

Abstract
Previous studies have indicated that lysine (K)-specific demethylase 3A (KDM3A) is associated with diverse diabetes-associated cardiovascular complications in response to high glucose levels. However, the effects of KDM3A on the pathological progression of cardiovascular injuries in response to high insulin levels remain unknown. The present study aimed to explore whether KDM3A knockdown may attenuate high insulin-induced vascular smooth muscle cell (VSMC) dysfunction, and to further investigate the underlying mechanisms. Primary VSMCs were isolated from the thoracic aorta of Sprague-Dawley rats. Lentiviral vectors encoding control-small interfering (si)RNA or KDM3A-siRNA were transduced into VSMCs for 72 h, and cells were subsequently incubated in medium containing 100 nM insulin for a further 5 days. Cellular proli feration, migration and apoptosis were measured by Cell Counting kit-8, Transwell chamber assay and flow cytometry, respectively. Reactive oxygen species (ROS) were detected using the dihydroethidium fluorescent probe. The mRNA expression levels of interleukin-6 and monocyte chemotactic protein-1 were measured by reverse transcription-quantitative polymerase chain reaction. Furthermore, the protein expression levels of KDM3A, mitogen-activated protein kinases (MAPKs), nuclear factor (NF)-κB/p65, B-cell lymphoma 2 (Bcl-2)-associated X protein and Bcl-2 were evaluated by western blotting. Lentivirus transduction with KDM3A-siRNA markedly reduced the elevated expression of KDM3A induced by high insulin stimulation in VSMCs. In addition, inhibition of KDM3A significantly ameliorated insulin-induced VSMC proliferation and migration, which was accompanied by decreased ROS levels, cell apoptosis and inflammatory cytokine levels. Furthermore, KDM3A gene silencing mitigated phosphorylation of MAPKs and NF-κB/p65 activation. In conclusion, KDM3A inhibition may exert numerous protective effects on high insulin-stimulated VSMCs, and the underlying mechanisms may be partly associated with inactivation of MAPK/NF-κB signaling pathways.
Keywords: lysine (K)-specific demethylase 3A, vascular smooth muscle cells, insulin, mitogen-activated protein kinase/nuclear factor-κB
Introduction
Diabetes mellitus (DM) is a severe metabolic disorder, which is associated with a high rate of hospitalization and mortality, due to diverse micro- and macrovascular complications (1). The coexistence of DM in patients with coronary artery disease (CAD) results in increased severity of vascular lumen stenosis and plaque burden (2,3). Furthermore, the risk of restenosis following percutaneous coronary intervention is increased in these patients (4,5). In healthy individuals, physiological concentrations of insulin facilitate vasodilatation and maintain blood circulation. However, in patients with type 2 DM, hyperinsulinemia, secondary to insulin resistance, is a prominent feature; therefore, the physiological functions of insulin are impaired. Previous clinical and epidemiological studies have indicated that hyperinsulinemia serves a critical role in the pathogenesis of DM-associated vascular diseases (6–8). In addition, high serum levels of insulin are regarded as an independent risk factor and poor prognostic indicator of CAD (9).
Vascular smooth muscle cells (VSMCs) have an impo rtant role in the pathogenesis of vascular occlusive diseases (10–12). VSMC proliferation, and migration of VSMCs from the media to the intima, is a critical step in atherosclerosis-associated intimal thickening and post-angioplasty restenosis (10,13). As well as hyperglycemic stress in type 2 DM, hyperinsulinemia may also induce VSMC dysfunction. It has previously been reported that abnormal serum insulin levels are capable of promoting the progression of atherosclerosis (14). In addition, increased proliferation and migration of VSMCs, as well as increased oxidative stress and inflammation, may be closely linked (15). Further attention should therefore be paid to elucidate the molecular mechanisms underlying hyperinsulinemia-induced VSMC dysfunction, in order to identify potential targets and novel therapeutic approaches for the treatment of DM-associated cardiovascular complications.
Lysine (K)-specific demethylase 3A (KDM3A), also known as Jumonji domain-containing 1A, contains a zinc finger structural domain and possesses histone demethylase activity (16). Previous studies have indicated that KDM3A participates in numerous pathophysiological processes, through the modulation of cellular proliferation, senility, metabolism and tumor occurrence (17–19). However, its potential functions in DM-associated cardiovascular complications have yet to be fully elaborated. Our previous study demonstrated that KDM3A expression was upregulated in type 2 diabetic vascular and heart tissue, whereas downregulation of KDM3A markedly promoted rehabilitation of diabetic vascular complications and attenuated diabetic myocardial injury; KDM3A inhibition also significantly alleviated high glucose-induced VSMC dysfunction in vitro (20). However, to the best of our knowledge, the effects of KDM3A on hyperinsulinemia-induced VSMC dysfunction remain unknown. The present study aimed to explore the potential protective effects and underlying mechanisms of KDM3A inhibition on VSMC proliferation and migration, as well as oxidative stress, inflammation and apoptosis in response to a high concentration of insulin in vitro.
Materials and methods
Primary VSMC isolation and culture
Primary VSMCs were isolated from the thoracic aorta of male Sprague-Dawley (SD) rats (weight, 150–180 g) according to the previously described tissue explant method (21). Ten male SD wild-type rats (SPF grade, 4–5 week) were obtained from Wuhan University Experiment Animal Center. Rats were given free access to standard rat chow and water until the time of the experiment and were housed in a temperature- and light-controlled environment (24°C; 12/12 h-light/dark cycle). In order to obtain the primary VSMCs, the SD rats were sacrificed. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 1% penicillin/streptomycin (Hyclone; GE Healthcare Life Sciences) at 37°C in an atmosphere containing 95% air and 5% CO2. VSMCs were identified by morphology and immunoreactivity to α-smooth muscle actin, as previously described (21). VSMCs at the 3rd and 5th passages were used for the subsequent experiments. The present study was approved by the Animal Care and Use Committee of Wuhan University (Wuhan, China), and experiments were conducted in accordance with Institutional Guidelines and the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85–23, revised 1996).
Small interfering (si)RNA transduction and experimental designs
siRNA specific to the rat KDM3A gene was constructed and purchased from Shanghai GeneChem Co., Ltd. (Shanghai, China). Our preliminary study indicated that a lentivirus encoding KDM3A-siRNA exhibited optimal infection efficiency with 40 multiplicity of infection (MOI) in rat VSMCs (20). In the present study, equal amounts of lenti-KDM3A-siRNA or lenti-control-siRNA (as a negative control; siRNA sequence, GCAGGTGTCACTAGGCTTA; Shanghai GeneChem Co., Ltd.) at 40 MOI were transduced into VSMCs once they reached 40–50% confluence. The lentiviruses were constructed by Shanghai GeneChem Co., Ltd. After 12 h, the lentivirus-containing serum-free medium was replaced with complete culture medium. A total of 72 h post-transduction, enhanced green fluorescent protein expression was observed under a fluorescent microscopy to confirm successful transduction of the lentiviruses. In addition, western blotting was used to directly detect KDM3A expression, and to thereby indicate the efficiency of lentivirus transduction.
To determine whether KDM3A participated in high insulin-induced VSMC dysfunction, VSMCs were initially divided into two groups: i) Control group, in which VSMCs were cultured in normal medium for 5 days and ii) high concentration insulin stimulation group (HI), in which VSMCs were cultured in medium containing 100 nM insulin (to imitate the hyperinsulinemic state) for 5 days. Secondly, to further determine the precise roles and underlying mechanism of KDM3A in VSMCs undergoing treatment with a high concentration of insulin, VSMCs were randomly divided into three groups: i) Normal medium + control siRNA group (N + C-siRNA), in which VSMCs incubated with lenti-control-siRNA for 12 h, and incubated in culture medium until 72 h post-tranfection, and were then cultured with normal medium for a further 5 days; ii) high concentration insulin stimulation + control siRNA group (HI + C-siRNA), in which VSMCs transduced with lenti-control-siRNA for 72 h were cultured in medium containing 100 nM insulin for 5 days; iii) high concentration insulin stimulation + KDM3A-siRNA group (HI + K-siRNA), in which VSMCs transduced with lenti-KDM3A-siRNA for 72 h were cultured in medium containing 100 nM insulin for 5 days.
Cell proliferation and migration assays
To determine the proliferative ability of VSMCs in the various groups, cells were digested and seeded in 96-well plates at 6×103 cells/well in 200 μl corresponding culture medium. Following 24 h incubation, 20 μl Cell Counting kit-8 (CCK-8) reagent (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was added to each well and the cells were incubated for a further 4 h. Subsequently, the optical density (OD) values of all wells were determined using a microplate spectrophotometer (Bio-Rad Laboratories, Inc., Hercules, CA, USA) at 450 nm.
A Transwell chamber assay (Corning Inc., Corning, NY, USA) was performed to measure the migratory activity of VMSCs. Briefly, VSMCs were digested and seeded in the upper chamber with 200 μl serum-free medium at 1×105. Subsequently, 600 μl DMEM containing 10% FBS was added to the lower chamber and was incubated for 12 h at 37°C. The non-migrating cells on the upper side of the membrane were gently removed using a cotton swab. The cells that had migrated to the underside of the membrane were fixed with methanol for 30 min and stained with 0.5% crystal violet in the dark for 30 min at 25°C. The number of migrated cells was counted under a light microscope (Olympus Corporation, Tokyo, Japan) at ×200 magnification in at least three random fields.
Reactive oxygen species (ROS) detection
Dihydroethidium (DHE) fluorescent probe (Beyotime Institute of Biotechnology, Haimen, China) was used to detect ROS generation in VSMCs. Briefly, VSMCs from each group were adjusted to a density of 1.5×105/well and were seeded into 24-well plates. After 24 h, the medium was removed and the DHE fluorescent probe, dissolved in serum-free medium, was added to all of the wells (10 μmol/l) for 30 min at 37°C. Subsequently, DHE fluorescence intensity was immediately observed using a fluorescence microscopy at an excitatory wavelength of 495 nm. Images from at least three random fields were captured and the mean fluorescence intensity was calculated with ImageJ software version 1.49 (National Institutes of Health, Bethesda, MD, USA).
Apoptosis assays
For the identification of apoptotic cells, Annexin V-allophycocyanin (APC)/7-aminoactinomycin (7-AAD) double staining was performed. Briefly, cells were harvested, washed twice with ice-cold PBS and were then stained using an apoptosis detection kit (Apoptosis Assay kit; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) according to the manufacturer’s protocol. Cells were resuspended in PBS and incubated with binding buffer containing 5 μl 7-AAD at room temperature for 15 min in the dark. Subsequently, 1 μl Annexin V-APC was added to the cell suspension and incubated for 15 min in the dark. Finally, cells were analyzed by fluorescence-activated cell sorting within 1 h using a FACSCalibur with CellQuest software version 5.1 (Bio-Rad Laboratories, Inc.). Cells exhibiting Annexin V-APC-and 7-AAD-negative staining were considered viable. Cells that stained positively for Annexin V-APC only were considered early apoptotic. Cells exhibiting Annexin V-APC- and 7-AAD-positive staining were considered late apoptotic or necrosis. The percentage of early apoptotic plus late apoptotic cells was calculated following flow cytometric analysis.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) assay
RT-qPCR was used to detect the mRNA expression levels of interleukin (IL)-6 and monocyte chemotactic protein (MCP)-1 in VSMCs. Total RNA was extracted from the samples and was purified using a RNA isolation kit (TRIzol; Applied Biosystems; Thermo Fisher Scientific, Inc.). Isolated RNA was reverse transcribed into cDNA using a cDNA synthesis kit (First-Strand Synthesis system; Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocol. cDNA was then amplified using an ABI Prism 7500 Sequence Detection system (Applied Biosy stems; Thermo Fisher Scientific, Inc.) in the presence of the appropriate proportions of primers and SYBR-Green Supermix (Bio-Rad Laboratories, Inc.). PCR cycling conditions were as follows: Predenaturation at 50°C for 120 sec, followed by 40 cycles of denaturation at 95°C for 15 sec, annealing at 60°C for 30 sec and extension at 60°C for 30 sec. The 4 types of deoxynucleo-tides in the single chain were connected to the primers according to the principles of Watson-Crick base pairing, and then extended to form complementary DNA double strand. GAPDH expression was used as an internal control. Gene expression levels were calculated using the comparative quantification method (2−ΔΔCq) (22). The primer sequences used in the present study were as follows: MCP-1, forward 5′-GTCGGCTGGAGAACTACAAGA-3′, reverse 5′-GTGCTGAAGTCCTTAGGGTTG-3′; IL-6, forward 5′-GTTGCCTTCTTGGGACTGATG-3′, reverse 5′-TACTGGTCTGTTGTGGGTGGT-3′; and GAPDH, forward 5′-ACAGCAACAGGGTGGTGGAC-3′ and reverse 5′-TTTGAGGGTGCAGCGAACTT-3′.
Western blotting
VSMCs were lysed using a RIPA Lysis buffer (P0013B; Beyotime Institute of Biotechnology), and nuclear and cytoplasmic proteins were extracted according to the manufacturer’s protocol (Beyotime Institute of Biotechnology). Following centrifugation at 14926 × g at 4°C for 5 min, the supernatant was collected and protein concentration was detected using a bicinchoninic acid protein assay (Beyotime Institute of Biotechnology). Protein samples (concentration, 4.5 μg/μl) were separated by 10% SDS-PAGE and were electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes (EMD Millipore, Billerica, MA, USA). The nonspecific binding sites in the membranes were blocked with 5% non-fat dried milk dissolved in Tris-buffered saline containing 0.1% Tween-20 for 90 min at 25°C. Subsequently, the PVDF membranes were incubated with primary antibodies (dilution, 1:1,000) against KDM3A (ab106456), GADPH (ab181602), B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax; ab32503) and Bcl-2 (ab32124) (all Abcam, Cambridge, MA, USA); extracellular signal-regulated kinase (ERK)1/2 (9102) and phosphorylated (p)-ERK1/2 (4370) (both Cell Signaling Technology, Inc., Danvers, MA, USA); c-Jun NH2-terminal protein kinase (JNK) (51151-1-AP; Proteintech Group, Inc., Chicago, IL, USA), p-JNK (ab131499; Abcam), p38 (9212) and p-p38 (4511) (both Cell Signaling Technology, Inc.), nuclear factor (NF)-κB/p65 (BS1257; Bioworld Technology, Inc., St. Louis Park, MN, USA) overnight at 4°C, followed by incubation with corresponding secondary antibodies (HRP-labeled goat anti-rabbit; dilution 1:50,000; BA1054; Boster, Wuhan, China) at room temperature for 60 min. The protein bands were visualized using an enhanced chemiluminescence system (Thermo Fisher Scientific, Inc.). GAPDH was used as the internal loading control. The blots were semi-quantified using BandScan 4.30 (Glyko, Novato, CA, USA).
Statistical analysis
Statistical analyses were conducted using SPSS statistical software (version 19.0; IBM Corp., Armonk, NY, USA). All continuous values are expressed as the mean ± standard deviation. Student’s t-test was used for between-group comparisons. One-way analysis of variance followed by Bonferroni post hoc test was used for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
High insulin stimulation promotes the proliferation and migration of VSMCs
The effects of high concentration insulin on the proliferation and migration of VSMCs were determined using a CCK-8 assay kit and Transwell chamber assay, respectively. As shown in Fig. 1A, VSMC proliferation was significantly increased >2-fold in the HI group compared with in the control group (P<0.05). Consistently, the migration of VSMCs in the HI group was markedly elevated compared with in the control group (P<0.05; Fig. 1B). These findings indicated that high insulin concentration may induce VSMC dysfunction by increasing proliferative and migratory abilities.
Figure 1.
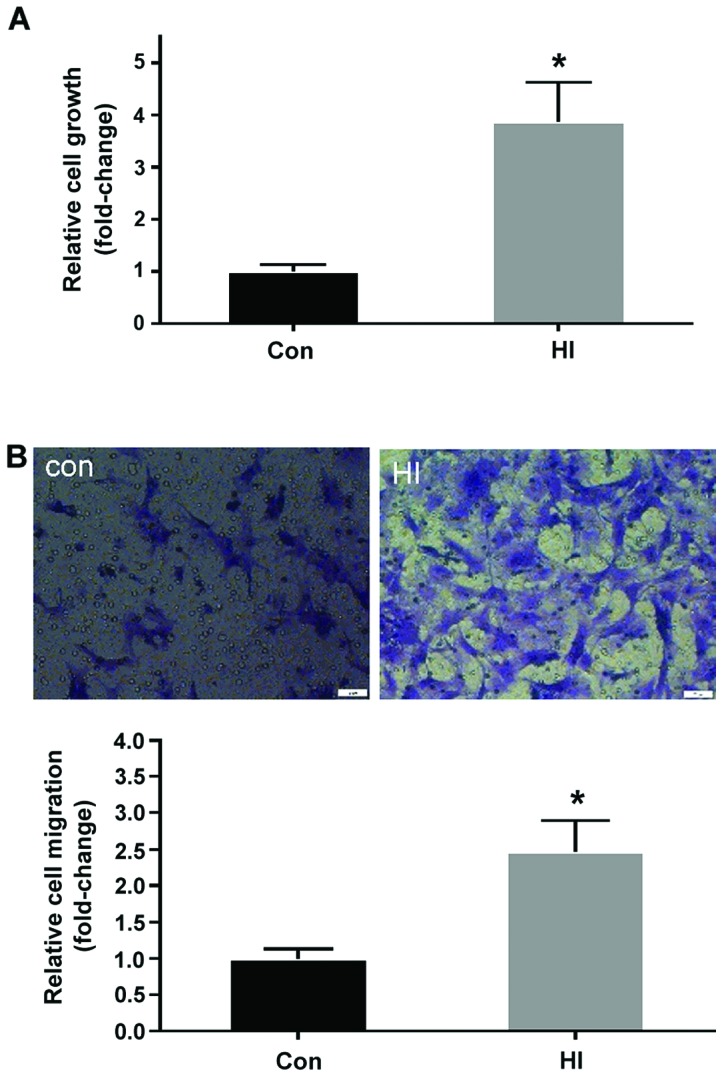
Treatment with a high insulin concentration significantly promotes proliferation and migration of VSMCs. (A) Effects of high insulin on VSMC proliferation (n=5). (B) Representative images of migrated VSMCs following 100 nM insulin stimulation (magnification, ×200) (upper panel). Relative cell migration was calculated following high insulin stimulation (n=3) (lower panel). All values are expressed as the mean ± standard deviation. *P<0.05 vs. the Con group. Con, control group; HI, high insulin group; VSMCs, vascular smooth muscle cells.
High insulin stimulation induces ROS generation, apoptosis and inflammation, accompanied by increased KDM3A expression in VSMCs
ROS generation, apoptosis and inflammation are considered the central contributors associated with the induction of VSMC dysfunction. To further investigate the effects of high insulin levels on ROS production in VSMCs, ROS concentrations were evaluated by measuring the intensity of DHE fluorescence. As shown in Fig. 2A, compared with in the control group, the mean fluorescent density of ROS in the HI group was significantly enhanced (P<0.05). The effects of insulin were also observed on apoptosis and inflammation. As shown in Fig. 2B, the percentage of apoptotic cells (2.32±0.46%) was very low in the control group, whereas the percentage of apoptotic cells was increased to 16.20±0.30% following treatment with 100 nM insulin (P<0.05). Furthermore, the ratio between proapoptotic Bax and anti-apoptotic Bcl-2 expression was significantly altered in response to high insulin levels. As shown in Fig. 2C, the expression levels of Bax were markedly increased, whereas Bcl-2 expression was decreased, in the HI group compared with in the control group (P<0.05). With regards to inflammation, the mRNA expression levels of IL-6 and MCP-1 were significantly increased in VSMCs following stimulation with high insulin levels compared with in the control group (P<0.05; Fig. 2D).
Figure 2.

Treatment with a high insulin concentration markedly increases ROS production, cell apoptosis and inflammation, accompanied by increased KDM3A expression. (A) Representative fluorescent images of dihydroethidium staining in the control and HI groups (magnification, ×100) (left panel). Fluorescent mean density was determined using ImageJ software (right panel). (B) Representative flow cytometry plots of apoptotic VSMCs following stimulation with 100 nM insulin (left panel). Relative apoptotic rates of VSMCs were determined in the control and HI groups (right panel). (C) Representative immunoblots for Bax and Bcl-2 expression in VSMCs stimulated with 100 nM insulin (upper panel). Densitometric analysis of western blotting (lower panel). (D) Effects of high insulin stimulation on mRNA expression levels of IL-6 and MCP-1 in VSMCs. (E) Effects of 100 nM insulin on KDM3A expression, as determined by western blotting (left panel). Densitometric analyses of western blotting (right panel). All values are expressed as the mean ± standard deviation, n=3. *P<0.05 vs. the Con group. Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2; Con, control group; HI, high insulin group; IL, interleukin; KDM3A, lysine (K)-specific demethylase 3A; MCP-1, monocyte chemotactic protein-1; ROS, reactive oxygen species; VSMCs, vascular smooth muscle cells
The present study aimed to further determine whether KDM3A is involved in high insulin-induced VSMC pathological injury. Western blotting was performed to detect KDM3A expression. As shown in Fig. 2E, KDM3A expression was markedly upregulated in high insulin-stimulated VSMCs compared with in the control group (P<0.05). Taken to gether these findings suggested that KDM3A may participate in the numerous VMSC injuries caused by high insulin levels. Subsequently, the precise effects of KDM3A, and the potential underlying mechanisms, were further explored.
KDM3A knockdown attenuates high insulin-induced VSMC dysfunction
To determine the role of KDM3A in high insulin-induced VSMC dysfunction, a lentivirus encoding KDM3A-siRNA was transduced into VSMCs prior to high insulin treatment. Fluorescent microscopy and western blotting were then performed to confirm that the lentivirus had been successful transduced. As shown in Fig. 3A, VSMCs transduced with lentivirus emitted strong green fluorescence; the transduction efficiency was ~90.1% at 40 MOI. Furthermore, the elevated expression of KDM3A in the HI + C-siRNA group was significantly abolished by lenti-KDM3A-siRNA transduction (Fig. 3B). These results suggested that lenti-KDM3A-siRNA transduction could suppress KDM3A expression in high insulin-induced VSMCs.
Figure 3.

Transduction with lentiviral vectors containing K-siRNA suppresses KDM3A protein expression in VSMCs. KDM3A knockdown attenuates high insulin-induced VSMC proliferation and migration. (A) Representative images of the efficiency of lentiviral transduction at 40 MOI in VSMCs. VSMCs were transduced with lentivirus for 72 h and observed by fluorescent microscopy (magnification, ×100). (B) Effects of lenti-K-siRNA on KDM3A protein expression, as determined by western blotting (upper panel). Densitometric analysis of western blotting (lower panel). (C) Effects of KDM3A knockdown on VSMC proliferation (n=5). (D) Representative images of the effects of KDM3A knockdown on VSMC migration (magnification, ×200) (upper panel). Relative cell migration following transduction with lentivirus (n=3) (lower panel). Data are expressed as the mean ± standard deviation, n=3. **P<0.05 vs. the HI + C-siRNA group; #P<0.05 vs. the N + C-siRNA group. C-siRNA, control siRNA; HI, high concentration insulin stimulation; KDM3A, lysine (K)-specific demethylase 3A; K-siRNA, KDM3A siRNA; N, normal medium; siRNA, small interfering RNA; VSMCs, vascular smooth muscle cells.
The effects of KDM3A inhibition on VSMC dysfunction, including the effects on proliferation and migration, following treatment with high insulin levels were investigated. As shown in Fig. 3C, the OD values at 450 nm in the HI + C-siRNA group were increased ~2-fold compared with in the N + C-siRNA group (P<0.05). However, KDM3A knockdown resulted in a 45.7% decrease in OD values compared with in the HI + C-siRNA group (P<0.05). In addition, the effects of KDM3A inhibition on cell migration were similar (Fig. 3D). Briefly, the mean number of migrated cells in the HI + C-siRNA group was significantly increased compared with in the N + C-siRNA group (P<0.05), whereas the number of migrated cells was reduced bŷ25.7% in KDM3A-silenced VSMCs compared with in the HI+C-siRNA group (P<0.05). These observations indicated that knockdown of KDM3A expression may attenuate high insulin-induced proliferation and migration of VSMCs .
KDM3A knockdown mitigates high insulin-induced ROS generation, apoptosis and inflammation
The present study clearly demonstrated that ROS generation, apoptosis and inflammation are crucial contributors to high insulin-induced VSMC dysfunction. Therefore, the effects of KDM3A knockdown on these pathological events were determined. As presented in Fig. 4A, DHE fluorescent density was markedly enhanced in the HI + C-siRNA group compared with in the N + C-siRNA group (P<0.05). However, transduction with lenti-KDM3A-siRNA led to a marked decrease in mean fluorescent density compared with in the HI + C-siRNA group (P<0.05). With regards to cellular apoptosis, the percentage of apoptotic cells was decreased by 48.4% in lenti-KDM3A-siRNA transduced VSMCs compared with in cells in the HI + C-siRNA group (P<0.05; Fig. 4B). Furthermore, KDM3A inhibition significantly downregulated Bax expression and upregulated Bcl-2 expression compared with in the HI + C-siRNA group (P<0.05; Fig. 4C). IL-6 and MCP-1 are commonly regarded as the hallmarks of inflammatory injury in VSMCs. The present study indicated that lenti-KDM3A-siRNA transduction markedly decreased the mRNA expression levels of IL-6 and MCP-1 by 32.2 and 34.3% compared with in the HI + C-siRNA group, respectively (P<0.05; Fig. 4D). These results further indicated that KDM3A knockdown may reverse excessive generation of ROS, apoptosis and inflammation, and thereby attenuate VSMC dysfunction caused by high insulin stimulation.
Figure 4.

KDM3A knockdown decreases reactive oxygen species generation, cell apoptosis and inflammation in high insulin-treated VSMCs. (A) Representative fluorescent images of dihydroethidium staining post-transduction with lenti-K-siRNA or lenti-C-siRNA (magnification, ×100) (left panel). Fluorescent mean density measured by ImageJ software (right panel). (B) Representative flow cytometry plots of the effects of KDM3A inhibition on high insulin-induced cellular apoptosis (left panel). Relative apoptosis rates post-transduction with lentiviruses (right panel). (C) Representative immunoblots for Bax and Bcl-2 expression post-transduction with lenti-K-siRNA or lenti-C-siRNA (upper panel). Densitometric analysis of western blotting (lower panel). (D) Effects of KDM3A inhibition on the mRNA expression levels of IL-6 and MCP-1 in VSMCs. Data are expressed as the mean ± standard deviation, n=3. **P<0.05 vs. the HI + C-siRNA group; #P<0.05 vs. the N + C-siRNA group. Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2; C-siRNA, control siRNA; HI, high concentration insulin stimulation; IL-6, interleukin-6; KDM3A, lysine (K)-specific demethylase 3A; MCP-1, monocyte chemotactic protein-1; K-siRNA, KDM3A siRNA; N, normal medium; siRNA, small interfering RNA; VSMCs, vascular smooth muscle cells
KDM3A knockdown inactivates mitogen-activated protein kinase (MAPK)/NF-κB signaling in high insulin-treated VSMCs
Activation of MAPK/NF-κB signaling pathways serves a key role in expediting VSMC dysfunction. To determine whether KDM3A knockdown affects MAPK/NF-κB activity and impacts high insulin-induced VSMCs pathology, western blotting was conducted to detect ERK1/2, JNK, p38 MAPK and nuclear NF-κB/p65 protein expression. As shown in Fig. 5, high concentration insulin significantly upregulated the expression of p-ERK1/2, p-JNK, p-p38 MAPK and nuclear NF-κB/p65 in VSMCs compared with in the N + C-siRNA group (P<0.05), whereas lenti-KDM3A-siRNA transduction into VSMCs markedly reduced the expression levels of p-ERK1/2, p-JNK, p-p38 MAPK and NF-κB/p65 by 26.01, 26.08, 33.56 and 24.00% compared with in the HI + C-siRNA group, respectively (P<0.05). The expression levels of total ERK1/2, JNK and p38 MAPK were not signifi-cantly altered. These findings indicated that the cytoprotecive effects of KDM3A knockdown were in part associated with inactivation of MAPK/NF-κB signaling pathways.
Figure 5.

Knockdown of KDM3A suppresses the expression levels of p-ERK1/2, p-JNK, p-p38 MAPK and NF-κB/p65 in vascular smooth muscle cells following high concentration insulin stimulation. (A) Representative immunoblots of p-ERK1/2, ERK1/2 and GAPDH (upper panel). Densitometric analysis (lower panel). (B) Representative immunoblots of p-JNK, JNK and GAPDH (upper panel). Densitometric analysis (lower panel). (C) Representative immunoblots of p-p38 MAPK, p38 MAPK and GAPDH (upper panel). Densitometric analysis (lower panel). (D) Representative immunoblots of nuclear NF-κB/p65 and GAPDH (upper panel). Densitometric analysis (lower panel). Values are expressed as the mean ± standard deviation, n=3. **P<0.05 vs. the HI + C-siRNA group; #P<0.05 vs. the N + C-siRNA group. ERK1/2, extracellular signal-regulated kinases 1/2; C-siRNA, control siRNA; HI, high concentration insulin stimulation; JNK, c-Jun NH2-terminal protein kinase; KDMA3, lysine (K)-specific demethylase 3A; K-siRNA, KDM3A siRNA; MAPK, mitogen-activated protein kinase; N, normal medium; NF-κB, nuclear factor-κB; p, phosphorylated; siRNA, small interfering RNA.
Discussion
Although it has previously been demonstrated that KDM3A inhibition may exert protective effects against hyperglycemia-induced VSMC lesions (20), the detailed involvement of KDM3A in hyperinsulinemia-induced VSMC damage remains to be investigated. The present study demonstrated that stimulation with a high concentration of insulin significantly increased proliferation and migration of VSMCs in vitro, alongside increased ROS generation, apoptosis and inflammation. In addition, these outcomes were associated with a marked upregulation of KDM3A. Therefore, the present study hypothesized, and subsequently confirmed, that KDM3A inhibition via lentivirus vectors could evidently ameliorate cellular proliferation and migration, and limit inflammation, oxidative stress and apoptotic cascade in high insulin-treated VSMCs. Mechanistic studies indicated that KDM3A knockdown markedly suppressed p-ERK1/2, p-JNK, p-p38 MAPK and NF-κB/p65 expression. To the best of our knowledge, the present study is the first to elaborate the effects of KDM3A on modulation of high insulin-induced VSMC dysfunction. These results indicated that KDM3A may be a potential target to attenuate hyperinsulinemia-induced vascular injury.
It is well established that DM is closely associated with adverse health outcomes, among which micro- and macrovascular complications are considered the most severe manifestations (23,24). Intensive treatment regimens that normalize glucose levels were previously considered effective therapeutic approaches; however, the risk of cardiovascular events is not efficiently controlled. In addition, whether micro- and/or macrovascular lesions may be attenuated by intensive glucose-lowering therapy remains controversial (25,26). Type 2 DM is comprised of complex metabolic disorders, including hyperglycemia, hyperlipidemia and hyperinsulinemia (21). Experimental studies have demonstrated that high concentrations of insulin may confer direct or indirect effects on amplifying cardiovascular stress (15). Abhijit et al reported that VSMCs exposed to hyperinsulinemia may exhibit increased ROS generation (27). In addition, Shiny et al and Martínez-Hervás et al provided evidence to suggest that hyperinsulinemia aggravates the inflammatory response and reduces VSMC survival, respectively (28,29). It is well documented that ROS and inflammation may induce VSMC proliferation and migration (15,27,28); however, the association between apoptosis and VSMC dysfunction remains debatable; a study has indicated that apoptosis may also trigger VSMC proliferation, leading to neointimal hyperplasia (30); however, the underlying mechanisms of this phenomenon are unclear (30). In line with previous studies, the present study indicated that stimulation with high concentration insulin promoted VSMC dysfunction and pathological injuries. Notably, due to the controversial information regarding intensive glucose-lowering therapy, as well as the roles of hyperinsulinemia in DM-associated cardiovascular complications irrespective of hyperglycemia (31), reversing the hyperinsulinemia-induced VSMC dysfunction in type 2 DM may be considered a promising therapeutic approach to protect against vascular complications.
MAPK cascades are evolutionary conserved signaling modules that consist of protein-serine/threonine kinase family proteins, including ERK1/2, JNK and p38 MAPK (32). NF-κB serves as an important downstream transcription factor of MAPK activation in VSMCs (33). MAPK/NF-κB signaling pathways may translate environmental information into appropriate responses via phosphorylating their substrate proteins and thus regulate various cellular processes, including proliferation, differentiation, inflammation, survival and apoptosis (34). These signaling pathways also participate in numerous pathological processes in VSMCs. Liu et al previously demonstrated that insulin-mediated proliferation of VSMCs could be suppressed by PD98059, a selective inhibitor of the ERK1/2 signaling pathway, thus suggesting that MAPKs have a regulatory impact on insulin-induced VSMC injuries (35). Oxidative stress and inflammation are vital factors associated with VSMC dysfunction; therefore, any factor that increases oxidative stress and inflammation may lead to VSMC injuries (15). ERK, JNK, p38 MAPK and NF-κB are all ROS-sensitive regulators (36). In addition, ROS generation may be mediated by activation of MAPK/NF-κB signal pathways (31). Inhibition of ERK1/2, JNK or p38 MAPK by specific inhibitors or siRNAs has been reported to attenuate ROS accumulation-induced VSMC injuries (36,37). Therefore, activation of MAPKs and ROS production may augment each other under pathological conditions. Furthermore, causal associations exist between activation of MAPK/NF-κB signaling pathways and inflammation. Our previous study indicated that ERK1/2 and NF-κB activities could increase IL-6 and MCP-1 expression in VSMCs (36); these findings were concordant with the findings published by Wronkowitz et al (38). In addition, close associations exist between MAPK activation and VSMC apoptosis. Gomez et al (30) indicated that p38 MAPK and JNK pathways initiated first apoptotic wave of VSMCs and contributed to vascular inflammation; these results were highly consistent with those of another study (39). The present study demonstrated that high concentration insulin could significantly exacerbate oxidative stress, inflammation and apoptosis of VSMCs. However, whether ERK1/2, JNK and p38 MAPK were involved in high insulin-induced inflammation, ROS accumulation and apoptosis in VSMCs remained ambiguous. In addition, the effects of MAPK subgroups may be divergent according to the species from which VSMCs were obtained and the external stimulus acting upon them. In the present study, stimulating rat VSMCs with 100 nM insulin led to VSMC dysfunction; in addition, oxidative stress, inflammation and apoptosis were concomitantly increased. Notably, mechanistic studies further revealed that the expression levels of p-ERK1/2, p-JNK, p-p38 MAPK and NF-κB/p65 were significantly upregulated. Therefore, the present study suggested that activation of MAPK/NF-κB signaling pathways may serve prominent roles in aggravating the deleterious effects of hyperinsulinemia on VSMCs.
KDM3A is a member of the 2-oxygluta rate/Fe(II)-dependent Jumonji C-domain-containing protein family, which selectively removes mono- and di-methyl groups from histone H3 lysine H3K9 (40). Although the biochemical features of KDM3A, as a histone demethylase, have been well characterized, its physiological significance remains incompletely understood. KDM3A was originally reported to be upregulated and serve an important role in the setting of hypoxia (41). To date, it has been revealed that KDM3A may increase cell proliferation, migration, invasion and angiogenesis in various cancer types, and elevated KDM3A may be a poor prognostic maker in patients with cancer (17–19,42). Furthermore, KDM3A has also been reported to be involved in stem cell renewal and VSMC differentiation (43). However, its potential functions in diabetic cardiovascular complications have rarely been studied. Our previous findings demonstrated that the expression of KDM3A was significantly increased in injured blood vessels following carotid artery balloon treatment in rats with type 2 DM. Furth ermore, downregulating KDM3A could markedly inhibit vascular remodeling and efficiently promote renovation of endothelium, even though blood sugar levels continued to rise. Our previous study also indicated that VSMC proliferation and migration were enhanced in the KDM3A-upregulated group under high glucose conditions; these effects were blunted by KDM3A gene knockdown (20). In another study using diabetic db/db mice, it was demonstrated that 2,4-pyridinedicarboxylic acid, an inhibitor of KDM3A, could inhibit myocardial fibrosis by increasing the density of cardiac microcirculation (unpublished data). Therefore, it may be hypothesized that KDM3A serves a vital role in diabetic cardiovascular complications. Since our previous study mainly focused on hyperglycemia, and since hyperinsulinemia is a prominent feature of type 2 DM that may exert adverse effects on the cardiovascular system, the present study aimed to determine whether KDM3A participated in hyperinsulinemia-induced VSMC injuries. The present study indicated that incubating VSMCs with 100 nM insulin markedly contributed to cellular dysfunction, alongside KDM3A upregulation. Furthermore, a high concentration of insulin activated MAPK/NF-κB signaling pathways, whereas KDM3A knockdown via its specific siRNA markedly reduced the expression levels of p-ERK1/2, p-JNK, p-p38 MAPK and NF-κB/p65. In addition, KDM3A knockdown attenuated oxidative stress, inflammation and apoptosis, and limited proliferation and migration of VSMCs. These findings indicated that KDM3A may act as an upstream regulatory factor of MAPK/NF-κB signa ling pathways in VSMCs undergoing stimulation with a high concentration of insulin, and its effects may be partly mediated through MAPK/NF-κB signaling pathways.
In conclusion, the present study is the first, to the best of our knowledge, to report the effects of KDM3A on aggravating hyperinsulinemia-induced VSMC injuries. Notably, the results indicated that silencing KDM3A expression could attenuate VSMC damage caused by high insulin levels, at least in part, through scavenging ROS, moderating inflammation and suppressing apoptotic activation, in a MAPK/NF-κB pathway-dependent manner. Therefore, KDM3A may be a promising therapeutic target to prevent DM-associated cardiovascular diseases.
Acknowledgments
The present study was supported by the National Natural Science Foundation of China (grant nos. 81200156 and 81570331).
References
- 1.Wegner M, Neddermann D, Piorunska-Stolzmann M, Jagodzinski PP. Role of epigenetic mechanisms in the development of chronic complications of diabetes. Diabetes Res Clin Pract. 2014;105:164–175. doi: 10.1016/j.diabres.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 2.van der Leeuw J, Beulens JW, van Dieren S, Schalkwijk CG, Glatz JF, Hofker MH, Verschuren WM, Boer JM, van der Graaf Y, Visseren FL, et al. Novel biomarkers to improve the prediction of cardiovascular event risk in type 2 diabetes mellitus. J Am Heart Assoc. 2016;5:5. doi: 10.1161/JAHA.115.003048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ndrepepa G, Colleran R, Luttert A, Braun S, Cassese S, Kufner S, Hieber J, Fusaro M, Laugwitz KL, Schunkert H, et al. Prognostic value of gamma-glutamyl transferase in patients with diabetes mellitus and coronary artery disease. Clin Biochem. 2016;49:1127–1132. doi: 10.1016/j.clinbiochem.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 4.Minha S, Bental T, Assali A, Vaknin-Assa H, Lev EI, Rechavia E, Battler A, Kornowski R. A comparative analysis of major clinical outcomes using drug-eluting stents versus bare metal stents in diabetic versus nondiabetic patients. Catheter Cardiovasc Interv. 2011;78:710–717. doi: 10.1002/ccd.23062. [DOI] [PubMed] [Google Scholar]
- 5.Myers GR, Weintraub WS. Coronary artery disease: revascularization strategies for patients with CAD and diabetes. Nat Rev Cardiol. 2010;7:364–366. doi: 10.1038/nrcardio.2010.82. [DOI] [PubMed] [Google Scholar]
- 6.Beckman JA, Paneni F, Cosentino F, Creager MA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part II. Eur Heart J. 2013;34:2444–2452. doi: 10.1093/eurheartj/eht142. [DOI] [PubMed] [Google Scholar]
- 7.El-Atat FA, Stas SN, McFarlane SI, Sowers JR. The relationship between hyperinsulinemia, hypertension and progressive renal disease. J Am Soc Nephrol. 2004;15:2816–2827. doi: 10.1097/01.ASN.0000133698.80390.37. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura A, Monma Y, Kajitani S, Noda K, Nakajima S, Endo H, Takahashi T, Nozaki E. Effect of glycemic state on postprandial hyperlipidemia and hyperinsulinemia in patients with coronary artery disease. Heart Vessels. 2016;31:1446–1455. doi: 10.1007/s00380-015-0757-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee KK, Fortmann SP, Fair JM, Iribarren C, Rubin GD, Varady A, Go AS, Quertermous T, Hlatky MA. Insulin resistance independently predicts the progression of coronary artery calcification. Am Heart J. 2009;157:939–945. doi: 10.1016/j.ahj.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Sun HJ, Zhao MX, Ren XS, Liu TY, Chen Q, Li YH, Kang YM, Wang JJ, Zhu GQ. Salusin-β promotes vascular smooth muscle cell migration and intimal hyperplasia after vascular injury via ROS/NFκB/MMP-9 pathway. Antioxid Redox Signal. 2016;24:1045–1057. doi: 10.1089/ars.2015.6475. [DOI] [PubMed] [Google Scholar]
- 11.Xu Z, Han Y, Liu J, Jiang F, Hu H, Wang Y, Liu Q, Gong Y, Li X. miR-135b5p and miR-499a-3p promote cell proliferation and migration in atherosclerosis by directly targeting MEF2C. Sci Rep. 2015;5:12276. doi: 10.1038/srep12276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Zhang J, Xu L, Xu C, Chen S, Yang J, Jiang H. Inhibition of neointimal hyperplasia in the rat carotid artery injury model by a HMGB1 inhibitor. Atherosclerosis. 2012;224:332–339. doi: 10.1016/j.atherosclerosis.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 13.Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen C, Cai Y, Huang H, Yang Y, Liu Y, et al. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation. 2014;130:1452–1465. doi: 10.1161/CIRCULATIONAHA.114.011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caccamo G, Bonura F, Bonura F, Vitale G, Novo G, Evola S, Evola G, Grisanti MR, Novo S. Insulin resistance and acute coronary syndrome. Atherosclerosis. 2010;211:672–675. doi: 10.1016/j.atherosclerosis.2010.03.033. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Yu C, Zhang B, Wang Y. The injurious effects of hyperinsulinism on blood vessels. Cell Biochem Biophys. 2014;69:213–218. doi: 10.1007/s12013-013-9810-6. [DOI] [PubMed] [Google Scholar]
- 16.Dubin SB. Characterization of amniotic fluid lamellar bodies by resistive-pulse counting: relationship to measures of fetal lung maturity. Clin Chem. 1989;35:612–616. [PubMed] [Google Scholar]
- 17.Tateishi K, Okada Y, Kallin EM, Zhang Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature. 2009;458:757–761. doi: 10.1038/nature07777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osawa T, Tsuchida R, Muramatsu M, Shimamura T, Wang F, Suehiro J, Kanki Y, Wada Y, Yuasa Y, Aburatani H, et al. Inhibition of histone demethylase JMJD1A improves anti-angiogenic therapy and reduces tumor-associated macrophages. Cancer Res. 2013;73:3019–3028. doi: 10.1158/0008-5472.CAN-12-3231. [DOI] [PubMed] [Google Scholar]
- 19.Parrish JK, Sechler M, Winn RA, Jedlicka P. The histone demethylase KDM3A is a microRNA-22-regulated tumor promoter in Ewing sarcoma. Oncogene. 2015;34:257–262. doi: 10.1038/onc.2013.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jing C, Xu L, Zhang J, Hu X, Chen S, Hu Q, Xu C, Jiang H. GW24-e2386 down-regulation of histone demethylase KDM3A attenuates balloon injury-induced neointimal hyperplasia in diabetic rats through modulation of epigenetic histone lysine 9 di-methylation. Heart. 2013;2013:A9–A10. [Google Scholar]
- 21.Qi H, Jing Z, Xiaolin W, Changwu X, Xiaorong H, Jian Y, Jing C, Hong J. Histone demethylase JMJD2A inhibition attenuates neointimal hyperplasia in the carotid arteries of balloon-injured diabetic rats via transcriptional silencing: inflammatory gene expression in vascular smooth muscle cells. Cell Physiol Biochem. 2015;37:719–734. doi: 10.1159/000430390. [DOI] [PubMed] [Google Scholar]
- 22.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 23.Cavicchioli MG, Guerbali CC, Ochiai C, Silva RM, Camara G, Petry TB. The contribution of diabetes education in the treatment of people with type 2 diabetes and risk of cardiovascular disease. Curr Atheroscler Rep. 2016;18:44. doi: 10.1007/s11883-016-0591-4. [DOI] [PubMed] [Google Scholar]
- 24.Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- 25.Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, Marre M, Cooper M, Glasziou P, Grobbee D, et al. ADVANCE Collaborative Group Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–2572. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 26.Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, Zieve FJ, Marks J, Davis SN, Hayward R, et al. VADT Investigators Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360:129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- 27.Abhijit S, Bhaskaran R, Narayanasamy A, Chakroborty A, Manickam N, Dixit M, Mohan V, Balasubramanyam M. Hyperinsulinemia-induced vascular smooth muscle cell (VSMC) migration and proliferation is mediated by converging mechanisms of mitochondrial dysfunction and oxidative stress. Mol Cell Biochem. 2013;373:95–105. doi: 10.1007/s11010-012-1478-5. [DOI] [PubMed] [Google Scholar]
- 28.Shiny A, Regin B, Mohan V, Balasubramanyam M. Coordinated augmentation of NFAT and NOD signaling mediates proliferative VSMC phenotype switch under hyperinsulinemia. Atherosclerosis. 2016;246:257–266. doi: 10.1016/j.atherosclerosis.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 29.Martínez-Hervás S, Vinué A, Núñez L, Andrés-Blasco I, Piqueras L, Real JT, Ascaso JF, Burks DJ, Sanz MJ, González-Navarro H. Insulin resistance aggravates atherosclerosis by reducing vascular smooth muscle cell survival and increasing CX3CL1/CX3CR1 axis. Cardiovasc Res. 2014;103:324–336. doi: 10.1093/cvr/cvu115. [DOI] [PubMed] [Google Scholar]
- 30.Gomez C, Martinez L, Mesa A, Duque JC, Escobar LA, Pham SM, Vazquez-Padron RI. Oxidative stress induces early-onset apoptosis of vascular smooth muscle cells and neointima formation in response to injury. Biosci Rep. 2015;35:35. doi: 10.1042/BSR20140122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ceriello A, Ihnat MA, Thorpe JE. Clinical review 2: the ‘metabolic memory’: is more than just tight glucose control necessary to prevent diabetic complications? J Clin Endocrinol Metab. 2009;94:410–415. doi: 10.1210/jc.2008-1824. [DOI] [PubMed] [Google Scholar]
- 32.Lv H, Yu Z, Zheng Y, Wang L, Qin X, Cheng G, Ci X. Isovitexin exerts anti-inflammatory and anti-oxidant activities on lipopolysaccharide-induced acute lung injury by inhibiting MAPK and NF-κB and activating HO-1/Nrf2 pathways. Int J Biol Sci. 2016;12:72–86. doi: 10.7150/ijbs.13188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Liu J, Pang X, Zhao J, Wang S, Wu D. Aldosterone induces C-reactive protein expression via MR-ROS-MAPK-NF-κB signal pathway in rat vascular smooth muscle cells. Mol Cell Endocrinol. 2014;395:61–68. doi: 10.1016/j.mce.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 34.Chen J, Xu J, Li J, Du L, Chen T, Liu P, Peng S, Wang M, Song H. Epigallocatechin-3-gallate attenuates lipopoly-saccharide-induced mastitis in rats via suppressing MAPK mediated inflammatory responses and oxidative stress. Int Immunopharmacol. 2015;26:147–152. doi: 10.1016/j.intimp.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 35.Liu K, Zhao W, Gao X, Huang F, Kou J, Liu B. Diosgenin ameliorates palmitate-induced endothelial dysfunction and insulin resistance via blocking IKKβ and IRS-1 pathways. Atherosclerosis. 2012;223:350–358. doi: 10.1016/j.atherosclerosis.2012.06.012. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, Chen J, Yang J, Xu CW, Pu P, Ding JW, Jiang H. Resveratrol attenuates oxidative stress induced by balloon injury in the rat carotid artery through actions on the ERK1/2 and NF-kappa B pathway. Cell Physiol Biochem. 2013;31:230–241. doi: 10.1159/000343364. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, Wang X, Wu T, Li B, Liu T, Wang R, Liu Q, Liu Z, Gong Y, Shao C. Isoliensinine induces apoptosis in triple-negative human breast cancer cells through ROS generation and p38 MAPK/JNK activation. Sci Rep. 2015;5:12579. doi: 10.1038/srep12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wronkowitz N, Görgens SW, Romacho T, Villalobos LA, Sánchez-Ferrer CF, Peiró C, Sell H, Eckel J. Soluble DPP4 induces inflammation and proliferation of human smooth muscle cells via protease-activated receptor 2. Biochim Biophys Acta. 2014;1842:1613–1621. doi: 10.1016/j.bbadis.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 39.Perez-Vizcaino F, Bishop-Bailley D, Lodi F, Duarte J, Cogolludo A, Moreno L, Bosca L, Mitchell JA, Warner TD. The flavonoid quercetin induces apoptosis and inhibits JNK activation in intimal vascular smooth muscle cells. Biochem Biophys Res Commun. 2006;346:919–925. doi: 10.1016/j.bbrc.2006.05.198. [DOI] [PubMed] [Google Scholar]
- 40.Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 41.Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK, Bührer C. Hypoxia upregulates the histone demethylase JMJD1A via HIF-1. Biochem Biophys Res Commun. 2008;372:892–897. doi: 10.1016/j.bbrc.2008.05.150. [DOI] [PubMed] [Google Scholar]
- 42.Uemura M, Yamamoto H, Takemasa I, Mimori K, Hemmi H, Mizushima T, Ikeda M, Sekimoto M, Matsuura N, Doki Y, et al. Jumonji domain containing 1A is a novel prognostic marker for colorectal cancer: in vivo identification from hypoxic tumor cells. Clin Cancer Res. 2010;16:4636–4646. doi: 10.1158/1078-0432.CCR-10-0407. [DOI] [PubMed] [Google Scholar]
- 43.Lockman K, Taylor JM, Mack CP. The histone demethylase, Jmjd1a, interacts with the myocardin factors to regulate SMC differentiation marker gene expression. Circ Res. 2007;101:e115–e123. doi: 10.1161/CIRCRESAHA.107.164178. [DOI] [PubMed] [Google Scholar]