

Abstract
Hepatic stellate cells (HSCs) are the main extracellular matrix (ECM)-producing cells in liver fibrosis. Activated HSCs stimulate the proliferation and migration of hepatocellular carcinoma (HCC) cells. Cysteine-rich 61 (CCN1/Cyr61) is an ECM protein. Our previous studies demonstrated that the expression of CCN1 was significantly higher in benign hepatic cirrhosis tissue and cancer-adjacent hepatic cirrhosis tissues. CCN1 is a target gene of β-catenin in HCC and promotes the proliferation of HCC cells. The present study aimed to examine whether CCN1 can activate HSCs and affect the function of activated HSCs in promoting the progression of HCC. CCN1 expression was determined during the progression of liver fibrosis in a mouse model. LX-2 cells, which were infected with adenoviruses AdCCN1 or AdRFP, and HepG2 cells were co-cultured or subcutaneously co-implanted into in nude mice. MTT assay, Crystal Violet staining, Boyden chamber, matrigel invasion and monolayer scratch assays were used to analyze the proliferation, migration and invasion capability of HepG2 cells. Xenograft sizes were measured and histological analyses were performed by hematoxylin and eosin, immunohistochemical, immunefluorescence and Sirius Red staining. It was demonstrated that the expression of CCN1 was continually increased in liver fibrosis and the that expression may be correlated with the progression of liver fibrosis. CCN1 affected the function of LX-2 and enhanced the effect of LX-2 on promoting the viability, migration and invasion of HepG2 cells in vitro. CCN1 enhanced the effect of LX-2 on promoting the growth of HepG2 xenografts in vivo. CCN1 also affected the function of activated HSCs and regulated the formation of the xenograft microenvironment, including fibrogenesis and angiogenesis, which are beneficial for the progression of HCC. These findings demonstrated that CCN1 may be involved in the progression of the hepatic cirrhosis-HCC axis through regulating HSCs.
Keywords: cysteine-rich 61, hepatic stellate cells, liver fibrosis, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common malignant tumor and the third most life-threatening type of cancer worldwide. Almost 78% of global HCC cases were reported in Asian countries each year (1). Since the 1990s, HCC has become the third leading cause of cancer-associated mortality in China. Liver fibrosis is associated with HCC, with 90% of HCC cases arising from the cirrhotic liver (2,3).
Liver fibrosis occurs as the result of the chronic wound-healing response of the liver and is associated with major alterations of extracellular matrix (ECM) (4). Hepatic stellate cells (HSCs) are the main ECM-producing cell in liver fibrosis. In the normal liver, HSCs are found in the space of Disse and are the major storage sites of vitamin A. Following chronic injury, the HSCs are activated and transdifferentiate into myofibroblast cells, acquiring contractile, proinflammatory and fibrogenic properties. The activated HSCs migrate and accumulate at sites of tissue repair, secreting large quantities of ECM and regulating ECM degradation (5).
Cysteine-rich 61 (CCN1/Cyr61), a member of the CCN family, is an ECM protein and regulates multiple cellular activities, including cell adhesion, migration, proliferation, survival, apoptosis and angiogenesis, through binding different integrins (6–11). Previous studies have demonstrated that CCN1 has complex roles in cutaneous wound healing fibrosis through binding to different integrins on the fibroblast membrane (12–14).
Several studies have focused on the roles of CCN1 in liver disease. A study by Kim et al (20) and our previous study showed that level of CCN1 was elevated in the cirrhotic liver in humans and in mice with carbon tetrachloride (CCl4)-induced liver fibrosis, with CCN1 protein located predominantly in hepatocytes (15). In our previous study, it was found that the expression of CCN1 was significantly higher in benign hepatic cirrhosis tissue and cancer-adjacent hepatic cirrhosis tissue, compared with that in normal liver tissue (15). These results are in accordance with those reported by Rashid et al (16). CCN1 is involved in macrophage infiltration and the hepatic proinflammatory response (17). In our previous study, it was also found that CCN1 was a target gene of β-catenin in HCC and promoted the proliferation of HepG2 cells (15). CCN1 triggers the senescence of activated HSCs and promotes the regression of liver fibrosis (18–20). Previous studies have demonstrated that CCN1 induces chol-angiocyte proliferation and ductular reactions, and identified CCN1/αvβ5/nuclear factor (NF)-κB/jagged 1 (JAG1) as a critical axis for biliary injury repair (21). CCN1 also suppresses hepatocarcinogenesis by inhibiting epidermal growth factor receptor (EGFR)-dependent hepatocyte compensatory proliferation (22).
In addition, several studies have found that certain integrin subunits are located on HSC membranes and mediate the proliferation, migration and fibrogenic activation of HSCs (16,23–25), which suggests that CCN1 may be involved in activated HSCs.
It is generally known that activated HSCs can promote the progression of HCC. Clinical investigations have found that activated HSCs in peritumoral tissues are associated with earlier recurrence rates, mortality rates and high recurrence rates (26). Activated HSCs are directly involved in hepatocarcinogenesis in a transforming growth factor (TGF)-β-dependent manner by inducing autocrine TGF-β signaling and nuclear β-catenin accumulation in neoplastic hepatocytes (27). Activated HSCs stimulate the proliferation, growth and migration of HCC cells in vitro and in vivo through cytokine secretion, whereas ECM regulates angiogenesis and tumor immunity inhibition (28–30). HCC cells also stimulate the growth and migration of human HSCs (31). HCC cell-activated HSC cross-talk in the liver promotes the progression of HCC (32). However, whether CCN1 can affect these functions of activated HSCs remains to be elucidated. The present study investigated whether CCN1 can activate HSCs and whether it enhances the effect of HSCs on promoting the progression of HCC.
Materials and methods
Ethics statement
All animals were purchased from the Experimental Animal Center of the Third Military Medical University (Chongqing, China). All animal protocols were approved by the Ethics Committee of the Third Military Medical University. Animal experiments were performed in accordance with the China Regulations for the Administration of Affairs Concerning Experimental Animals and the guidelines of the U.S. National Institutes of Health.
Cell lines
The human hepatic stellate cells LX-2 or human HCC cells HepG2 were cultured in T75 flasks in DMEM (Dulbecco's modified Eagle medium, high glucose, HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% fetal calf serum (FCS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 100 U/ml penicillin and 10 μg/ml streptomycin at 37°C in a humidified 5% carbon dioxide atmosphere. The HepG2 cells were used to investigate the effects of CCN1 on HSCs in promoting the viability and migration of the HCC cells. HepG2-LX2 cells were co-cultured in 6-well plates and Transwell inserts.
Establishment of the mouse liver fibrosis model and histological analyses
A total of 100 BALB/c male mice were purchased from the Experimental Animal Center of the Third Military Medical University (Chongqing, China). Liver fibrosis was induced in male mice (~4 weeks of age; 17–18 g) via intraperitoneal injection of 1 ml/kg body weight of CCl4 (diluted in olive oil, 1:4; 1 ml/kg body weight) twice a week. The control animals received an equal volume of olive oil. All mice were maintained in barrier environment, received free access to sterile feed and water, temperature 24–26°C, 12 h light/dark cycle. Experimental animals were maintained in the EVC Animal Care Systems. The mice were treated for 5 weeks and were sacrificed 3 days following injection. Following the final injection of CCl4, mice were sacrificed for another 3 weeks, 3 mice weekly (weeks 6–8). The liver tissues were sectioned (1.2×1.2×0.5 cm) and fixed in 10% formaldehyde, and were embedded in paraffin. The tissue sections (4 μm) were stained with hematoxylin and eosin (H&E), Sirius red and immunohistochemical staining. The tissue structure was observed using hematoxylin and eosin. The staining procedure was performed as described previously (15).
Analysis of the expression of CCN1 was performed on the paraffinized sections of liver tissue using immunohistochemical staining. The staining procedure was performed as described previously (15). The sections were evaluated using a standard bright field microscope. Digital images were captured using the NIS-Element Imaging Analysis system (Nikon, Tokyo, Japan). Positive staining was defined when >10% of cells exhibited brown staining. The degree of the staining was digitalized automatically using the NIS-Element Imaging Analysis system when the positive and negative points were defined. The mean density of five randomly selected microscopic fields was calculated, reflecting the relative expression level of CCN1. The results are presented as the mean ± standard deviation.
The fibrotic regions were observed using Sirius red staining. The numbers of fibers were assessed using the NIS-Element Imaging Analysis in six randomly selected regions from each tissue section.
Transient infection and collection of conditioned medium (CM) from LX-2
AdCCN1 and AdRFP were acquired from Professor Tong-Chuan He (Molecular Oncology Laboratory, Department of Surgery, University of Chicago Medical Center, Chicago, IL, USA). The LX-2 cells were seeded in T75 flasks. At 70% confluence, the cells were infected with adenovirus AdCCN1 or AdRFP, respectively. Following infection for 24 h at 37°C, the LX-2 cells were harvested to analyze the expression of CCN1 using western blot assays. Following infection with AdCCN1 or AdRFP for 20 h, respectively, the LX-2 cells were washed twice with serum-free DMEM, and then incubated for another 24 h with 12 ml serum-free DMEM; the media were collected as CCN1-CM and control-CM, respectively (CM was 10X concentrated). Furthermore, half of CCN1-CM was concentrated to 10X concentration by Amicon Ultra centrifugal filter (UFC800324; Merck Millipore) following the introductions.
Measurement of cell viability
The LX-2 cells were seeded into 96-well plates (2,000 cells/well) in DMEM supplemented with 10% FCS. Following infection with AdCCN1 or AdRFP for 20 h, respectively, the LX-2 cells were washed twice with serum-free DMEM and then incubated for another 0-72 h with DMEM supplemented with 10% FCS. The viability of cells was measured using an MTT assay.
The HepG2 cells were seeded into 96-well plates (2,000 cells/well) in DMEM supplemented with 10% FCS. After 24 h, the seeded HepG2 cells were washed with serum-free DMEM and were cultured in the CM from the different LX-2 cells or the control medium, following which an MTT assay was performed to analyze the viability of the cells.
The HepG2 cells were cultured alone or were co-cultured with the differently treated LX-2 cells in a 6-well plate (1×103 cells/well) with Transwell inserts. After 5 days, colony formation of the HepG2 cells was assayed using crystal violet staining.
Migration assays
The migration of the HepG2 cells was assessed using two assays. For the analysis of cell invasion capability, a Matrigel invasion assay was performed in 6-well Transwell inserts with 8-μm pore size filters coated with Matrigel (diluted 1:6, BD Biosciences, Franklin Lakes, NJ, USA). The HepG2 cells were harvested, resuspended in serum-free DMEM and placed in the upper compartment (5×105 cells/well). To the lower compartment, DMEM supplemented with CM from the activated LX-2 cells or control medium was added. Following incubation at 37°C for 30 h, the filters were collected and cells adhering to the lower surface were fixed in formalin and stained with crystal violet. The numbers of cells in 12 randomly selected fields in each well were counted.
Cell migration capability was assessed using a Boyden chamber assay. The HepG2 cells were seeded into 6-well plates at 1×106 cells/well. The HepG2 cells were harvested following incubation for 30 h, resuspended in serum-free DMEM and placed in the upper compartment at 5×105 cells/well. The lower compartment contained DMEM supplemented with CM from the activated LX-2 cells or control medium. Following incubation for 24 h, the filters were collected and cells adhering to the lower surface were fixed in formalin and stained with crystal violet. Cells in 12 randomly selected fields in each well were counted.
The LX-2 cells and HepG2 cells migration capabilities were assessed using a monolayer scratch assay. Following adherence, the LX-2 cells were infected with AdCCN1 or AdRFP, followed by the same treatments as described for the viability experiment described above. The HepG2 cells were then washed with serum-free DMEM and were cultured in CM from the activated LX-2 cells or control medium. Subsequently, the cell layer was scratched using a sterile toothpick. The migration into the space was measured at 0 and 48 h (LX-2 cells) or 72 h (HepG2 cells).
Western blot analysis
The LX-2 cells were seeded into T75 flasks at 60% confluence in DMEM supplemented with 10% FCS. After 24 h, the LX-2 cells were infected with AdCCN1 or AdRFP for 72 h. The HepG2 cells were washed with serum-free DMEM, and were cultured in CM from the LX-2 cells or control medium. Following incubation for 36 h, the HepG2 cells were harvested for subsequent analysis.
The cell lysates were prepared with cell lysis buffer containing a protease inhibitor cocktail. Protein concentrations of all samples were determined by Bicinchoninic acid Protein Assay Kit (Beyotime Institute of Biotechnology, Haimen, China). Total protein for each sample (50 μg) was loaded onto an 8% SDS-PAGE gel for electrophoresis and transferred onto a PVDF membrane. The membrane was then incubated with antibodies against phosphorylated-β-catenin (cat. no 9566, 1:1,000), total-β-catenin (cat. no. 8480, 1:1,000) (both from Cell Signaling Technology, Inc., Danvers, MA, USA), cyclin D1 (cat. no. 60186-1-Ig, 1:1,000), VEGF (cat. no. 19003-1-AP, 1:1,000), CD34 (cat. no. 14486-1-AP, 1:1,000), CD31 (cat. no. 66065-1-Ig; 1:1,000) (all from ProteinTech Group, Inc.), survivin (cat. no. ab76424; 1:1,000 dilution; Abcam, Cambridge, UK), and c-myc (cat. no. MA1-980, 1:1,000; Thermo Fisher Scientific, Inc.), respectively at 4°C overnight. The blots were then incubated with horseradish peroxidase-conjugated secondary antibodies (cat. nos. SA00001-1 and SA00001-2, both 1:5,000; ProteinTech Group, Inc.) at room temperature incubation for 2 h, following which the immune-reactive signals were detected using an ECL kit (EMD Millipore, Billerica, MA, USA).
Subcutaneous tumor models in nude mice
Four groups of mice (n=5 per group) were used in the following experiments, respectively. Following the infection with AdCCN1 or AdRFP for 24 h, LX-2 cells were harvested. The flank of 4-week-old nude mice were injected subcutaneously with HepG2 cells alone, or with HepG2 cells + LX-2 cells, HepG2 cells + LX-2-RFP cells (LX-2 cells infected with AdRFP), or HepG2 cells + LX-2-CCN1 cells (LX-2 cells infected with AdCCN1) at 5.0×106 cells per mouse, respectively.
The length and width of tumor masses were dynamically measured and the tumor volume was estimated using the formula: axb2×0.5, where a and b represent the maximal and minimal diameters, respectively. The mice were sacrificed on day 26, and growth curves were plotted using the tumor volumes for each experimental group at the set points. Tumor masses were removed and fixed in 4% formaldehyde. Paraffin-embedded consecutive sections (5 μm) were cut for histological analysis, and total proteins of the liver tissues were collected for western blot analysis.
The tissue structure of the subcutaneous tumor tissue was observed using H&E staining. Collagenous fibers in the subcutaneous tumor were observed using Sirius red staining. Immunohistochemical staining was performed for Ki67.
The microvessels in the subcutaneous tumor tissues were observed by immunofluorescence staining for CD31. The sections were visualized and images were captured using a Leica light and fluorescence microscope (Leica Microystems, GmbH, Wetzlar, Germany). The protein expression of VEGF was detected using western blot analysis in the subcutaneous tumor tissues.
Statistical analysis
Data are expressed as the mean ± standard deviation. Statistical analysis of the two mean values was performed using Student's t-test. One-way analysis of variance was performed to compare multiple mean values. All calculations were performed using the SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Dynamic expression of CCN1 is observed in mouse fibrotic liver tissues
To examine the role of CCN1 in the liver fibrosis-carcinoma axis, the present study observed the dynamic expression of CCN1 protein in the liver fibrosis mouse model. As shown in Fig. 1A–C, liver fibrosis continually progressed and the expression of CCN1 continually increased as CCl4 was injected. However, when there was no CCl4 injection, the degree of hepatic fibrosis was alleviated, and the expression of CCN1 was also reduced (Fig. 1). These findings suggested that changes in the expression of CCN1 may be associated with the severity of fibrosis during the progression of liver fibrosis.
Figure 1.

CCN1 is expressed in mouse fibrotic liver tissue. Liver fibrosis was induced in mice by intraperitoneal injection of 1 ml/kg body weight of CCl4 twice a week. (A) Liver tissue sections were stained with H&E (magnification, ×400), Sirius red (magnification, ×100) and with immunohistochemical staining for CCN1 (magnification, ×400). (B) Liver sections were stained with Sirius red to reveal collagen deposition. The numbers of fibers were assessed via NIS-Element Imaging analysis of six randomly selected regions. Data are presented as the mean ± standard deviation. (C) Analysis of the expression of CCN1 was performed on paraffinized sections of mice liver tissues via immunohistochemical staining. The degree of staining was digitalized automatically using the NIS-Element Imaging Analysis system. The mean density of random five microscopic fields was calculated as the relative expression level of CCN1. Data are presented as the mean ± standard deviation; 6, 7 and 8 weeks refer to the weeks 1, 2 and 3, respectively, following the final injection of CCl4. CCN1, cysteine-rich 61; CCl4, carbon tetrachloride; H&E, hematoxylin and eosin.
CCN1 activates HSCs and affects cell function
CCN1 was not expressed in LX-2 cells prior to infection with AdCCN1 (Fig. 2A). LX-2-CCN1 cells, comprising LX-2 cells overexpressing CCN1, expressed the markers of HSC activation and fibrosis, including α-SMA and collagen I (Fig. 2B). LX-2-CCN1 also exhibited cell proliferation correlated with signaling molecules, including cyclin D1, and angiogenesis molecules, including VEGF, CD34 and CD31 (Fig. 2C). However, the expression of β-catenin did not alter when phosphorylated-β-catenin was downregulated (Fig. 2C). CCN1 also promoted the viability and the migration of LX-2 cells. However, viability and migration were inhibited in the LX-2-CCN1 cells treated by function-inhibiting monoclonal anti-CCN1, which was obtained following recombined CCN1 protein treatment (Fig. 2D–F).
Figure 2.



CCN1 activates HSCs and affects cell function. LX-2 cells were infected with adenovirus AdCCN1 or AdRFP, respectively. (A) Following infection for 24 h, western blot analysis was performed to detect the expression levels of CCN1 in LX-2 cells, LX-2-RFP cells and LX-2-CCN1 cells. (B) Following infection for 72 h, α-SMA and collagen I, which are markers of HSC activation and fibrosis, were detected in the LX-2-CCN1 cells. (C) Expression levels of p-β-catenin, β-catenin, cyclin D1, VEGF, CD31 and CD34 were analyzed. CCN1 antibody (0.5 μg/ml) was used to neutralize CCN1 in control groups. (D) Viability of LX-2-CCN1 cells was analyzed using MTT assays. (E) Following treatment with RCCN1 (0.6 μg/ml), LX-2 cells were analyzed using MTT assays. CCN1 antibody (0.5 mg/ml) was used in assays. The final concentrations were 0.5 and 2.5 μg/ml. (F) Monolayer scratch assay was used to analyze the migration of LX-2-CCN1 cells. *P<0.05 and **P<0.01. HSCs, hepatic stellate cells; CCN1, cysteine-rich 61; VEGF, vascular endothelial growth factor; SMA, smooth muscle actin; p-, phosphorylated; RCCN1, recombined CCN1; OD, optical density.
CCN1 enhances the effect of activated HSCs on promoting the viability of HCC cells in vitro
Activated HSCs acquire fibrogenic properties but also can promote the progression of HCC. The present study aimed to determine whether CCN1 enhances the functions of activated HSCs. First, the effect of CCN1 on activated HSCs in promoting the viability of HCC cells was examined. The HepG2 cells were incubated with CM collected from the three treatment groups of LX-2 cells. The MTT assay revealed that the effect of CM from LX-2-CCN1 cells on increasing HepG2 cell viability was more marked compared with cells incubated with CM from LX-2 and LX-2-RFP cells (Fig. 3A and B). In addition, the HepG2 cells and three LX-2 cell groups were co-cultured in a Transwell insert plate. The HepG2 cells co-cultured with LX-2-CCN1 cells formed more clones than those cultured with the other LX-2 groups (Fig. 3C).
Figure 3.

CCN1 enhances the function of hepatic stellate cells in promoting the viability of HCCs. (A) Western blot analysis was used to detect expression levels of CCN1 in the original CM and 10X concentration CM collected from LX-2 cells infected with AdCCN1 or AdRFP. Expression levels were also detected following treatment with 2.5 and 5 μg. HepG2 cells were cultured with or without CM. (B) Viability of HepG2 cells, analyzed using MTT assays. *P<0.05 and **P<0.01 vs. the CM-LX-2-RFP group. (C) HepG2 cells were cultured alone or were co-cultured with different LX-2 cells in a 6-well plate with Transwell inserts. After 5 days, colony formation of HepG2 cells was examined using crystal violet staining (magnification, ×100). (D) Expression of p-β-catenin, β-catenin, survivin, cyclin D1 and c-myc in HepG2 cells were analyzed using western blot analysis. CCN1, cysteine-rich 61; CM, conditioned medium; HCCs, hepatocellular carcinoma cells; p-, phosphorylated; RCCN1, recombined CCN1.
To gain insight into the molecular mechanisms underlying the effects described above, the present study analyzed the activation of several correlated signaling molecules, which are known to be important in HCC. The results of the western blot analysis revealed that stimulation with CM from LX-2-CCN1 cells reduced the expression of phosphorylated-β-catenin in the HepG2 cells, whereas the level of total-β-catenin remained unchanged (Fig. 3D). The expression levels of cyclin D1 and c-myc, which are target genes of β-catenin, were upregulated (Fig. 3D). This indicated that stimulation of CM from LX-2-CCN1 cells significantly induced β-catenin signaling transduction in HepG2 cells. The western blot analysis revealed that there was a significant increase in the expression of survivin in the HepG2 cells treated with CM from LX-2-CCN1 cells (Fig. 3D).
CCN1 enhances the function of activated HSCs in promoting the migration and invasion of HCC cells
In the present study, HepG2 cells were incubated with CM collected from the three groups of LX-2 cells, following which the migratory activity of the HepG2 cells was analyzed. The Boyden chamber and Matrigel invasion assays demonstrated that CM from the LX-2-CCN1 cells significantly stimulated the migratory potential of HepG2 cells (Fig. 4A and B). These data were confirmed by scratch-healing assays, which revealed significantly faster wound healing in the HepG2 cells treated with CM from LX-2-CCN1 cells (Fig. 4C). In order to understand the molecular mechanism of the above, the present study analyzed the activation of ERK, and the expression of E-cadherin and MMP-9, which are known to be important in HCC metastasis and invasion. The results of the western blot analysis showed that stimulation with CM from the LX-2-CCN1 cells enhanced the expression of phosphorylated-ERK and MMP-9 in HepG2 cells., whereas the expression of E-cadherin was reduced (Fig. 4D).
Figure 4.

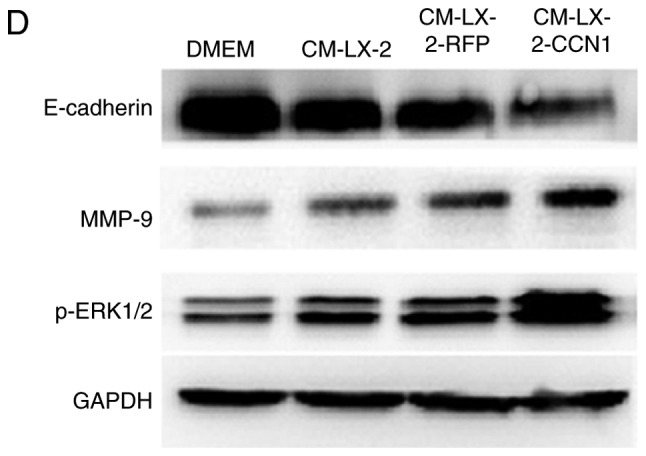
CCN1 enhances the function of hepatic stellate cells in promoting the migration and invasion of hepatocellular carcinoma cells. HepG2 cells were cultured with or without CM collected from LX-2 cells infected with AdCCN1 or AdRFP. (A) Boyden chamber (magnification, ×100), (B) Matrigel invasion (magnification, ×100) and (C) monolayer scratch assays were used to analyze the migration and invasion capabilities of the HepG2 cells. *P<0.05. (D) Expression of p-ERK1/2, MMP-9 and E-cadherin in the HepG2 cells were analyzed using western blot analysis. CCN1, cysteine-rich 61; CM, conditioned medium; ERK, extracellular signal-regulated kinase; MMP, matrix metalloproteinase; p-, phosphorylated.
CCN1 enhances the function of HSCs in promoting the proliferation of HCC cells in vivo
The present study also examined the effect of CCN1 on HSCs in promoting the proliferation of HCC cells in vivo. The HepG2 cells were implanted subcutaneously into nude mice, either alone or in combination with three different LX-2 cells (LX-2, LX-2-RFP and LX-2-CCN1). As shown in Fig. 5A, the implantation of LX-2 cells alone did not result in tumor formation, but all of the mice developed tumors at the site of implantation in the other groups. The tumor size was significantly larger in the group subjected to subcutaneous co-implantation of LX-2 and HepG2 cells, compared with that in the group subjected to single subcutaneous implantation of HepG2 cells. The injection of HepG2 cells + LX-2-CCN1 cells promoted more marked tumor growth, compared with that in the group injected with HepG2 cells + LX-2 or HepG2 cells + LX-2-RFP (Fig. 5B).
Figure 5.



CCN1 enhances the function of hepatic stellate cells in promoting the proliferation of hepatocellular carcinoma cells in vivo. (A) Nude mice and tumor tissues of animals following injection at 26 days. (B) Tumor sizes were measured at a 4-day interval from day 6 post-injection. The graph shows the tumor volumes for each treatment group. *P<0.05 and **P<0.01 vs. the CM-LX-2-RFP group. (C) H&E staining of tumor tissues showed hyperplasia of fibrous connective tissues, inflammatory cell infiltration and multinuclear tumor cells in subcutaneous tumor samples (magnification, ×200). Immunohistochemical detection of Ki67 in subcutaneous tumor samples (magnification, ×200). Collagenous fibers in subcutaneous tumor were observed using Sirius red staining (magnification, ×200). (D) Microvessels in subcutaneous tumor tissues were observed using immunofluorescence staining for CD31 (magnification, ×200). Data were quantified as the number of microvessels (CD31-positive endothelial cells). *P<0.05. (E) Protein expression of VEGF in subcutaneous tumor tissues was detected using western blot analysis. CCN1, cysteine-rich 61; H&E, hematoxylin and eosin; VEGF, vascular endothelial growth factor.
H&E staining of the subcutaneous tumor tissue revealed that the tissue structure was more complex in the HepG2 cells + LX-2-CCN1 cells, compared with that in the other groups. There was more marked hyperplasia in multinuclear tumor cells, fibrous connective tissue and infiltration of inflammatory cells in the HepG2 + LX-2-CCN1 cells group, compared with the other groups (Fig. 5C).
To determine whether CCN1 promotes the proliferation of HCC xenografts, the present study examined the expression of Ki-67, a nuclear protein necessary for tumor cell proliferation, in subcutaneous tumors with immunohistochemical staining. Subcutaneous tumors from the HepG2 + LX-2-CCN1 group exhibited more marked Ki-67-positive staining, compared with those from other groups (Fig. 5C).
According to the results of the Sirius red staining, collagenous fiber was significantly increased in the subcutaneous tumors of the HepG2 + LX-2-CCN1 co-transplantation group, compared with than that in the other groups (Fig. 5C).
Microvessel density and protein expression of VEGF in subcutaneous tumors
The microvessel density in the subcutaneous tumor tissues was examined by immunofluorescent staining for CD31 which is a specific endothelial marker. As shown in Fig. 5D, CD31-positive cells and the number of microvessels were higher in the subcutaneous tumors of HepG2 cells + LX-2-CCN1 cells, compared with those in other groups. In addition, the expression of VEGF, an important molecule for angiogenesis, was examined. The western blot assay showed that the expression of VEGF was upregulated in the subcutaneous tumors of the HepG2 cells + LX-2-CCN1 cells (Fig. 5E). It has been reported that CCN1 can induce the expression of VEGF, EGF and other growth factors, which is expressed by HSCs induced by CCN1, promotes the formation of blood vessels (6). Therefore, the above data indicated that co-transplantation of HepG2 cells and LX-2-CCN1 cells significantly promoted tumor angiogenesis.
Discussion
It is known that CCN1 is an extracellular matrix protein, and its involvement in tumors progression through binding different integrins is complex (7,8,33). Previous studies have demonstrated that CCN1 has various effects on the fibrosis of cutaneous wound healing through binding different integrins on the fibroblast membrane. CCN1 can stimulate skin fibroblast migration, adhesion and proliferation through binding integrins αvβ5 and αvβ3 (12–14). However, CCN1 also restricts fibrosis through inducing skin fibroblast senescence and apoptosis through binding integrin α6β1 (34).
Increasing studies have shown that CCN1 can regulate several types of liver cell. According to a study by Bian et al, CCN1 was expressed in the hepatocytes of mice with non-alcoholic fatty liver disease, and was involved in macrophage infiltration and the hepatic proinflammatory response (17). In our previous study, it was found that the protein expression of CCN1 was not detected in the liver tissues of healthy individuals, but its expression level was elevated in hepatic cirrhosis tissue and markedly increased in cancer-adjacent hepatic cirrhosis tissue (15). It was also found that CCN1 is a target gene of β-catenin in hepatocelluar carcinoma and promotes the proliferation of HepG2 cells (15). Kim et al demonstrated that CCN1 triggers cellular senescence through the accumulation of reactive oxygen species in activated HSCs, which limits fibrogenesis and promotes the regression of liver fibrosis induced by diverse injuries. It was suggested that chronic persistent liver injuries might overwhelm the antifibrotic activities of CCN1 despite the elevated accumulation of CCN1 (20).
Previous studies have demonstrated that CCN1 induces cholangiocyte proliferation and ductular reaction, and identified CCN1/αvβ5/NF-κB/JAG1 as a critical axis for biliary injury repair (21). CCN1 also suppresses hepatocarcinogenesis by inhibiting EGFR-dependent hepatocyte compensatory proliferation (22). These results indicate that CCN1 has various effects on the liver inflammation-fibrosis-carcinoma axis.
In liver fibrosis, activated HSCs are the main ECM-producing cells and are important in promoting the progression of HCC (35). Several integrin subunits are located on the HSC membrane, mediating the proliferation, migration and fibrogenic activation of HSCs (16,23–25). Therefore, CCN1 may have various effects on activated HSCs through binding to different integrins at different stages of liver disease. In addition, proteomic analyses of ECM from the LX-2 human hepatic stellate cell line and human foreskin fibroblasts revealed that certain components were found in both, but they exhibited different connectivities within each protein-protein interaction network (16). This indicates that the role of CCN1 in HSCs may be different from those of skin fibroblasts.
In the present study, it was shown that CCN1 was not expressed in the liver tissues of normal mice, but was overexpressed in the mouse model of liver fibrosis, with the expression tendency closely associated with the severity of liver fibrosis. Rashid et al (16) and Kim et al (20) also found that CCN1 accumulated at higher levels in the livers of patients with cirrhosis and murine models of hepatic injury and fibrosis. These results indicated that CCN1 may be involved in the process of hepatic fibrosis. CCN1 is an immediate-early gene and is transcriptionally activated on stimulation by serum growth factors within minutes (6). Therefore, CCN1 may be a suitable marker for diagnosing and predicting prognosis of liver fibrosis.
The present study also found that CCN1 activated LX-2 cells and affected their function. CCN1 promoted the viability and migration of LX-2 cells, induced the fiber differentiation of LX-2 cells and increased the expression of proliferation-correlated signaling molecules and angiogenesis molecules in LX-2-CCN1 cells. The animal experiments performed in the present study revealed that the expression of CCN1 was directly correlated with the progression of liver fibrosis in mice. On the basis of these findings, it was hypothesized that CCN1 can promote HSC fibrosis, which differs from the results reported by Kim et al (20). CCN1 may have complex effects through binding different integrins on the HSC membrane during liver inflammation-fibrosis-cirrhosis-cancer progression.
The activated LX-2 cells promoted the proliferation of HepG2 cells in vitro and in vivo, and CM from the LX-2-CCN1 cells stimulated the expression of cyclin D1, c-myc and survivin, and the activity of β-catenin signaling in HepG2 cells. These proteins are all factors promoting cell proliferation. β-catenin is a key molecule in the canonical Wnt signaling pathway. Aberrant activation of canonical Wnt/β-catenin signaling has been shown to contribute to the development of HCC (36,37). Cyclin D1 and c-myc are known tumor-associated signaling molecules (38,39). Survivin is an important inhibitor of apoptosis and is overexpressed in several types of tumor; it can promote the invasion, metastasis, growth and survival of malignant cells, and confer resistance to specific chemotherapeutic drugs (39,40). The present study also found that CM from LX-2-CCN1 cells promoted the migration and invasion of HepG2 cells, stimulated the expression of phosphorylated-ERK1/2 and MMP-9, and inhibited the expression of E-cadherin. Phosphorylated-ERK and MMP-9 are invasion-associated signaling molecules and are crucial in the progression of HCC (29,41). The findings indicated that the malignant phenotype of HepG2 cells was promoted by CM from LX-2-CCN1, which demonstrated that CCN1 enhanced the function of activated HSCs in promoting the progression of HCC.
Activated HSCs promote the progression of HCC in part through regulating the formation of the tumor microenvironment, including regulating ECM and angiogenesis (28–30). In the present study, it was found that CCN1 enhanced this function. Histological analysis of the subcutaneous tumors revealed that the tissue structure was more complex in the HepG2 + LX-2-CCN1 cell group, compared with that in the other groups. The hyperplasia in multinuclear tumor cells, fibrous connective tissue and infiltration of inflammatory cells were more marked in the HepG2 + LX-2-CCN1 cell group. In addition, increased numbers of collagenous fibers and microvessels were found in the subcutaneous tumors of the HepG2 + LX-2-CCN1 cell group. This indicated that CCN1 has an effect on activated HSC-regulated tumor microenvironment formation, which is conducive to the progression of HCC. These findings indicated that CCN1 was involved in the progression of the hepatic cirrhosis-HCC axis through regulating HSCs.
In conclusion, the results of the present study indicated that CCN1/Cyr61 activated LX-2 cells and affected the cell function. CCN1 enhanced the function of HSCs in promoting the progression of HCC. Therefore, CCN1 may be important during the progression of the hepatic cirrhosis-HCC axis through regulating HSCs.
Acknowledgments
This study was supported by The National Natural Science Foundation of China (grant no. 30672393), the Southwest Hospital Routine Management Project of Science and Technology Innovation Program (grant no. SWH2016JCYB-13) and the Chengdu Military General Hospital Routine Management Project of Science (grant no. 2016KC04).
Glossary
Abbreviations
- CCN1/Cyr61
cysteine-rich 61
- HSCs
hepatic stellate cells
- HCC
hepatocellular carcinoma
- CM
conditioned medium
References
- 1.Song P, Tang W, Tamura S, Hasegawa K, Sugawara Y, Dong J, Kokudo N. The management of hepatocellular carcinoma in Asia: A guideline combining quantitative and qualitative evaluation. Biosci Trends. 2010;4:283–287. [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Schütte K, Bornschein J, Malfertheiner P. Hepatocellular carcinoma-epidemiological trends and risk factors. Dig Dis. 2009;27:80–92. doi: 10.1159/000218339. [DOI] [PubMed] [Google Scholar]
- 4.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li JT, Liao ZX, Ping J, Xu D, Wang H. Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J Gastroenterol. 2008;43:419–428. doi: 10.1007/s00535-008-2180-y. [DOI] [PubMed] [Google Scholar]
- 6.Lau LF. CCN1/CYR61: The very model of a modern matricellular protein. Cell Mol Life Sci. 2011;68:3149–3163. doi: 10.1007/s00018-011-0778-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitz P, Gerber U, Jüngel E, Schütze N, Blaheta R, Bendas G. Cyr61/CCN1 affects the integrin-mediated migration of prostate cancer cells (PC-3) in vitro. Int J Clin Pharmacol Ther. 2013;51:47–50. doi: 10.5414/CPP51047. [DOI] [PubMed] [Google Scholar]
- 8.Haseley A, Boone S, Wojton J, Yu L, Yoo JY, Yu J, Kurozumi K, Glorioso JC, Caligiuri MA, Kaur B. Extracellular matrix protein CCN1 limits oncolytic efficacy in glioma. Cancer Res. 2012;72:1353–1362. doi: 10.1158/0008-5472.CAN-11-2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jun JI, Kim KH, Lau LF. The matricellular protein CCN1 mediates neutrophil efferocytosis in cutaneous wound healing. Nat Commun. 2015;6:7386. doi: 10.1038/ncomms8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chintala H, Krupska I, Yan L, Lau L, Grant M, Chaqour B. The matricellular protein CCN1 controls retinal angiogenesis by targeting VEGF, Src homology 2 domain phosphatase-1 and Notch signaling. Development. 2015;142:2364–2374. doi: 10.1242/dev.121913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang F, Hao F, An D, Zeng L, Wang Y, Xu X, Cui MZ. The matricellular protein Cyr61 is a key mediator of platelet-derived growth factor-induced cell migration. J Biol Chem. 2015;290:8232–8242. doi: 10.1074/jbc.M114.623074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov. 2011;10:945–963. doi: 10.1038/nrd3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grzeszkiewicz TM, Kirschling DJ, Chen N, Lau LF. CYR61 stimulates human skin fibroblast migration through integrin alpha vbeta 5 and enhances mitogenesis through integrin alpha vbeta 3, independent of its carboxyl-terminal domain. J Biol Chem. 2001;276:21943–21950. doi: 10.1074/jbc.M100978200. [DOI] [PubMed] [Google Scholar]
- 14.Chen N, Chen CC, Lau LF. Adhesion of human skin fibroblasts to Cyr61 is mediated through integrin alpha 6beta 1 and cell surface heparan sulfate proteoglycans. J Biol Chem. 2000;275:24953–24961. doi: 10.1074/jbc.M003040200. [DOI] [PubMed] [Google Scholar]
- 15.Li ZQ, Ding W, Sun SJ, Li J, Pan J, Zhao C, Wu WR, Si WK. Cyr61/CCN1 is regulated by Wnt/β-catenin signaling and plays an important role in the progression of hepatocellular carcinoma. PLoS One. 2012;7:e35754. doi: 10.1371/journal.pone.0035754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rashid ST, Humphries JD, Byron A, Dhar A, Askari JA, Selley JN, Knight D, Goldin RD, Thursz M, Humphries MJ. Proteomic analysis of extracellular matrix from the hepatic stellate cell line LX-2 identifies CYR61 and Wnt-5a as novel constituents of fibrotic liver. J Proteome Res. 2012;11:4052–4064. doi: 10.1021/pr3000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bian Z, Peng Y, You Z, Wang Q, Miao Q, Liu Y, Han X, Qiu D, Li Z, Ma X. CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. J Lipid Res. 2013;54:44–54. doi: 10.1194/jlr.M026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borkham-Kamphorst E, Schaffrath C, Van de Leur E, Haas U, Tihaa L, Meurer SK, Nevzorova YA, Liedtke C, Weiskirchen R. The anti-fibrotic effects of CCN1/CYR61 in primary portal myofibroblasts are mediated through induction of reactive oxygen species resulting in cellular senescence, apoptosis and attenuated TGF-β signaling. Biochim Biophys Acta. 2014;1843:902–914. doi: 10.1016/j.bbamcr.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 19.Borkham-Kamphorst E, Steffen BT, Van de Leur E, Haas U, Tihaa L, Friedman SL, Weiskirchen R. CCN1/CYR61 overexpression in hepatic stellate cells induces ER stress-related apoptosis. Cell Signal. 2016;28:34–42. doi: 10.1016/j.cellsig.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 20.Kim KH, Chen CC, Monzon RI, Lau LF. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol. 2013;33:2078–2090. doi: 10.1128/MCB.00049-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim KH, Chen CC, Alpini G, Lau LF. CCN1 induces hepatic ductular reaction through integrin αvβ(5)-mediated activation of NF-κB. J Clin Invest. 2015;125:1886–1900. doi: 10.1172/JCI79327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen CC, Kim KH, Lau LF. The matricellular protein CCN1 suppresses hepatocarcinogenesis by inhibiting compensatory proliferation. Oncogene. 2016;35:1314–1323. doi: 10.1038/onc.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang G, Brigstock DR. Integrin expression and function in the response of primary culture hepatic stellate cells to connective tissue growth factor (CCN2) J Cell Mol Med. 2011;15:1087–1095. doi: 10.1111/j.1582-4934.2010.01072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patsenker E, Popov Y, Wiesner M, Goodman SL, Schuppan D. Pharmacological inhibition of the vitronectin receptor abrogates PDGF-BB-induced hepatic stellate cell migration and activation in vitro. J Hepatol. 2007;46:878–887. doi: 10.1016/j.jhep.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 25.Zhou X, Murphy FR, Gehdu N, Zhang J, Iredale JP, Benyon RC. Engagement of alphavbeta3 integrin regulates proliferation and apoptosis of hepatic stellate cells. J Biol Chem. 2004;279:23996–24006. doi: 10.1074/jbc.M311668200. [DOI] [PubMed] [Google Scholar]
- 26.Ju MJ, Qiu SJ, Fan J, Xiao YS, Gao Q, Zhou J, Li YW, Tang ZY. Peritumoral activated hepatic stellate cells predict poor clinical outcome in hepatocellular carcinoma after curative resection. Am J Clin Pathol. 2009;131:498–510. doi: 10.1309/AJCP86PPBNGOHNNL. [DOI] [PubMed] [Google Scholar]
- 27.Mikula M, Proell V, Fischer AN, Mikulits W. Activated hepatic stellate cells induce tumor progression of neoplastic hepatocytes in a TGF-beta dependent fashion. J Cell Physiol. 2006;209:560–567. doi: 10.1002/jcp.20772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao W, Zhang L, Yin Z, Su W, Ren G, Zhou C, You J, Fan J, Wang X. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int J Cancer. 2011;129:2651–2661. doi: 10.1002/ijc.25920. [DOI] [PubMed] [Google Scholar]
- 29.Santamato A, Fransvea E, Dituri F, Caligiuri A, Quaranta M, Niimi T, Pinzani M, Antonaci S, Giannelli G. Hepatic stellate cells stimulate HCC cell migration via laminin-5 production. Clin Sci (Lond) 2011;121:159–168. doi: 10.1042/CS20110002. [DOI] [PubMed] [Google Scholar]
- 30.Amann T, Bataille F, Spruss T, Mühlbauer M, Gäbele E, Schölmerich J, Kiefer P, Bosserhoff AK, Hellerbrand C. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009;100:646–653. doi: 10.1111/j.1349-7006.2009.01087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sancho-Bru P, Juez E, Moreno M, Khurdayan V, Morales-Ruiz M, Colmenero J, Arroyo V, Brenner DA, Ginès P, Bataller R. Hepatocarcinoma cells stimulate the growth, migration and expression of pro-angiogenic genes in human hepatic stellate cells. Liver Int. 2010;30:31–41. doi: 10.1111/j.1478-3231.2009.02161.x. [DOI] [PubMed] [Google Scholar]
- 32.Coulouarn C, Corlu A, Glaise D, Guénon I, Thorgeirsson SS, Clément B. Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res. 2012;72:2533–2542. doi: 10.1158/0008-5472.CAN-11-3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jandova J, Beyer TE, Meuillet EJ, Watts GS. The matrix protein CCN1/CYR61 is required for α(V)β(5)-mediated cancer cell migration. Cell Biochem Funct. 2012;30:687–695. doi: 10.1002/cbf.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–685. doi: 10.1038/ncb2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang DY, Friedman SL. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology. 2012;56:769–775. doi: 10.1002/hep.25670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nejak-Bowen KN, Monga SP. Beta-catenin signaling, liver regeneration and hepatocellular cancer: Sorting the good from the bad. Semin Cancer Biol. 2011;21:44–58. doi: 10.1016/j.semcancer.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monga SP. Role of Wnt/β-catenin signaling in liver metabolism and cancer. Int J Biochem Cell Biol. 2011;43:1021–1029. doi: 10.1016/j.biocel.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Awuah PK, Monga SP. Cell cycle-related kinase links androgen receptor and β-catenin signaling in hepatocellular carcinoma: Why are men at a loss. Hepatology. 2012;55:970–973. doi: 10.1002/hep.24774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z, Guo Y, Li J, Xu J, Liu B. Cotransfection of survivin and CD44v3 short hairpin RNAs affects proliferation, apoptosis, and invasiveness of colorectal cancer. Dig Dis Sci. 2013;58:1590–1601. doi: 10.1007/s10620-012-2539-z. [DOI] [PubMed] [Google Scholar]
- 40.Chen L, Liang L, Yan X, Liu N, Gong L, Pan S, Lin F, Zhang Q, Zhao H, Zheng F. Survivin status affects prognosis and chemosensitivity in epithelial ovarian cancer. Int J Gynecol Cancer. 2013;23:256–263. doi: 10.1097/IGC.0b013e31827ad2b8. [DOI] [PubMed] [Google Scholar]
- 41.Jia YL, Shi L, Zhou JN, Fu CJ, Chen L, Yuan HF, Wang YF, Yan XL, Xu YC, Zeng Q, et al. Epimorphin promotes human hepatocellular carcinoma invasion and metastasis through activation of focal adhesion kinase/extracellular signal-regulated kinase/matrix metalloproteinase-9 axis. Hepatology. 2011;54:1808–1818. doi: 10.1002/hep.24562. [DOI] [PubMed] [Google Scholar]