

Abstract
The selective inhibition of transcription of a chosen gene by an artificial agent has numerous applications. Usually, these agents are designed to bind a specific nucleotide sequence in the promoter or within the transcribed region of the chosen gene. However, since optimal binding sites might not exist within the gene, it is of interest to explore the possibility of transcription inhibition when the agent is designed to bind at other locations. One of these possibilities arises when an additional transcription initiation site (e.g. secondary promoter) is present upstream from the primary promoter of the target gene. In this case, transcription inhibition might be achieved by inducing the formation of an RNA-DNA hybrid (R-loop) upon transcription from the secondary promoter. The R-loop could extend into the region of the primary promoter, to interfere with promoter recognition by RNA polymerase and thereby inhibit transcription. As a sequence-specific R-loop-inducing agent, a peptide nucleic acid (PNA) could be designed to facilitate R-loop formation by sequestering the non-template DNA strand. To investigate this mode for transcription inhibition, we have employed a model system in which a PNA binding site is localized between the T3 and T7 phage RNA polymerase promoters, which respectively assume the roles of primary and secondary promoters. In accord with our model, we have demonstrated that with PNA-bound DNA substrates, transcription from the T7 promoter reduces transcription from the T3 promoter by 30-fold, while in the absence of PNA binding there is no significant effect of T7 transcription upon T3 transcription.
Graphical abstract
1. INTRODUCTION
Sequence-specific agents have been employed for the selective inhibition of gene function. One approach, termed “anti-gene”, targets DNA to interfere with transcription, as distinguished from “anti-sense”, in which the nascent RNA product is targeted (reviewed in [1]). The anti-gene approach has typically employed triplex-forming oligonucleotides (TFOs), which bind double-stranded DNA via Hoogsteen (or reverse-Hoogsteen) base pairing in the promoter or within the transcribed region of the gene, to inhibit transcription at initiation or during the elongation stage (reviewed in [2–5]). However, stable triplex formation requires a sufficiently long DNA sequence, in which one strand contains only (or almost only) purines, and the complementary strand, contains only (or almost only) pyrimidines (reviewed in [6]). The requirement for such a predominantly homopurine-homopyrimidine sequence severely limits the choice of target sequences.
A number of artificial nucleic acid analogs have been developed in recent years to create anti-gene and anti-sense agents with enhanced binding properties for DNA or RNA. One of the most successful of such analogs is peptide nucleic acid (PNA), in which the negatively charged sugar-phosphate backbone of natural nucleic acids is replaced by an uncharged peptide-like backbone ([7]; reviewed in [8]). PNA is able to invade double-stranded DNA to bind the complementary sequence within one of the DNA strands, thus rendering the opposite DNA strand unpaired. Although certain versions of PNAs can invade DNA sequences with mixed purine and pyrimidine content [9–11], the most stable PNA-DNA complexes are formed with homopurine-homopurine sequences, because in that case two PNA moieties can bind the same complementary DNA strand, one by Watson-Crick base pairing, and the other by Hoogsteen base pairing (reviewed in [8]). Thus, although in principle, the use of PNA relaxes the limitations on choice of target sequence, the targeting of homopurine-homopyrimidine sequences can enhance its effectiveness. In addition to the triplex-forming potential, there could be other features that render a particular sequence preferable for PNA binding; for example, A/T-richness (that facilitates DNA opening, and consequent PNA invasion), decreased protein associations (that makes the sequence more accessible), and the presence of multiple adjacent copies of the target sequence (that may facilitate cooperative binding of several PNA molecules).
However, the preferable target sequences might not exist within the chosen gene, while being available elsewhere in its vicinity. Thus, it is of interest to explore possibilities of transcription inhibition using PNA binding sites that occur outside of the target gene of interest. One of these possibilities is PNA-induced R-loop formation. R-loops are formed when in the course of transcription the nascent RNA re-hybridizes with the template DNA strand to form an RNA-DNA duplex (that in some cases could be hundreds of base pairs long) and renders the non-template DNA strand unpaired (reviewed in [12–21]). (R-loops also can form “in trans”, but this requires specific proteins that promote RNA invasion into DNA (reviewed in [16, 22]).) R-loop formation is facilitated by any factors that provide an “advantage” to the hybrid between the nascent RNA and the DNA template strand over the hybrid between the template and the non-template DNA strands. These factors include sequences for which thermodynamic stability of the RNA-DNA duplex is greater than that of the DNA-DNA duplex ([23–26] and references therein), breaks in the non-template DNA strand [27, 28], negative supercoiling (that facilitates DNA unwinding) [27, 29], unusual structure formation [30, 31], or sequestering of the non-template DNA strand by some ligand [32]. In accord with that, we have shown that PNA binding to the non-template DNA strand can induce R-loop formation [32].
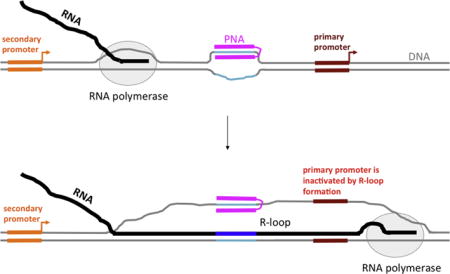
Importantly, an R-loop could extend a significant distance from the causative sequence or structure that initiates its formation, providing an opportunity for R-loop-mediated “action at a distance” for a DNA-binding agent. This opportunity could be realized in the presence of additional secondary promoter(s) upstream from the primary promoter of the target gene. Such upstream secondary promoters occur, for example, in the c-Myc (reviewed in [33]) and DHFR ([34, 35] and references therein) genes. Note that both of these genes are implicated in carcinogenesis (review in [36, 37]), and consequently are potential targets for cancer therapies. In this case, transcription inhibition might be achieved by PNA-induced R-loop formation upon transcription from the secondary promoter. The R-loop might extend into the region of the primary promoter, disrupting recognition by RNA polymerase and thus inhibiting transcription (Fig. 1). (Note that in this figure, the PNA-binding site is a homopurine-homopyrimidine sequence, and the PNA contains duplex- and triplex-forming moieties, both of which bind DNA target sequence. However, similar effects upon transcription are expected for the case of a mixed-sequence PNA, which would only form the duplex. Also, several adjacent PNA binding sites could be present.) Interestingly, the most upstream promoter (P0) of the c-Myc gene has highly enhanced activity in certain cancer cells [38, 39]; thus, using this promoter as an “R-loop inducer” in these cells might be expected to be very efficient, providing an opportunity for selective inhibition of c-Myc expression in cancer cells.
Fig. 1. Model for inhibition of primary promoter by PNA-induced R-loop formation upon transcription from an upstream initiation site.
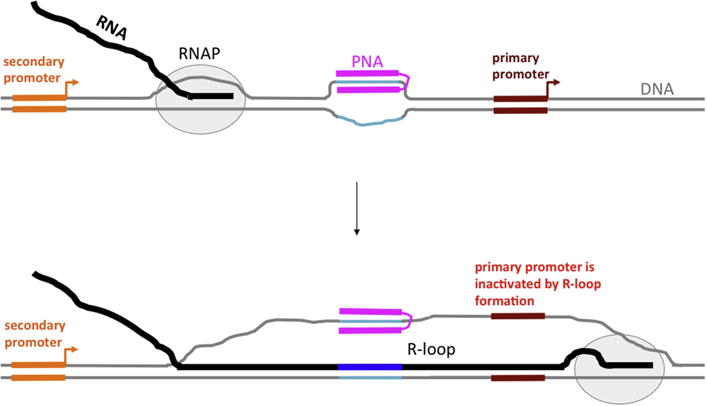
DNA is shown in gray, except for PNA binding sequence, which is shown in turquoise; RNA is shown in black, except for sequence transcribed from PNA-binding insert, which is shown in dark blue; PNA is shown in magenta. PNA contains duplex- and triplex-forming moieties, connected by a flexible linker. Primary and secondary promoters are shown in brown-red and orange, respectively. Bent arrows indicate the start and the direction of transcription. RNA polymerase (RNAP) is shown as a gray oval.
To demonstrate the possibility of transcription inhibition by an R-loop generated from an upstream secondary promoter, we have employed a model system, in which the T3 and T7 phage RNA polymerase promoters assume roles of the primary and secondary transcription promoters, respectively; and a PNA binding site is localized between them. In accordance with our prediction, in the presence of PNA, transcription initiated from the “secondary” T7 promoter strongly exacerbates inhibition of transcription from the “primary” T3 promoter.
2. MATERIALS AND METHODS
2.1. PNA
Triplex-forming PNA that hybridizes to the sequence AAGAAGAA) is the same as that we have used previously [32, 40] (see Fig. 2A and its legend for details). In this type of PNA (usually referred as bis-PNA) the parts forming Watson-Crick and Hoogsteen base pairs with the target DNA strand are connected together by a linker. This connection strongly improves DNA-hybridizing capabilities of PNA in comparison with the situation in which the Watson-Crick and Hoogsteen parts are represented by two separate PNA molecules [41].
Fig. 2. PNA-DNA hybrids and DNA substrates.
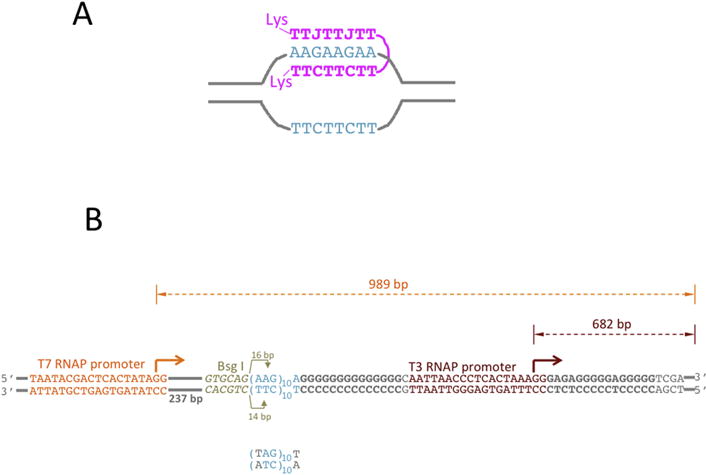
A: PNA-DNA hybrid. PNA is shown in magenta; The DNA sequence that binds PNA is shown in turquoise, the rest of DNA is shown in gray lines. The TTC-moiety of PNA forms Watson-Crick base pairing with the complementary DNA strand, while the TTJ-moiety forms Hoogsteen base pairing with that same DNA strand. J is pseudoisocytosine, which is a cytosine analog that forms non-protonated Hoogsteen base pairing with G, thus relaxing the pH-dependence for triplex formation. CTT and JTT PNA moieties are connected by a flexible linker, which is comprised of three 8-amino-3,6-dioxaoctonic acid residues. Each PNA end contains positively charged lysine (Lys) residue that increases PNA solubility and additionally strengthens PNA-DNA interaction. B: DNA substrates for transcription. PNA binding sequence is shown in turquoise. It contains ten AAG repeats, which provide binding sites for about three PNA molecules. The mutated PNA binding sequence is shown below. It contains A-T inversion in each repeat (shown in gray), which disrupts PNA-DNA binding. Immediately upstream from the PNA binding sequence, there is a Bsg I restrictase recognition sequence (shown in yellowish-gray italic). Bsg I cleaves an arbitrary sequence at exact distance (shown by yellowish-gray arrows) from its recognition site. The Bsg I cleavage site is localized roughly in the middle of PNA binding sequence; and, consequently, the cleavage is inhibited upon PNA binding. This cleavage inhibition could be used to monitor PNA binding. T3 (“primary”) and T7 (“secondary”) promoters are shown in brown-red and orange, respectively. Bent arrows show the start and the direction of transcription. The distance from the starts to the end of the template (shown by double-arrowed dashed lines) correspond to the size of respective full-size (run-off) transcripts. The rest of DNA is shown in gray. G-rich sequences downstream from the PNA binding site and from the T3 promoter are shown in bold gray.
2.2. DNA substrates
The DNA substrates are shown in Fig. 2B. The DNA insert containing Bsg I recognition sequence (yellowish-gray), the PNA binding sequence (turquoise), the T3 RNAP promoter (brown-red) and the flanking sequences (gray) was obtained by annealing of two complementary DNA oligonucleotides (IDT) and cloned into the BamHI/XhoI site of the pUCGTG-TS plasmid [42], 237 bp downstream from the T7 RNAP promoter (orange). All plasmids were purified using standard Qiagen maxiprep protocol, except that cell lysis time was reduced to several seconds. To obtain linearized substrates, the plasmids were digested by Dra III restriction enzyme and restriction products were purified from agarose gels, as described in [26, 28, 43]. As a template for the “spiking transcript” (used to eliminate effects of loading errors, see below), the plasmid pWT-C [43] linearized by BamHI was used. This substrate produces a run-off product of 234 nt without any other detectable transcription products. That product is much shorter than the run-off products from the T3 and T7 promoters (682 nt and 989 nt, respectively) and clearly resolved from them during gel-electrophoresis.
2.3. PNA-DNA hybridization
PNA-DNA hybridization was performed in buffer: 9 mM TrisHCl pH~8.5, 0.08 mM EDTA. DNA concentration was 10 ng/μl, PNA concentration was 10 μM. In PNA(−) controls, volumes were adjusted by water, so that concentrations of all other components were the same. Both PNA(+) and PNA(−) mixtures were incubated for 2 hours at 37°C; and then stored at −80°C.
2.4. Bsg I restriction reaction
Bsg I restriction was performed in total volume 6 μl, which contained 2 units of Bsg I restriction enzyme, 1x CutSmart buffer supplemented with 80 μM of S-Adenosyl methionine (SAM) (all from NEB), 2 μl of PNA-DNA hybridization mixture described in previous subsection (which corresponds to 20 ng of DNA or PNA-DNA hybrids) for 5 hours at 37°C.
After that, 2 μl of 8.4× standard glycerol gel-loading solution supplemented with 80 mM EDTA were added to the restriction reaction mixture; and the samples were analyzed on 1% agarose gels.
2.5. Transcription
The in vitro transcription experiment was performed in two steps:
At the first step, T7 RNAP transcription was performed without radioactive labeling of the transcript (i.e. in the absence of a radioactive NTP). For that, 1 μl of substrate mixture (obtained as described in PNA-DNA hybridization subsection) was added to 11 μl of a mixture containing 36 mM TrisHCl (pH 7.9), 5.5 mM MgCl2, 9.1 mM NaCl, 1.8 mM spermidine, 4.5 mM DTT, 0.18 mM of each non-radioactive (“cold”) NTP, 1.5 units/μl of RNasin, 1.8 units/μl of T7 RNAP (both from Promega corp, Madison, WI), and the resulting mixture was incubated for 30 min at 37°C.
At the second step, 4 μl of the T7 transcription mixture, obtained from the first step were added to 11 μl of the mixture with the same composition as described above, except that it contained 1.7 units/μl of T3 RNAP (Promega corp, Madison, WI) instead of T7 RNAP, and in addition, it was supplemented with 10 μCi of radioactive (α-32P) CTP; and incubated for 30 min at 37°C.
Spiking transcripts were obtained by transcription from 10 ng of the spiking DNA template (see the DNA substrates subsection) under the same conditions as T7 transcription at the first step described above, except the reaction was supplemented with 10 μCi of radioactive (α-32P) CTP.
All transcription reactions were stopped by adding 0.4 μl of 0.5 M EDTA. The spiking transcription mixture was diluted by addition 60 μl buffer of buffer containing 1.7 mM TrisHCl (pH 7.9) and 1.7 mM EDTA; and 1 μl of the obtained spiking mixture was added to each of the samples.
After stopping the transcription reaction and adding the spiking transcript, 1.5 μl of sample were mixed with 2 μl of formamide loading buffer, heated at 85°C for ~ 5 min and analyzed by gel-electrophoresis in a 5% sequencing gel.
The gels were quantitated by phosphoimaging. To eliminate effect of possible loading errors upon comparison of transcription yield for different samples, all radioactive signals of interest were normalized on the spiking transcript signal in the same lane. The rationale for this is that loading errors should be the same for the spiking transcripts and the transcript of interest within the same loading aliquot; and, since the same amounts of spiking transcript were added to each sample, this normalization should cancel the effect of loading errors.
2.6. RNase H treatment
In some experiments the samples were treated by RNase H between the first and the second steps of transcription experiment (see above).
In this case, 3 μl of the T7 transcription mixture, which obtained at the first step (and after that was stored at −80°C prior to following treatment) were supplemented with 2 units of RNase H, 0.4 μl of 10 × RNase H buffer (both from NEB) and water, so that the final volume became 4 μl. The reaction mixture was incubated for 2 hours at 37°C, and then the second (T3) step of transcription experiment was performed as described above.
3. RESULTS
3.1. Experimental design
The DNA substrates used in this work were designed to explore the mechanism for transcription blockage shown in Fig. 1.
For that purpose, these substrates (Fig. 2B) contained following elements:
T3 RNAP promoter (brown-red) models “primary” promoter. The yield of transcription from this promoter is the main parameter of interest in the current work. The yield of transcription from this promoter was quantified from the intensity of radioactive signal that corresponds to the run-off product from this promoter, which length (682 nt) corresponds to the distance between the T3 transcription start site and the end of the linearized DNA template.
T7 RNAP promoter (orange) models the “secondary” promoter, that is localized upstream from the T3 (i.e., “primary”) promoter, and is expected to modulate its activity. The run-off product from this promoter is 989 nt, which is well-separated on gels from the run-off product from the T3 promoter.
PNA-binding (AAG)n/(TTC)n sequence motif (turquoise) is localized upstream from the T3 RNAP promoter, and downstream from the T7 RNAP promoter. PNA (Fig. 2A; magenta) is designed to bind to the non-template DNA strand. According to our model, PNA binding facilitates R-loop formation upon transcription from the T7 promoter [32], which would extend into the region of the T3 promoter thus inhibiting T3 transcription. Note that in Fig. 2A only one PNA molecule is shown that binds an 8 nt DNA sequence. In our construct (Fig. 2B) the AAG/TTC-stretch is 30 bp long, providing binding sites for up to three PNA molecules. For a negative control for the PNA-binding sequence, the AAG/TTC sequence motif was replaced by TAG/ATC (Fig. 2B, bottom), which has the same G/C content, but is not complementary to PNA. Adjacent to the PNA-binding sequence, a recognition site for the Bsg I restriction endonuclease was positioned in such a way that its cleavage site is localized within the PNA-binding sequence; consequently, its action would be inhibited when the PNA is bound, so we can monitor the efficiency of PNA binding by inhibition of Bsg I cleavage. We observed essentially complete inhibition of Bsg I cleavage in the samples with PNA-binding sequence pre-incubated with PNA, indicating nearly 100% sequence-specific PNA binding (Fig.S1).
In addition to the above-mentioned essential elements of our design, we placed G-rich homopurine-homopyrimidine stretches (bold gray) both upstream and downstream from the T3 RNAP promoter. These sequences are known to produce extra-stable RNA-DNA hybrids ([44]; [28] and references therein) and thus should further stabilize the R-loop generated by PNA.
3.2. In the presence of PNA binding, transcription from T7 (“secondary”) promoter strongly inhibits transcription from T3 (“primary”) promoter
Fig. 3 and Fig. 4 show typical results and their quantitation, respectively, for the T3 RNAP transcription experiment for substrates with and without the PNA binding sequence (Fig. 3; lanes 1–4, and 5–8, respectively); either in the presence, or in the absence of PNA (Fig. 3, lanes 1, 2, 5, 6, and 3, 4, 7, 8, respectively); and with or without transcription of the substrates with T7 RNAP (Fig. 3, lanes 1, 3, 5, 7 and 2, 4, 6, 8, respectively). In the absence of PNA binding and T7 transcription, the difference in the yield of T3 RNAP transcription for substrates with and without PNA-binding sequence was insignificant (Fig. 3, lanes 4 and 8, respectively; and respective quantitation in Fig. 4). Addition of PNA to substrates with the PNA-binding site substantially (about 8-fold) reduces the yield of T3 RNAP transcription (Fig. 3, lane 2 vs. lane 4; and respective quantitation in Fig. 4). This happens despite the fact that the PNA-binding sequence is localized 15 bp upstream from the T3 RNAP promoter (see Fig. 2B). Apparently, through this distance PNA binding is still able to partially inhibit T3 transcription initiation either by steric interference with RNAP binding or by propagation of the PNA-induced DNA structure distortion into the promoter region.
Fig. 3. T3 RNAP transcription in the presence or absence of the PNA binding sequence, PNA, and preceding T7 transcription.
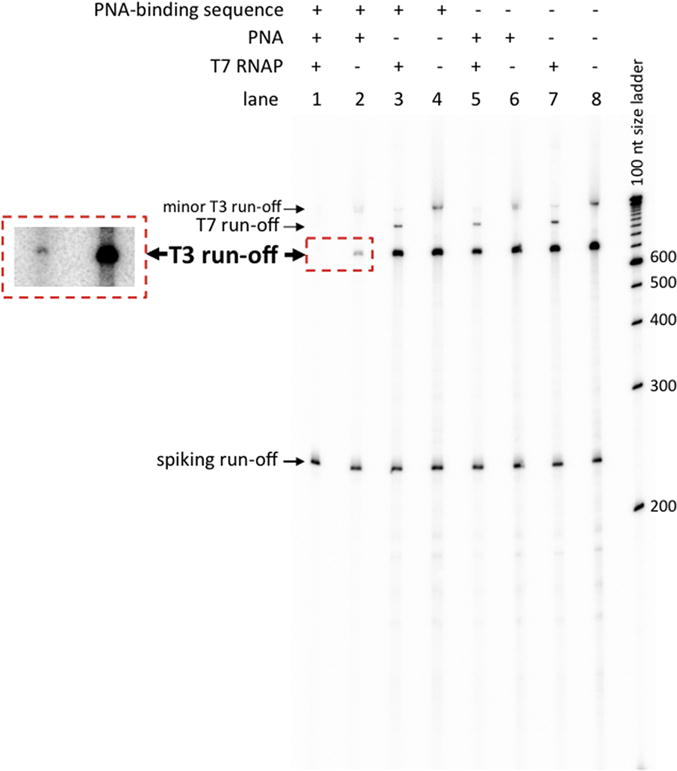
The major T3 run-off transcription product is marked by large bold letters. For lanes 1 and 2, an area of the same gel at higher exposure containing this product is shown in the box with red dashed borders. A longer smeary T3 transcription product(s) with much smaller intensity is also seen, which does not affect interpretation of our results. (It might correspond to promoter-independent initiation from the ends of substrate.) In the cases, in which T7 transcription was performed prior to T3 transcription, a signal that corresponds to T7 transcription run-off is also seen, that is much lower than that for the T3 transcript, because T7 transcription was initially performed in the absence of radioactively labeled nucleotide triphosphate (see Transcription subsection in Materials and Methods). Spiking transcript run-off is seen in each lane of the gel. To verify the sizes of transcription products, a 100 nt step-size denatured DNA ladder (the most-right lane in the gel) was used.
Fig. 4. Quantitation of yields for T3 RNAP transcription in the presence or absence of PNA binding sequence, PNA, and preceding T7 transcription.
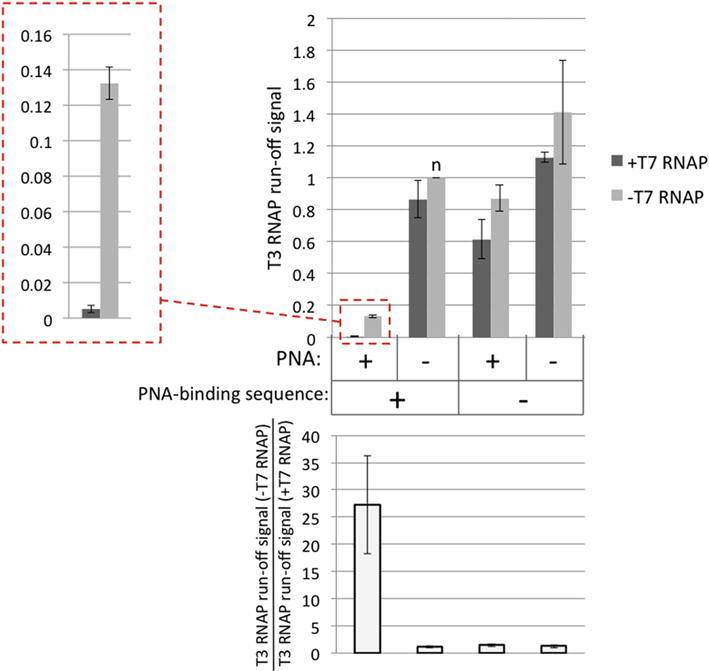
The heights of the columns and the error bars correspond to the mean values and the standard deviations, respectively, obtained from two independent experiments. As a measure of transcription yield, a radioactive signal that corresponds to main T3 run-off transcription product (shown in Fig. 3 in bold letters) was used. All signals were normalized to the spiking run-off signal in the same lane, and also normalized to the signal corresponding to the substrate containing PNA-binding sequence, in the absence of PNA and T7 transcription (the column marked “n” in the top panel of the Figure); so, the height of this column is exactly 1. In the top panel, the darker and the lighter-gray columns correspond to the experiment in the presence and in the absence of T7 transcription, respectively. An enlarged image for the first two columns is shown at the left in the box with dashed red borders. The bottom panel shows the ratio between the yields of T3 transcripts in the absence and in the presence of T7 transcription (i.e. the ratio of heights for respective lighter-gray and darker-gray columns in the top panel).
In the absence of the PNA-binding sequence, the addition of PNA causes slight (about 1.5-fold) decrease in the transcription yield (Fig. 3, lane 6 vs. lane 8; and respective quantitation in Fig. 4), which is close to experimental error and might be due to some minor off target effect of PNA upon transcription.
From the perspective of our model, the most important results concern the effect of T7 transcription upon the yield of T3 transcription. In the absence of either the PNA-binding sequence, or PNA, the effect of T7 transcription upon the yield of T3 transcription is only weakly pronounced, and is comparable to experimental error (Fig. 3, lanes 3–8; and respective quantitation in Fig. 4).
The situation changes dramatically in the presence of both PNA and the PNA-binding sequence (Fig. 3, lanes1, 2; Fig. 4, in the box with red dashed borders). In this case, T7 transcription causes 20–30-fold decrease in T3 transcription. This strongly supports our model, in which PNA binding causes R-loop formation during T7 transcription, and that this R-loop is extended into the area of T3 promoter to effectively suppress T3 transcription.
Note that in the presence of PNA binding (lane 1), the T7 RNAP run-off product practically disappears. This is in accordance with our previous observations [32] that PNA binding to the non-template strand within a transcribed region strongly decreases the intensity of the run-off product. That can be explained by our model [32] that R-loop formation in the wake of RNAP destabilizes the transcription complex and renders it more prone to spontaneous pausing or termination some distance downstream from the R-loop-initiating region (which in this case is the PNA-binding sequence). Thus, after R-loop formation, most of the transcription complexes wouldn’t reach the end of the DNA substrate (over 700 bp downstream from the R-loop initiation site) to produce run-off transcripts. Additional transcription blockages could occur when RNAPs encounter R-loops formed during previous rounds of transcription. These two mechanisms of transcription blockage could lead to the observed dramatic decrease in the yield of runoff transcript. The Supplementary Results and Discussion and Figs S3 and S4 provide more detailed explanation and data in its support.
3.3. RNase H abolishes the inhibitory effect of T7 (“secondary”) transcription upon T3 (“primary”) transcription
To determine whether RNA-DNA hybrids created by T7 transcription in the presence of bound PNA are indeed responsible for the T3 transcription inhibition, we treated T7 RNAP-transcribed samples by RNase H (that specifically digests RNA within RNA-DNA hybrids) prior to T3 transcription.
Results of the respective experiments are shown in Fig. 5, and their quantitation is presented in Fig. 6. Only the substrate with PNA-binding sequence was used in these experiments.
Fig. 5. Effect of RNaseH treatment upon T3 RNAP transcription.
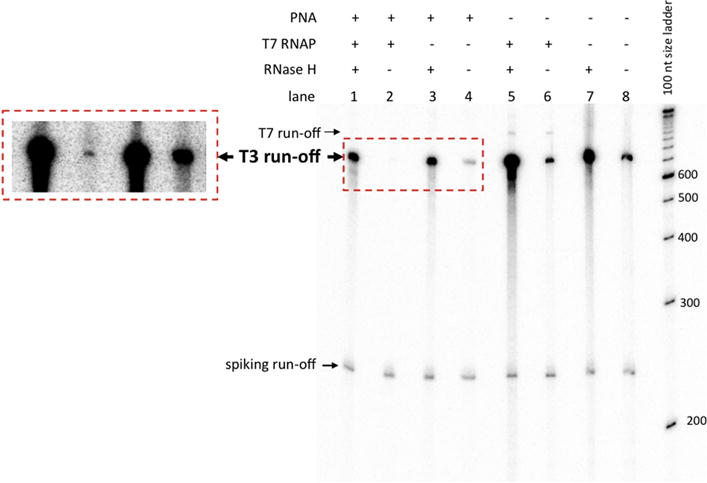
The transcription products are the same as in Fig. 3. Only the substrate containing the PNA-binding sequence was used in this experiment. At the left, for lanes 1–4, an area of the same gel at higher exposure containing T3 run-off product is shown in the box with red dashed borders.
Fig. 6. Quantitation for the results from Fig. 5 and its repetition.
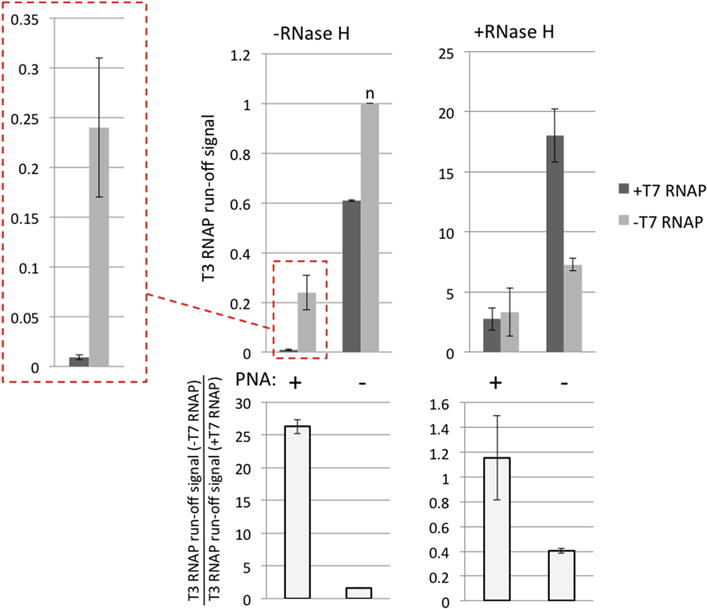
Quantitation was performed in the same way, as for Fig. 4. Note that the height of the column marked “n” (top-left panel) that corresponds to PNA(−), T7(−), RNaseH(−) conditions was used for normalization of both RNase H(−) and RNase H (+) data.
It is seen that for the PNA-bound substrate in the absence of RNase H treatment, T7 transcription reduces the yield of T3 transcription about 27-fold (Fig. 5, lane 2 vs. lane 4; Fig. 6, in the box with red dashed borders), similar as shown in Figs 3,4.
However, treatment with RNase H after T7 transcription dramatically increases the yield of T3 transcription (Fig. 5, lane 1 vs. lane 2; quantitation in Fig. S2) and abolishes the difference between the T7-transcribed and T7-non-transcribed samples (Fig. 5, lane 1 vs. lane 3; Fig. 6, +RNase H panel, PNA + columns).
This confirms our model that RNA-DNA hybrids created by T7 transcription in the presence of bound PNA cause T3 transcription inhibition.
Note that for PNA(+) T7(−), PNA(−) T7(+) and PNA(−)T7(−) samples (Fig. 5, lanes 3, 4; lanes 5, 6; and lanes 7, 8, respectively) treatment by RNase H also increases the yield of T3 transcription, although this increase is much smaller than that for PNA(+)T7(+) samples (Fig. 5, lanes 1, 2) (see Fig.S2 for quantitation). This effect is most likely caused by the presence of the G-rich homopurine-homopyrimidine stretch immediately downstream from the T3 promoter (See Fig. 2B and Experimental design subsection of the Results, point (iv)). As mentioned above, this type of sequence is prone to R-loop formation; and its location close to the promoter could render R-loop formation efficient even for relatively short stretches. In support of this interpretation, we have previously shown that the presence of a G-rich stretch closely downstream from the promoter causes strong transcription blockage, which is abolished in the presence of RNase H, confirming R-loop formation as the cause of blockage [45]. Thus, in addition to PNA-dependent R-loops produced from the T7 promoter, R-loops produced from the T3 promoter are also likely to appear in our system. However, the results in Fig.S2 indicate that their effect upon transcription in our system is much less than the effect of PNA-induced R-loops produced from T7 promoter; thus, they do not affect interpretation of our results. Note that since the G-rich stretches are localized far away from T7 promoter (Fig. 2B), they wouldn’t be expected to produce a high yield of R-loop formation during T7 transcription[45]. However, they might additionally stabilize the PNA-induced R-loops produced by T7 transcription. This was our rationale for placing these sequences into our transcription substrates. In ongoing work we intend to determine the extent to which these sequences contribute to transcription blockage by PNA-induced R-loops.
4. DISCUSSION
We have explored a novel mode for transcription inhibition by PNA-induced R-loop formation, for which the general scheme is shown in Fig. 1 in our in vitro model system. The unique feature of this mode is that R-loop formation is induced by transcription from a “secondary” transcription start site localized upstream from the transcription promoter of interest.
We studied a model system, for which the T3 and T7 phage promoters, respectively, mimic the “primary” and “secondary” promoters.
We demonstrated that if PNA is bound to the non-template DNA strand between the T7 and T3 promoters, transcription from the T7 promoter prior to T3 transcription causes strong (about 30-fold) decrease in the yield of transcription from the T3 promoter, and this decrease is abolished when the transcription substrates are treated with RNase H before starting T3 transcription. In contrast, in the absence of PNA binding T7 transcription has no substantial effect upon T3 transcription. These results indicate that the observed 30-fold transcription blockage is caused by R-loop formation, which requires both the presence of PNA and T7 transcription, strongly supporting the scheme of blockage shown in the Fig. 1.
Several parameters of the model might be adjusted in future studies to simplify the system. First, in the present system PNA binding per se in the absence of T7 transcription causes a substantial (about 8-fold) decrease in the yield of T3 transcription. Note that the 30-fold decrease in the yield of T3 transcription for the PNA-bound substrate, due to T7 transcription, is measured relative to the PNA-bound substrate in the absence of T7 transcription (i.e., the composite effect of PNA binding and T7 transcription resulted in a 200–300-fold decrease in T3 transcription). Thus, the “intrinsic” effect of PNA upon transcription does not affect the interpretation of our results with respect to the R-loop-mediated mechanism shown in Fig. 1. We could eliminate this “intrinsic” effect of PNA by setting the PNA-binding site farther upstream from the T3 promoter. Second, in our substrates the T3 promoter is flanked by G-rich stretches. We placed those stretches to further stabilize the R-loop produced by transcription from the T7 promoter, but they could form unusual DNA structures (e.g., quadruplexes (reviewed in [46])) that could affect transcription; also, a G-rich stretch localized downstream from the T3 promoter could trigger additional R-loop formation upon transcription from that promoter. This additional stabilization may not be necessary, and consequently, these G-rich stretches could be eliminated. However, since these stretches are present in all of our substrates, their contributions do not affect our conclusions.
As mentioned in the Introduction, the presence of additional transcription start sites upstream from the major promoter occurs in many genes, notably in c-Myc and DHFR genes, of which both are implicated in carcinogenesis [33–37]. Thus, this mode of transcription inhibition could be of practical interest.
In addition, PNA-mediated DNA opening could initiate promoter-independent transcription [47]. In this case PNA invasion into DNA could serve both to create an artificial secondary promoter and to induce R-loop formation. Though R-loop-forming properties of artificial promoters created by PNA invasion have not yet been studied, it is likely that R-loops would form upon transcription from such an artificial promoter, provided that the DNA region opened by PNA is sufficiently large.
An advantage of R-loop-based approaches for gene inhibition is that R-loops can extend significantly beyond the sequence or structure in which their formation was initiated [48, 49], to provide an opportunity for “action at a distance”. R-loop formation is facilitated by sequences, for which the RNA-DNA duplex is more thermodynamically stable than the DNA-DNA duplex [49, 50]. However, topological considerations predict, that once a stable R-loop is initiated, transcription must continue in an “R-loop mode” [51], in which it could extend into sequences that are not intrinsically “R-loop-prone”. On the other hand, theoretical considerations and experimental evidence suggests that in the “R-loop mode” there is an increased probability for spontaneous termination of transcription[28, 32, 51]; consequently, there may be some limitation on the distance, beyond which an R-loop could efficiently “reach” from its initiation site before transcription arrest. It will be interesting to explore the relationship between the distance between the R-loop initiating sequence (e.g. PNA-binding site) and the target downstream promoter and transcription inhibition from the target promoter.
Numerous observations in vitro and in living cells indicate that R-loops form much more efficiently if the “R-loop-prone sequence” is localized close to 5′-end of the transcript (e.g., near the transcription start site) [27, 45, 52, 53].
However, the requirement for an R-loop-initiating sequence or structure to be in vicinity to the 5′-end of transcript for efficient R-loop formation may be alleviated for PNA-induced R-loops in comparison with R-loops induced in the intact R-loop-prone sequences: According to the “thread back” model for R-loop formation (e.g., [27]), the nascent RNA “tail” must “thread” into the transiently unpaired DNA region formed by DNA “breathing”. Since the disruption of DNA base pairing is energetically unfavorable under physiological conditions, this unpaired region is likely to be very short (probably, only a few base pairs), and it would exist only for a short period before collapsing back into the double helix. Thus, it would be challenging for a long RNA “tail” to thread into it. That is why the probability of R-loop formation is much higher for a shorter RNA tail. In contrast to DNA “breathing”, PNA binding produces a relatively long, permanently unpaired DNA region, which provides an opportunity for stable “nucleation” of an RNA-DNA hybrid, and creates much more “room” for the RNA “tail” to “thread through”. Thus, R-loop formation could be efficiently initiated despite the presence of a long nascent RNA “tail”, i.e. far downstream from the transcription start site. In accord with that, in our case the distance between the T7 transcription start site and the PNA-binding site is over 200 bp. However, in the presence of PNA binding, transcription from the T7 promoter very efficiently inhibits transcription from the T3 promoter, suggesting efficient R-loop formation despite a long distance between the T7 promoter and the PNA binding site.
Normally the R-loops formed in cells are eventually removed by special nucleases or helicases (reviewed in [14]); consequently, the probability that a given transcribed sequence harbors an R-loop depends upon a balance between the rate of R-loop formation and the rate of its removal. Since PNA binding to the non-template strand dramatically facilitates R-loop formation, it is likely that in its presence the probability for the target promoter sequence to be occupied by an R-loop would be great enough to strongly reduce transcription in vivo.
An important effect of R-loop formation in vivo is that it renders the displaced non-template sensitive to DNA-modifying enzymes (e.g. activation-induced deaminase (AID) (reviewed in [54, 55])). DNA modifications in the non-template strand, as well as intermediates of their processing by DNA repair machinery (e.g. abasic sites and single-strand DNA breaks) would additionally stabilize the R-loop, and also would interfere with promoter recognition by RNAP, thus exacerbating transcription inhibition.
For the type of PNA used in this work, invasion into the intact relaxed DNA duplex occurs efficiently only at low ionic strength, substantially below the physiological level. This dependence upon ionic conditions occurs because PNA invasion is facilitated by transient DNA opening (“breathing”); and an increase in ionic strength stabilizes the DNA duplex, thus suppressing DNA “breathing” [56]. However, once formed, the PNA-DNA complex remains stable under high salt concentrations (e.g., [40, 57]). The inhibition of PNA invasion by high salt concentration can be alleviated by negative supercoiling, which facilitates DNA breathing [56]. Also, PNA invades efficiently into transcribed DNA even under physiological ionic conditions (especially when it is targeted to the non-template strand), presumably due to the DNA opening in the course of transcription [58].
In our model experiments, we have “pre-hybridized” the PNA to DNA at low ionic strength to achieve near 100% invasion, and then have used these PNA-DNA hybrids in the transcription reaction. This facilitates the analysis of the specific effect of bound PNA upon transcription, rather than the composite effects that include both the effects of bound PNA upon transcription per se, and the efficiency of PNA invasion into DNA during transcription.
In future studies, it will be interesting to test the effect of PNA when it is added directly into the transcription mixture under physiological salt conditions, to more adequately model the in vivo situation. More recent PNA modifications with superior DNA-binding properties have been shown to have increased ability to invade DNA under physiological ionic conditions [11].
The documentation of PNA-mediated targeting to genomic DNA in vivo [59–61] supports the feasibility of our approach for biological applications. In addition, reagents other than PNA, which are able to sequence-specifically sequester the non-template DNA strand could be used in our approach. For example, the CRISPR/Cas system containing catalytically inactive Cas9 (reviewed in [62]) could hybridize RNA oligonucleotides to the non-template DNA strand to render the template DNA strand unpaired. In principle, this approach could operate in a similar way as PNA binding, provided that the unpaired template DNA strand localized within the CRISPR/Cas/DNA complex could be accessible for the transcribing RNAP.
5. CONCLUSIONS
We have shown that in the presence of PNA binding to the non-template DNA strand within the DNA region localized between two head-to-tail-oriented promoters, transcription from the upstream promoter can strongly inhibit transcription from the downstream promoter. This inhibition is due to R-loop formation upon transcription from the upstream promoter that extends into the region of the downstream promoter to inhibit transcription initiation. This novel mode of transcription inhibition could have applications for artificially targeted gene regulation.
Supplementary Material
Highlights.
Analysis of transcription interaction between two collinear head-to-tail promoters
PNA bound to the non-template strand between the promoters inhibits transcription
R-loops from upstream promoter inhibit transcription from downstream promoter
Results lead to suggested approach for artificial gene expression regulation
Acknowledgments
We thank the members of Hanawalt lab for helpful discussions, and the anonymous reviewers for helpful comments and suggestions, including the suggestion that the CRISPR/cas system might also work in our approach.
FUNDING
This research was supported by a grant (CA077712) from the National Cancer Institute to P.C.H., a Stanford Cancer Institute 2016 Fellowship Award (PTA 1164311–123-GHTDS) to B.P.B., and a VPUE Summer Research grant to A.D.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Helene C. The anti-gene strategy: control of gene expression by triplex-forming-oligonucleotides. Anticancer Drug Des. 1991;6:569–584. [PubMed] [Google Scholar]
- 2.Praseuth D, Guieysse AL, Helene C. Triple helix formation and the antigene strategy for sequence-specific control of gene expression. Biochim Biophys Acta. 1999;1489:181–206. doi: 10.1016/s0167-4781(99)00149-9. [DOI] [PubMed] [Google Scholar]
- 3.Vasquez KM, Glazer PM. Triplex-forming oligonucleotides: principles and applications. Q Rev Biophys. 2002;35:89–107. doi: 10.1017/s0033583502003773. [DOI] [PubMed] [Google Scholar]
- 4.Rogers FA, Lloyd JA, Glazer PM. Triplex-forming oligonucleotides as potential tools for modulation of gene expression. Curr Med Chem Anticancer Agents. 2005;5:319–326. doi: 10.2174/1568011054222300. [DOI] [PubMed] [Google Scholar]
- 5.Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB. The triple helix: 50 years later, the outcome. Nucleic Acids Res. 2008;36:5123–5138. doi: 10.1093/nar/gkn493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank-Kamenetskii MD, Mirkin SM. Triplex DNA structures. Annu Rev Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- 7.Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen PE. PNA Technology. Mol Biotechnol. 2004;26:233–248. doi: 10.1385/MB:26:3:233. [DOI] [PubMed] [Google Scholar]
- 9.He G, Rapireddy S, Bahal R, Sahu B, Ly DH. Strand invasion of extended, mixed-sequence B-DNA by gammaPNAs. J Am Chem Soc. 2009;131:12088–12090. doi: 10.1021/ja900228j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lohse J, Dahl O, Nielsen PE. Double duplex invasion by peptide nucleic acid: a general principle for sequence-specific targeting of double-stranded DNA. Proc Natl Acad Sci U S A. 1999;96:11804–11808. doi: 10.1073/pnas.96.21.11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rapireddy S, Bahal R, Ly DH. Strand invasion of mixed-sequence, double-helical B-DNA by gamma-peptide nucleic acids containing G-clamp nucleobases under physiological conditions. Biochemistry. 2011;50:3913–3918. doi: 10.1021/bi2002554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 13.Chedin F. Nascent Connections: R-Loops and Chromatin Patterning. Trends Genet. 2016;32:828–838. doi: 10.1016/j.tig.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sollier J, Cimprich KA. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015;25:514–522. doi: 10.1016/j.tcb.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richard P, Manley JL. R Loops and Links to Human Disease. J Mol Biol. 2016 doi: 10.1016/j.jmb.2016.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costantino L, Koshland D. The Yin and Yang of R-loop biology. Curr Opin Cell Biol. 2015;34:39–45. doi: 10.1016/j.ceb.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santos-Pereira JM, Aguilera A. R loops: new modulators of genome dynamics and function. Nat Rev Genet. 2015;16:583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- 18.Lin Y, Wilson JH. Transcription-induced DNA toxicity at trinucleotide repeats: double bubble is trouble. Cell Cycle. 2011;10:611–618. doi: 10.4161/cc.10.4.14729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groh M, Gromak N. Out of balance: R-loops in human disease. PLoS Genet. 2014;10:e1004630. doi: 10.1371/journal.pgen.1004630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Usdin K, Kumari D. Repeat-mediated epigenetic dysregulation of the FMR1 gene in the fragile X-related disorders. Front Genet. 2015;6:192. doi: 10.3389/fgene.2015.00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim N, Jinks-Robertson S. Transcription as a source of genome instability, N. at Rev Genet. 2012;13:204–214. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang F, Doudna JA. CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys. 2017 doi: 10.1146/annurev-biophys-062215-010822. [DOI] [PubMed] [Google Scholar]
- 23.Roy D, Yu K, Lieber MR. Mechanism of R-loop formation at immunoglobulin class switch sequences. Mol Cell Biol. 2008;28:50–60. doi: 10.1128/MCB.01251-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniels GA, Lieber MR. RNA:DNA complex formation upon transcription of immunoglobulin switch regions: implications for the mechanism and regulation of class switch recombination. Nucleic Acids Res. 1995;23:5006–5011. doi: 10.1093/nar/23.24.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol. 2003;4:442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- 26.Belotserkovskii BP, Liu R, Tornaletti S, Krasilnikova MM, Mirkin SM, Hanawalt PC. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc Natl Acad Sci U S A. 2010;107:12816–12821. doi: 10.1073/pnas.1007580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol Cell Biol. 2010;30:146–159. doi: 10.1128/MCB.00897-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belotserkovskii BP, Neil AJ, Saleh SS, Shin JH, Mirkin SM, Hanawalt PC. Transcription blockage by homopurine DNA sequences: role of sequence composition and single-strand breaks. Nucleic Acids Res. 2013;41:1817–1828. doi: 10.1093/nar/gks1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masse E, Drolet M. Escherichia coli DNA topoisomerase I inhibits R-loop formation by relaxing transcription-induced negative supercoiling. J Biol Chem. 1999;274:16659–16664. doi: 10.1074/jbc.274.23.16659. [DOI] [PubMed] [Google Scholar]
- 30.Grabczyk E, Mancuso M, Sammarco MC. A persistent RNA.DNA hybrid formed by transcription of the Friedreich ataxia triplet repeat in live bacteria, and by T7 RNAP in vitro. Nucleic Acids Res. 2007;35:5351–5359. doi: 10.1093/nar/gkm589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev. 2004;18:1618–1629. doi: 10.1101/gad.1200804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belotserkovskii BP, Hanawalt PC. PNA binding to the non-template DNA strand interferes with transcription, suggesting a blockage mechanism mediated by R-loop formation. Mol Carcinog. 2015;54:1508–1512. doi: 10.1002/mc.22209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcu KB, Bossone SA, Patel AJ. myc function and regulation. Annu Rev Biochem. 1992;61:809–860. doi: 10.1146/annurev.bi.61.070192.004113. [DOI] [PubMed] [Google Scholar]
- 34.Blume SW, Meng Z, Shrestha K, Snyder RC, Emanuel PD. The 5′-untranslated RNA of the human dhfr minor transcript alters transcription pre-initiation complex assembly at the major (core) promoter. J Cell Biochem. 2003;88:165–180. doi: 10.1002/jcb.10326. [DOI] [PubMed] [Google Scholar]
- 35.Martianov I, Ramadass A, Serra Barros A, Chow N, Akoulitchev A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature. 2007;445:666–670. doi: 10.1038/nature05519. [DOI] [PubMed] [Google Scholar]
- 36.Prochownik EV. c-Myc: linking transformation and genomic instability. Curr Mol Med. 2008;8:446–458. doi: 10.2174/156652408785747988. [DOI] [PubMed] [Google Scholar]
- 37.Schweitzer BI, Dicker AP, Bertino JR. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990;4:2441–2452. doi: 10.1096/fasebj.4.8.2185970. [DOI] [PubMed] [Google Scholar]
- 38.Hoover RG, Kaushal V, Lary C, Travis P, Sneed T. c-myc transcription is initiated from P0 in 70% of patients with multiple myeloma. Curr Top Microbiol Immunol. 1995;194:257–264. doi: 10.1007/978-3-642-79275-5_30. [DOI] [PubMed] [Google Scholar]
- 39.Eick D, Polack A, Kofler E, Lenoir GM, Rickinson AB, Bornkamm GW. Expression of P0- and P3-RNA from the normal and translocated c-myc allele in Burkitt’s lymphoma cells. Oncogene. 1990;5:1397–1402. [PubMed] [Google Scholar]
- 40.Belotserkovskii BP, Liu R, Hanawalt PC. Peptide nucleic acid (PNA) binding and its effect on in vitro transcription in friedreich’s ataxia triplet repeats. Mol Carcinog. 2009;48:299–308. doi: 10.1002/mc.20486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Egholm M, Christensen L, Dueholm KL, Buchardt O, Coull J, Nielsen PE. Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA. Nucleic Acids Res. 1995;23:217–222. doi: 10.1093/nar/23.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tornaletti S, Patrick SM, Turchi JJ, Hanawalt PC. Behavior of T7 RNA polymerase and mammalian RNA polymerase II at site-specific cisplatin adducts in the template DNA. J Biol Chem. 2003;278:35791–35797. doi: 10.1074/jbc.M305394200. [DOI] [PubMed] [Google Scholar]
- 43.Belotserkovskii BP, De Silva E, Tornaletti S, Wang G, Vasquez KM, Hanawalt PC. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J Biol Chem. 2007;282:32433–32441. doi: 10.1074/jbc.M704618200. [DOI] [PubMed] [Google Scholar]
- 44.Roberts RW, Crothers DM. Stability and properties of double and triple helices: dramatic effects of RNA or DNA backbone composition. Science. 1992;258:1463–1466. doi: 10.1126/science.1279808. [DOI] [PubMed] [Google Scholar]
- 45.Belotserkovskii BP, Soo Shin JH, Hanawalt PC. Strong transcription blockage mediated by R-loop formation within a G-rich homopurine-homopyrimidine sequence localized in the vicinity of the promoter. Nucleic Acids Res. 2017;45:6589–6599. doi: 10.1093/nar/gkx403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maizels N. Dynamic roles for G4 DNA in the biology of eukaryotic cells. Nat Struct Mol Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 47.Mollegaard NE, Buchardt O, Egholm M, Nielsen PE. Peptide nucleic acid.DNA strand displacement loops as artificial transcription promoters. Proc Natl Acad Sci U S A. 1994;91:3892–3895. doi: 10.1073/pnas.91.9.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang FT, Yu K, Hsieh CL, Lieber MR. Downstream boundary of chromosomal R-loops at murine switch regions: implications for the mechanism of class switch recombination. Proc Natl Acad Sci U S A. 2006;103:5030–5035. doi: 10.1073/pnas.0506548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roy D, Lieber MR. G clustering is important for the initiation of transcription-induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol Cell Biol. 2009;29:3124–3133. doi: 10.1128/MCB.00139-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wongsurawat T, Jenjaroenpun P, Kwoh CK, Kuznetsov V. Quantitative model of R-loop forming structures reveals a novel level of RNA-DNA interactome complexity. Nucleic Acids Res. 2012;40:e16. doi: 10.1093/nar/gkr1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belotserkovskii BP, Hanawalt PC. Anchoring nascent RNA to the DNA template could interfere with transcription. Biophys J. 2011;100:675–684. doi: 10.1016/j.bpj.2010.12.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen L, Chen JY, Zhang X, Gu Y, Xiao R, Shao C, Tang P, Qian H, Luo D, Li H, Zhou Y, Zhang DE, Fu XD. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol Cell. 2017;68:745–757 e745. doi: 10.1016/j.molcel.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dumelie JG, Jaffrey SR. Defining the location of promoter-associated R-loops at near-nucleotide resolution using bisDRIP-seq. Elife. 2017;6 doi: 10.7554/eLife.28306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamperl S, Cimprich KA. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst) 2014 doi: 10.1016/j.dnarep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larson ED, Maizels N. Transcription-coupled mutagenesis by the DNA deaminase AID. Genome Biol. 2004;5:211. doi: 10.1186/gb-2004-5-3-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bentin T, Nielsen PE. Enhanced peptide nucleic acid binding to supercoiled DNA: possible implications for DNA “breathing” dynamics. Biochemistry. 1996;35:8863–8869. doi: 10.1021/bi960436k. [DOI] [PubMed] [Google Scholar]
- 57.Cherny DY, Belotserkovskii BP, Frank-Kamenetskii MD, Egholm M, Buchardt O, Berg RH, Nielsen PE. DNA unwinding upon strand-displacement binding of a thymine-substituted polyamide to double-stranded DNA. Proc Natl Acad Sci U S A. 1993;90:1667–1670. doi: 10.1073/pnas.90.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Larsen HJ, Nielsen PE. Transcription-mediated binding of peptide nucleic acid (PNA) to double-stranded DNA: sequence-specific suicide transcription. Nucleic Acids Res. 1996;24:458–463. doi: 10.1093/nar/24.3.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schleifman EB, McNeer NA, Jackson A, Yamtich J, Brehm MA, Shultz LD, Greiner DL, Kumar P, Saltzman WM, Glazer PM. Site-specific Genome Editing in PBMCs With PLGA Nanoparticle-delivered PNAs Confers HIV-1 Resistance in Humanized Mice. Mol Ther Nucleic Acids. 2013;2:e135. doi: 10.1038/mtna.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schleifman EB, Glazer PM. Peptide nucleic acid-mediated recombination for targeted genomic repair and modification. Methods Mol Biol. 2014;1050:207–222. doi: 10.1007/978-1-62703-553-8_17. [DOI] [PubMed] [Google Scholar]
- 61.Chin JY, Kuan JY, Lonkar PS, Krause DS, Seidman MM, Peterson KR, Nielsen PE, Kole R, Glazer PM. Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids. Proc Natl Acad Sci U S A. 2008;105:13514–13519. doi: 10.1073/pnas.0711793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahas A, Neal Stewart C, Jr, Mahfouz MM. Harnessing CRISPR/Cas systems for programmable transcriptional and post-transcriptional regulation. Biotechnol Adv. 2017 doi: 10.1016/j.biotechadv.2017.11.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.