

Abstract
NMR spectroscopy of membrane proteins involved in electron transport is difficult due to the presence of both the lipids and paramagnetic centers. Here we report the solution NMR study of the NADPH-cytochrome P450 oxidoreductase (POR) in its reduced and oxidized states. We interrogate POR, first, in its truncated soluble form (70 kDa), which is followed by experiments with the full-length protein incorporated in a lipid nanodisc (240 kDa). To overcome paramagnetic relaxation in the reduced state of POR as well as the signal broadening due to its high molecular weight, we utilized the methyl-TROSY approach. Extrinsic 13C-methyl groups were introduced by modifying the engineered surface-exposed cysteines with methyl-methanethiosulfonate (MMTS). Chemical shift dispersion of the resonances from different sites in POR was sufficient to monitor differential effects of the reduction-oxidation process and conformation changes in the POR structure related to its function. Despite the high molecular weight of the POR-nanodisc complex, the surface-localized 13C-methyl probes were sufficiently mobile to allow for signal detection at 600 MHz without perdeuteration. This work demonstrates a potential of the solution methyl-TROSY in analysis of structure, dynamics, and function of POR, which may also be applicable to similar paramagnetic and flexible membrane proteins.
Keywords: NADPH-cytochrome P450 oxidoreductase, diflavin oxidoreductase, POR, CYPOR, CPR, lipid nanodisc, NMR spectroscopy, electron transport
Introduction
NADPH-cytochrome P450 oxidoreductase (POR, CYPOR, or CPR) mediates the transfer of electrons from NADPH to cytochromes P450 and other microsomal proteins, including heme oxygenase (Barnaba et al. 2017, Scott et al. 2016). The POR/P450 system is localized in the endoplasmic reticulum and is involved in the synthesis of steroid hormones, fatty acids, and prostaglandins as well as xenobiotic degradation. Cytochrome P450 hemoproteins act as monooxygenases—inserting molecular oxygen in C-H, N-H, and S-H bonds of their substrates (Hannemann et al. 2007). In humans, there are about 50 microsomal P450s but only one POR (Waskell and Kim 2015). POR is an integral membrane protein anchored on the cytoplasmic side of the endoplasmic reticulum membrane with a short N-terminal membrane-binding domain (Figure 1). The C-terminal cytosolic part, residues 57-678, consists of two flexibly joined soluble domains that incorporate flavin cofactors, FAD and FMN, along with a binding site for NADPH.
Figure 1.
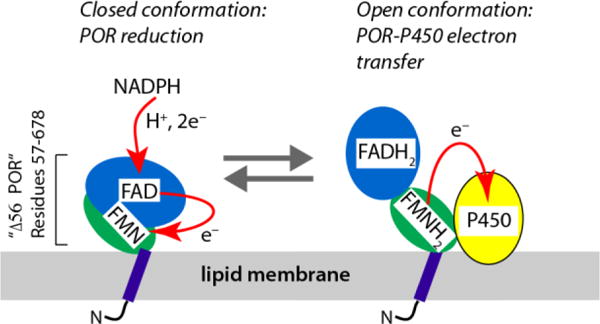
Structural organization of NADPH-cytochrome P450 oxidoreductase, POR, and the electron-transport cycle. Note: the redox forms of the flavin cofactors only aim to indicate the overall reduced/oxidized status of POR molecule; the exact redox state of each flavin and the complete sequence of the reaction cycle are complicated with more intermediate forms (Waskell and Kim 2015).
X-ray diffraction studies of the cytosolic region (using a construct lacking 56 N-terminal residues—“Δ56 POR” in the following text) identified two conformations that are required for transfer of electrons from NADPH to the redox partners of POR (Waskell and Kim 2015): the “closed” conformation to enable initial electron transfer (ET) from NADPH to the enzyme-bound flavin cofactors, and the “open” conformation to allow ET from FMN to the POR partners such as cytochromes P450 and heme oxygenase (Wang et al. 1997, Sugishima et al. 2014). In its functional cycle, POR must repeatedly undergo the open-closed transitions to allow the uninterrupted ET through the POR/P450 system. This process, however, has not been structurally resolved. The two major difficulties associated with POR function are that (1) the protein productively interacts with the cytochrome P450 only when integrated in the lipid bilayer (precluding crystallization) (Waskell and Kim 2015, Aigrain et al. 2011), and (2) the ET process involves unpaired electrons in a form of flavin semiquinones (Waskell and Kim 2015) leading to signal loss in NMR spectra due to paramagnetic relaxation.
The ET in POR was initially followed utilizing 31P and 13C flavin NMR signals of POR homologs in solution (van Schagen and Muller 1981, Evrard et al. 1999). Using heteronuclear 15N NMR spin relaxation and measurements of residual dipolar couplings, Vincent and co-workers extensively characterized the conformational state of the isolated cytosolic portion of the protein, Δ56 POR, in its oxidized state but did not report any measurements on the reduced samples (Vincent et al. 2012). Interactions of a similar soluble construct Δ66 POR with heme oxygenase was also documented by NMR in the oxidized state (Spencer et al. 2014) as well as interaction of the isolated FMN-binding domain with the cytochrome c (Huang et al. 2015) and cytochrome P450 17A1 (Estrada et al. 2016).
In this paper, we utilized extrinsic 13C-methyl labeling to record NMR signals from both oxidized and reduced POR samples. Surface localization of the methyl probes sufficiently attenuated paramagnetic relaxation enhancement, making it possible to map a range of sites in POR and evaluate their response to both reduction and conformational changes. Most remarkably, when the full-length POR was integrated in a lipid nanodisc, the NMR signal detection was still possible in both oxidized and reduced POR without perdeuteration.
Results and Discussion
Extrinsic methyl labeling
To introduce the 13C-labeled methyl groups on the surface of the POR molecule, we utilized a reaction of 13C-methyl-methanethiosulfonate (13C-MMTS) with solvent-exposed cysteine thiol groups (Bruice and Kenyon 1985, Bruice et al. 1976). The resulting 13C-methylthiocysteine residue (MTC) has a similar geometry to methionine and gives rise to a strong signal in the methyl-TROSY NMR experiment (Tugarinov et al. 2003, Religa et al. 2011). To ensure specific localization of the labels, we used a cysteine-less rat POR construct where all seven wild-type cysteines were mutated out (Xia et al. 2011), while new cysteines were introduced at chosen locations. In the following text, the wild-type cysteine-less POR construct will be referred to as “POR”.
Localization of the probes
Figure 2 demonstrates four sites on a model of POR corresponding to mutations E127C, Q157C, S308C, and Q517C (upon methylation producing MTC127, MTC157, MTC308, and MTC517, respectively). The MTC127 occurs at the membrane-facing region of the FMN domain and is relatively distant from the FMN cofactor, while MTC157 is farther from the membrane and closer to the FMN, therefore sensitive to the its redox state (Supporting Table 1). The MTC517 is in the FAD-binding domain: it will be sensitive to the redox state of both flavins as well as the domain closing. It is also located near a putative interface of the POR-P450 complex (Hamdane et al. 2009). The MTC308 is localized at the remote end of the FAD-binding domain unlikely to be significantly affected by the enzyme redox status, open/closing transition or binding of the P450. In the full-length protein, however, the MTC308 may experience a dramatic change in its distance to the membrane upon closing transition thus becoming a possible reporter of the conformational changes in POR. The membrane distance for MTC127 is expected to be relatively invariable in the open and closed POR conformations.
Figure 2.
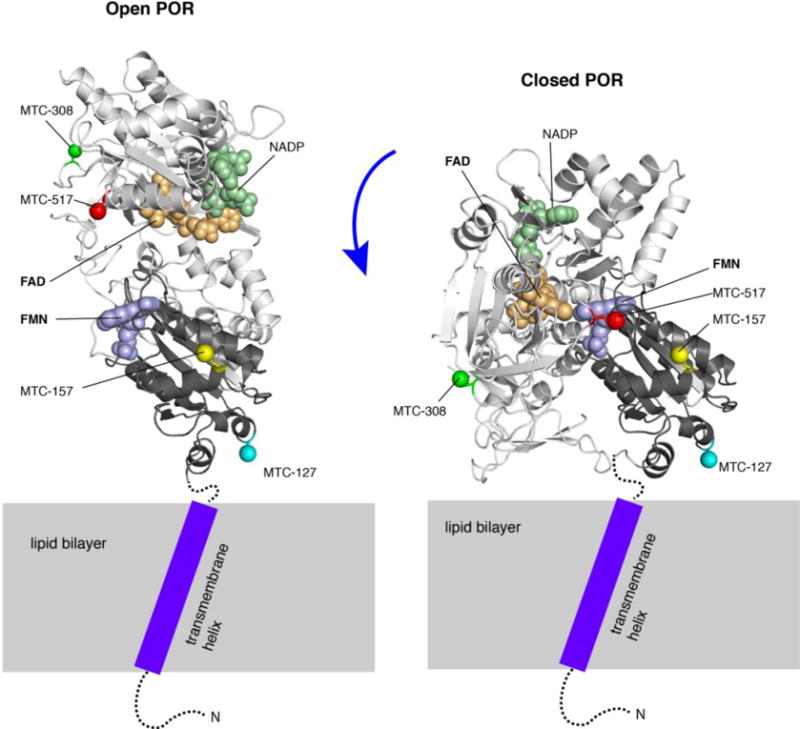
Localization of the methyl probes shown on a model of the membrane-bound open (left) and closed (right) conformations of POR (based on Δ56 POR crystal structures 3SE9 and 1AMO). Cartoon model of FAD-binding domain is colored light grey; FMN-binding domain—black; FAD, FMN, and NADP are shown as spheres in wheat, pale violet, and pale green, respectively. The lipid bilayer is schematically shown as a gray rectangle. The cytosolic unstructured portions of the N-terminal peptide (residues 1-56 absent from the crystal structures) are depicted as a dotted line. A purple rectangle stands for a transmembrane region of the N-terminal peptide containing a tilted helix (based on the solid-state NMR data (Huang et al. 2014)). The γ-atoms of residues 127, 157, 308, and 517 are shown as colored spheres. Note: the particular orientations of the cytosolic portion of POR relative to the membrane surface was never structurally resolved, and only chosen in this model to emphasize differential membrane proximity of the labeled sites.
Redox states
To create the oxidized and reduced states of POR in the NMR tube, we used potassium ferricyanide as a general electron acceptor (oxidant) or NADPH as a cognate electron donor for POR (reductant) (Waskell and Kim 2015). Figure 3.A demonstrates typical absorbance spectra characterizing redox states of the POR flavins recorded immediately after NMR data acquisition. Oxidized POR flavins give rise to the prominent absorption bands with maxima at 380 nm and 460 nm (red trace). Reduction with NADPH manifests itself in a characteristic decrease of the flavin peaks and appearance of the 550-650 nm semiquinone absorbance band (blue trace). Since the exact redox state of the reduced POR by excess NADPH is complicated (Vermilion and Coon 1978), we compared our reduced spectrum with those of the dithionite-reduced POR (we could not use dithionite in our experiments as it would strip off the 13C-methyl labels). The spectral shape observed in our NADPH-reduced samples is similar to the two-electron reduced POR when both FAD and FMN are semiquinones (Rwere et al. 2016). This di-semiquinone state was stably maintained in our samples for, at least, four hours needed for a typical NMR experiment (Figure 3.B). The UV-visible absorption spectra were recorded after every NMR measurement to verify the sample redox state.
Figure 3.
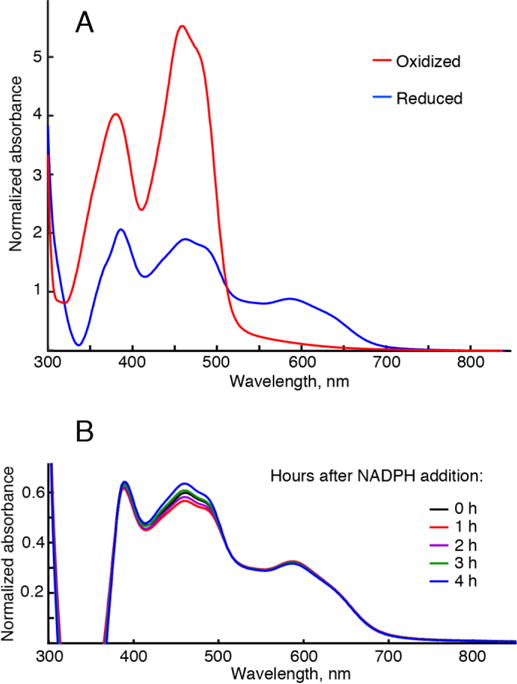
UV-visible absorbance spectra of the POR flavins. (A) Representative spectra of Q157C/Q517C Δ56 POR labeled with 13C-MMTS oxidized with ferricyanide, red trace, and reduced with excess NADPH, blue-trace. Vertical axis is an absorbance normalized by the protein concentration. Reduced trace (blue) is distorted in the wavelength range shorter than 400 nm due to incomplete compensation of the NADPH absorbance by a reference NADPH solution. (B) The time evolution of absorbance in a typical NMR sample reduced with NADPH. The sample was prepared the same way as if making a POR sample for NMR, but it was sealed in the optical cell instead of a Shigemi tube to allow for the repeated absorbance measurements. Consecutive recordings of absorption spectra were taken at fixed time intervals given in the figure.
Resonance assignment
To make assignments of the MTC methyls at specific sites in POR, we created single-cysteine mutants of Δ56 POR, modified their thiols with 13C-MMTS, and acquired 1H-13C HMQC spectra. Figure 4 shows that these sites are spectrally resolved and their surface localization attenuated paramagnetic relaxation—MTC signals are detected even in the reduced POR. Each single-cysteine mutant spectrum contained two peaks: the specific resonance (labeled in the figure) and the broad peak seen in the center of the spectral region. Supporting Figure 1 shows the same spectral plane of the cysteine-less Δ56 POR incubated with 13C-MMTS under the same conditions and concentrations that were used to produce samples for Figure 2. Lack of any significant signals confirms that MMTS labeling is highly specific for cysteines and the label is not retained non-specifically by the protein or flavins. Therefore, we conclude that the “middle peak” must originate from a reaction with the same cysteine that gives rise to the specific resonance.
Figure 4.
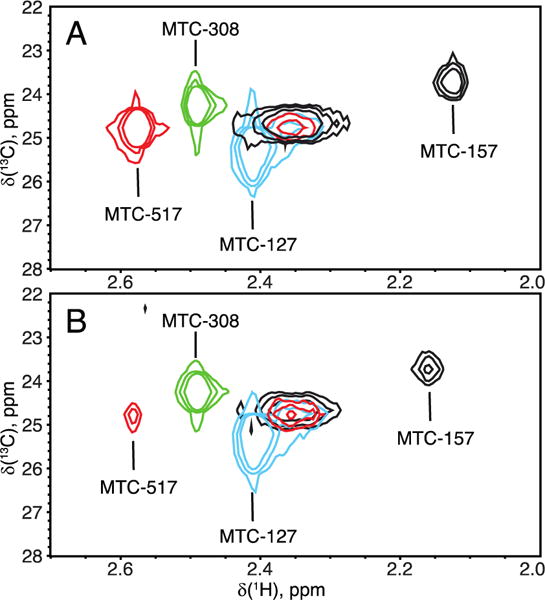
Overlay of 1H-13C HMQC spectra of the four single-cysteine mutants of Δ56 POR in the oxidized (A) and reduced (B) states. Contour levels were adjusted to account for the differences in NMR sample concentration. Line widths of all peaks are summarized in Supporting Tables 2 and 3.
Similarity of the chemical shifts of the middle peaks in four samples implies the similarity of the magnetic and hence structural environment. One hypothesis would be that each sample contains a fraction of POR in such a form that magnetic environments of any of the four cysteines are highly similar, which might be a result of (1) hydrolytic degradation or (2) unfolding followed by non-specific aggregation. To test this hypothesis, we performed size-exclusion chromatography (SEC) of the NMR sample to separate Δ56 POR (70 kDa) from possible degradation products or large unfolded aggregates.
Figure 5.A demonstrates HMQC spectra of a dual-cysteine mutant Q157C/Q517C recorded before and immediately after the SEC and reveals no significant changes of relative intensities of all three peaks. This result indicates that the signal in the center of MTC region originates from a monomeric fully folded Δ56 POR molecule. The proportion of spectral intensity observed as the middle peak widely varies among different mutants and different preparations of the same mutant (Supporting Table 4). We also observed that over the life of the NMR sample this resonance diminished with time faster than the “specific” peaks. This fact might be explained by a hypothesis that the middle peak originates from a conformation of a MTC side chain extended into solvent, while the specific peaks correspond to side chains interacting with the protein surface. In this scenario, the similar chemical shifts could be explainable as well as the faster loss of the label. In agreement with this explanation, the middle peak is relatively narrower in carbon dimension than the specific peaks, which would be expected for the methyls of extended side chains. However, the proton dimension appears to be inhomogeneously broadened indicating multiple states with close chemical shifts.
Figure 5.
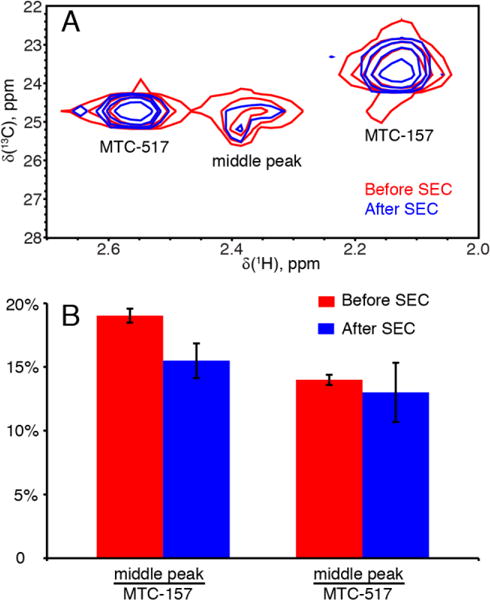
“Middle peak” species is not removed by size-exclusion chromatography. (A) 1H-13C HMQC spectra obtained from Δ56 POR sample before (red) and after (blue) size-exclusion chromatography, SEC, without any additional concentrating steps. (B) Relative intensity of the middle peak as a percentage of MTC517 and MTC157 intensities before (red) and after (blue) the size-exclusion chromatography.
Effect of reduction
Despite the presence of the middle peak, the specific resonance assignments are unambiguous, and the signals are well resolved to allow the use of multiple-cysteine mutants to significantly cut costs and experimental time. Figure 6 demonstrates an overlay of the reduced and oxidized states of the dual-cysteine Q157C/Q517C Δ56 POR highlighting the paramagnetic effects at these two sites in the POR reduced with NADPH. As expected from the site-to-flavin distances (Supporting Table 1), the MTC517 is most affected by paramagnetic relaxation enhancement, which manifests itself in a decrease of the peak intensity. The residues MTC517 and MTC157 experience chemical shift perturbations that may originate from both pseudocontact shifts and changes in the protein conformation (Bertini et al. 2002). Considering distant localization of MTC157 from the interdomain interface (Figure 2), its chemical shift perturbation observed in Figure 6 must originate purely from a pseudocontact shift, which signifies that the magnetic susceptibility tensor of the semiquinone is anisotropic. The chemical shift perturbation of MTC517 is more difficult to interpret because this site in the FAD-binding domain comes into close proximity of the FMN domain in the closed POR structure (Figure 2, right panel). As it could be anticipated from examination of site-to-flavin distances for MTC127 and MTC308 residues (Supporting Table 1), they were found to be quite insensitive to reduction of the corresponding POR mutant proteins (Supporting Figure 2).
Figure 6.
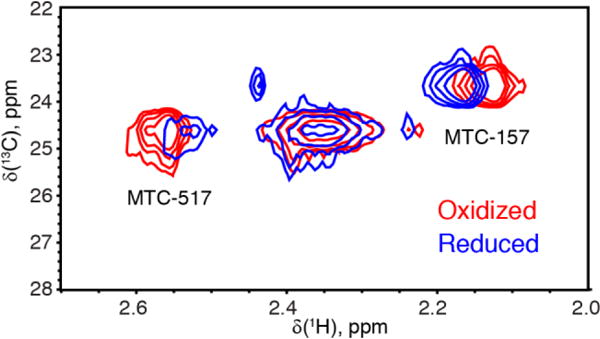
Effect of reduction with NADPH. 1H-13C HMQC spectra at 600 MHz of the Q157C/Q517C Δ56 POR in the oxidized (red) and reduced (blue) forms.
Conformational transition
To gain an additional insight into the opening-closing transition in POR, we employed a conformationally restrained Δ56 POR construct where the loop connecting FAD- and FMN-binding domains was shortened by removal of the four-residue sequence TGEE at the positions 236-239 (Hamdane et al. 2009). We will refer to this protein as “ΔTGEE” to contrast it with the “full-loop” parent construct. This modification causes conformational strain in the closed form making POR to adopt an “open” structure captured by the X-ray crystallography (Figure 2, left panel). The open form was hypothesized to correspond to the conformation where FMN-binding domain is accessible for interaction with the cytochrome P450 (Waskell and Kim 2015). Figure 7.A demonstrates the effect of loop deletion on the chemical shifts of the two most sensitive residues, MTC157 and MTC517, in the oxidized Δ56 POR. The perturbation of MTC517 by this mutation may only occur due to an opening transition because the distance from MTC517 to the loop deletion site is around 45 Å. Since, it is not straightforward to convert the chemical shift change into the amplitude of a structural rearrangement, we will restrict ourselves to a statement that the oxidized ΔTGEE Δ56 POR conformation in solution is distinct from the conformation of oxidized full-loop Δ56 POR.
Figure 7.

Effect of the loop deletion (236-TGEE-239) on chemical shifts of Q157C/Q517C Δ56 POR in the oxidized (A) and reduced (B) states. Absorbance spectra of the oxidized (C) and reduced (D) NMR samples (recorded immediately after acquisition).
Examination of the reduced form of ΔTGEE Δ56 POR (Figure 7.B, blue contours) reveals that peaks of MTC157 and MTC517 are each split in two (MTC517—unresolved). One resonance remains close to the position of oxidized ΔTGEE (as in panel A, blue contours) while a new resonance is observed at the position near the chemical shift of a reduced full-loop POR (red contours). The observed splitting of the MTC157 and MTC517 resonances must result from a population inhomogeneity and slow exchange kinetics between the two states. Two forms of POR giving rise to these peaks may due to (1) incomplete reduction of flavins or (2) a slow-exchange closed-open conformational equilibrium in the ΔTGEE POR (or both). Similar flavin (460 nm) and semiquinone (580 nm) absorbance in Figure 7.D indicates that the ΔTGEE POR is reduced similarly to the full-loop POR, which makes the first explanation less likely.
Based on X-ray diffraction analysis, the four-residue deletion from the hinge region was expected to destabilize the closed state by not allowing it to form native interdomain non-covalent bonds (POR “cannot properly close”) (Hamdane et al. 2009). Fewer interdomain bonds in the closed state would imply the weaker force constant for the opening-closing transition, which must result in the slower frequency of conformational fluctuations. The peak separation of MTC517 signals is approx. 0.4 ppm in 15N dimension (corresponding to Δω=150 s−1), and for MTC157—0.025 ppm in 1H dimension (Δω=94 s−1). To observe the two conformer signals with this separation requires the exchange rate constant, kex, to be much smaller than 100 s−1 (kex ≪ Δω) (Kaplan and Fraenkel 1980, Palmer 2004, Kay 2005).
It is remarkable that the new resonances of the ΔTGEE POR have chemical shifts similar to the ones of the reduced full-loop POR (Figure 7.B). This implies that both proteins adopt similar conformations in the NADP(H)-saturated reduced state, despite they were in distinct oxidized conformations. Yet, we must emphasize qualitative nature of our argument: at this time, the measured chemical shift perturbations cannot be converted into structural restraints because the magnetic susceptibility tensors of FAD and FMN semiquinones (in the context of POR) are not known.
Full-length POR in lipid nanodics
NMR studies of membrane proteins integrated in the lipid bilayers are very challenging due the presence of the lipid environment. Full-length cytochrome P450 proteins and a small reductase, cytb5, were studied in the lipid membranes by solid state NMR spectroscopy (Durr et al. 2007, Yamamoto et al. 2013, Yamamoto et al. 2013). The N-terminal transmembrane peptide of POR was reported by Huang and co-workers to form a transmembrane helix crossing the bilayer with a tilt of 13 degrees from the bilayer normal (Huang et al. 2014). Solution NMR studies of membrane proteins were made possible utilizing soluble lipid bilayer mimics such as bicelles (Whiles et al. 2002, Prosser et al. 2006, Poget and Girvin 2007, Raschle et al. 2010) and, in particular, lipid nanodiscs (Nasr et al. 2017, Hagn et al. 2013). A complex of the cytochrome P450 2B4 with cytb5 in the peptide-based lipid nanodiscs was successfully assembled by Ramamoorthy’s laboratory for solution NMR measurements (Zhang et al. 2016). Yet, the full-length POR, due to its large molecular weight, remained recalcitrant to NMR analysis either in the solution and solid states.
To create a sample of the full-length membrane-bound POR for solution NMR experiments, we incorporated the full-length Q157C/Q517C POR construct in the 10-nm lipid nanodiscs following the established protocols (Das and Sligar 2009). Figure 8 demonstrates the MTC spectral regions of 0.1 mM oxidized (panel A) and reduced (panel B) POR-nanodisc samples obtained at 600 MHz overlaid with the corresponding Δ56 spectrum (cf. Figure 6). Similarity of chemical shifts of the MTC157 and MTC517 in soluble and membrane-bound forms indicates that their magnetic environments were not significantly perturbed—in agreement with their distant location from the N-terminal transmembrane domain. Linewidths are roughly doubled in the POR-nanodisc spectrum relatively to Δ56 construct reflecting slow tumbling of the POR-nanodisc complex (Supporting Tables 3 and 4). However, the total mass of the complex (240 kDa) is a factor of 3.4 greater than that of the Δ56 POR (70 kDa). The relatively smaller increase in the line width may be explained by a flexible attachment of the cytosolic POR domain to its transmembrane region, which provides additional local mobility.
Figure 8.
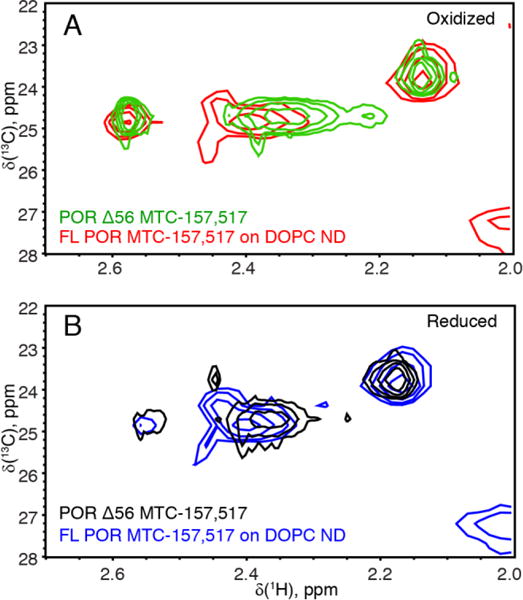
Protonated POR-nanodisc methyl-TROSY (1H-13C HMQC) spectra. (A) The air-oxidized full-length Q157C/Q517C POR in a lipid nanodisc (red) overlaid with the Δ56 POR (green). (B) The same samples reduced with the excess NADPH under anaerobic conditions: the full-length POR-nanodisc (blue); the Δ56 POR (black). A peak in the lower right corner is a product of the full-length POR degradation (as judged by increasing intensity with the age of this sample). Spectra were recorded at room temperature on a 600 MHz spectrometer with a cryogenic probe.
It was reported earlier that the redox potential of POR is modulated by a lipid composition of the bilayer (Das and Sligar 2009) (see also a review by Barnaba et al. (Barnaba et al. 2017)). In our experiments, MTC157 and MTC517 signals were sensitive to reduction with NADPH in both the Δ56 POR and the POR-nanodisc samples (Figure 8). Therefore, using methyl-TROSY detection, one could monitor the redox reaction in POR and, possibly, even measure the redox potentials of individual flavins by following the MTC signals from FAD- and FMN-binding domains in a titration of POR with NADPH. Liu and coworkers recently reported that activity of POR and coupling in POR-P450 complex in nanodiscs are enhanced when nanodiscs are made with the natural lipid preparations extracted from endoplasmic reticulum (ER) relatively to the synthetic lipid mixtures mimicking ER composition (Liu et al. 2017). This finding indicates that POR makes specific protein-lipid interactions modulating enzymatic activity of POR. Our methyl-TROSY approach may be directly applied for detection of such lipid-interaction sites in the full-length POR incorporated in a nanodisc by itself or in a complex with the cytochrome P450.
Finally, we need to emphasize that in our experiments, the POR protein, the nanodisc lipids, and the matrix scaffold protein were all fully protonated, yet we observe a sufficient signal despite the 240 kDa molecular weight of a POR-nanodisc assembly. In a separate nanodisc preparation, we confirmed that hydrophilic MMTS does not bind to the nanodiscs in the absence of POR (Supporting Figure 3). These observations indicate that the full-length POR in lipid nanodiscs is fully amenable for investigation of its conformational transitions and the redox cycle by the solution methyl-TROSY NMR experiments.
Summary
In this report, we demonstrated that the methyl-TROSY detection in combination with the extrinsic methyl labeling of the POR surface residues enables solution NMR analysis of the membrane-bound full-length POR without a need for (expensive) perdeuteration. Surface localization of methyl labels achieves a dual sensitivity gain by (1) having extrinsic methyl groups sufficiently removed from unpaired electrons of the semiquinones, and (2) maximizing local mobility to compensate for slow tumbling of the POR-nanodisc complex. Chemical shifts of surface-localized methyl group demonstrate site-specific sensitivity to reduction and conformational transitions in POR. This study opens a new avenue of research into the POR functional cycle at the membrane surface using solution NMR and highlights the power of the methyl-TROSY approach to membrane proteins and their assemblies.
Materials and Methods
Protein construct
Mutation sites for methyl labeling were selected to be solvent-exposed, polar and located in different regions of a protein molecule, such as near the cleft between FMN- and FAD- domains or anticipated POR-P450 or POR-membrane interaction interfaces. The full-length cysteine-less construct of rat POR and its Δ56 POR variant lacking 56 N-terminal residues (a transmembrane domain with the linker) was subjected to site-directed mutagenesis to introduce surface-exposed cysteines. The codons for E127, Q157, S308, and Q517 were converted to cysteine codons using QuikChange II Site-Directed Mutagenesis Kit (Agilent) following the standard protocol. To facilitate protein purification, 6-His-tag was included at the N-terminus of the protein construct.
Protein expression and purification
Δ56 POR
Protein expression and purification was performed as described earlier (Xia et al. 2011, Hamdane et al. 2009). Specifically, kanamycin resistant pET28a plasmid containing POR genes carrying His-Tag was transformed into E.coli C41 (DE3) strain. Cell culture was allowed to grow in Luria Broth media at 37°C for 3 hours. Then protein overexpression was induced by addition of IPTG to 0.5 mM. The cell mixture was grown in the dark for 20 hours at room temperature and pelleted at 5000 g for 10 min. The cell pellet was resuspended in PBS buffer pH 7.4 (1 mM KH2PO4, 10 mM Na2HPO4, 137 mM NaCl, and 2.7 mM KCl) and sonicated on ice using Branson Sonifier 450. Supernatant obtained after cell lysate was centrifuged at 5000 g for 15 min and passed through 5mL His60 Ni Superflow resin [Clontech Lab] equilibrated with PBS buffer pH 7.4. Protein was eluted with 0.5 M imidazole in PBS buffer pH 7.4. Eluate was dialyzed vs PBS pH 7.4 with addition of 2 mM TCEP and passed through 2′,5′-ADP Sepharose 4B [GE Healthcare Life Science] gravity column equilibrated with PBS buffer pH 7.4 (2mM DTT was used instead of TCEP). Protein was eluted using with PBS pH 7.4 buffer containing 20mM 2′(3′)-AMP [Santa Cruz Biotechnology]. Purified sample was characterized with SDS-PAGE, UV-vis spectroscopy and MALDI-TOF spectrometry.
Full-length POR
Ampillicin resistant pOR263 plasmid containing full-length POR genes and carrying His-Tag was transformed into E.coli C41 (DE3) cells. Cell culture was grown on Terrific Broth media at 37°C for 3 hours. Protein overexpression was induced by addition of IPTG to 0.5 mM final concentration and cell mixture was grown in the dark for 20 hours at 18°C. Cell culture was centrifuged at 5000 g and the cell pellet that was resuspended in 25 mM phosphate buffer pH 7.7, 100 mM NaCl, 10% glycerol with addition of one tablet of EDTA-free Complete Roche protease inhibitor (1836170) (using one tablet per each 1000 ml). Cells were lysed in the presence of 30 μg/ml lysozyme at 4°C for an hour followed by sonication on ice. The cell lysate was centrifuged at 5000g for 15min and obtained supernatant was centrifuged at 30,000 g for one hour. Pellet from second centrifugation was resuspended in 25 mM phosphate buffer pH 7.7, 100 mM NaCl, 0.3% Triton X-100, 10% glycerol, 2 mM TCEP, 1 mM PMSF with addition of the protease inhibitor and incubated overnight at 4°C to allow for detergent extraction of POR. Further purification was carried out according to the procedure described above for Δ56 version of a protein. After the final step, to replace Triton X-100 in the POR sample with sodium cholate (necessary for nanodisc assembly), the final eluate was passed through Ni Superflow column, which was washed with 25mM sodium cholate [Alfa Aesar] and eluted with 0.5 M imidazole and 25mM sodium cholate.
Protocol for the POR-nanodisc complex assembly
Nanodisc assembly was performed according to the established protocols with minor modifications (Das and Sligar 2009, Bayburt and Sligar 2010, Nath et al. 2007). Briefly:
To mimic the endoplasmic reticulum membrane, we used a negatively charged fluid lipid mix: 15% 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol), DOPG, and 85% of 1,2-dioleoyl-sn-glycero-3-phosphocholine, DOPC. Lipids were received from Avanti Polar Lipids in chloroform and stored at −20°C.
To prepare nanodiscs with POR, we estimated amount of POR required for the assembly and back-calculated necessary quantities of the matrix scaffold protein MSP1D1 and the lipids. Molar excess of MSP1D1 over POR (8:1) was chosen to ensure that most POR-nanodisc complexes will have one POR molecule per nanodisc. Molar ratio of lipids to MSP1D1 was set to 65:1 following the original published protocols from http://sligarlab.life.uiuc.edu/nanodisc/protocols.html.
Lipids in chlorophorm were mixed and dried in a glass vial under the stream of nitrogen, followed by overnight incubation under a house vacuum. A dry lipid film was dissolved in 100 mM sodium cholate, 100 mM phosphate, 50 mM NaCl pH 7.4 on a vortex.
The solution was sonicated to clarity and combined with aliquots of concentrated matrix scaffold protein MSP1D1 (His-tag removed) and His-tagged full-length Q157C/Q517C POR. A buffer aliquot was also added to adjust the cholate concentration to final concentration of 20 mM in the assembly mixture.
To trigger the nanodisc assembly, the detergent was gradually removed by dialysis in the presence of Amberlite XAD-2 beads [Sigma-Aldrich] added to the solvent outside of the dialysis bag (2 g of damp beads per each milliliter of assembly reaction mixture). Dialysis was performed at 4°C overnight.
To remove excess empty nanodiscs (not including POR molecules), the dialysate was passed through His60 Ni-Superflow resin. The POR-nanodisc complexes were bound to the resin and then eluted with 0.5 M imidazole.
To separate nanodiscs from lipid aggregates, the sample was centrifuged 5 min at 15,000 g and then passed through the Superose 6 Increase 10/300 column. The peak fractions corresponding to the molecular weight near 200-300 kDa were collected and concentrated.
MALDI-TOF mass-spectrometry
MALDI-TOF mass spectra were recorded on a Voyager-DE ProBioSpectrometryTM workstation PerSeptive Biosystems. The matrix was a 1:1 mixture of dihydroxybenzoic acid (DHB, TCI) α-Cyano-4- hydroxycinnamic acid (α-CHCA, Fluka) (Signor and Boeri Erba 2013). Before matrix-protein mixture was applied, MALDI-TOF plate was covered with the thin layer of saturated α-CHCA matrix solution to stimulate microcrystal formation.
Labeling with 13C-MMTS
Site-directed labeling with 13CH3-methyl-methanthiosulfanate (13C-MMTS) was performed as described (Religa et al. 2011). Purified protein samples were incubated with 5 mM dithiothreitol (DTT) overnight in phosphate-buffered saline (PBS pH 7.4: 1 mM KH2PO4, 10 mM Na2HPO4, 137 mM NaCl, and 2.7 mM KCl,) and dialyzed using Slide-A-Lyzer Dialysis Cassettes, 10,000 MWCO [Thermo Scientific] against a degassed buffer containing 50 mM potassium phosphate pH 7.5 and 1 mM EDTA at 4°C. The dialyzate was concentrated with Vivaspin®2 Centrifugal Concentrator 10,000 MWCO [GE Healthcare Life] to 300μL. The 100 mM stock solution of 13C-MMTS [Thermo Scientific] in DMSO was added in achieve 50% molar excess of 13C-MMTS to a protein and incubated overnight at 4°C. Unreacted 13C-MMTS was removed by dialysis against PBS. The sample was concentrated and degassed under vacuum for one hour. D2O was added to 10% for the NMR spectrometer lock. Protein concentrations were determined by flavin absorbance at 460 nm using the extinction coefficient of 21,600 M−1 cm−1.
Prepration of reduced and oxidized POR
Sample oxidation and reduction was carried out by adding 4-fold molar excess of K3[Fe(CN)6] or NADPH, respectively. The concentrations of NADPH and K3[Fe(CN)6] solutions were determined by specific absorption at ε(340 nm) = 6,220 M−1 cm−1 and ε(420 nm) = 1,040 M−1 cm−1, respectively (Fruscione et al. 2008, Appleby and Morton 1959). Preparation of reduced NMR samples was performed anaerobically in a glovebag [Glas-Col] filled with nitrogen gas.
NMR spectroscopy
2D 13C-1H Heteronuclear Multiple-Quantum Correlation (HMQC) NMR spectra were recorded on a 600 MHz Varian VNMR-S spectrometer with the Cold Probe and a 600 MHz Bruker Avance III with a CryoProbe. Shigemi tubes were used for all samples. Spectra were processed with NMRPipe (Delaglio et al. 1995) and Sparky (Goddard and Kneller). NMR chemical shifts of the full loop and ΔTGEE POR mutants were deposited with BMRB, accession number 27202.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R15 GM126528-01 to ELK and by R01 GM097031 to JJK, and also—by the Regular Research Grant 2016 from Committee on Research (COR), Marquette University. ARG acknowledges Eisch Research Fellowship during the academic year 2016–2017. Authors acknowledge NMR instrument center of Biochemistry Department of the Medical College of Wisconsin for providing access to the Bruker 600 MHz instrument. The authors are grateful to Dr. Blake Hill (Medical College of Wisconsin, Milwaukee) and Dr. Kevin MacKenzie (Baylor College of Medicine, Houston) for acquisition of high-field NMR data on the POR-nanodisc sample at the NMR facility of the Baylor College of Medicine.
Footnotes
orcid.org/0000-0003-3819-3912
References
- Barnaba C, Gentry K, Sumangala N, Ramamoorthy A. The catalytic function of cytochrome P450 is entwined with its membrane-bound nature. F1000Research. 2017;6:662. doi: 10.12688/f1000research.11015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EE, Wolf CR, Otyepka M, Humphreys SC, Reed JR, Henderson CJ, McLaughlin LA, Paloncýová M, Navrátilová V, Berka K, Anzenbacher P, Dahal UP, Barnaba C, Brozik JA, Jones JP, Estrada DF, Laurence JS, Park JW, Backes WL. The Role of Protein-Protein and Protein-Membrane Interactions on P450 Function. Drug Metabolism and Disposition. 2016;44:576–590. doi: 10.1124/dmd.115.068569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannemann F, Bichet A, Ewen KM, Bernhardt R. Cytochrome P450 systems - biological variations of electron transport chains. Biochimica Et Biophysica Acta-General Subjects. 2007;1770:330–344. doi: 10.1016/j.bbagen.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Waskell L, Kim J-JP. Cytochrome P450. Heidelberg New York Dordrecht London: Springer Cham; 2015. Electron Transfer Partners of Cytochrome P450; pp. 33–68. (Structure, Mechanism, and Biochemistry P. R. Ortiz de Montellano). [Google Scholar]
- Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Three-dimensional structure of NADPH-cytochrome P450 reductase: prototype for FMN- and FAD-containing enzymes. Proc Natl Acad Sci U S A. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugishima M, Sato H, Higashimoto Y, Harada J, Wada K, Fukuyama K, Noguchi M. Structural basis for the electron transfer from an open form of NADPH-cytochrome P450 oxidoreductase to heme oxygenase. Proceedings of the National Academy of Sciences. 2014;111:2524–2529. doi: 10.1073/pnas.1322034111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aigrain L, Pompon D, Truan G. Role of the interface between the FMN and FAD domains in the control of redox potential and electronic transfer of NADPH-cytochrome P450 reductase. Biochemical Journal. 2011;435:197–206. doi: 10.1042/BJ20101984. [DOI] [PubMed] [Google Scholar]
- van Schagen CG, Muller F. A 13C nuclear-magnetic-resonance study on free flavins and Megasphaera elsdenii and Azotobacter vinelandii flavodoxin. 13C-enriched flavins as probes for the study of flavoprotein active sites. Eur J Biochem. 1981;120:33–39. doi: 10.1111/j.1432-1033.1981.tb05666.x. [DOI] [PubMed] [Google Scholar]
- Evrard A, Zeghouf M, Fontecave M, Roby C, Coves J. 31P nuclear magnetic resonance study of the flavoprotein component of the Escherichia coli sulfite reductase. Eur J Biochem. 1999;261:430–437. doi: 10.1046/j.1432-1327.1999.00274.x. [DOI] [PubMed] [Google Scholar]
- Vincent B, Morellet N, Fatemi F, Aigrain L, Truan G, Guittet E, Lescop E. The closed and compact domain organization of the 70-kDa human cytochrome P450 reductase in its oxidized state as revealed by NMR. J Mol Biol. 2012;420:296–309. doi: 10.1016/j.jmb.2012.03.022. [DOI] [PubMed] [Google Scholar]
- Spencer ALM, Bagai I, Becker DF, Zuiderweg ERP, Ragsdale SW. Protein/Protein Interactions in the Mammalian Heme Degradation Pathway. Heme Oxygenase-2, Cytochrome P450 Reductase, and Bileverdin Reductase. Journal of Biological Chemistry. 2014;289:29836–29858. doi: 10.1074/jbc.M114.582783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Zhang M, Rwere F, Waskell L, Ramamoorthy A. Kinetic and structural characterization of the interaction between the FMN binding domain of cytochrome P450 reductase and cytochrome c. J Biol Chem. 2015;290:4843–4855. doi: 10.1074/jbc.M114.582700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada DF, Laurence JS, Scott EE. Cytochrome P450 17A1 Interactions with the FMN Domain of Its Reductase as Characterized by NMR. J Biol Chem. 2016;291:3990–4003. doi: 10.1074/jbc.M115.677294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruice TW, Kenyon GL. Alkyl Alkanethiolsulfonate Sulfhydryl-Reagents - Beta-Sulfhydryl-Modified Derivatives of L-Cysteine as Substrates for Trypsin and Alpha-Chymotrypsin. Bioorganic Chemistry. 1985;13:77–87. [Google Scholar]
- Bruice TW, Maggio ET, Kenyon GL. Reversible Derivatization of Cysteinyl Sulfhydryls to Generate New Substrates for Trypsin and Chymotrypsin. Federation Proceedings. 1976;35:1475–1475. [Google Scholar]
- Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE. Cross-correlated relaxation enhanced 1H-13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc. 2003;125:10420–10428. doi: 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- Religa TL, Ruschak AM, Rosenzweig R, Kay LE. Site-directed methyl group labeling as an NMR probe of structure and dynamics in supramolecular protein systems: applications to the proteasome and to the ClpP protease. J Am Chem Soc. 2011;133:9063–9068. doi: 10.1021/ja202259a. [DOI] [PubMed] [Google Scholar]
- Xia C, Hamdane D, Shen AL, Choi V, Kasper CB, Pearl NM, Zhang H, Im SC, Waskell L, Kim JJ. Conformational changes of NADPH-cytochrome P450 oxidoreductase are essential for catalysis and cofactor binding. J Biol Chem. 2011;286:16246–16260. doi: 10.1074/jbc.M111.230532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdane D, Xia C, Im SC, Zhang H, Kim JJ, Waskell L. Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J Biol Chem. 2009;284:11374–11384. doi: 10.1074/jbc.M807868200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Yamamoto K, Zhang M, Popovych N, Hung I, Im SC, Gan ZH, Waskell L, Ramamoorthy A. Probing the Transmembrane Structure and Dynamics of Microsomal NADPH-cytochrome P450 oxidoreductase by Solid-State NMR. Biophysical Journal. 2014;106:2126–2133. doi: 10.1016/j.bpj.2014.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermilion JL, Coon MJ. Purified liver microsomal NADPH-cytochrome P-450 reductase. Spectral characterization of oxidation-reduction states. J Biol Chem. 1978;253:2694–2704. [PubMed] [Google Scholar]
- Rwere F, Xia CW, Im S, Haque MM, Stuehr DJ, Waskell L, Kim JJP. Mutants of Cytochrome P450 Reductase Lacking Either Gly-141 or Gly-143 Destabilize Its FMN Semiquinone. Journal of Biological Chemistry. 2016;291:14639–14661. doi: 10.1074/jbc.M116.724625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I, Luchinat C, Parigi G. Magnetic susceptibility in paramagnetic NMR. Progress in Nuclear Magnetic Resonance Spectroscopy. 2002;40:249–273. doi: 10.1016/j.pnmrs.2019.06.003. [DOI] [PubMed] [Google Scholar]
- Kaplan JI, Fraenkel G. NMR of Chemically Exchanging Systems. Academic Press; 1980. [Google Scholar]
- Palmer AG. NMR Characterization of the Dynamics of Biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- Kay LE. NMR studies of protein structure and dynamics. Journal of Magnetic Resonance. 2005;173:193–207. doi: 10.1016/j.jmr.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Durr UH, Waskell L, Ramamoorthy A. The cytochromes P450 and b5 and their reductases–promising targets for structural studies by advanced solid-state NMR spectroscopy. Biochim Biophys Acta. 2007;1768:3235–3259. doi: 10.1016/j.bbamem.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Durr UH, Xu J, Im SC, Waskell L, Ramamoorthy A. Dynamic interaction between membrane-bound full-length cytochrome P450 and cytochrome b5 observed by solid-state NMR spectroscopy. Sci Rep. 2013;3:2538. doi: 10.1038/srep02538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Gildenberg M, Ahuja S, Im SC, Pearcy P, Waskell L, Ramamoorthy A. Probing the transmembrane structure and topology of microsomal cytochrome-p450 by solid-state NMR on temperature-resistant bicelles. Sci Rep. 2013;3:2556. doi: 10.1038/srep02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiles JA, Deems R, Vold RR, Dennis EA. Bicelles in structure-function studies of membrane-associated proteins. Bioorganic Chemistry. 2002;30:431–442. doi: 10.1016/s0045-2068(02)00527-8. [DOI] [PubMed] [Google Scholar]
- Prosser RS, Evanics F, Kitevski JL, Al-Abdul-Wahid MS. Current Applications of Bicelles in NMR Studies of Membrane-Associated Amphiphiles and Proteins‚Ć,‚Ä°. Biochemistry. 2006;45:8453–8465. doi: 10.1021/bi060615u. [DOI] [PubMed] [Google Scholar]
- Poget SF, Girvin ME. Solution NMR of membrane proteins in bilayer mimics: Small is beautiful, but sometimes bigger is better. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2007;1768:3098–3106. doi: 10.1016/j.bbamem.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle T, Hiller S, Etzkorn M, Wagner G. Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Current Opinion in Structural Biology. 2010;20:471–479. doi: 10.1016/j.sbi.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasr ML, Baptista D, Strauss M, Sun ZYJ, Grigoriu S, Huser S, Pluckthun A, Hagn F, Walz T, Hogle JM, Wagner G. Covalently circularized nanodiscs for studying membrane proteins and viral entry. Nature Methods. 2017;14:49–52. doi: 10.1038/nmeth.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagn F, Etzkorn M, Raschle T, Wagner G. Optimized Phospholipid Bilayer Nanodiscs Facilitate High-Resolution Structure Determination of Membrane Proteins. J Am Chem Soc. 2013;135:1919–1925. doi: 10.1021/ja310901f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Huang R, Ackermann R, Im S-C, Waskell L, Schwendeman A, Ramamoorthy A. Reconstitution of the Cytb5–CytP450 Complex in Nanodiscs for Structural Studies using NMR Spectroscopy. Angewandte Chemie. 2016:n/a–n/a. doi: 10.1002/anie.201600073. [DOI] [PubMed] [Google Scholar]
- Das A, Sligar SG. Modulation of the cytochrome P450 reductase redox potential by the phospholipid bilayer. Biochemistry. 2009;48:12104–12112. doi: 10.1021/bi9011435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KC, Hughes JMX, Hay S, Scrutton NS. Liver microsomal lipid enhances the activity and redox coupling of colocalized cytochrome P450 reductase-cytochrome P450 3A4 in nanodiscs. Febs Journal. 2017;284:2302–2319. doi: 10.1111/febs.14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010;584:1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Atkins WM, Sligar SG. Applications of Phospholipid Bilayer Nanodiscs in the Study of Membranes and Membrane Proteins. Biochemistry. 2007;46:2059–2069. doi: 10.1021/bi602371n. [DOI] [PubMed] [Google Scholar]
- Signor L, Boeri Erba E. Matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometric analysis of intact proteins larger than 100 kDa. J Vis Exp. 2013 doi: 10.3791/50635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruscione F, Sturla L, Duncan G, Van Etten JL, Valbuzzi P, De Flora A, Di Zanni E, Tonetti M. Differential role of NADP(+) and NADPH in the activity and structure of GDP-D-mannose 4,6-dehydratase from two chlorella viruses. Journal of Biological Chemistry. 2008;283:184–193. doi: 10.1074/jbc.M706614200. [DOI] [PubMed] [Google Scholar]
- Appleby CA, Morton RK. Lactic Dehydrogenase and Cytochrome B2 of Bakers Yeast - Enzymic and Chemical Properties of the Crystalline Enzyme. Biochemical Journal. 1959;73:539–550. doi: 10.1042/bj0730539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: http://www.cgl.ucsf.edu/home/sparky/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.