosphorylated by JNK following cellular stress, which results in dissociation from BAD and subsequent translocation of BAD to mitochondria (Sunayama et al., 2005). As JNK also protects cells by phosphorylation of BAD Thr201 (Yu et al., 2004), the role of JNK in the regulation of BAD activity may depend on cell type or cellular stimuli.
Figure 9.
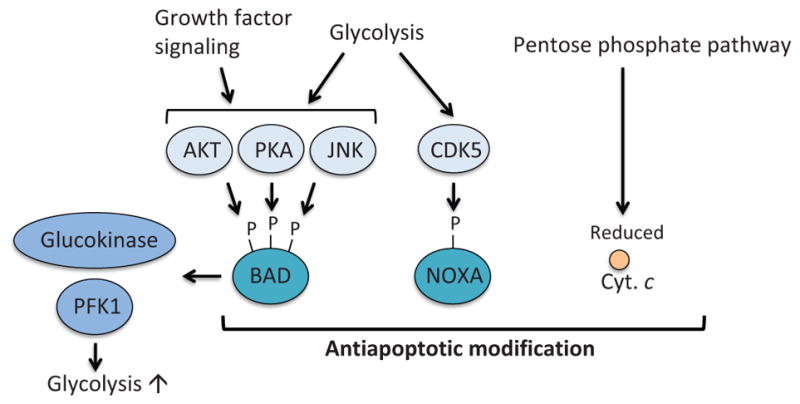
Metabolism-mediated modification of pro-apoptotic proteins. BAD is phosphorylated by AKT, PKA, and JNK that are regulated by glycolysis and growth factor signaling. Phosphorylation of BAD results in suppression of the antiapoptotic functions of BAD. Moreover, phosphorylated BAD contributes to activation of glucokinase and PFK1, therefore enhancing the glycolysis pathway. Atypical cyclin-dependent kinase, CDK5, is activated by glycolysis. Active CDK5 phosphorylates and inhibits the BH3-only protein NOXA. The pentose phosphate pathway produces NADPH, which also controls redox state of cytochrome c and suppresses apoptosome formation and apoptosis. PKA; protein kinase A, JNK; c-Jun NH2-terminal kinase, CaMKII; calcium-calmodulin-dependent kinase II.
In addition to inducing MOMP, the proapoptotic BH3-only protein BAD also regulates glycolysis (Danial et al., 2003, 2008) (Fig. 9). By forming a large complex with PKA, PP1, WAVE-1, and glucokinase (hexokinase IV) on the outer mitochondrial membrane, BAD activates glucokinase and, therefore, promotes glycolysis (Danial et al., 2003). Insulin secretion from pancreatic β-cells is dependent on phosphorylation at Ser155 in the BAD BH3 domain and is impaired in BAD-deficient mice (Danial et al., 2008). A phosphomimetic BH3-peptide of BAD can restore glucokinase activity and protect pancreatic β-cells in a mouse model of diabetes (Gimenez-Cassina et al., 2014; Ljubicic et al., 2015; Szlyk et al., 2014). BAD was also shown to regulate glycolysis via activation of phosphofructokinase-1 (PFK1), a rate-limiting glycolytic enzyme, upon JNK1-mediated phosphorylation at Thr201 (Deng et al., 2008). Genetic ablation or pharmacological inhibition of JNK1 suppresses glycolysis induced by growth factor stimulation (Deng et al., 2008). These studies clearly indicate that BAD has an additional role in regulating glucose metabolism apart from its established proapoptotic activity. It remains to be investigated whether we could exploit the dual functions of BAD to kill cancer cells by modulating metabolism.
3.4 NOXA
The BH3-only protein NOXA (also known as its gene name PMAIP1) interacts with MCL-1 and promotes its proteasomal degradation (Chen et al., 2005; Czabotar et al., 2007) (Figs. 3 and 5). It has been shown that NOXA is regulated by glucose metabolism (Lowman et al., 2010) (Fig. 9). Lowman et al. showed in hematological cancer cells that the atypical cyclin-dependent kinase CDK5 is activated in a glucose-dependent manner and phosphorylates NOXA at Ser13, which leads to sequestration of NOXA in a stable multiprotein complex within the cytosol and suppression of proapoptotic activity of NOXA. Conversely, glucose deprivation results in hypophosphorylation of this residue, which leads to activation of NOXA. In support of this notion, glucose-deprivation or treatment with 2-deoxyglucose (2-DG), which inhibits hexokinase, induces NOXA-dependent apoptosis (Alves et al., 2006; Ramírez-Peinado et al., 2011). However, it was also shown in T-cell lymphoma that 2-DG-induced cell death was not completely blocked by pan-caspase inhibitor q-VD-OPH, suggesting that nonapoptotic cell death may also play a role upon glucose deprivation (Zagorodna et al., 2012).
3.5 Caspase-2
Caspase-2 is the most evolutionarily conserved caspase, sharing significant structural homology with the initiator caspase of Drosophila, Dronc. However, the critical role of caspase-2 was initially questioned by the very minor phenotype of caspase-2-deficient mice (Bergeron et al., 1998). Unlike transgenic mice lacking other caspases (e.g., caspase-8, caspase-9), which exhibit severe developmental anomalies (Bergeron et al., 1998; Hakem et al., 1998; Kuida et al., 1998; O’Reilly et al., 2002; Varfolomeev et al., 1998), caspase-2 knockout mice develop normally without notable defect except an increase in the number of oocytes in the ovary (Bergeron et al., 1998). Interestingly, the critical role of caspase-2 as an initiator caspase and its regulation by glucose metabolism were discovered by a series of biochemical studies utilizing frog egg extracts (Fig. 10). Caspase-2 activation is metabolically suppressed by phosphorylation of the prodomain at Ser135 (Ser164 in human and mouse) by calcium-calmodulin-dependent kinase II (CaMKII) (Nutt et al., 2005). CaMKII phosphorylation of caspase-2 takes place in the presence of G6P or NADPH, and can be inhibited by the G6P dehydrogenase inhibitor, dehydroisoandrosterone (DHEA), indicating that phosphorylation is positively regulated by the pentose phosphate pathway (Nutt et al., 2005). Subsequent studies showed that 14-3-3ζ binds the phosphorylated form of caspase-2 and prevents caspase-2 activation resulting from dephosphorylation by protein phosphatase 1 (PP1) (Nutt et al., 2009). 14-3-3ζ itself is also metabolically regulated by acetylation, a process which promotes release of 14-3-3ζ from caspase-2 (Andersen et al., 2011). However, in the presence of G6P, Sirtuin1 removes this acetyl group from 14-3-3ζ, keeping 14-3-3ζ bound to capsase-2 (Andersen et al., 2011). Additionally, the activity of CaMKII is also regulated by the pentose phosphate pathway. Two phosphatases, PP1 and PP2A/B55β, can inactivate CaMKII by direct dephosphorylation, which is suppressed in the presence of G6P (Huang et al., 2014; McCoy et al., 2013a). More recently, it was found that free coenzyme A (CoA), a product derived from enhanced acetyl-CoA synthesis in the presence of G6P, directly activates CaMKII (McCoy et al., 2013b). Activation of caspase-2 is also regulated by the mTOR pathway, which is discussed later (see Section 5.4).
Figure 10.

Caspase-2 is metabolically regulated. In a state of glucose abundance, CoA-activated CaMKII phosphorylates caspase-2, which creates a binding site for 14-3-3ζ and keeps caspase-2 inactive. In periods of low glycolytic flux, 14-3-3ζ is acetylated, which triggers release of this adaptor protein from caspase-2, allowing for PP1-mediated dephosphorylation of caspase-2. PP1 and PP2A also dephosphorylate and inactivate CaMKII. The dephosphorylated form of caspase-2 can then be recruited to the PIDDsome for activation.
3.6 Cytochrome c
Cytochrome c is a key protein that initiates the intrinsic apoptosis pathway (Fig. 2). Interestingly, cytochrome c itself undergoes antiapoptotic modification by glucose metabolism (Vaughn and Deshmukh, 2008) (Fig. 9). As described earlier, the pentose phosphate pathway generates NADPH, which controls redox state of cells through production of glutathione. Vaughn and Deshmukh (2008) showed that NADPH derived from the pentose phosphate pathway controls redox state of cytochrome c. In the reduced form, cytochrome c is not capable of inducing efficient apoptosome formation. Conversely, in the oxidized form, which can be created by ROS, cytochrome c can readily trigger apoptosome assembly. These results indicate that glucose metabolism negatively regulates apoptosis even downstream of MOMP through the production of NADPH. In this regard, it is noteworthy that nucleotides can directly bind to cytochrome c and prevents its interaction with APAF-1 (Chandra et al., 2006). Thus, it is possible that NADPH derived from the pentose phosphate pathway also directly contributes to the metabolic inhibition of cytochrome c.
4. P53 AND GLUCOSE METABOLISM
The tumor suppressor p53 is a transcription factor that induces cell cycle arrest, cell death, and senescence in response to various cellular stresses (Vousden and Prives, 2009). p53-inducible apoptosis genes include BAX, APAF-1, NOXA (PMAIP1), PUMA (BBC3), and PIDD (PIDD1), (for review, see Bourdon et al., 2003; Moll, 2007). Among various cellular stresses, DNA damage is the most extensively studied p53-inducing stimulus. However, recent studies indicate that changes in glucose metabolism can also modulate p53 induction. Responding to low glucose, for example, AMP-activated protein kinase (AMPK) phosphorylates and activates p53, which triggers cell cycle arrest (Jones et al., 2005). In turn, p53 also regulates glucose metabolism by suppressing glycolysis, while promoting mitochondrial respiration (Fig. 11). As p53 is genetically mutated or functionally inactivated in the majority of cancers, it is therefore expected for these cells to become highly glycolytic following loss of normal p53 activity (Muller and Vousden, 2013; Soussi and Wiman, 2015). Moreover, some of the tumor-associated p53 mutations are known as gain-of-function mutations, conferring new oncogenic properties to p53 protein (Lang et al., 2004; Olive et al., 2004; Oren and Rotter, 2010). Therefore, p53-mediated glucose metabolism is likely dysregulated in most cancers, thereby contributing toward the manifestation of the Warburg effect. Here, we will review the role p53 plays to regulate glucose metabolism and how critical it is for tumor suppression.
Figure 11.

Metabolic enzymes targeted by p53. p53 regulates basal expression of AIF and SCO2 and facilitates oxidative phosphorylation. The expression of GLUT1, GLUT4, and HK2 is negatively regulated by p53, whereas TIGAR expression is induced by p53. The net result of p53-mediated regulation of these glycolytic enzymes is the suppression of glycolysis. In addition, p53 directly binds and inhibits G6PD activity and downregulates the pentose phosphate pathway.
4.1 GLUT1/GLUT4 and HK2
Glucose uptake mediated by GLUTs is the first step of glucose metabolism. p53 limits this first step by directly binding to the GLUT1 and GLUT4 promoters, consequently attenuating their gene expression levels (Schwartzenberg-Bar-Yoseph et al., 2004). p53 that has a tumor-associated mutation in the DNA-binding domain fails to suppress the promoter activity of GLUT1 and GLUT4 (Schwartzenberg-Bar-Yoseph et al., 2004), thereby increasing cellular glucose levels. Mutant p53, but not wild-type p53, also promotes GLUT1 translocation to the plasma membrane and enhances glucose uptake (Zhang et al., 2013).
p53 also downregulates the gene expression of hexokinase-2 (HK2), an enzyme that converts glucose to G6P. The loss of the p53 gene results in an increase in HK2 mRNA levels by transcript stabilization (Wang et al., 2014). Moreover, tumor-associated mutant p53 enhances HK2 gene expression through two p53 response elements of the HK2 promoter region (Mathupala et al., 1997).
4.2 TIGAR (TP53-Induced Glycolysis and Apoptosis Regulator)
TIGAR (TP53-Induced Glycolysis and Apoptosis Regulator) was identified by microarray analysis as a novel p53-target gene inducible by DNA damage and ribosomal stress (Bensaad et al., 2006). Through a domain similar to fructose-2,6-bisphosphatase (FBPase-2), TIGAR converts fructose-2,6-bisphosphate to fructose-6-bisphosphate (Fig. 6). Fructose-2,6-bispho-sphate functions as a potent allosteric activator of PFK1, a rate-limiting enzyme of glycolysis. Therefore, TIGAR inhibits glycolysis, thereby redirecting cellular glucose metabolism to the pentose phosphate pathway shunt. As a result, TIGAR promotes generation of NADPH and ribose-5-phosphate (Bensaad et al., 2006).
The role of TIGAR in cancer is complex. As a p53-inducible gene, it is presumably a tumor suppressor. Indeed, via the production of the antioxidant NADPH, TIGAR reduces the cellular level of ROS that can cause genome instability (Bensaad et al., 2006). Interestingly, a more recent study showed that TIGAR knockout mice develop normally, but exhibit a defect in intestinal regeneration mainly due to ROS accumulation and the lack of ribose phosphate production (Cheung et al., 2013). In contrast, a mouse intestinal adenoma model showed that TIGAR is critical for tumor development—TIGAR knockout tumors were smaller than TIGAR wild type tumors, and mice with TIGAR knockout tumors exhibited enhanced survival (Cheung et al., 2013), indicating that TIGAR can facilitate tumorigenesis. Supporting the oncogenic function, TIGAR is often overexpressed in various cancer cell lines and tumor tissues (Bensaad et al., 2006; Cheung et al., 2013; Wanka et al., 2012b). Likewise, high expression levels of TIGAR protect glioma cells from cell death induced by hypoxia or ROS (Wanka et al., 2012b). Taken together, it is suggested that although TIGAR suppresses glycolysis and reduces ROS levels, activation of the pentose phosphate pathway by TIGAR could also benefit cancer cells for survival and proliferation by providing NADPH and nucleotides. Given that p53 is genetically or functionally inactivated in the majority of cancers, it is possible that the oncogenic dysregulation of TIGAR may be controlled by a p53-independent mechanism.
4.3 Apoptosis-Inducing Factor and Synthesis of Cytochrome c Oxidase 2
p53 promotes mitochondrial oxidative phosphorylation by inducing expression of at least two gene products which are essential for the mitochondrial ETC complexes: AIF and SCO2. AIF was initially found as a protein involved in caspase-independent apoptosis. Like cytochrome c, AIF resides in the mitochondrial intermembrane space. Upon MOMP, AIF translocates to the nucleus and induces DNA fragmentation independently of caspases (Joza et al., 2001; Susin et al., 1999). In addition to its procell death function, AIF contributes to maintaining ETC integrity. The loss of AIF compromises the mitochondrial respiratory chain, particularly complexes I and III, resulting in defective oxidative phosphorylation (Brown et al., 2006; Hangen et al., 2015; Vahsen et al., 2004). A recent study showed that the role of AIF in mitochondrial respiration is mediated by CHCHD4, a protein required for ETC biogenesis (Hangen et al., 2015). Interestingly, unlike other p53-targeted proapoptotic genes, such as PUMA or NOXA, AIF is induced at the basal level of p53 and its expression is not further enhanced upon cellular stress (Stambolsky et al., 2006). AIF-deficient cells exhibit increased lactate production while showing reduced oxidative phosphorylation, indicating enhanced glucose-dependency for energy production (Vahsen et al., 2004). Thus, it appears that AIF mediates the balance between glycolysis and oxidative phosphorylation controlled by p53.
SCO2 is required for the assembly of cytochrome c oxidase complex in the ETC complex IV (Matoba et al., 2006). In p53-deficient cells, SCO2 expression is downregulated and mitochondrial respiration is impaired (Matoba et al., 2006; Wanka et al., 2012a). Moreover, depletion of SCO2 is sufficient to promote glycolysis even in the presence of wild type p53 (Matoba et al., 2006), suggesting that SCO2 is a key regulator for oxidative phosphorylation. Taken together, these results further strengthen a link between the loss of p53 functions and enhanced glycolysis in cancer cells.
4.4 Glucose-6-Phosphate Dehydrogenase
Besides its roles as a transcription factor, p53 also functions in a transcription-independent manner (Green and Chipuk, 2006; Green and Kroemer, 2009). Surprisingly, p53 directly binds and inhibits Glucose-6-Phosphate Dehydrogenase (G6PD), an enzyme that catalyzes oxidation of G6P in the pentose phosphate pathway (Jiang et al., 2011) (Fig. 6). Compared to p53-proficient cells, p53-deficient MEFs or p53-depleted cells by RNAi exhibit higher glucose consumption and NADPH production (Jiang et al., 2011), suggesting the upregulation of the pentose phosphate pathway. Importantly, tumor-associated p53 mutants fail to inhibit G6PD (Jiang et al., 2011). Therefore, these results indicate that p53 functions as a tumor suppressor not only by regulating gene expression but also by directly inhibiting the pentose phosphate pathway.
5. REGULATION OF APOPTOSIS BY LIPID METABOLISM
Apoptosis induction is not solely dependent on protein–protein interaction, as lipids also play important roles. Accumulating evidence reveals the implication of lipids in a number of physiological settings which affect tumorigenesis. Several types of lipids including cholesterol (Cafforio et al., 2005; Lucken-Ardjomande et al., 2008), cardiolipin (Kuwana et al., 2002; Lutter et al., 2000; Montero et al., 2010; Ott et al., 2002), and prostaglandins (Huang et al., 2011; Lalier et al., 2011; Li et al., 2010), are known to affect apoptosis machinery. We will evaluate the recent evidence of how cancer cells manipulate this pathway to evade apoptosis, with particular emphasis on ceramide and fatty acids.
5.1 Introduction of Sphingolipid Pathway
Implication of sphingolipid metabolism in apoptosis has been discussed in depth for over a decade (Hannun and Obeid, 2008; Stancevic and Kolesnick, 2010). Sphingolipids are a complex class of lipids involved in a range of cellular processes associated with several disease states. While providing structural integrity to membranes, sphingolipid metabolism regulates major cellular processes such as apoptosis, proliferation, and senescence. Cancer associated modification of enzyme expression, activity, and subcellular localization in sphingolipid metabolism promotes suppression of apoptosis and decreased sensitivity to chemotherapeutic agents.
The sphingolipid pathway generates the bioactive lipid metabolite ceramide, which consists of a sphingosine backbone forming an amide bond with the carboxyl group of a fatty acid. Ceramide acts as the key metabolic intermediate of the sphingolipid pathway, and can be produced or utilized through several other pathways (Fig. 12). Ceramide metabolism is dynamic, with flux of ceramide into any of the described pathways being reversible at the level of at least the first metabolite with the exception of de novo synthesis where ceramide cannot be saturated back to dihydroceramide (Fig. 12). However, it is important to note that tissue specificity and responses to particular stimuli will alter the flux through the pathway.
Figure 12.

Signaling lipid pathways. Ceramide is synthesized by three pathways; the de novo pathway, the sphingomyelinase (SMase) pathway, and the salvage pathway (see detail in main text).
5.2 Pro-apoptotic Functions of Ceramide
Pro-apoptotic functions of ceramide have long been studied among the lipid metabolites (Fig. 13). One of the earliest studies that directly showed the involvement of ceramide in apoptosis was by Obeid et al. (1993); a synthetic cell membrane-permeable analog of ceramide induced chromatin fragmentation, a characteristic of apoptosis, in leukemic cells. Thereafter, a number of studies have described the molecular mechanisms of ceramide-induced apoptosis. For example, overexpression of BCL-2 or BCL-xL suppresses ceramide-induced apoptosis (El-Assaad et al., 1998; Zhang et al., 1996), whereas ceramide can inhibit BCL-2 by PP2A-mediated dephosphorylation at S70 (Ruvolo et al., 1999). Overexpression of BAX sensitizes cancer cells to ceramide (Haefen et al., 2002), whereas BAX-deficient HCT116 cells showed reduced sensitivity to ceramide-induced apoptosis (Haefen et al., 2002). Interestingly, BAX and caspase activation induced by ceramide are significantly suppressed following p53 knockdown (Kim et al., 2002). More recent studies showed that ceramide enhances BAX-induced MOMP through direct interaction with BAX and formation of a platform for BAX activation to take place on the outer mitochondrial membrane (Ganesan et al., 2010; Lee et al., 2011).
Figure 13.
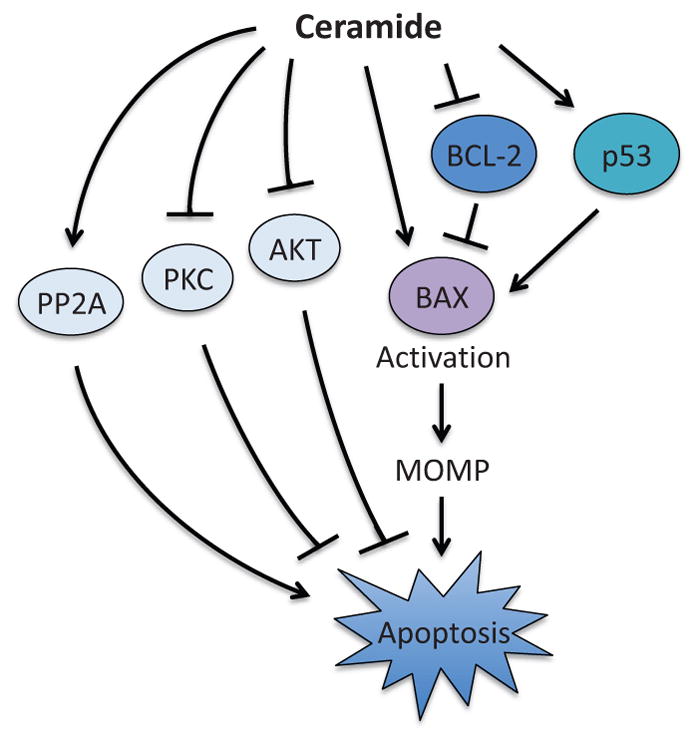
Pro-apoptotic function of ceramide. Ceramide induces MOMP through the regulation of BAX by direct binding and suppression of BCL-2 in a p53-dependent manner. Ceramide also promotes apoptosis through the regulation of PP2A, AKT, and PKC. However, the precise molecular mechanisms are unknown. PP2A; protein phosphatase 2A, PKC; protein kinase C, MOMP; mitochondrial outer membrane permeabilization.
In addition to regulating BCL-2 family proteins, ceramide is also known to impact the induction of apoptosis through the regulation of the kinase signaling pathways. Ceramide suppresses the activity of the multifunctional kinase, protein kinase C (PKC), while ceramide-induced apoptosis is suppressed by PKC activator, phorbol 12-myristate 13-acetate (Lee et al., 1996; Obeid et al., 1993). It has also been shown that ceramide promotes apoptosis through inhibition of AKT activity (Zhou et al., 1998). Conversely, the expression of constitutively active AKT could override ceramide-induced apoptosis (Zhou et al., 1998). Ceramide was also shown to activate the protein phosphatase PP2A, although the molecular mechanism still remains elusive (Lin et al., 2007).
5.3 Role of Pathways That Regulate Ceramide Production
Several apoptotic stimuli, such as tumor necrosis factor (TNF), Fas ligand, growth factor withdrawal, and DNA-damaging reagents, can increase ceramide production (Ogretmen and Hannun, 2004), suggesting that ceramide might mediate both the intrinsic and the extrinsic pathways of apoptosis induced by various stresses. Ceramide can be produced through several different enzymatic reactions (Fig. 12). Hereafter, we will review the proapoptotic function of ceramide, focusing on each of the ceramide production pathways.
5.3.1 Sphingomyelinase (SMase) Pathway
Sphingomyelinase (SMase)-mediated ceramide production has long been linked to apoptosis. In this pathway, direct hydrolysis of sphingomyelin (SM) by SMase produces ceramide, which is reversible by the action of SM synthase (Fig. 12). It has been shown that ultraviolet, ionizing radiation, TNF stimulation, and chemotherapeutics activate SMase (Hannun and Obeid, 2008). Moreover, SMase activity is required for BAX activation and MOMP induced by ultraviolet or BCL-2 inhibitors (e.g., ABT263) (Beverly et al., 2013; Kashkar et al., 2005). In addition, targeted overexpression of SMase on mitochondria is sufficient to induce BAX activation (Birbes et al., 2005). Likewise, in vitro incubation of SMase and isolated mitochondria induces BAX activation on mitochondria fraction (Birbes et al., 2005). The critical role of the SMase-mediated ceramide synthesis pathway in the induction of apoptosis was further strengthened by in vivo studies. Lymphoblast lines derived from patients with Niemann–Pick disease, a genetic disorder characterized with extremely low SMase activity, generated little ceramide and failed to induce apoptosis in response to ionizing radiation; exogenous expression of SMase could rescue this phenotype (Santana et al., 1996). Supporting this observation, SMase knockout mice displayed a defect in inducing ceramide production and apoptosis in thymus and lung following ionizing radiation or in liver after intravenous injection of anti-Fas antibody (Lin et al., 2000; Santana et al., 1996). Furthermore, a mouse xenograft study demonstrated that tumors grow much more rapidly in the absence of SMase (Garcia-Barros et al., 2003). Interestingly, when tumor cells that express SMase were implanted in SMase knockout mice, the xenograft tumors still exhibited resistance to ionizing radiation (Garcia-Barros et al., 2003), indicating that the SMase pathway may impact tumor survival not only by directly affecting ceramide production within tumor cells but also in someway regulating tumor microenvironment. Nevertheless, thymocytes and splenocytes derived from SMase knockout mice and wild type mice showed equal sensitivity to Fas ligand-induced apoptosis (Lin et al., 2000). Thus, it is suggested that the apoptotic role of SMase may operate in a cell type-specific or context-dependent manner. Of note, SMase-generated ceramide can also lead to activation of the endolysosomal protease cathepsin D, which may play a role in TNF-induced apoptosis (Heinrich et al., 2004).
5.3.2 De Novo Synthesis
De novo synthesis of ceramide begins with the production 3-ketosphinganine through the enzymatic condensation of serine and palmitoyl-CoA by serine palmitoyltransferase (SPT) (Hannun and Obeid, 2002) (Fig. 12). Subsequent reduction by 3-ketosphinganine reductase produces sphinganine, which reacts with (dihydro)ceramide synthase (CerS; six mammalian CerS isoforms have been identified) to produce the saturated ceramide precursor dihydroceramide (Pewzner-Jung et al., 2006). The final step in the de novo synthesis pathway involves the introduction of a 4, 5-trans double bond catalyzed by dihydroceramide desaturase, forming ceramide (Causeret et al., 2000).
The de novo ceramide synthesis pathway can be activated in response to various cellular stresses. An earlier study demonstrated that daunorubicin, a topoisomerase II inhibitor which elicits a DNA damage response, induces apoptosis following ceramide generation, which can be inhibited by fumonisin B1, a potent and specific inhibitor of CerS (Bose et al., 1995). Likewise, activation of the B-cell receptor, which is known to trigger activation-induced cell death, also leads to an elevation of ceramide levels in B-cells through the de novo synthesis pathway (Kroesen et al., 2001). The molecular mechanism of how the de novo pathway is activated following such cellular stimuli remains unclear. However, at least one of the CerS isoforms, CerS6, is induced by p53 (Hoeferlin et al., 2013), which may explain why DNA damage often activates ceramide production. Interestingly, activation of SPT has also been reported upon DNA damage and hypoxia in leukemia cells and neuroblastoma cells, respectively (Kang et al., 2010; Perry et al., 2000).
Interestingly, while BAK and BAX are critical for MOMP, BAK activation is required for promoting the activity of CerS (Siskind et al., 2010), indicating that a “feed-forward model” is in effect. Ceramide production is abrogated in BAK-deficient cells, but not in BAX-deficient cells (Siskind et al., 2010), illustrating a MOMP-independent role of BAK.
5.3.3 Salvage Pathway
Salvage of free sphingosine by CerS isoforms procures ceramide via a reaction which can also be reversed by the action of ceramidase; free sphingosine can further be metabolized by two sphingosine kinases (SphK1 and SphK2) to sphingosine-1-phosphate (S1P) (Fig. 12). S1P can either re-enter the sphingolipid metabolic pool by dephosphorylation by sphingosine-1-phosphate phosphatase (S1PP) or leave the pool through irreversible breakdown to phosphoethanolamine and hexadecenal by S1P lyase (S1PL).
The role of S1P is somewhat “enigmatic,” as its pro- and anti-apoptotic functions have been documented. An anti-apoptotic function of S1P was first reported in a study showing that S1P inhibits apoptosis induced by serum deprivation, TNF, or Fas stimulation (Cuvillier et al., 1996). The study also showed that PKC, directly or indirectly, activates SphK, resulting in upregulation of cellular S1P levels (Cuvillier et al., 1996). In support with this notion, apoptosis induced by PKC inhibitors can be suppressed in the presence of S1P (Cuvillier et al., 1996). Moreover, overexpression of SphK confers cells resistance to apoptosis induced by serum deprivation, ceramide, or Fas ligand through upregulation of cellular S1P levels (Olivera et al., 1999). In fact, overexpression of SphK1 is noted in several types of cancer (Datta et al., 2014; Li et al., 2009; Liu et al., 2010, 2013; Meng et al., 2014; Ruckhäberle et al., 2008; Zhang et al., 2014).
In addition to apoptosis, S1P also mediates activation of the NF-κB pathway regulating cell survival, inflammatory, and immune responses. It was demonstrated earlier that TNFα triggers activation of SphK and generation of S1P, while TNFα-induced NF-κB activation can be suppressed by the SphK inhibitor N,N-dimethylsphingosine (Xia et al., 1998). TNF receptor-associated factor 2 (TRAF2), a key ubiquitin E3 ligase for the activation of the canonical NF-κB pathway, directly interacts with SphK, which is required for TNF-induced NF-κB activation (Xia et al., 2002). Interestingly, a more recent study has shown that S1P is a critical cofactor for the catalytic activity of TRAF2 (Alvarez et al., 2010). As S1P is produced by SphK on the plasma membrane, the activation of TRAF2 by S1P directly links sphingolipid metabolism to the NF-κB pathway.
Surprisingly, S1P also crosses the plasma membrane through S1P transporters and functions as a receptor ligand (Lee et al., 1998; Maceyka et al., 2012). To date, five isoforms of S1P receptors (S1PR1–S1PR5) have been identified (Nagahashi et al., 2014; Rosen et al., 2013). Acting on the G-protein-coupled transmembrane receptors, S1P can activate various cellular signaling pathways (Spiegel and Milstien, 2003). Recently, it was shown that through S1PR1, S1P activates the ERK and the PI3K pathways, suppressing BIM expression and enhancing the accumulation of MCL-1 protein, respectively (Rutherford et al., 2013). Importantly, a significant correlation between S1PR1 expression levels and hyper-activation of the ERK pathway, and an association between high S1PR1 levels and resistance to apoptosis were documented in ER-positive breast tumors (Rutherford et al., 2013).
As opposed to the pro-survival functions of S1P, a recent in vitro reconstitution study demonstrated that S1P and hexadecenal can directly activate BAK and BAX, respectively, to promote MOMP (Chipuk et al., 2012). When considered alongside evidence of how the sphingolipid pathway is dysregulated in cancer cells, this work has dramatic implications for the field of cancer biology. Decreased activity of the SMase enzymes that catalyze this process has been observed in several types of cancer (Cheng et al., 2007; Hertervig et al., 1997; Wu et al., 2004). In colon and liver cancer cells, alternative splicing of SMase produces an isoform greatly reduced in its enzymatic activity, dramatically decreasing the amount of ceramide produced (Cheng et al., 2007; Rhein et al., 2012). Expression of S1PL, which catalyzes degradation of S1P to produce hexadecenal, is also significantly reduced in colon cancer (Oskouian et al., 2006). Moreover, the loss of S1PL confers cells resistance to DNA damage-induced apoptosis (Colié et al., 2009). These studies clearly suggest that sphingolipid metabolism plays a vital role in the mitochondrial pathway of apoptosis.
5.4 Fatty Acids
Composed of a hydrocarbon chain with a carboxylic acid at one end, fatty acids play an important role in the regulation of apoptosis. Since fatty acids contribute to plasma membrane constitution in dividing cells, acquisition of fatty acids is critical for proliferating cells, such as cancer cells. In general, cells acquire fatty acids from exogenous sources via CD36-mediated uptake and/or de novo lipogenesis involving fatty acid synthase (FASN) (Coburn et al., 2000; Febbraio et al., 1999; Glatz et al., 2010; Kuhajda, 2006; Menendez and Lupu, 2007). The de novo fatty acids synthesis pathway starts from citrate produced by the Krebs cycle in mitochondria (Fig. 6). Once cytosolic, citrate is converted to acetyl-CoA by ATP citrate lyase. Two molecules of acetyl-CoA are used to produce one molecule of malonyl-CoA by acetyl-CoA carboxylase. FASN catalyzes the synthesis of the long-chain saturated fatty acid palmitate from acetyl-CoA and malonyl-CoA with the reducing power from NADPH.
Accumulating evidence strongly suggests that FASN plays a key role in cancer metabolism (Currie et al., 2013; Menendez and Lupu, 2007). Upregulation of FASN expression has been reported in several cancer types, including breast, prostate, ovarian, lung, melanoma, and lymphoma (Gelebart et al., 2012; Orita et al., 2008; Rahman et al., 2012; Ueda et al., 2010; Zecchin et al., 2010). Given the dependence of such cancer cells on FASN for fatty acid production, genetic or pharmacological inhibition of FASN has been shown to be highly effective to specifically kill cancer cells, but not normal cells, by inducing apoptosis (De Schrijver et al., 2003; Kuhajda, 2006; Pizer et al., 1996; Zhou et al., 2003). To date, several FASN inhibitors have been developed (e. g., cerulenin, C75, and orlistat) (Kridel et al., 2004; Kuhajda et al., 2000). However, it has been noted that despite their in vitro efficacy, cerulenin and C75 have serious side effect in vivo: anorexia and body weight loss (Loftus et al., 2000). The side effect appears to be accounted for by a neuronal effect that reduces appetite, which hampers the use of the inhibitors for cancer patients. In an effort to avoid the adverse effect, the next generation of FASN inhibitors is currently being developed (Turrado et al., 2012).
Although the attenuation of fatty acid production is clearly an important consequence of FASN blockage, the toxicity of FASN inhibition may also be mediated, in part, by the accumulation of malonyl-CoA (Bandyopadhyay et al., 2006; Pizer et al., 2000). As FASN produces fatty acids by catalyzing the condensation reaction between malonyl-CoA and acetyl-CoA, inhibition of FASN will result in accumulation of these substrates (Pizer et al., 2000) (Fig. 6). Interestingly, 5-(tetradecyloxy)-2-furoic acid (TOFA), an inhibitor of acetyl-CoA carboxylase (ACC), also prevents fatty acid production by blocking ACC that catalyzes the conversion of acetyl-CoA to malonyl-CoA (Fig. 6) but does not show the same cytotoxicity as FASN inhibitors (Pizer et al., 2000). Of note, while FASN inhibitors increase cellular malonyl-CoA levels, TOFA does not do so and rather reduces the level of malonyl-CoA (Pizer et al., 2000). Therefore, in addition to the reduced fatty acid production, the cytotoxicity of FASN inhibitors may also be mediated by elevated malonyl-CoA levels. Malonyl-CoA can function as a physiological inhibitor of the outer mitochondrial membrane enzyme CPT-1, which catalyzes β-oxidation in mitochondria (Bandyopadhyay et al., 2006). Through this pathway, the accumulation of malonyl-CoA enhances an increased cellular level of ceramide, contributing to apoptosis induction in part (Bandyopadhyay et al., 2006).
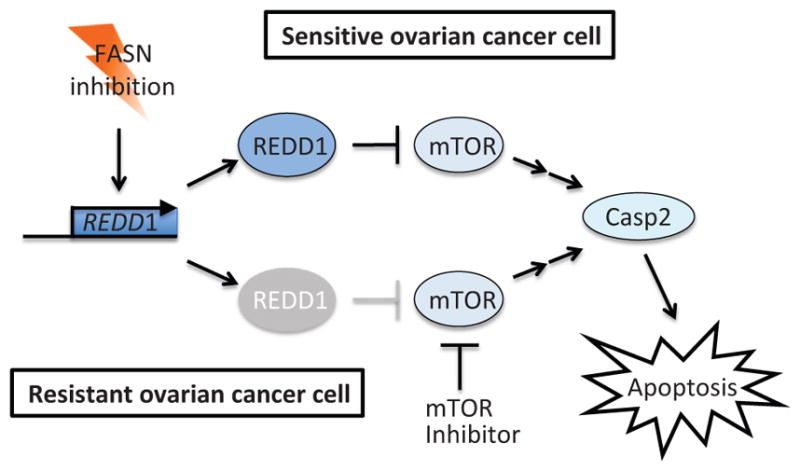
Recently, it was found that FASN inhibitors induce caspase-2 activation by upregulation of REDD1, a known hypoxia-inducible mTOR pathway suppressor (Knowles et al., 2008; Yang et al., 2015). Since REDD1 is an inhibitor of the mTOR pathway, this finding positions caspase-2 downstream of the mTOR pathway (Fig. 14). FASN inhibitors readily induce REDD1 expression followed by caspase-2 activation in FASN inhibitor-sensitive ovarian cancer cells. However, in FASN inhibitor-resistant cells, the expression of REDD1 is attenuated and caspase-2 activation is abrogated (Yang et al., 2015). Importantly, pharmacological inhibition of mTOR could sensitize the resistant cells to FASN inhibition (Yang et al., 2015), suggesting the efficacy of combination therapy with an mTOR inhibitor and a FASN inhibitor in this type of cancer.
Figure 14.

Activation of caspase-2 by FASN inhibitors is mediated by the mTOR pathway. Upon FASN inhibition, the mTOR inhibitory protein REDD1 is induced and stimulates caspase-2 activation and apoptosis in FASN-sensitive ovarian cancer cells. On the other hand, in FASN-resistant ovarian cancer cells, REDD1 induction is impaired, but mTOR inhibitor treatment can substitute function of REDD1 and restore caspase-2-mediated sensitization of cells to apoptosis.
While many cancer cells are “addicted” to FASN for fatty acid production, a crosstalk between FASN and the receptor tyrosine kinase HER2 has been noted (Fig. 15). HER2 overexpression is observed in 20 and 15% of breast and ovarian cancer, respectively, and is associated with poor clinical outcomes (Berchuck et al., 1990; Seshadri et al., 1993; Slamon et al., 1987, 1989; Tan et al., 2011). FASN positively regulates HER2 oncogene expression (Menendez et al., 2004). Conversely, HER2 directly phosphorylates and activates FASN, while it also promotes FASN gene expression (Jin et al., 2010; Kumar-Sinha et al., 2003). Pharmacological or RNAi-mediated inhibition of FASN attenuates HER2 expression in breast cancer cells (Menendez et al., 2004). Furthermore, cotreatment with the HER2 inhibitor trastuzumab and cerulenin showed synergistic effects in causing apoptotic cell death in HER2-positive breast cancer cells (Menendez et al., 2004; Vazquez-Martin et al., 2007). Whether a similar crosstalk is present between FASN and other oncoproteins remains elusive.
Figure 15.
Regulation of FASN activity and expression by HER2. In HER2-positive cancer cells, HER2 overexpression increases FASN expression through the PI3K/AKT and MEK/ERK pathways. HER2 also directly phosphorylates and enhances the enzymatic activity of FASN. FASN in turn upregulates HER2 expression, thereby establishing an autoregulatory loop. HER2; human epidermal growth factor receptor 2, FASN; fatty acid synthase, ERK; extracellular-signal-regulated kinase 2.
6. CONCLUDING REMARKS
Metabolic reprogramming in cancer cells has been investigated over 60 years since Otto Warburg first described glycolytic characteristics of cancer cells. Roles of lipid metabolites, including ceramide and fatty acids, in cell survival have also been noted since the early 1990’s. Whereas it still remains under much debate whether such altered metabolism is a cause or a consequence of the carcinogenic process, metabolic changes certainly provide the advantage for excessive proliferation and survival in cancer cells. Importantly, inactivation of p53 would prompt a metabolic shift toward glycolysis for energy production. Given the high frequency of p53 inactivation observed in cancer cells, either genetically or functionally, there clearly is a crosstalk between metabolism and the apoptotic pathway. Over the last decade, we have gained significant insight into how various metabolites impact MOMP by directly interacting with BCL-2 family proteins or indirectly modulating signaling pathways that regulate BCL-2 family proteins. Further studies are required to fully uncover the link between apoptotic defects and unique metabolic preferences in cancer cells. Identification of such a relationship and its underlying molecular mechanism will help us develop a novel therapeutic strategy that targets the altered metabolic traits to selectively eliminate cancer cells by promoting apoptosis.
Acknowledgments
We would like to thank Sally Kornbluth and Owen Wilkins for valuable discussion. We are grateful for our grant support: NCI Career Development Award R00 CA140948 (to M.K.), American Cancer Society Institutional Research Grant IRG-82-003-30 (to M.K.), Hitchcock Foundation Pilot Grant (to M.K.), and Cancer Center core grant P30 CA23108 (to the Norris Cotton Cancer Center).
References
- Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423–432. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–7464. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, Milstien S, Spiegel S. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves NL, Derks IAM, Berk E, Spijker R, van Lier RAW, Eldering E. The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity. 2006;24:703–716. doi: 10.1016/j.immuni.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Andersen JL, Thompson JW, Lindblom KR, Johnson ES, Yang CS, Lilley LR, Freel CD, Moseley MA, Kornbluth S. A biotin switch-based proteomics approach identifies 14-3-3ζ as a target of Sirt1 in the metabolic regulation of caspase-2. Mol Cell. 2011;43:834–842. doi: 10.1016/j.molcel.2011.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Zhan R, Wang Y, Pai SK, Hirota S, Hosobe S, Takano Y, Saito K, Furuta E, Iiizumi M, Mohinta S, Watabe M, Chalfant C, Watabe K. Mechanism of apoptosis induced by the inhibition of fatty acid synthase in breast cancer cells. Cancer Res. 2006;66:5934–5940. doi: 10.1158/0008-5472.CAN-05-3197. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res. 2005;94:29–86. doi: 10.1016/S0065-230X(05)94002-5. [DOI] [PubMed] [Google Scholar]
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29:2570–2581. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Berchuck A, Kamel A, Whitaker R, Kerns B, Olt G, Kinney R, Soper JT, Dodge R, Clarke-Pearson DL, Marks P. Overexpression of HER-2/neu is associated with poor survival in advanced epithelial ovarian cancer. Cancer Res. 1990;50:4087–4091. [PubMed] [Google Scholar]
- Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beverly LJ, Howell LA, Hernandez-Corbacho M, Casson L, Chipuk JE, Siskind LJ. BAK activation is necessary and sufficient to drive ceramide synthase-dependent ceramide accumulation following inhibition of BCL2-like proteins. Biochem J. 2013;452:111–119. doi: 10.1042/BJ20130147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birbes H, Luberto C, Hsu YT, El Bawab S, Hannun YA, Obeid LM. A mitochondrial pool of sphingomyelin is involved in TNFalpha-induced Bax translocation to mitochondria. Biochem J. 2005;386:445–451. doi: 10.1042/BJ20041627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995;82:405–414. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- Bourdon JC, Laurenzi VD, Melino G, Lane D. p53: 25 years of research and more questions to answer. Cell Death Differ. 2003;10:397–399. doi: 10.1038/sj.cdd.4401243. [DOI] [PubMed] [Google Scholar]
- Brown D, Yu BD, Joza N, Benit P, Meneses J, Firpo M, Rustin P, Penninger JM, Martin GR. Loss of Aif function causes cell death in the mouse embryo, but the temporal progression of patterning is normal. Proc Natl Acad Sci USA. 2006;103:9918–9923. doi: 10.1073/pnas.0603950103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchakjian MR, Kornbluth S. The engine driving the ship: metabolic steering of cell proliferation and death. Nat Rev Mol Cell Biol. 2010;11:715–727. doi: 10.1038/nrm2972. [DOI] [PubMed] [Google Scholar]
- Cafforio P, Dammacco F, Gernone A, Silvestris F. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis. 2005;26:883–891. doi: 10.1093/carcin/bgi036. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K, Horne D, Feunteun J, Lenoir G, Medema R, Vainchenker W, Kroemer G. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004;23:4362–4370. doi: 10.1038/sj.onc.1207572. [DOI] [PubMed] [Google Scholar]
- Causeret C, Geeraert L, Van der Hoeven G, Mannaerts GP, Van Veldhoven PP. Further characterization of rat dihydroceramide desaturase: tissue distribution, subcellular localization, and substrate specificity. Lipids. 2000;35:1117–1125. doi: 10.1007/s11745-000-0627-6. [DOI] [PubMed] [Google Scholar]
- Chandel NS, vander Heiden MG, Thompson CB, Schumacker PT. Redox regulation of p53 during hypoxia. Oncogene. 2000;19:3840–3848. doi: 10.1038/sj.onc.1203727. [DOI] [PubMed] [Google Scholar]
- Chandra D, Bratton SB, Person MD, Tian Y, Martin AG, Ayres M, Fearnhead HO, Gandhi V, Tang DG. Intracellular nucleotides act as critical prosurvival factors by binding to cytochrome C and inhibiting apoptosome. Cell. 2006;125:1333–1346. doi: 10.1016/j.cell.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Wu J, Hertervig E, Lindgren S, Duan D, Nilsson A, Duan RD. Identification of aberrant forms of alkaline sphingomyelinase (NPP7) associated with human liver tumorigenesis. Br J Cancer. 2007;97:1441–1448. doi: 10.1038/sj.bjc.6604013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, Athineos D, Lee P, Ridgway RA, Lambie W, Nixon C, Strathdee D, Blyth K, Sansom OJ, Vousden KH. TIGAR Is Required for Efficient Intestinal Regeneration and Tumorigenesis. Dev Cell. 2013;25(5):463–477. doi: 10.1016/j.devcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CW, Kanies C, Kim KW, Fang WB, Parkhurst C, Xie M, Henry T, Yang E. Protein phosphatase 2A dephosphorylation of phosphoserine 112 plays the gatekeeper role for BAD-mediated apoptosis. Mol Cell Biol. 2003;23:6350–6362. doi: 10.1128/MCB.23.18.6350-6362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipuk JE, McStay GP, Bharti A, Kuwana T, Clarke CJ, Siskind LJ, Obeid LM, Green DR. Sphingolipid metabolism cooperates with BAK and BAX to promote the mitochondrial pathway of apoptosis. Cell. 2012;148:988–1000. doi: 10.1016/j.cell.2012.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn CT, Knapp FF, Febbraio M, Beets AL, Silverstein RL, Abumrad NA. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J Biol Chem. 2000;275:32523–32529. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- Colié S, Van Veldhoven PP, Kedjouar B, Bedia C, Albinet V, Sorli SC, Garcia V, Djavaheri-Mergny M, Bauvy C, Codogno P, Levade T, Andrieu-Abadie N. Disruption of sphingosine 1-phosphate lyase confers resistance to chemotherapy and promotes oncogenesis through Bcl-2/Bcl-xL upregulation. Cancer Res. 2009;69:9346–9353. doi: 10.1158/0008-5472.CAN-09-2198. [DOI] [PubMed] [Google Scholar]
- Coloff JL, Macintyre AN, Nichols AG, Liu T, Gallo CA, Plas DR, Rathmell JC. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011a;71:5204–5213. doi: 10.1158/0008-5472.CAN-10-4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coloff JL, Mason EF, Altman BJ, Gerriets VA, Liu T, Nichols AN, Zhao Y, Wofford JA, Jacobs SR, Ilkayeva O, Garrison SP, Zambetti GP, Rathmell JC. Akt requires glucose metabolism to suppress puma expression and prevent apoptosis of leukemic T cells. J Biol Chem. 2011b;286:5921–5933. doi: 10.1074/jbc.M110.179101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie E, Schulze A, Zechner R, Walther TC, Farese RV. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DCS, Fairlie WD, Hinds MG, Colman PM. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci USA. 2007;104:6217–6222. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- Dang CV. Links between metabolism and cancer. Genes Dev. 2012a;26:877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. MYC on the path to cancer. Cell. 2012b;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, Gygi SP, Korsmeyer SJ. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 2003;424:952–956. doi: 10.1038/nature01825. [DOI] [PubMed] [Google Scholar]
- Danial NN, Walensky LD, Zhang CY, Choi CS, Fisher JK, Molina AJA, Datta SR, Pitter KL, Bird GH, Wikstrom JD, Deeney JT, Robertson K, Morash J, Kulkarni A, Neschen S, Kim S, Greenberg ME, Corkey BE, Shirihai OS, Shulman GI, Lowell BB, Korsmeyer SJ. Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat Med. 2008;14:144–153. doi: 10.1038/nm1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Loo SY, Huang B, Wong L, Tan SSL, Tan TZ, Lee SC, Thiery JP, Lim YC, Yong WP, Lam Y, Kumar AP, Yap CT. SPHK1 regulates proliferation and survival responses in triple-negative breast cancer. Oncotarget. 2014;5:5920–5933. doi: 10.18632/oncotarget.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Ranger AM, Lin MZ, Sturgill JF, Ma YC, Cowan CW, Dikkes P, Korsmeyer SJ, Greenberg ME. Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell. 2002;3:631–643. doi: 10.1016/s1534-5807(02)00326-x. [DOI] [PubMed] [Google Scholar]
- De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res. 2003;63:3799–3804. [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- del Peso L, González-García M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Deng H, Yu F, Chen J, Zhao Y, Xiang J, Lin A. Phosphorylation of Bad at Thr-201 by JNK1 promotes glycolysis through activation of phosphofructokinase-1. J Biol Chem. 2008;283:20754–20760. doi: 10.1074/jbc.M800024200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Venkatachalam MA, Wang J, Patel Y, Saikumar P, Semenza GL, Force T, Nishiyama J. Up-regulation of apoptosis inhibitory protein IAP-2 by hypoxia. Hif-1-independent mechanisms. J Biol Chem. 2001;276:18702–18709. doi: 10.1074/jbc.M011774200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Wang J. Hypoxia selection of death-resistant cells. A role for Bcl-X(L) J Biol Chem. 2004;279:9215–9221. doi: 10.1074/jbc.M312225200. [DOI] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell. 2002;13:2276–2288. doi: 10.1091/mbc.01-12-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Assaad W, El-Sabban M, Awaraji C, Abboushi N, Dbaibo GS. Distinct sites of action of Bcl-2 and Bcl-xL in the ceramide pathway of apoptosis. Biochem J. 1998;336(Pt 3):735–741. doi: 10.1042/bj3360735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Erler JT, Cawthorne CJ, Williams KJ, Koritzinsky M, Wouters BG, Wilson C, Miller C, Demonacos C, Stratford IJ, Dive C. Hypoxia-mediated downregulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol. 2004;24:2875–2889. doi: 10.1128/MCB.24.7.2875-2889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SF, Silverstein RL. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol. 2013;23:620–633. doi: 10.1016/j.tcb.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Jänicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2002;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Debatin KM. HIF-1-regulated glucose metabolism: a key to apoptosis resistance? Cell Cycle. 2007;6:790–792. doi: 10.4161/cc.6.7.4084. [DOI] [PubMed] [Google Scholar]
- Ganesan V, Perera MN, Colombini D, Datskovskiy D, Chadha K, Colombini M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis. 2010;15:553–562. doi: 10.1007/s10495-009-0449-0. [DOI] [PubMed] [Google Scholar]
- Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–1159. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- Gelebart P, Zak Z, Anand M, Belch A, Lai R. Blockade of Fatty Acid Synthase Triggers Significant Apoptosis in Mantle Cell Lymphoma. PLoS One. 2012;7:e33738. doi: 10.1371/journal.pone.0033738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez-Cassina A, Garcia-Haro L, Choi CS, Osundiji MA, Lane EA, Huang H, Yildirim MA, Szlyk B, Fisher JK, Polak K, Patton E, Wiwczar J, Godes M, Lee DH, Robertson K, Kim S, Kulkarni A, Distefano A, Samuel V, Cline G, Kim YB, Shulman GI, Danial NN. Regulation of hepatic energy metabolism and gluconeogenesis by BAD. Cell Metab. 2014;19:272–284. doi: 10.1016/j.cmet.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatz JFC, Luiken JJFP, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev. 2010;90:367–417. doi: 10.1152/physrev.00003.2009. [DOI] [PubMed] [Google Scholar]
- Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001;15:1406–1418. doi: 10.1101/gad.889901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Chipuk JE. p53 and metabolism: Inside the TIGAR. Cell. 2006;126:30–32. doi: 10.1016/j.cell.2006.06.032. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Große L, Wurm CA, Brüser C, Neumann D, Jans DC, Jakobs S. Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 2016;35:402–413. doi: 10.15252/embj.201592789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Haefen C, Wieder T, Gillissen B, Stärck L, Graupner V, Dörken B, Daniel PT. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene. 2002;21:4009–4019. doi: 10.1038/sj.onc.1205497. [DOI] [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Hangen E, Feraud O, Lachkar S, Mou H, Doti N, Fimia GM, Lam NV, Zhu C, Godin I, Muller K, Chatzi A, Nuebel E, Ciccosanti F, Flamant S, Benit P, Perfettini JL, Sauvat A, Bennaceur-Griscelli A, Ser-Le Roux K, Gonin P, Tokatlidis K, Rustin P, Piacentini M, Ruvo M, Blomgren K, Kroemer G, Modjtahedi N. Interaction between AIF and CHCHD4 Regulates Respiratory Chain Biogenesis. Mol Cell. 2015;58:1001–1014. doi: 10.1016/j.molcel.2015.04.020. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. The Ceramide-centric Universe of Lipid-mediated Cell Regulation: Stress Encounters of the Lipid Kind. J Biol Chem. 2002;277:25847–25850. doi: 10.1074/jbc.R200008200. [DOI] [PubMed] [Google Scholar]
- Heinrich M, Neumeyer J, Jakob M, Hallas C, Tchikov V, Winoto-Morbach S, Wickel M, Schneider-Brachert W, Trauzold A, Hethke A, Schütze S. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ. 2004;11:550–563. doi: 10.1038/sj.cdd.4401382. [DOI] [PubMed] [Google Scholar]
- Hertervig E, Nilsson A, Nyberg L, Duan RD. Alkaline sphingomyelinase activity is decreased in human colorectal carcinoma. Cancer. 1997;79:448–453. [PubMed] [Google Scholar]
- Hoeferlin LA, Fekry B, Ogretmen B, Krupenko SA, Krupenko NI. Folate stress induces apoptosis via p53-dependent de novo ceramide synthesis and up-regulation of ceramide synthase 6. J Biol Chem. 2013;288:12880–12890. doi: 10.1074/jbc.M113.461798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Yang CS, Wojton J, Huang NJ, Chen C, Soderblom EJ, Zhang L, Kornbluth S. Metabolic control of Ca2+/calmodulin-dependent protein kinase II (CaMKII)-mediated caspase-2 suppression by the B55β/protein phosphatase 2A (PP2A) J Biol Chem. 2014;289:35882–35890. doi: 10.1074/jbc.M114.585844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O’Sullivan B, He Z, Peng Y, Tan AC, Zhou L, Shen J, Han G, Wang XJ, Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS, Li CY. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17:860–866. doi: 10.1038/nm.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K, Yamanaka K, Kamura T, Minato N, Conaway RC, Conaway JW, Klausner RD, Pause A. Identification of the von Hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc Natl Acad Sci USA. 1999;96:12436–12441. doi: 10.1073/pnas.96.22.12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, Mckinnon PJ, Cleveland JL, Zambetti GP. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310–316. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Yuan LX, Boulbes D, Baek JM, Wang YN, Gomez-Cabello D, Hawke DH, Yeung SC, Lee MH, Hortobagyi GN, Hung MC, Esteva FJ. Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overexpressing breast cancer cells. Breast Cancer Res. 2010;12:R96. doi: 10.1186/bcr2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanford WL, Cho SK, Li CYJ, Sasaki T, Elia AJ, Cheng HYM, Ravagnan L, Ferri KF, Zamzami N, Wakeham A, Hakem R, Yoshida H, Kong YY, Mak TW, Zúñiga-Pflücker JC, Kroemer G, Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci USA. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MS, Ahn KH, Kim SK, Jeon HJ, Ji JE, Choi JM, Jung KM, Jung SY, Kim DK. Hypoxia-induced neuronal apoptosis is mediated by de novo synthesis of ceramide through activation of serine palmitoyltransferase. Cell Signal. 2010;22:610–618. doi: 10.1016/j.cellsig.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Kashkar H, Wiegmann K, Yazdanpanah B, Haubert D, Krönke M. Acid sphingomyelinase is indispensable for UV light-induced Bax conformational change at the mitochondrial membrane. J Biol Chem. 2005;280:20804–20813. doi: 10.1074/jbc.M410869200. [DOI] [PubMed] [Google Scholar]
- Kilic M, Kasperczyk H, Fulda S, Debatin KM. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene. 2007;26:2027–2038. doi: 10.1038/sj.onc.1210008. [DOI] [PubMed] [Google Scholar]
- Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27:7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kim SS, Chae HS, Bach JH, Lee MW, Kim KY, Lee WB, Jung YM, Bonventre JV, Suh YH. P53 mediates ceramide-induced apoptosis in SKN-SH cells. Oncogene. 2002;21:2020–2028. doi: 10.1038/sj.onc.1205037. [DOI] [PubMed] [Google Scholar]
- Knowles LM, Yang C, Osterman A, Smith JW. Inhibition of fatty-acid synthase induces caspase-8-mediated tumor cell apoptosis by up-regulating DDIT4. J Biol Chem. 2008;283:31378–31384. doi: 10.1074/jbc.M803384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 2004;64:2070–2075. doi: 10.1158/0008-5472.can-03-3645. [DOI] [PubMed] [Google Scholar]
- Kroesen BJ, Pettus B, Luberto C, Busman M, Sietsma H, de Leij L, Hannun YA. Induction of apoptosis through B-cell receptor cross-linking occurs via de novo generated C16-ceramide and involves mitochondria. J Biol Chem. 2001;276:13606–13614. doi: 10.1074/jbc.M009517200. [DOI] [PubMed] [Google Scholar]
- Kuhajda FP. Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. 2006;66:5977–5980. doi: 10.1158/0008-5472.CAN-05-4673. [DOI] [PubMed] [Google Scholar]
- Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc Natl Acad Sci USA. 2000;97:3450–3454. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Kumar-Sinha C, Ignatoski KW, Lippman ME, Ethier SP, Chinnaiyan AM. Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer Res. 2003;63:132–139. [PubMed] [Google Scholar]
- Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell. 2009;138:838–854. doi: 10.1016/j.cell.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Lalier L, Cartron PF, Olivier C, Logé C, Bougras G, Robert JM, Oliver L, Vallette FM. Prostaglandins antagonistically control Bax activation during apoptosis. Cell Death Differ. 2011;18:528–537. doi: 10.1038/cdd.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El-Naggar AK, Lozano G. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–872. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Lavrik IN, Golks A, Baumann S, Krammer PH. Caspase-2 is activated at the CD95 death-inducing signaling complex in the course of CD95-induced apoptosis. Blood. 2006;108:559–565. doi: 10.1182/blood-2005-07-007096. [DOI] [PubMed] [Google Scholar]
- Lee H, Rotolo JA, Mesicek J, Penate-Medina T, Rimner A, Liao WC, Yin X, Ragupathi G, Ehleiter D, Gulbins E, Zhai D, Reed JC, Haimovitz-Friedman A, Fuks Z, Kolesnick R. Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PLoS One. 2011;6:e19783. doi: 10.1371/journal.pone.0019783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Hannun YA, Obeid LM. Ceramide inactivates cellular protein kinase Calpha. J Biol Chem. 1996;271:13169–13174. doi: 10.1074/jbc.271.22.13169. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Van Brocklyn JR, Thangada S, Liu CH, Hand AR, Menzeleev R, Spiegel S, Hla T. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Li F, Huang Q, Chen J, Peng Y, Roop DR, Bedford JS, Li C-Y. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal. 2010;3:ra13–ra13. doi: 10.1126/scisignal.2000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Li W, Yu CP, Xia JT, Zhang L, Weng GX, Zheng HQ, Kong QL, Hu LJ, Zeng MS, Zeng YX, Li M, Li J, Song LB. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res. 2009;15:1393–1399. doi: 10.1158/1078-0432.CCR-08-1158. [DOI] [PubMed] [Google Scholar]
- Lim JH, Lee ES, You HJ, Lee JW, Park JW, Chun YS. Ras-dependent induction of HIF-1alpha785 via the Raf/MEK/ERK pathway: a novel mechanism of Ras-mediated tumor promotion. Oncogene. 2004;23:9427–9431. doi: 10.1038/sj.onc.1208003. [DOI] [PubMed] [Google Scholar]
- Lin CF, Chen CL, Chiang CW, Jan MS, Huang WC, Lin YS. GSK-3beta acts downstream of PP2A and the PI 3-kinase-Akt pathway, and upstream of caspase-2 in ceramide-induced mitochondrial apoptosis. J Cell Sci. 2007;120:2935–2943. doi: 10.1242/jcs.03473. [DOI] [PubMed] [Google Scholar]
- Lin T, Genestier L, Pinkoski MJ, Castro A, Nicholas S, Mogil R, Paris F, Fuks Z, Schuchman EH, Kolesnick RN, Green DR. Role of acidic sphingomyelinase in Fas/CD95-mediated cell death. J Biol Chem. 2000;275:8657–8663. doi: 10.1074/jbc.275.12.8657. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisztwan J, Imbert G, Wirbelauer C, Gstaiger M, Krek W. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 1999;13:1822–1833. doi: 10.1101/gad.13.14.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zheng H, Zhang Z, Wu Z, Xiong H, Li J, Song L. Overexpression of sphingosine kinase 1 is associated with salivary gland carcinoma progression and might be a novel predictive marker for adjuvant therapy. BMC Cancer. 2010;10:495. doi: 10.1186/1471-2407-10-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SQ, Su YJ, Qin MB, Mao YB, Huang JA, Tang GD. Sphingosine kinase 1 promotes tumor progression and confers malignancy phenotypes of colon cancer by regulating the focal adhesion kinase pathway and adhesion molecules. Int J Oncol. 2013;42:617–626. doi: 10.3892/ijo.2012.1733. [DOI] [PubMed] [Google Scholar]
- Ljubicic S, Polak K, Fu A, Wiwczar J, Szlyk B, Chang Y, Alvarez-Perez JC, Bird GH, Walensky LD, Garcia-Ocaña A, Danial NN. Phospho-BAD BH3 mimicry protects β cells and restores functional β cell mass in diabetes. Cell Rep. 2015;10:497–504. doi: 10.1016/j.celrep.2014.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science. 2000;288:2379–2381. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]
- Lowman XH, McDonnell MA, Kosloske A, Odumade OA, Jenness C, Karim CB, Jemmerson R, Kelekar A. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol Cell. 2010;40:823–833. doi: 10.1016/j.molcel.2010.11.035. [DOI] [PubMed] [Google Scholar]
- Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. 2002;277:23111–23115. doi: 10.1074/jbc.M202487200. [DOI] [PubMed] [Google Scholar]
- Lucken-Ardjomande S, Montessuit S, Martinou JC. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ. 2008;15:484–493. doi: 10.1038/sj.cdd.4402280. [DOI] [PubMed] [Google Scholar]
- Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–761. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- Lüthi AU, Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 2007;14:641–650. doi: 10.1038/sj.cdd.4402103. [DOI] [PubMed] [Google Scholar]
- Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22:50–60. doi: 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, Chandel NS, Thompson CB, Robey RB, Hay N. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004a;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Robey RB, Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol Cell Biol. 2004b;24:730–740. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra R, Brosius FC. Glucose uptake and glycolysis reduce hypoxia-induced apoptosis in cultured neonatal rat cardiac myocytes. J Biol Chem. 1999;274:12567–12575. doi: 10.1074/jbc.274.18.12567. [DOI] [PubMed] [Google Scholar]
- Manzl C, Krumschnabel G, Bock F, Sohm B, Labi V, Baumgartner F, Logette E, Tschopp J, Villunger A. Caspase-2 activation in the absence of PIDDosome formation. J Cell Biol. 2009;185:291–303. doi: 10.1083/jcb.200811105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzl C, Peintner L, Krumschnabel G, Bock F, Labi V, Drach M, Newbold A, Johnstone R, Villunger A. PIDDosome-independent tumor suppression by Caspase-2. Cell Death Differ. 2012;19:1722–1732. doi: 10.1038/cdd.2012.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997;272:22776–22780. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–760. doi: 10.1016/j.molcel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- McCoy F, Darbandi R, Chen SI, Eckard L, Dodd K, Jones K, Baucum AJ, Gibbons JA, Lin SH, Colbran RJ, Nutt LK. Metabolic regulation of CaMKII protein and caspases in Xenopus laevis egg extracts. J Biol Chem. 2013a;288:8838–8848. doi: 10.1074/jbc.M112.437186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy F, Darbandi R, Lee HC, Bharatham K, Moldoveanu T, Grace CR, Dodd K, Lin W, Chen SI, Tangallapally RP, Kurokawa M, Lee RE, Shelat AA, Chen T, Green DR, Harris RA, Lin SH, Fissore RA, Colbran RJ, Nutt LK. Metabolic activation of CaMKII by coenzyme A. Mol Cell. 2013b;52:325–339. doi: 10.1016/j.molcel.2013.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, Lupu R. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene over-expression in cancer cells. Proc Natl Acad Sci USA. 2004;101:10715–10720. doi: 10.1073/pnas.0403390101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XD, Zhou ZS, Qiu JH, Shen WH, Wu Q, Xiao J. Increased SPHK1 expression is associated with poor prognosis in bladder cancer. Tumour Biol. 2014;35:2075–2080. doi: 10.1007/s13277-013-1275-0. [DOI] [PubMed] [Google Scholar]
- Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, Ratcliffe PJ. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll UM. P53 Has a Direct Pro-apoptotic Action at the Mitochondria. In: Hainaut P, Wiman KG, editors. 25 Years of P53 Research. Springer; Netherlands, Dordrecht: 2007. pp. 165–181. [Google Scholar]
- Montero J, Mari M, Colell A, Morales A, Basañez G, Garcia-Ruiz C, Fernández-Checa JC. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim Biophys Acta. 2010;1797:1217–1224. doi: 10.1016/j.bbabio.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PAJ, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- Nagahashi M, Takabe K, Terracina KP, Soma D, Hirose Y, Kobayashi T, Matsuda Y, Wakai T. Sphingosine-1-phosphate transporters as targets for cancer therapy. Biomed Res Int. 2014;2014:651727. doi: 10.1155/2014/651727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt LK, Buchakjian MR, Gan E, Darbandi R, Yoon SY, Wu JQ, Miyamoto YJ, Gibbons JA, Gibbon JA, Andersen JL, Freel CD, Tang W, He C, Kurokawa M, Wang Y, Margolis SS, Fissore RA, Kornbluth S. Metabolic control of oocyte apoptosis mediated by 14-3-3zeta-regulated dephosphorylation of caspase-2. Dev Cell. 2009;16(6):856–866. doi: 10.1016/j.devcel.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt LK, Margolis SS, Jensen M, Herman CE, Dunphy WG, Rathmell JC, Kornbluth S. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell. 2005;123:89–103. doi: 10.1016/j.cell.2005.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly LA, Ekert P, Harvey N, Marsden V, Cullen L, Vaux DL, Hacker G, Magnusson C, Pakusch M, Cecconi F, Kuida K, Strasser A, Huang DCS, Kumar S. Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ. 2002;9:832–841. doi: 10.1038/sj.cdd.4401033. [DOI] [PubMed] [Google Scholar]
- Obeid LM, Linardic CM, Karolak LA, Hannun YA. Programmed cell death induced by ceramide. Science. 1993;259:1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4:604–616. doi: 10.1038/nrc1411. [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–860. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Olivera A, Kohama T, Edsall L, Nava V, Cuvillier O, Poulton S, Spiegel S. Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J Cell Biol. 1999;147:545–558. doi: 10.1083/jcb.147.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson M, Vakifahmetoglu H, Abruzzo PM, Högstrand K, Grandien A, Zhivotovsky B. DISC-mediated activation of caspase-2 in DNA damage-induced apoptosis. Oncogene. 2009;28:1949–1959. doi: 10.1038/onc.2009.36. [DOI] [PubMed] [Google Scholar]
- Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orita H, Coulter J, Tully E, Kuhajda FP, Gabrielson E. Inhibiting fatty acid synthase for chemoprevention of chemically induced lung tumors. Clin Cancer Res. 2008;14:2458–2464. doi: 10.1158/1078-0432.CCR-07-4177. [DOI] [PubMed] [Google Scholar]
- Oskouian B, Sooriyakumaran P, Borowsky AD, Crans A, Dillard-Telm L, Tam YY, Bandhuvula P, Saba JD. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc Natl Acad Sci USA. 2006;103:17384–17389. doi: 10.1073/pnas.0600050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell. 2007;128:533–546. doi: 10.1016/j.cell.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry DK, Carton J, Shah AK, Meredith F, Uhlinger DJ, Hannun YA. Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J Biol Chem. 2000;275:9078–9084. doi: 10.1074/jbc.275.12.9078. [DOI] [PubMed] [Google Scholar]
- Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J Biol Chem. 2006;281:25001–25005. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- Pizer ES, Thupari J, Han WF, Pinn ML, Chrest FJ, Frehywot GL, Townsend CA, Kuhajda FP. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000;60:213–218. [PubMed] [Google Scholar]
- Pizer ES, Wood FD, Heine HS, Romantsev FE, Pasternack GR, Kuhajda FP. Inhibition of fatty acid synthesis delays disease progression in a xenograft model of ovarian cancer. Cancer Res. 1996;56:1189–1193. [PubMed] [Google Scholar]
- Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB. Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J Biol Chem. 2001;276:12041–12048. doi: 10.1074/jbc.M010551200. [DOI] [PubMed] [Google Scholar]
- Rahman MT, Nakayama K, Rahman M, Katagiri H, Katagiri A, Ishibashi T, Ishikawa M, Iida K, Nakayama N, Otsuki Y, Nakayama S, Miyazaki K. Fatty acid synthase expression associated with NAC1 is a potential therapeutic target in ovarian clear cell carcinomas. Br J Cancer. 2012;107:300–307. doi: 10.1038/bjc.2012.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez-Peinado S, Alcázar-Limones F, Lagares-Tena L, El Mjiyad N, Caro-Maldonado A, Tirado OM, Munoz-Pinedo C. 2-deoxyglucose induces Noxa-dependent apoptosis in alveolar rhabdomyosarcoma. Cancer Res. 2011;71:6796–6806. doi: 10.1158/0008-5472.CAN-11-0759. [DOI] [PubMed] [Google Scholar]
- Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, Thompson CB. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol. 2003;23:7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhein C, Tripal P, Seebahn A, Konrad A, Kramer M, Nagel C, Kemper J, Bode J, Mühle C, Gulbins E, Reichel M, Becker CM, Kornhuber J. Functional implications of novel human acid sphingomyelinase splice variants. PLoS One. 2012;7:e35467. doi: 10.1371/journal.pone.0035467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribe EM, Jean YY, Goldstein RL, Manzl C, Stefanis L, Villunger A, Troy CM. Neuronal caspase 2 activity and function requires RAIDD, but not PIDD. Biochem J. 2012;444:591–599. doi: 10.1042/BJ20111588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Stevens RC, Hanson M, Roberts E, Oldstone MBA. Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu Rev Biochem. 2013;82:637–662. doi: 10.1146/annurev-biochem-062411-130916. [DOI] [PubMed] [Google Scholar]
- Ruckhäberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, Grösch S, Geisslinger G, Holtrich U, Karn T, Kaufmann M. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat. 2008;112:41–52. doi: 10.1007/s10549-007-9836-9. [DOI] [PubMed] [Google Scholar]
- Rutherford C, Childs S, Ohotski J, McGlynn L, Riddick M, MacFarlane S, Tasker D, Pyne S, Pyne NJ, Edwards J, Palmer TM. Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis. 2013;4:e927. doi: 10.1038/cddis.2013.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvolo PP, Deng X, Ito T, Carr BK, May WS. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J Biol Chem. 1999;274:20296–20300. doi: 10.1074/jbc.274.29.20296. [DOI] [PubMed] [Google Scholar]
- Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–4015. [PubMed] [Google Scholar]
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J, García-Sáez AJ. Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 2016;35:389–401. doi: 10.15252/embj.201593384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana P, Peña LA, Haimovitz-Friedman A, Martin S, Green DR, McLoughlin M, Cordon-Cardo C, Schuchman EH, Fuks Z, Kolesnick R. Acid sphingomye-linase-deficient human lymphoblasts and mice are defective in radiation-induced apopto-sis. Cell. 1996;86:189–199. doi: 10.1016/s0092-8674(00)80091-4. [DOI] [PubMed] [Google Scholar]
- Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627–2633. doi: 10.1158/0008-5472.can-03-0846. [DOI] [PubMed] [Google Scholar]
- Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3:169–179. doi: 10.1016/s1097-2765(00)80308-1. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123:3664–3671. doi: 10.1172/JCI67230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010a;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010b;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today. 2007;12:853–859. doi: 10.1016/j.drudis.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med. 2002;8:S62–S67. doi: 10.1016/s1471-4914(02)02317-1. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- Seshadri R, Firgaira FA, Horsfall DJ, McCaul K, Setlur V, Kitchen P. Clinical significance of HER-2/neu oncogene amplification in primary breast cancer. The South Australian Breast Cancer Study Group J Clin Oncol. 1993;11:1936–1942. doi: 10.1200/JCO.1993.11.10.1936. [DOI] [PubMed] [Google Scholar]
- Shi Y. Apoptosome: the cellular engine for the activation of caspase-9. Structure. 2002;10:285–288. doi: 10.1016/s0969-2126(02)00732-3. [DOI] [PubMed] [Google Scholar]
- Shiozaki EN, Chai J, Shi Y. Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. Proc Natl Acad Sci USA. 2002;99:4197–4202. doi: 10.1073/pnas.072544399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, Kanki JP, Green DR, D’Andrea AA, Look AT. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell. 2008;133:864–877. doi: 10.1016/j.cell.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siskind LJ, Mullen TD, Romero Rosales K, Clarke CJ, Hernandez-Corbacho MJ, Edinger AL, Obeid LM. The BCL-2 protein BAK is required for long-chain ceramide generation during apoptosis. J Biol Chem. 2010;285:11818–11826. doi: 10.1074/jbc.M109.078121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- Soussi T, Wiman KG. TP53: an oncogene in disguise. Cell Death Differ. 2015;22:1239–1249. doi: 10.1038/cdd.2015.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- Stambolsky P, Weisz L, Shats I, Klein Y, Goldfinger N, Oren M, Rotter V. Regulation of AIF expression by p53. Cell Death Differ. 2006;13:2140–2149. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- Stancevic B, Kolesnick R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010;584:1728–1740. doi: 10.1016/j.febslet.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J Cell Biol. 2005;170:295–304. doi: 10.1083/jcb.200409117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Szlyk B, Braun CR, Ljubicic S, Patton E, Bird GH, Osundiji MA, Matschinsky FM, Walensky LD, Danial NN. A phospho-BAD BH3 helix activates glucoki-nase by a mechanism distinct from that of allosteric activators. Nat Struct Mol Biol. 2014;21:36–42. doi: 10.1038/nsmb.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabi-lization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- Tan DSP, Iravani M, McCluggage WG, Lambros MBK, Milanezi F, Mackay A, Gourley C, Geyer FC, Vatcheva R, Millar J, Thomas K, Natrajan R, Savage K, Fenwick K, Williams A, Jameson C, El-Bahrawy M, Gore ME, Gabra H, Kaye SB, Ashworth A, Reis-Filho JS. Genomic analysis reveals the molecular heterogeneity of ovarian clear cell carcinomas. Clin Cancer Res. 2011;17:1521–1534. doi: 10.1158/1078-0432.CCR-10-1688. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Tinel A, Janssens S, Lippens S, Cuenin S, Logette E, Jaccard B, Quadroni M, Tschopp J. Autoproteolysis of PIDD marks the bifurcation between pro-death caspase-2 and pro-survival NF-kappaB pathway. EMBO J. 2007;26:197–208. doi: 10.1038/sj.emboj.7601473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel A, Tschopp J. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science. 2004;304:843–846. doi: 10.1126/science.1095432. [DOI] [PubMed] [Google Scholar]
- Turrado C, Puig T, García-Cárceles J, Artola M, Benhamú B, Ortega-Gutiérrez S, Relat J, Oliveras G, Blancafort A, Haro D, Marrero PF, Colomer R, López-Rodríguez ML. New synthetic inhibitors of fatty acid synthase with anticancer activity. J Med Chem. 2012;55:5013–5023. doi: 10.1021/jm2016045. [DOI] [PubMed] [Google Scholar]
- Ueda SM, Yap KL, Davidson B, Tian Y, Murthy V, Wang TL, Visvanathan K, Kuhajda FP, Bristow RE, Zhang H, Shih IM. Expression of Fatty Acid Synthase Depends on NAC1 and Is Associated with Recurrent Ovarian Serous Carcinomas. J Oncol. 2010;2010:285191. doi: 10.1155/2010/285191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unruh A, Ressel A, Mohamed HG, Johnson RS, Nadrowitz R, Richter E, Katschinski DM, Wenger RH. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene. 2003;22:3213–3220. doi: 10.1038/sj.onc.1206385. [DOI] [PubMed] [Google Scholar]
- Vahsen N, Candé C, Brière JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schägger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Mol Cell Biol. 2001;21(17):5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- Vaughn AE, Deshmukh M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat Cell Biol. 2008;10:1477–1483. doi: 10.1038/ncb1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting “HER2 super-expression” occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol. 2007;31:769–776. [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, Zhang Y, You MJ, Koochekpour S, Saleem M, Huang H, Lu J, Deng Y. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014;8:1461–1474. doi: 10.1016/j.celrep.2014.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanka C, Brucker DP, Bähr O, Ronellenfitsch M, Weller M, Steinbach JP, Rieger J. Synthesis of cytochrome C oxidase 2: a p53-dependent metabolic regulator that promotes respiratory function and protects glioma and colon cancer cells from hypoxia-induced cell death. Oncogene. 2012a;31:3764–3776. doi: 10.1038/onc.2011.530. [DOI] [PubMed] [Google Scholar]
- Wanka C, Steinbach JP, Rieger J. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J Biol Chem. 2012b;287:33436–33446. doi: 10.1074/jbc.M112.384578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Cheng Y, Nilsson A, Duan RD. Identification of one exon deletion of intestinal alkaline sphingomyelinase in colon cancer HT-29 cells and a differentiation-related expression of the wild-type enzyme in Caco-2 cells. Carcinogenesis. 2004;25:1327–1333. doi: 10.1093/carcin/bgh140. [DOI] [PubMed] [Google Scholar]
- Xia P, Gamble JR, Rye KA, Wang L, Hii CS, Cockerill P, Khew-Goodall Y, Bert AG, Barter PJ, Vadas MA. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci USA. 1998;95:14196–14201. doi: 10.1073/pnas.95.24.14196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Wang L, Moretti PAB, Albanese N, Chai F, Pitson SM, D’Andrea RJ, Gamble JR, Vadas MA. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem. 2002;277:7996–8003. doi: 10.1074/jbc.M111423200. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Wang HG. The protein kinase PKB/Akt regulates cell survival and apoptosis by inhibiting Bax conformational change. Oncogene. 2001;20:7779–7786. doi: 10.1038/sj.onc.1204984. [DOI] [PubMed] [Google Scholar]
- Yang CS, Matsuura K, Huang NJ, Robeson AC, Huang B, Zhang L, Kornbluth S. Fatty acid synthase inhibition engages a novel caspase-2 regulatory mechanism to induce ovarian cancer cell death. Oncogene. 2015;34:3264–3272. doi: 10.1038/onc.2014.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Yu C, Minemoto Y, Zhang J, Liu J, Tang F, Bui TN, Xiang J, Lin A. JNK suppresses apoptosis via phosphorylation of the proapoptotic Bcl-2 family protein BAD. Mol Cell. 2004;13:329–340. doi: 10.1016/s1097-2765(04)00028-0. [DOI] [PubMed] [Google Scholar]
- Zagorodna O, Martin SM, Rutkowski DT, Kuwana T, Spitz DR, Knudson CM. 2-deoxyglucose-induced toxicity is regulated by Bcl-2 family members and is enhanced by antagonizing Bcl-2 in lymphoma cell lines. Oncogene. 2012;31:2738–2749. doi: 10.1038/onc.2011.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecchin KG, Rossato FA, Raposo HF, Melo DR, Alberici LC, Oliveira HC, Castilho RF, Coletta RD, Vercesi AIBE, Graner E. Inhibition of fatty acid synthase in melanoma cells activates the intrinsic pathway of apoptosis. Lab Invest. 2010;91:232–240. doi: 10.1038/labinvest.2010.157. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, Hu W, Feng Z. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang J, Alter N, Reed JC, Borner C, Obeid LM, Hannun YA. Bcl-2 interrupts the ceramide-mediated pathway of cell death. Proc Natl Acad Sci USA. 1996;93:5325–5328. doi: 10.1073/pnas.93.11.5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang Y, Wan Z, Liu S, Cao Y, Zeng Z. Sphingosine kinase 1 and cancer: a systematic review and meta-analysis. PLoS One. 2014;9:e90362. doi: 10.1371/journal.pone.0090362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Altman BJ, Coloff JL, Herman CE, Jacobs SR, Wieman HL, Wofford JA, Dimascio LN, Ilkayeva O, Kelekar A, Reya T, Rathmell JC. Glycogen synthase kinase 3alpha and 3beta mediate a glucose-sensitive antiapoptotic signaling pathway to stabilize Mcl-1. Mol Cell Biol. 2007;27:4328–4339. doi: 10.1128/MCB.00153-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Coloff JL, Ferguson EC, Jacobs SR, Cui K, Rathmell JC. Glucose metabolism attenuates p53 and Puma-dependent cell death upon growth factor deprivation. J Biol Chem. 2008;283:36344–36353. doi: 10.1074/jbc.M803580200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, Simons JW, Semenza GL. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. [PubMed] [Google Scholar]
- Zhou H, Summers SA, Birnbaum MJ, Pittman RN. Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem. 1998;273:16568–16575. doi: 10.1074/jbc.273.26.16568. [DOI] [PubMed] [Google Scholar]
- Zhou W, Simpson PJ, McFadden JM, Townsend CA, Medghalchi SM, Vadlamudi A, Pinn ML, Ronnett GV, Kuhajda FP. Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells. Cancer Res. 2003;63:7330–7337. [PubMed] [Google Scholar]