

Abstract
The accumulation of misfolded proteins (MP), both unique and common, for different diseases is central for many chronic degenerative diseases. In certain patients MP accumulation is systemic (e.g. TTR amyloid) and in others this is localized to a specific cell type (e.g. Alzheimer’s disease). In neurodegenerative diseases, NDs, it is noticeable that the accumulation of MP progressively spreads throughout the nervous system. Our main hypothesis of this article is that MPs are not only markers but also active carriers of pathogenicity. Here, we discuss studies from comprehensive molecular approaches aimed at understanding MP conformational variations (polymorphism) and their bearing on spreading of MPs, MP toxicity, as well as MP targeting in imaging and therapy. Neurodegenerative disease (ND) represents a major and growing societal challenge, with millions of people worldwide suffering from Alzheimer’s or Parkinson’s diseases alone. For all NDs, current treatment is palliative without addressing the primary cause, and is not curative. Over recent years particularly the shape-shifting properties of misfolded proteins and their spreading pathways have been intensively researched. The difficulty in addressing ND has prompted most major pharma companies to severely downsize their nervous system disorder research. Increased academic research is pivotal for filling this void and to translate basic research into tools for medical professionals. Recent discoveries of targeting drug design against MPs and improved model systems to study structure, pathology spreading and toxicity strongly encourage future studies along these lines to provide an opportunity for selective imaging, prognostic diagnosis, and therapy.
Keywords: ALZHEIMER’S DISEASE, AMYLOIDOSIS, BIOCHEMISTRY, CHRONIC DISEASES, PATHOLOGY
Protein misfolding and amyloid diseases
Amyloid disease is characterized by the abnormal accumulation of aggregated misfolded protein (MP) in diseased tissue. Disease pathology can be localized or systemic. Localized amyloidosis occurs where the MP accumulates in the tissue where the MP is produced while systemic amyloidosis is caused by MPs synthesized distant from the site of deposition. Aggregated MPs in amyloidosis appear as ordered aggregates with fibrillar structure. To be pathologically defined as an amyloid fibril, it must be Congophilic i.e. have affinity for the histological dye Congo red and show characteristic colors and patterns known as birefringence when the Congo red-stained amyloid deposits are viewed through crossed polarizers. In addition to the described Congophilia and birefringence, the identity of the core protein defines the disease [1]. Each form of amyloidosis is hence defined by the molecular identity of the amyloid fibril protein that deposits in tissues and organs and gives rise to the disease condition (Fig. 1).
Figure 1.
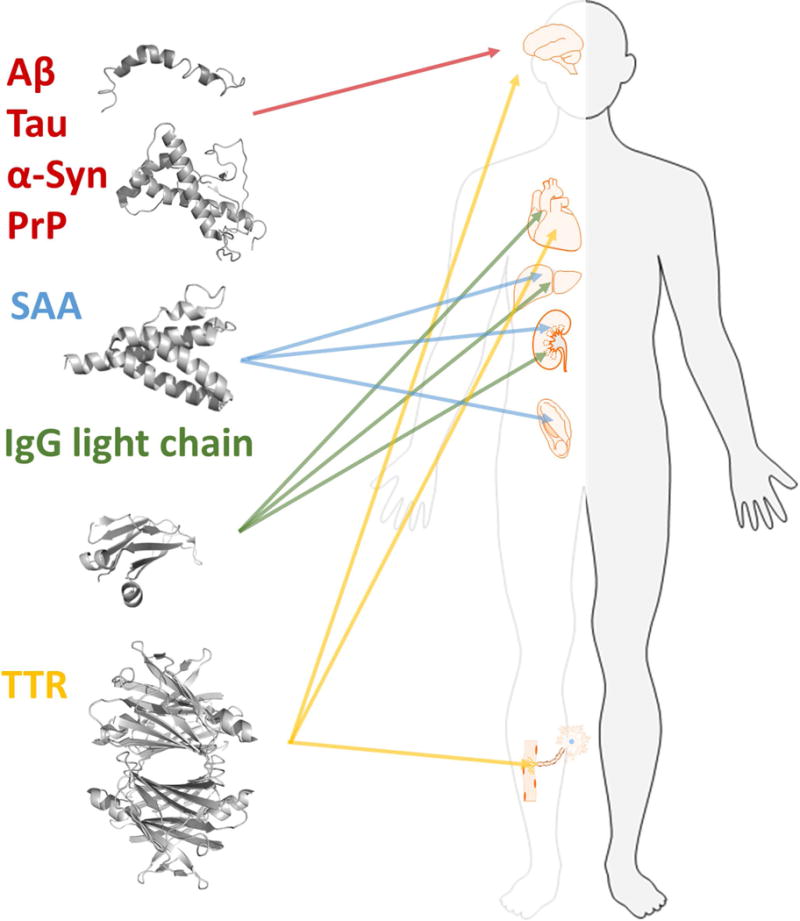
Multiple different proteins are associated with amyloid disease and form deposits in various organs. Each form of amyloidosis is defined by the molecular identity of the amyloid fibril protein that deposits and gives rise to the disease condition. The figure illustrates the proteins discussed in this article and the main organs affected by the respective protein. Aβ, Tau, α-syn, and PrP in brain, SAA in kidney, spleen, and liver. IgG light chains are found in most tissues except for the CNS but mainly in kidney, heart, and liver. TTR in heart, peripheral nerves, eye and CNS.
MP precursors are often intrinsically disordered proteins (IDPs) but also originate from stable globular proteins. Invariably disease severity is correlated with MPs accumulation. The associated MPs appear specific for each disease: Alzheimer’s disease (AD) neuropathology comprises senile amyloid plaque formed from the peptide Aβ and neurofibrillary tangles derived from the hyperphosphorylated form of microtubule-associated Tau protein. Parkinson’s disease and multiple system atrophy are distinct from AD as they are associated with alpha-synuclein (α-syn) aggregates, which occur in distinct neuronal and glial cell populations in characteristic brain regions. Similar to AD, however, they can occur concomitantly with Tau inclusions. In systemic amyloidosis proteins such as serum amyloid A (SAA), immunoglobulin (IgG) light chains or transthyretin (TTR) can form deposits within vital organs, e.g. liver, heart, kidneys, peripheral nerves or blood vessels (Fig. 1). While the amyloid precursor proteins in different forms of amyloidosis are unrelated in sequence or natively folded structure, all amyloid fibrils are β-sheet rich. They comprise repetitive segments of approximately 4.7 Å spaced β-strands along the direction of the main fibril axis with inter-sheet spacings occurring at approximately 10 Å in the direction perpendicular to it. This generic structural arrangement, termed cross-β, is shared by all amyloid fibrils forming a backbone which can otherwise differ in the fold of the non-homologous polypeptide chains within the fibril. A particular remarkable phenomenon, however, is that the same polypeptide chain can adopt multiple modified fibril states, referred to as structural polymorphism [2]. This is in apparent contrast to the Anfinsen “one sequence one fold” postulate [3, 4].
Assembly polymorphisms in organic chemistry
The polymorphic nature of amyloid fibrils was recognized already in the original description of the fibrillary ultrastructure of amyloid by Calkins and Cohen almost 60 years ago [5], and was elaborated on in some detail in 1965. “The Fibril is made up of filaments of varying lengths but with reproducible diameter of 75 ± 9 Å. It occurs most commonly in lateral aggregates of 4 but may be found in side-to-side groups of 1–8 filaments” [6]. While this may have been perceived as being new at the time it was not an unprecedented phenomenon in organic chemistry. Crystallographic polymorphism of organic small molecules has been known for a very long time. Here various packing patterns in organic molecule crystals impose physico-chemical diversity. For example the simple organic molecule benzamide was described in 1832 by Wöhler and von Liebig [7] to display crystallographic polymorphism, however it took until 2005 for the polymorphic forms to be revealed in atomic detail [8, 9]. Similar phenomena are described for D-mannitol, presenting three different crystal polymorphs with various physicochemical characteristics. An interesting observation of D-mannitol crystals is that seeding with preformed crystals of various types can, in addition to nucleating further growth of the initial templated polymorph, also trigger cross-nucleation into competing polymorphs. Hence, templated crystal growth can be altered by growth conditions and the alternative forms appear at interfaces determined by competing growth rates [10, 11].
Amyloid fibril polymorphism
Amyloid fibril polymorphism describes the variations in fibril structures formed by a given polypeptide chain. Variations between sequence different polypeptide chains, such as Aβ(1–40) and Aβ(1–42), may not therefore classify, strictly speaking, as polymorphisms. The polymorphism of a fibril sample can be analyzed easily with high-resolution microscopic techniques, such as electron and atomic force microscopy [12, 13]. The persistence length and fibril bending, the cross-over distance and the fibril width/height at or in between the cross-overs can be useful parameters in determining the fibril morphology. Less useful may be the fibril length, which can vary depending on sample pretreatment. For example, conditions that fragment long fibrils will affect the length but not necessarily alter the fibril morphology. Based on the measurement of the aforementioned parameters such as fibril width and cross-over distance, it is a common observation that fibril samples are polymorphic; that is, multiple fibril morphologies are present in one sample. Such polymorphic samples can, in some cases, produce uniform spectroscopic signatures, indicating that the fibril morphologies present in this sample consist of similarly structured building blocks or protomers. Hence, uniform spectral characteristics do not always guarantee a monomorphic sample. Conversely, it is possible that observation of spectroscopic heterogeneity does not necessarily imply separated polymorphs in a sample as a fibril morphology may consist of more than a single conformation of the fibril-forming polypeptide chain, as was suggested previously for several Aβ(1–40) derived fibril samples [14, 15].
The intra-sample polymorphism is not only typical for the cross-β fibrils that were formed in a test tube from chemically synthetized or recombinantly expressed polypeptide chains. It is also seen when analyzing amyloid fibrils extracted from patients or animal diseased with amyloidosis. A comparative analysis of fibrils extracted from different forms of amyloidosis revealed polymorphic fibril morphologies based on electron microcopy and this result was independent of the type of amyloidosis analyzed, the amyloid-forming protein. and the organ involved in the amyloid deposition [16]. Changing the conditions of fibril formation in vitro can alter the distribution of fibril polymorphs present in a sample [17]. It may also alter the extent of polymorphism of a sample, but it may also trade one fibril ensemble for another [18]. Generally speaking, the polymorphism of cross-β fibrils can arise from three types of variations: first, variations in the number of protofilaments constructing the mature fibril; second, variations in the relative arrangement of the protofilaments, and third, variations in the protofilament substructure [19]. Theoretical assembly polymorphism where protofilament assemblies generate different fibril surface structures despite having identical monomeric protomers is illustrated in Fig.2.
Figure 2.
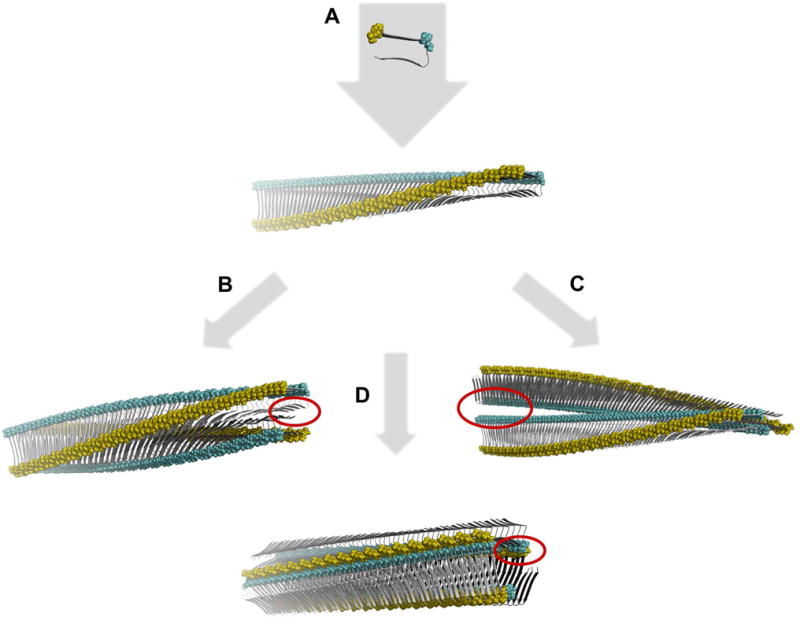
Schematic illustration of theoretical assembly polymorphism of identical monomeric building blocks. A. First a beta-strand monomeric protomer conformation structure is assembled in a linear monomorphic in register parallel beta-sheet polymeric single filament fibril with a twisted sheet. The single filament is then assembled in a face-to-face (B) or back-to-back (C) configuration rendering two different assembly polymorphs. Alternately three filaments assemble in a polygonal configuration offering yet an alternative structure (D). All assemblies generate distinctly different surface structures as illustrated by the yellow and cyan colored residues and unique interface cavities in each polymorph (red circles). The building blocks (top) are based on the 2BEG structure [20]. The 3D rendered structures were generated using PyMOL and coordinates were calculated by the program CreateFibril v 2.5 [21].
Variations in the protofilament substructure importantly involve differences in the peptide fold, such as variations in the β-strand composition, as well as variations in the packing of multiple polypeptide chains into a protofilament. Nevertheless, the three types of variations can to some extent be intertwined. For example, variations in the relative arrangement of the protofilaments, which involve altered interaction surfaces between the protofilaments, may be accommodated with at just minor re-adjustments in the peptide fold. Yet, all three levels of polymorphism need to be distinguished from additional sources of heterogeneity in a fibril sample, such as the variable bending and variable twisting of fibril structures, which can even vary within a single fibril particle [13]. Ultimately, it was found that the fibrils can participate in the formation of different fibril network super-structures. Star-like and bundle-like arrangements as well as amorphous fibril meshworks have been found in the amyloid deposits observed in cellular models of Aβ and serum amyloid A fibril formation [22, 23] and by electron microscopic analysis of histological specimens in diseased tissue [24–26]. All levels of polymorphisms contribute to variations of microscopically identifiable structures and can collectively be called aggregate morphotypes. Morphotypes can be defined as a specific subtype of amyloid aggregates of a distinct protein distinguishable from other aggregates of the same protein on the basis of morphologic or staining-specific characteristics.
Basis for protein conformational polymorphism
The diversity of native, rather well behaved, protein structures exemplifies the dramatic structural polymorphism of polypeptides. The SCOPe classification released in 2016 presents 1221 unique folds [27]. The native state is an ensemble of conformational states with a narrow distribution of conformations. Protein misfolding is by definition a process where aberrant conformations are attained by the protein molecule that are distinct from its native structure. This misfolded conformation is retained by intermolecular stabilization of identical protein chains.
Misfolded proteins (molten-globule like) are inherently comprised of a high degree of disorder. This plasticity provides a broad distribution of conformational polymorphism when such proteins assemble into multimers. With this in mind it is obvious that the theoretical analysis of amyloid fibril conformational polymorphism is a daunting task.
Methods to study polymorphic amyloids
Max Perutz proposed a polar-zipper model for poly-L-glutamine fibrils [28]. This model elegantly described the aggregation of poly-Q-containing proteins in disease. The Perutz paper, in addition to describing a molecular model for the poly-Q aggregates, also proposed a model for recruitment of vital regulating neuronal proteins into such a pathological aggregate, a proposed pathophysiological model which now is accepted for many neurodegenerative diseases including Huntington’s disease. The Eisenberg group has built significantly on this work using micro- and nanocrystals grown from short peptide fragments of amyloid-forming proteins to demonstrate various packing patterns into cross-β sheets [29] and revealed a plethora of so called steric zippers formed through assembly polymorphism [30].
Solid-state NMR is a recent technique which can provide information from signal fingerprints directly from spectra reflecting protein conformation, through secondary structures derived from chemical-shift assignments, to full 3D structures of amyloid fibrils. Data from this technique has over the last few years provided the most direct insight into how different secondary structures can be in different polymorphs of the same protein, for Aβ, α-synuclein, and for yeast prions.
While solid-state NMR has advantages in detecting residue-specific conformational features such as the distribution of β-strand structure along the sequence, it is challenging to determine the full quaternary structure. Electron microscopy and in particular cryo-EM have recently become potential methods to determine global features of the fibril architecture. Very recently, it even produced several reconstructions at 3.4–4.0 Å resolution that sufficed to construct a molecular model for the fibril [31, 32]. Electron tomography is a methodology that can visualize larger areas of a sample, such as a fibril deposit, at high resolution enabling the definition of multiple network structures or the interactions of fibrils with lipid membranes, such as cellular surfaces and lipid inclusions [22, 23]. However, it is more challenging to discern the fibril morphology with this methodology [22].
Fluorescence spectroscopy is a powerful complement to high resolution structural biology methods for studying amyloid structures. Luminescent conjugated oligo- and polythiophenes (LCOs and LCPs) represent a promising class of amyloid imaging agents, recognizing and differentiating a broader subset of protein aggregates than conventional amyloid dyes [33–43]. It was recently shown by combinatorial ssNMR, modeling, and fluorescence that the main molecular binding site for the LCO p-FTAA appears to overlap with that described for Congo red [44, 45] (see more below). LCOs and LCPs contain a twistable thiophene backbone and upon conformational restriction of this backbone, different emission profiles are observed from the oligo-thiophene derivatives. Hence in comparison to Congo red, which is a rigid molecule, oligo-thiophene based fluorescent dyes offer an optical fingerprint corresponding to a distinct conformational state of a specific molecule. Two LCOs can also be used in combination to increase the resolution and differentiate various polymorphs by their binding properties and relate this to identified polymorphs by AFM and TEM [46]. The conformational-sensitive optical properties of LCOs and LCPs have proven to be important for studying protein misfolding and aggregation. This property is especially useful as a marker for studies of amyloid polymorphism to translate in vivo signatures towards defined detailed in vitro studies.
Properties of polymorphic fibrils and oligomers
35 different proteins in humans are classified as amyloid proteins by the International Society of Amyloidosis [1]. Herein we discuss a subset of these proteins (Fig. 1) for which amyloid polymorphism has been described in the literature.
Aβ
Aβ is a proteolytic fragment from AβPP and is intimately linked biochemically and genetically to Alzheimer’s disease. Aβ is highly amyloidogenic and forms a multitude of aggregated states. Initially the monomer undergoes a conformational change and multimerizes forming dimers, trimers and hexamers. This process of forming hexamers appear to be significantly more prevalent for Aβ1–42 compared to 1–40 and is possibly linked to its higher cytotoxicity [47]. Oligomer formation with malleable heterogeneous protein folds as well as interactions between fibrils and transient exchanges forming oligomers on their surfaces is a dynamic energetically complex process (Fig. 3). This structural continuum is present at many levels of size distributions from single molecules through nanoscopic fibrils, to multi-million molecular assemblies forming microscopic aggregates (Fig. 3). If we focus on the structural polymorphism of the fibrils and do not include possible variations within oligomers, protofibrils, and their interactions, several high resolution structures of in vitro produced fibrils of Aβ1–40 and Aβ1–42 are reported in the literature. It is apparent that the conformational plasticity in forming these various types of fibril polymorphs implies comparable conformational stabilities of these various polymorphs within the energy landscape describing the conformational space of an aggregation-prone polypeptide chain (Fig. 3). Minute differences in assembly conditions allowing kinetic selectivity may be one reason for the propagation of certain fibril polymorphs over others.
Figure 3.
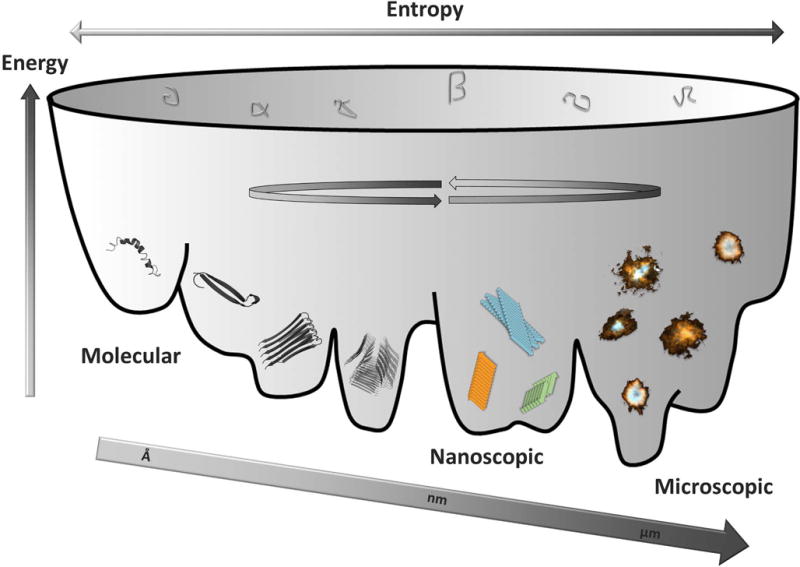
Illustration of the variable conformational landscape of Aβ molecular self-assembly from monomeric molecules, through oligomers into polymorphic nanoscopic fibrils into microscopic assemblies (plaque) with various morphologies found within brains of Alzheimer’s disease patients. Structures were generated in PyMol using PDB codes (from left to right) 1Z0Q, 2OTK, 2BEG.
In comparing fibrils formed in vivo versus in vitro it was shown that mature fibril polymorphs i.e. laterally associated multifilamentous bundled fibril types are formed with Aβ1–40 to a larger extent than with Aβ1–42 under the same experimental conditions. From LCO staining properties using two different LCOs in combination (qFTAA and hFTAA), this fibril type appeared prevalent in plaque cores in particular in transgenic tg-AβPP mice rich in Aβ1–40 [48]. Direct comparisons were not made with isolated Aβ fibrils from tissues here so the association was indirect. Spectroscopic changes of the LCO fluorescence response correlated in vitro with concomitant formation of bundled fibrils by AFM and TEM. A recent study of Aβ polymorphism in human samples using the same technology revealed a distribution of spectroscopic signatures from Aβ plaque cores [49]. This strongly suggests that a cloud-like diversity of Aβ conformations exist within each patient, representative of the variations of Aβ fibrils within a single sample [18]. The conformational cloud was weighted towards a certain type of mature or immature fibril distributions depending on the predisposing mutations in the PS1 and APP genes for fAD cases [49]. However, it is not directly known what causes these variations in the qFTAA staining properties; i.e. whether they are influenced, for example, by covalent modifications or the presence of non-fibril amyloid plaque components that account for different architectures of the biological deposit [22].
Aβ1–42 is dramatically more toxic than Aβ1–40. Even though tg-AβPP mice do not show neurodegeneration as a response to overexpression of Aβ peptides, it is notable that APP/PS1 mice producing more Aβ1–42 than Aβ1–40 have a significantly shorter lifespan than APP23 mice where the opposite 42/40 distribution is present [50]. Tg-Drosophila directly expressing Aβ1–42 peptides show severe neurodegeneration [51–53]. Importantly even single amino acid substitutions have profound impact on decreasing toxicity. It was recently shown that 1–41 and 1–43 are much less toxic than 1–42 suggesting that a specific conformational assembly with increased neurotoxicity is formed for Aβ1–42 [54]. Interestingly these neuronal deposits of Aβ1–42 appear predominantly intracellular [52] with an immature polymorph as deduced by LCO fluorescence (unpublished results). Notably Aβ1–40 is non-toxic in the fly likely due to efficient intracellular clearance [54].
Propagation of Aβ polymorphs by seeding
The notion of seeded nucleation reminiscent of prion-transmission has been intensively discussed in the literature over the past years and includes fibrils of Aβ, Tau, a-syn, PrP, SAA and others [2, 55–57]. As one example let us consider Aβ. Nucleation dependent polymerization of amyloid fibrils is reminiscent of crystal growth and was discussed early on in amyloid disease research [58]. Importantly, this mechanism appears protein sequence selective and can impose polymorphic templating similar to the case of crystal growth of organic molecules discussed earlier in this article. For research in the case of transmission of Aβ pathology the low neurotoxicity makes fibril formation of microscopically detectable aggregates the only readout available in mice [50, 59]. Many things can influence fibrillation of Aβ (Fig. 3), but by all measures in vitro the most efficient kinetic seeds are small fibril fragments and early stages of Aβ amyloidosis in vivo [50]. This implies that the high surface area of amyloid structures makes them active species for conversion. Furthermore, in the case of pure preparations of synthetic or recombinant Aβ fibrillized in vitro these are extremely poor at seeding plaque pathology in transgenic AD model mice. By contrast, pathology isolated from human AD brain or transgenic AD model mice readily seeds pathology in transgenic AD model mice. The resultant histopathology depends on both the source of the seed and on the host mouse model [59]. Interestingly not only does the pathology development speed up, but the amyloid conformation, the polymorph, appears to propagate from mouse to mouse [60] and from human to mouse [49]. Given that human cases of AD contain a variety of amyloid polymorphs [61] even cloud-like broad distributions within individuals [49], propagation of individual amyloid polymorphs should be considered during disease progression.
High resolution structural models of Aβ fibril folding polymorphs
Solid-state nuclear magnetic resonance (NMR) or electron cryo microscopy (cryo-EM) studies performed by different research groups recently provided data on the structure of Aβ1–42 peptide in fibrils [32, 62–65]. These groups observed different arrangements, indicating that the peptide can attain variable stable folds and protofilament structures (Fig. 2). Overall Aβ1–42 shows an intertwined horseshoe or kringle fold, however, with very distinct intermonomer interfaces. While the first structures have been obtained from solid-state NMR (Fig. 2A–B), Cryo-EM (Fig. 4 C–D) is recently also used demonstrating its exceptional potential to achieve high resolution structures of protein fibrils.
Figure 4.
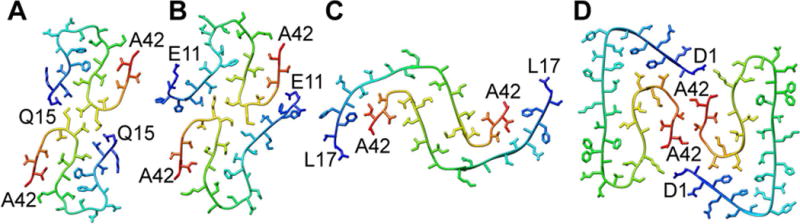
Polymorphism of Aβ1–42 fibrils formed in vitro. (A, B) Two in vitro formed structures were almost identical [62, 65], (C) while a different peptide fold and protofilament structure was observed with a third sample of Aβ1–42 fibrils assembled in vitro [63] and (D) in a recent study combining cryo-EM with NMR [32]. Images show the cross-sections. Structures were generated in PyMol using the pdb codes: (a) 5KK3 (b) 2NAO (c) 5AEF (d) 5OVQ.
High-resolution 3D structures are also available for Aβ1–40. Importantly, the folds are different from that of Aβ1–42. Hence, the C-terminal extended residues have profound influence on the folding of the protein. A point deletion within the Aβ1–40 protein termed the Osaka variant E22Δ has yet another distinct fold [66]. Several Aβ1–40 structural models are available, which include β-archs, and polymorphs showing dimers and trimers of the filament cross sections (Fig. 5) [67, 68]. The trimeric cross section (Fig. 5B) was obtained in fibril samples seeded with brain-derived amyloid fibrils isolated from an Alzheimer´s disease patient [69]. The C-terminus is hidden within the fibril (Fig. 5B) whereas it is located close to the surface of dimeric polymorph (Fig. 5A). A highly similar polymorph was previously described for Aβ1–40 also in pure in vitro samples showing that it is a polymorph attainable also in the absence of brain derived seeds [67]. Importantly the two different folding polymorphs were described as showing ultrastructure by TEM displaying striated and twisted ribbon morphologies, respectively.
Figure 5.
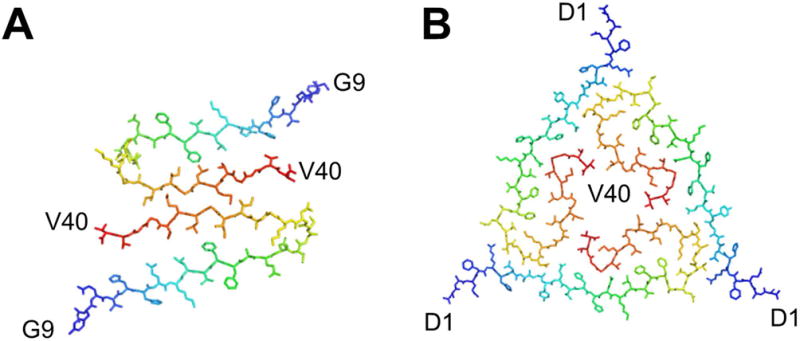
Polymorphism of Aβ1–40 fibrils formed purely in vitro in A [67] and seeded by fibrils from an Alzheimer’s disease patient in B [69]. Structures were generated in PyMol using the PDB codes (a) 2LMO (b) 2M4J.
α-synuclein
Parkinson’s disease is associated with Lewy body inclusions in dopaminergic neurons. Lewy body cores are composed of fibrils of the synaptic chaperone protein α-synuclein. In comparison with the much smaller Aβ, α-synuclein is 3-fold larger containing 120 residues. Differences in the fibril structures seem more dramatic due to its size which augments its conformational space compared to Aβ. Typical β-strand signatures of α-synuclein were found to be located in totally different parts of the protein for example, as shown in Figure 6. Indeed, for this protein, a wide variety of different pure polymorphs could be produced. It has been shown also for two different polymorphs that they induce different behavior not only with respect to structure, but also to function [70–72]. Although a first structural model has been developed [73], full 3D structure determination is still difficult due to its size.
Figure 6.
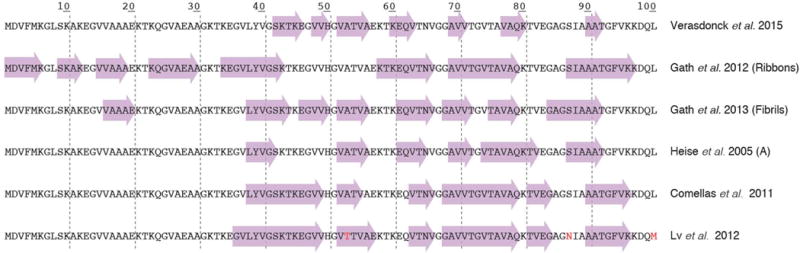
Secondary structure elements of different α-synuclein polymorphs as revealed by solid-state NMR studies. While certain studies revealed very similar elements, others, as the “ribbons” polymorph, show a highly distinct pattern. Data are from references [70, 74–79].
Tau
Tau is a microtubule stabilizing chaperone with its activity being regulated by phosphorylation and other posttranslational modifications. Misfolded hyperphosphorylated aggregated Tau is associated with various NDs including AD, frontotemporal dementia, corticobasal dementia, chronic traumatic encephalopathy, and progressive supranuclear palsy. Various isoforms of Tau are linked to different disease states. The mechanism behind this selectivity is unknown [80]. Recent structures of Tau fibrils by cryo-EM from AD brain reveal two ultrastructural polymorphs: Paired helical and straight filaments that differ in their protofilament packing [31]. Importantly, both fibril morphologies were found in the brain tissue of the same patient. These polymorphs are best described as assembly polymorphs rather than conformational polymorphs since the constituent fold of each horse shoe shaped protomer is highly similar (Fig. 7).
Figure 7.
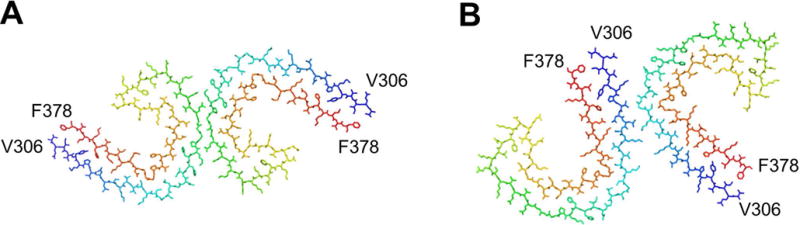
Assembly polymorphism of the core of Tau fibrils comprising residues 306–378 purified from Alzheimer’s disease brain. A. Protofilament structure from paired helical filaments and B. from straight filaments. Structures were generated in PyMol using the PDB codes (a) 5O3L (b) 5O3T from [31].
PrP
Demonstration that PrP in its native (PrPC) versus its misfolded infectious conformer (PrPSc) was identical in chemical composition with post translational modifications and disulfide bond intact but varied in conformation was the initial definition of an MP-driven disease [57, 81]. Prions composed of PrP thus constitute the canonical example of the strain phenomenon. Here, disease phenotype is intimately linked to the structure of prions. Although clinical samples are highlighted as variations in glycosylation patterns, hydrogen-deuterium exchange experiments of different prions purified from mouse brains show that various lengths of PrP comprise the fibril core in different prion strains [82]. Most importantly for the topic of this review, the structure can directly be interrogated using molecular probes. LCP fluorescence spectroscopy [43] and lifetime imaging (FLIM) makes it clear that the conformation of PrP within the strain aggregates are different [83]. Several aspects of a prion, directly linked to its conformation, make up its infectivity. Most often this infectivity is assayed based on neurotoxicity and not on amyloid fibrillation propensity. It is clear that fibril forming prion strains appear less neuroinvasive and less neurotoxic compared to diffuse aggregate assemblies particularly with synaptic localization patterns [84, 85]. Importantly, hyperstable prions such as those of synthetic origin, are less infectious than natural prions with lower conformational stability [86]. These findings suggest that identification of conformational stabilization by exogenous hyperstabilization molecules could be a viable treatment avenue [87]. In particular this strategy should be applicable to prions because of the rapidity of CJD in causing neurodegeneration to which such an aggressive shock-and-lock countermeasure is desirable. This approach was demonstrated to work in tg-mouse studies [45]. It would be interesting to propose that such a strategy is tested in human disease.
TTR
Amyloid from the plasma transport protein transthyretin (TTR) occurs both as a sporadic wild-type and dominantly inherited disease from missense mutations (>120 different mutations currently known) in the TTR gene. Extraction of amyloid fibrils deposited in the hearts of patients suffering from ATTR amyloidosis revealed structural variations and thus the polymorphism of the TTR-derived amyloid fibrils in this patient [88]. Moreover, there is evidence that TTR can undergo different proteolytic processing in different patients [89]. Fibril type A, the predominant fibril type, occurs in one set of patients and is composed of a mixture of length TTR 1–127 and TTR C-terminal sequence approximately residues 50–127. Fibril type B occurs in another set of patients, predominantly V30M patients, in which the fibril is derived from entirely full length TTR. Type A fibrils often target the heart while type B fibrils occur predominantly with early neurological symptoms. The differences between the two types of fibrils is observed morphologically in the microscope and by the brightness of the Congo red-green birefringence of histological sections analyzed by polarization microscopy. This implies that binding of molecular probes differ between the fibril types. It was recently shown that imaging of TTR in tissue reveals differences in LCP and LCO spectra indicating differences in structure. The differences were attributed to a mutation Y114H comprising a significant proportion of the amyloid protein [90]. Amyloid fibril structures from microcrystals of TTR peptides downstream and upstream of this site [91] suggest that a structural difference due to the Y114H mutation could induce a packing variation and thereby a new polymorph to explain these differences in probe fluorescence.
Oligomers of TTR have been described and often show the morphology of curvilinear protofibrils. These can form either at ambient conditions imposing a destabilizing mutation L55P from familial amyloid polyneuropathy (FAP) [92] or for wild type TTR under mildly denaturing conditions (pH 3, 4 °C) [93]. These protofibrillar structures are exceptional in that they appear to be hindered from further maturation over the time course of months. The process appears to be kinetically hindered at the stage of protofibrils (comprising approx. 200 monomers) because they are constantly undergoing monomer exchange at the surface [93]. Such an elusive and dynamic species would naturally be very hard to investigate and to target. Development of probes which target and specifically distinguish these states spectroscopically have been reported [94, 95], although the same probes also bind the native tetramer conformation at the thyroxine (T4) binding site. While the relationship of these protofibril species to oligomers in vivo is not known, soluble TTR oligomers have recently been detected in plasma of patients carrying heritable FAP mutations in the TTR gene [96]. Importantly this recent paper proposes a minimally invasive test for diagnosis and treatment follow up of FAP. Targeting of these species was performed using a peptide-based detection agent comprising a β-strand sequence of TTR. The structural basis for recognition appear to be a non-native β-strand motif determined from alanine scanning of the probe peptide.
Light chains
Systemic immunoglobulin light chain amyloidosis (AL) occurs as a secondary complication of a plasma cell dyscrasia which leads to abnormally high levels of free IgG light chains in the blood [97]. Normally, light chains are part of antibodies but due to an imbalance in the antibody production of an aberrantly proliferating plasma cell clone, light chains that are not complexed with a heavy chain are released into the blood. A fraction of the patients affected by this serum protein disturbance eventually develop AL amyloidosis. Amyloid fibrils primarily affect heart, kidneys and liver but they may occur in many other organs as well. The cardiac forms of systemic AL amyloidosis are devastating and can be life threatening if not treated by chemotherapy and heart transplantation. Systemic AL amyloidosisis is unique amongst all protein misfolding diseases as each patient essentially suffers from a patient-specific and sequence different LC [98]. While there are evidently different variants of systemic AL amyloidosis it is difficult to attribute these differences purely to morphological variations of the fibril and aggregate structures as there are substantial variations in the amino acid sequence of different fibril proteins as well as posttranslational modifications. As further support for the presence of chemical and potentially polymorphic AL deposits found in vivo, LCP staining and spectroscopy revealed a substantial variation of emission spectra from amyloid among AL patients [40]. Furthermore, ample evidence has been provided for morphological polymorphism in this disease as the amyloid fibrils that could be extracted from any one patient were polymorphic and each patient comprised a variety of different fibril architectures in the body that were all formed from the same sequence fibril protein [88]. Similar to observations made with fibrils extracted from systemic ATTR amyloidosis (see above), it was found that different patients were associated with different ensembles of fibril morphologies. Interestingly, however, within the same patient consistent fibril morphologies were seen in the different tissues (heart muscle, heart fat and abdominal fat) [16]. Detailed fibril structures by cryo-EM are currently available for a 12-residue peptide fragment of an amyloidogenic light chain [99]. Fibrils of the peptide fragment exhibit a significant morphological polymorphism as judged by electron microscopy.
SAA
Systemic AA amyloidosis occurs as a secondary complication of chronic inflammatory states in humans and animals (birds and mammals) [1]. Its associated amyloid deposits consist of truncated SAA protein and are usually found in spleen, liver, and kidneys. The time of onset of this disease can be drastically reduced in inflamed mice upon injection of amyloid-containing spleen extracts from AA amyloidotic mice, reminiscent of a transmission of prions [100]. Interestingly, AA amyloidosis in mice seeds promiscuously and can be induced using a variety of molecular agents acting as seeds [101, 102]. Extraction of amyloid fibrils from the spleens of AA amyloidotic mice revealed multiple fibril morphologies, demonstrating the in vivo polymorphism of AA amyloid fibrils. Consistent observations were made with the fibrils extracted from the AA amyloidotic tissues of deceased wild-living goats and island foxes (Urocyon littoralis). In all animals, multiple fibril morphologies were found per individual. Interestingly, comparison of the spleens and liver of the same animal revealed the same fibril morphologies to be present in both organs [16]. In contrast to different cases of human AL amyloidosis, each being associated with a genetically different fibril precursor protein, different AA amyloidotic mice showed the same polymorphs [88]. A recent study additionally shows that two isolates of AA amyloid fibrils induced two different amyloid deposit structures in identical recipient AA mice [103]. Isolates showed a variety of tissue deposition seeding activity. Whereas Congo red-positive amyloid was detected in both recipient mouse cohorts the pattern of LCO staining was different. Here the pattern of LCO binding in the recipient mice reflected the pattern of the initial isolate amyloid exemplifying for the first time that this progression reminiscent of templating of an amyloid strain polymorph is inducible outside of the brain, here in a systemic amyloidosis. Moreover, there are two variants of AA amyloidosis in humans, termed glomerular and vascular, depending on the deposition pattern of the AA amyloid in the kidneys [104]. These two variants of the disease are usually associated with different clinical presentations. Proteinuria is mainly confined to the glomerular AA amyloidosis, and with a different C-terminal truncation of the SAA protein. Glomerular amyloidosis occurs when the SAA is truncated after position 76, while the protein is variably truncated approximately at positions 50 and 94 in the vascular variant of the disease [104].
How do we target polymorphic amyloid fibrils and oligomers?
Amyloid fibrils of most proteins are highly polymorphic, both in vitro and in vivo. So how can such species be effectively targeted? A guide for structure based drug design at high resolution would require a model of monomorphic fibrils. In a recent study structural analysis followed by molecular modelling and molecular dynamics simulations fostered additional design of new molecules [45]. Herein, initially the yeast prion HET-s(218−289) amyloid fibril was used as a model because an atomic-resolution structure is available. From here on at least one binding site has been determined for LCP/LCOs to overlap at the atomistic level with that of Congo red (Fig. 8A). Extension of this work has revealed how positive and negative design by protein engineering of the binding site and anionic LCO/LCP molecule determines specificity and spectroscopic properties of the LCP/LCO. The molecule encounters an accessible elongated groove as the primary binding site. To optimally bind the amyloid, the anionic LCP/LCO must pair with specific regioregular arrangements of positively charged amino acids on the amyloid surface that match its negative charges [105]. While these are initial studies performed on yeast prions, they can offer available polymorphic test beds for amyloid-targeting probes. In yeast prions, polymorphism of the amyloid core has been shown to be highly context-dependent as the amyloid core varies whether the globular domains decorating the spine are present or not [106–108]. This offers an opportunity by protein engineering to exploit these systems for targeted drug design decoys for small molecule amyloid drugs and imaging agents.
Figure 8.
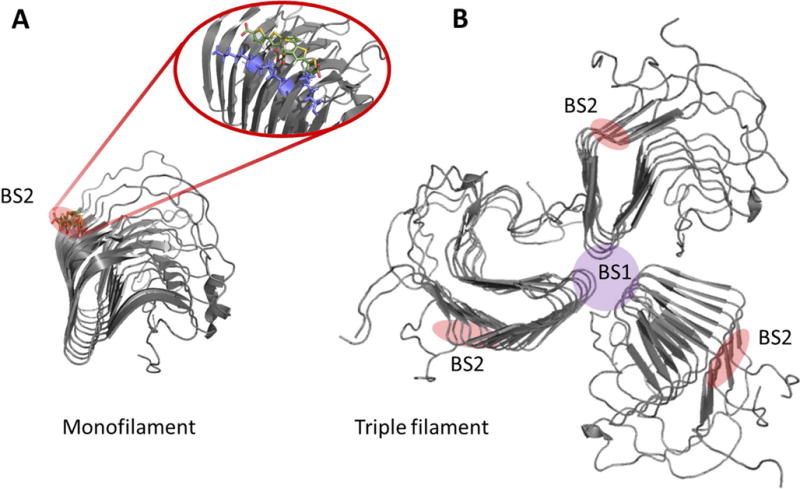
HET-s as a high resolution amyloid target testbed for amyloid probes. A. Structural model of HET-s (218–289) monofilament in complex with the LCO p-FTAA overlapping with the Congo red binding site. Pdb code 2MUS was used to create the structure in PyMol. B. Hypothetical trimeric HET-s filament where the BS2 site is retained and a novel binding site (BS1) suggestively forms at the filament interface. PDB: 2RNA [21, 45, 109]. Note: The BS2 and BS1 nomenclature comes from the Lockhart binding site model proposed for Aβ fibrils [110].
MP polymorphism - opportunities for diagnosis, prognosis, and intervention
The interaction of the cotton dye Congo red with fibrillar amyloid was an early diagnostic tool for systemic amyloids that was soon applied to staining fibrillar amyloid-containing pathology of Alzheimer’s disease and other neurodegenerative diseases. Thioflavin S (ThS), a sulfonated fluorescent polymeric benzothiazole, was also reactive with amyloid fibrils in tissue. A related dye the cationic Thioflavine T (ThT) containing a single benzothiazole moiety, displayed minimal background for monitoring in vitro fibril formation due to the large hypochromic shift in its excitation spectrum upon fibril binding [111]. These molecules were not suitable for in vivo detection of Alzheimer’s disease (AD) pathologies in animal models or in humans due to their low affinity, lack of blood-brain barrier penetration, and metabolic lability. They also did not differentiate between Aβ and tau pathologies, an essential distinction in AD.
The first Aβ-selective imaging ligand with nM affinity successfully used in living human patients to selectively detect Aβ pathology by positron emission tomography (PET) was 11C – labeled Pittsburgh Compound B (PIB) [112], which has become a gold standard for non-invasive Aβ brain imaging. Numerous amyloid ligands, many targeting Aβ, with a variety of structures have since been synthesized and studied [113]. Imaging tools fostered a revolution in thinking about AD by tracking the longitudinal progression of Aβ pathology development in living individuals decades before clinical signs were observable. Subsequently, other Aβ imaging molecules were developed incorporating the longer half-life 18F- isotopic PET label. Amyvid™ [florbetapir, Neuraceq™ [florbetaben], and Vizamyl™ [flutemetamol]) all compete for the same binding site on Aβ as PIB.
Targeting Aβ
Multiple independent binding sites for different ligands – the Lockhart model
With the clinical utility of 11C-PIB binding established and multiple ligands for Aβ being developed, it was important to determine whether different ligands were seeing the same structures. Using a series of fluorescent and radioligand binding studies, Lockhart, Ye, and colleagues defined three distinguishable high affinity binding sites on synthetic fibrils which bound different classes of chemical structures [114, 115]. Their evidence suggesting partial interaction between some of the sites has received support [116]. Although the molecular structure of those sites remains to be experimentally determined, the ability to use displacement of radioligand binding from synthetic Aβ fibrils and brain tissue sections to operationally identify candidate molecules for optimization was a logical approach. One of the sites, BS1, was assigned to ThT. Another, more abundant site in Aβ fibrils was BS2 which was associated with binding of Congo red – like molecules. BS3 was least abundant and was associated with PIB binding. 3H-X34, a highly fluorescent analog of Congo red, binds to the BS2 Aβ site and is not effectively competed for by either ThT or PIB [117]. ThT, Congo red, and X34 seem to be pan-amyloid specific, interacting with amyloid fibrils formed from a variety of proteins. The corresponding structural site for BS2 on Het-s fibrils is illustrated in Fig. 8A. Whereas a BS1 site is hypothetically located at the interface of protomers comprising individual filaments (Fig. 8B). Such a model would jive well with the abundance of BS2 sites compared to BS1 sites. 3H-X34 binds with a constant stoichiometry of 1:1 or 1:2 ligand/Aβ monomer, while ThT and 3H-PIB binding depend on the conditions of fibrillation. This indication that binding of some amyloid ligands was sensitive to fibril conformation is a potentially valuable tool to assess phenotypes, or strains, encoded in the structure of the amyloid fibrils. However, it is frustrating in development of quantitative imaging probes that the complications from various conformational and assembly polymorphisms alter the target.
One of the first examples of phenotypic expression of fibril polymorphism in the form of ligand binding was the observation that PIB bound abundantly and with high affinity to human AD Aβ pathology. By contrast, PIB bound weakly and in very low amounts to natural and engineered animal models of AD [118–120]. PIB binding to synthetic Aβ peptide amyloid fibrils is weaker and occupies a smaller number of sites which depend strongly on the conditions of fibrillation. Binding is detectable mainly because larger amounts of fibrils can be assayed. PIB binds to a minority of all Aβ pathology in brain sections from AD patients [121]. Interestingly, a very small number of AD subjects whose histology on autopsy showed full Aβ and tau pathology lack in vitro 3H-PIB binding [122, 123]. A similar lack of PIB binding is observed in AD subjects expressing the Arctic mutation in the amyloid precursor protein [124].
Targeting Tau
Selective tau ligand binding has been more challenging to achieve with trials of tau PET ligand imaging in patients only recently reaching the clinic. In many ways tau is a much more challenging target than Aβ. There are six tau isoforms generated by alternative splicing of the tau gene as well as differential isoform expression depending on the specific disease state. Tau is also subject to multiple types of posttranslational modification including phosphorylation, acetylation, and O-linked N-acetyl-glycosylation to mention just a few. Development of ligands using recombinant tau fibrils assembled in vitro often doesn’t result in tau-selective compounds in human disease tissue This demonstrates the requirement for a different strategy of screening putative ligands. In addition, tau pathologies in different neurodegenerative diseases bind different tau ligands. Table 1 compares tauopathy-dependence of several ligands which don’t compete with each other for binding tau pathology, apparently recognizing different tau species. The fibril structural basis for this selectivity remains to be determined. A common binding site on tau tangles in multiple tau diseases is recognized by the PBB3 tau ligand [125] which is selective for tau over Aβ and more generally recognizes tau pathology in several of the tauopathies where other tau ligands showed differential reactivity.
Table 1.
Tau ligands distinguish between tau pathologies in different diseases.
| Ligand | Tau | ||||
|---|---|---|---|---|---|
| AD | PSP | FTD | CBD | P301S (mouse) | |
| Congo Red | + | + | + | + | + |
| FDDNP | + | + | − | − | − |
| T807* (AV-1451) | + | − | − | − | − |
| THK-523* | + | − | − | − | − |
| PBB3 | + | + | + | + | + |
AD = Alzheimer’s disease; PSP = progressive supranuclear palsy; FTD = frontotemporal dementia; CBD = corticobasal degeneration; P301S = tau P301S mutant expressed in a tg-mouse model of FTD/CBD.
T807 and THK-523 bind to distinct sites on tau.
Recently a new class of thiophene-based fluorescent ligands that selectively detect tau pathology in AD was reported [126]. As shown in Fig. 9, the pyridinyl-butadienyl-motif in PBB3 was replaced by a bi-thiophenevinyl moiety rendering a set of compounds denoted bi-thiophene-vinyl-benzothiazoles/benzothiazoliums (bTVBTs). When utilizing the bTVBTs in combination with a thiophene-based ligand, q-FTAA-CN [127] selective towards aggregated Aβ species, the two pathological hallmarks in AD could easily be distinguished (Fig. 9). Thus, minor chemical alterations of the thiophene backbone can be utilized to develop high-affinity ligands towards distinct disease-associated protein aggregates.
Figure 9.
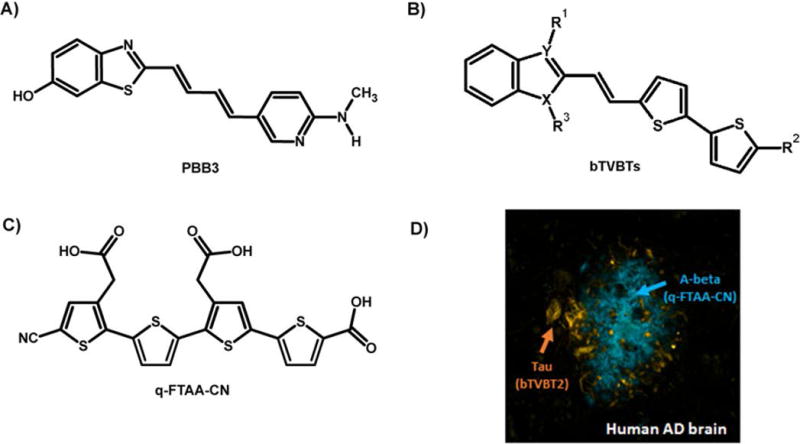
Chemical structure of fluorescent ligands for protein aggregates. A) The chemical structure of PBB3, a high-affinity ligand for tau deposits in human AD brain. B) Chemical structure of novel bi-thiophenevinyl-benzothiazoles/benzoimidazoles (bTVBTs). C) The chemical structure of q-FTAA-CN, a high-affinity ligand for Aβ deposits in human AD brain. D) Image of a human brain section with AD-pathology that was stained with tau-4 (yellow-red) and q-FTAA-CN (blue).
Targeting α-Synuclein
Specific high affinity ligands for misfolded α-synuclein pathology suitable for brain imaging have been difficult to identify [128, 129]. SIL23 is not sufficiently selective in the presence of Aβ or tau pathology. It is useable as a tool to find ligands with appropriate properties for in vivo brain imaging [130]. α-synuclein pathology is characteristic of multiple neurodegenerative diseases including Parkinson’s disease, dementia with Lewy bodies, and a series of distinct diseases with α-synuclein inclusions grouped together as multiple system atrophy [80]. The pathology spreads in a prion-like fashion and different polymorphic forms of α-synuclein are observed along with different clinical presentations reminiscent of conformational strains [131]. It is likely that multiple folding pathways and products are present and disease-relevant as described above.
Not a bug, but a feature
While frustrating for developers of ligands, the sensitivity of ligand binding to variations in fibrils offers the potential to link polymorphic structures, also known as “strains”, with different diseases, disease stages, and progression rates. The appropriate ligand(s) could provide important diagnostic and prognostic information by imaging and/or analysis of pathology biomarkers in biofluids. There are hints of this potential. Aging of plaques in a transgenic mouse model changes the ratio of binding of different oligothiophenes [48]. The fluorescent properties of poly- and oligothiophene probes are also sensitive to probe backbone conformation [132]. This allows the monitoring of protein conformational states by these probes and can distinguish prion strains in tissue sections [83, 133].
In humans the Aβ pathology in rapidly progressive AD cases has been shown to have different conformational stability than typical sporadic AD [134]. Different fibril structures have been determined for the typical and the rapidly progressive forms AD (by ssNMR) which both differ from fibril structure in the posterior cortical atrophy AD variant [61]. It would be useful to have ligands that could report on the rate of progression.
The utility of probes of informative polymorphic forms of MP pathology can potentially extend beyond imaging applications. Exosomes produced by neurons and other brain cell types in AD and PD patients have been shown to contain Aβ and tau pathology forms [135, 136]. Exosomes are thought to be one way MP pathology spreads in the brain [137]. These small vesicles are present in CSF and in blood and could potentially serve as surrogate biomarkers for disease pathology development if they include the informative polymorphic fibril forms in accessible biofluids, or to determine when brain imaging with selected PET ligands would be most useful.
Combinatorial treatments targeted at stopping spread and toxicity of MPs
Unfortunately, the difficulty in addressing ND has prompted most major pharma companies to severely downsize their nervous system disorder research [138]. Increased academic research is essential for filling this void. There are many strategies to prevent MP proliferation to mitigate disease. Several are aimed at lowering MP protomer substrate for further growth. This has been achieved through stabilizing the native state (TTR) [139], inhibition of proteases (Aβ), antibodies for increased clearance (Aβ), iRNA (TTR), reducing inflammation (SAA), destroying the producing cell (light chain). These strategies and their outcomes have recently been reviewed [140, 141]. Other strategies have been to target the MP aggregated states. To stop neurodegeneration an important target is to stop the propagation of MP states. A major challenge for drug development is to target shape-shifting polymorphic MPs. Since individual patients display a distribution of MP polymorphs observed as microscopic morphotypes, a particular clinical disease progression, as recently shown for AD [49] (Fig 10) was suggested to be dominated by various polymorphs that propagate efficiently [61]. The diversity of various MP polymorphs render specific targeting complicated. Given this structural polymorphism it will by extension efficient treatment will require an individualized medicine approach and may be specific for different stages of disease (Fig. 10). In theory this strongly suggests that different MPs may be most efficiently targeted by combinations of sequence-specific (e.g. antibody MP clearance↑) and conformational inhibitors (seeding↓ and propagation↓) (Fig. 10).
Figure 10.
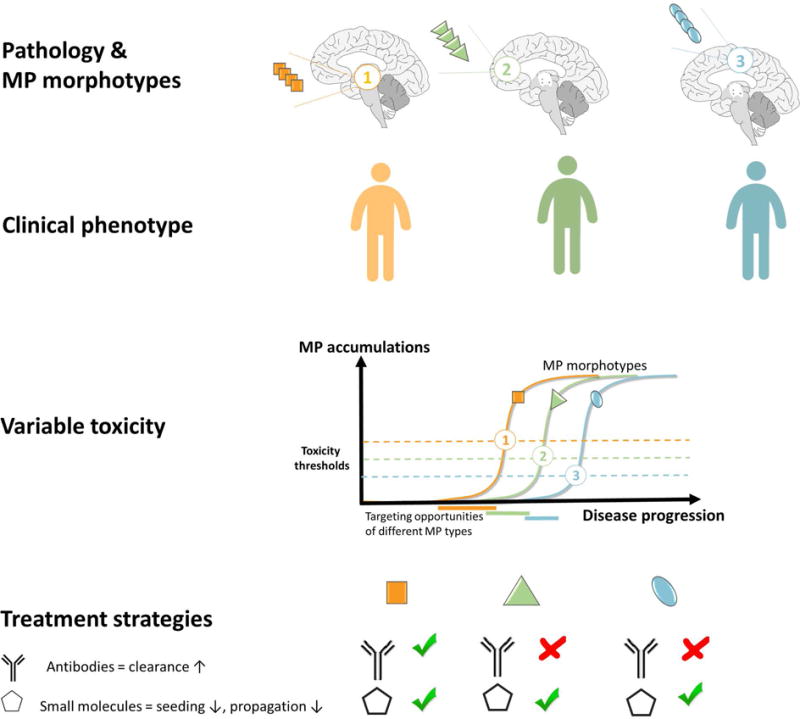
Theoretical model of how 3 different MP morphotypes propagate either in different individuals or at various stages of neurodegenerative disease and display diverse toxicity thresholds depending on polymorphic “strain”, physiology/cell-type and/or genotype. This model proposes various windows of opportunity for intervention for treatments targeted for increased clearance (e.g. antibody) and for targeted small molecules specific for each morphotype. In theory each tailored treatment will show various efficacy at different stages of disease progression.
Small molecule fibril stabilizers [87] prevent fibril fragmentation and/or prevent growth of fibrils either blocking end-elongation or secondary surface-catalyzed fibril formation (Fig. 10). Direct targeting the MP aggregate with thiophene-based small molecules was recently shown to work via this mechanism to prevent prion disease in mice [45]. Similarly, it was recently shown that binding of curcumin to Aβ promotes fibrillation and shifts the Aβ equilibria towards mature fibrils at the expense of neurotoxic oligomers and prevented neurodegeneration in Drosophila [51]. Targeting can be achieved by finding common and divergent structural motifs within MP model structures and to target these by diversifying the chemistry of the ligand to match the MP structures. Combinatorial chemistry using fragments from various MP binding motifs [142] and testing cocktails with small molecules and biologics are foreseen as promising approaches (Fig. 10). Published proof-of-concept experiments referenced above are highly encouraging that this approach is viable.
Acknowledgments
Our work was funded by The Swedish Research Council (2015-04521, 2015-05868), the Göran Gustafsson Foundation, The Swedish Alzheimer Foundation, Linköping University, NIH/NINDS (R21 NS080576), BrightFocus Foundation (A2014044S), the French ANR (ANR-14-CE09-0024B), and the Deutsche Forschungsgemeinschaft (FA 456/15-1).
Dr. Hammarström reports grants from The Swedish Research Council, grants from The Göran Gustafsson Foundation, grants from The Swedish Alzheimer Foundation. Dr. Nyström reports a grant from the Swedish Alzheimer Foundation, Dr. Nilsson reports a grant from the Swedish Research Council, Dr. LeVine reports grants from NIH/NINDS, grants from BrightFocus Foundation, Dr. Böckmann reports a grant from the French ANR, Dr. Fändrich reports a grant from the Deutsche Forschungsgemeinschaft, during the conduct of the study; Dr. Hammarström, Dr. Nyström and Dr. Nilsson are shareholders of Furcifer AB, a company that founded Ebba Biotech AB, who makes some of the amyloid ligands described in the paper available to researchers globally. In addition, Dr. Nilsson has a patent issued.
Footnotes
PROF. PER HAMMARSTRÖM (Orcid ID : 0000-0001-5827-3587)
References
- 1.Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJM, Westermark P. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid-Journal of Protein Folding Disorders. 2016;23:209–13. doi: 10.1080/13506129.2016.1257986. [DOI] [PubMed] [Google Scholar]
- 2.Riek R, Eisenberg DS. The activities of amyloids from a structural perspective. Nature. 2016;539:227–35. doi: 10.1038/nature20416. [DOI] [PubMed] [Google Scholar]
- 3.Anfinsen CB. The formation and stabilization of protein structure. Biochem J. 1972;128:737–49. doi: 10.1042/bj1280737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–30. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 5.Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature. 1959;183:1202–3. doi: 10.1038/1831202a0. [DOI] [PubMed] [Google Scholar]
- 6.Shirahama T, Cohen AS. Structure of amyloid fibrils after negative staining and high-resolution electron microscopy. Nature. 1965;206:737–8. doi: 10.1038/206737a0. [DOI] [PubMed] [Google Scholar]
- 7.Wöhler F, von Liebig J. Untersuchungen über das Radikal der Benzoesäure. J Ann Pharm. 1832;3:249–82. [Google Scholar]
- 8.David WI, Shankland K, Pulham CR, Blagden N, Davey RJ, Song M. Polymorphism in benzamide. Angew Chem Int Ed Engl. 2005;44:7032–5. doi: 10.1002/anie.200501146. [DOI] [PubMed] [Google Scholar]
- 9.Thun J, Seyfarth L, Senker J, Dinnebier RE, Breu J. Polymorphism in benzamide: solving a 175-year-old riddle. Angew Chem Int Ed Engl. 2007;46:6729–31. doi: 10.1002/anie.200701383. [DOI] [PubMed] [Google Scholar]
- 10.Tao J, Jones KJ, Yu L. Cross-nucleation between D-Mannitol polymorphs in seeded crystallization. Cryst Growth Des. 2007;7:2410–4. [Google Scholar]
- 11.Yu L. Survival of the fittest polymorph: how fast nucleater can lose to fast grower. Crystengcomm. 2007;9:847–51. [Google Scholar]
- 12.Goldsbury CS, Wirtz S, Muller SA, Sunderji S, Wicki P, Aebi U, Frey P. Studies on the in vitro assembly of A beta 1–40: Implications for the search for A beta fibril formation inhibitors. Journal of Structural Biology. 2000;130:217–31. doi: 10.1006/jsbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- 13.Meinhardt J, Sachse C, Hortschansky P, Grigorieff N, Fandrich M. A beta(1–40) Fibril Polymorphism Implies Diverse Interaction Patterns in Amyloid Fibrils. Journal of Molecular Biology. 2009;386:869–77. doi: 10.1016/j.jmb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopez del Amo JM, Schmidt M, Fink U, Dasari M, Fandrich M, Reif B. An Asymmetric Dimer as the Basic Subunit in Alzheimer’s Disease Amyloid beta Fibrils. Angew Chem Int Edit. 2012;51:6136–9. doi: 10.1002/anie.201200965. [DOI] [PubMed] [Google Scholar]
- 15.Sachse C, Fandrich M, Grigorieff N. Paired beta-sheet structure of an A beta(1–40) amyloid fibril revealed by electron microscopy. P Natl Acad Sci USA. 2008;105:7462–6. doi: 10.1073/pnas.0712290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Annamalai K, Liberta F, Vielberg MT, et al. Common Fibril Structures Imply Systemically Conserved Protein Misfolding Pathways In Vivo. Angew Chem Int Ed Engl. 2017;56:7510–4. doi: 10.1002/anie.201701761. [DOI] [PubMed] [Google Scholar]
- 17.Kodali R, Williams AD, Chemuru S, Wetzel R. A beta(1–40) Forms Five Distinct Amyloid Structures whose beta-Sheet Contents and Fibril Stabilities Are Correlated. Journal of Molecular Biology. 2010;401:503–17. doi: 10.1016/j.jmb.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klement K, Wieligmann K, Meinhardt J, Hortschansky P, Richter W, Fandrich M. Effect of different salt ions on the propensity of aggregation and on the structure of Alzheimer’s A beta(1–40) amyloid fibrils. Journal of Molecular Biology. 2007;373:1321–33. doi: 10.1016/j.jmb.2007.08.068. [DOI] [PubMed] [Google Scholar]
- 19.Fandrich M, Meinhardt J, Grigorieff N. Structural polymorphism of Alzheimer A beta and other amyloid fibrils. Prion. 2009;3:89–93. doi: 10.4161/pri.3.2.8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luhrs T, Ritter C, Adrian M, et al. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–7. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smaoui MR, Poitevin F, Delarue M, Koehl P, Orland H, Waldispuhl J. Computational assembly of polymorphic amyloid fibrils reveals stable aggregates. Biophys J. 2013;104:683–93. doi: 10.1016/j.bpj.2012.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han S, Kollmer M, Markx D, Claus S, Walther P, Fandrich M. Amyloid plaque structure and cell surface interactions of beta-amyloid fibrils revealed by electron tomography. Sci Rep-Uk. 2017;7 doi: 10.1038/srep43577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kollmer M, Meinhardt K, Haupt C, et al. Electron tomography reveals the fibril structure and lipid interactions in amyloid deposits. P Natl Acad Sci USA. 2016;113:5604–9. doi: 10.1073/pnas.1523496113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bergstrom J, Gustavsson A, Hellman U, et al. Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. J Pathol. 2005;206:224–32. doi: 10.1002/path.1759. [DOI] [PubMed] [Google Scholar]
- 25.Gondo T, Ishihara T, Kawano H, et al. Localized Amyloidosis in Squamous-Cell Carcinoma of Uterine Cervix - Electron-Microscopic Features of Nodular and Star-Like Amyloid Deposits. Virchows Arch A. 1993;422:225–31. doi: 10.1007/BF01621806. [DOI] [PubMed] [Google Scholar]
- 26.Wisniewski HM, Wegiel J, Wang KC, Lach B. Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 1992;84:117–27. doi: 10.1007/BF00311383. [DOI] [PubMed] [Google Scholar]
- 27.SCOPe S-SCoP. Berkeley BLaU. UC Berkeley; 2017. [Google Scholar]
- 28.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–8. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–8. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sawaya MR, Sambashivan S, Nelson R, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–7. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 31.Fitzpatrick AWP, Falcon B, He S, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–+. doi: 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gremer L, Scholzel D, Schenk C, et al. Fibril structure of amyloid-beta(1–42) by cryo-electron microscopy. Science. 2017;358:116–9. doi: 10.1126/science.aao2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aslund A, Sigurdson CJ, Klingstedt T, et al. Novel Pentameric Thiophene Derivatives for in Vitro and in Vivo Optical Imaging of a Plethora of Protein Aggregates in Cerebral Amyloidoses. Acs Chemical Biology. 2009;4:673–84. doi: 10.1021/cb900112v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berg I, Nilsson KPR, Thor S, Hammarstrom P. Efficient imaging of amyloid deposits in Drosophila models of human amyloidoses. Nat Protoc. 2010;5:935–44. doi: 10.1038/nprot.2010.41. [DOI] [PubMed] [Google Scholar]
- 35.Hammarstrom P, Simon R, Nystrom S, Konradsson P, Aslund A, Nilsson KPR. A Fluorescent Pentameric Thiophene Derivative Detects in Vitro-Formed Prefibrillar Protein Aggregates. Biochemistry. 2010;49:6838–45. doi: 10.1021/bi100922r. [DOI] [PubMed] [Google Scholar]
- 36.Nilsson KP, Aslund A, Berg I, et al. Imaging distinct conformational states of amyloid-beta fibrils in Alzheimer’s disease using novel luminescent probes. ACS Chem Biol. 2007;2:553–60. doi: 10.1021/cb700116u. [DOI] [PubMed] [Google Scholar]
- 37.Nilsson KP, Hammarstrom P, Ahlgren F, et al. Conjugated polyelectrolytes–conformation-sensitive optical probes for staining and characterization of amyloid deposits. Chembiochem. 2006;7:1096–104. doi: 10.1002/cbic.200500550. [DOI] [PubMed] [Google Scholar]
- 38.Nilsson KP, Herland A, Hammarstrom P, Inganas O. Conjugated polyelectrolytes: conformation-sensitive optical probes for detection of amyloid fibril formation. Biochemistry. 2005;44:3718–24. doi: 10.1021/bi047402u. [DOI] [PubMed] [Google Scholar]
- 39.Nilsson KPR. Small organic probes as amyloid specific ligands - Past and recent molecular scaffolds. Febs Letters. 2009;583:2593–9. doi: 10.1016/j.febslet.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 40.Nilsson KPR, Ikenberg K, Aslund A, et al. Structural Typing of Systemic Amyloidoses by Luminescent-Conjugated Polymer Spectroscopy. American Journal of Pathology. 2010;176:563–74. doi: 10.2353/ajpath.2010.080797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nilsson KPR, Joshi-Barr S, Winson O, Sigurdson CJ. Prion Strain Interactions Are Highly Selective. J Neurosci. 2010;30:12094–102. doi: 10.1523/JNEUROSCI.2417-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sigurdson CJ, Nilsson KPR, Hornemann S, et al. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. P Natl Acad Sci USA. 2009;106:304–9. doi: 10.1073/pnas.0810680105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sigurdson CJ, Peter K, Nilsson R, et al. Prion strain discrimination using luminescent conjugated polymers. Nat Methods. 2007;4:1023–30. doi: 10.1038/nmeth1131. [DOI] [PubMed] [Google Scholar]
- 44.Back M, Appelqvist H, LeVine H, Nilsson KPR. Anionic Oligothiophenes Compete for Binding of X-34 but not PIB to Recombinant A beta Amyloid Fibrils and Alzheimer’s Disease Brain-Derived A beta. Chem-Eur J. 2016;22:18335–8. doi: 10.1002/chem.201604583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrmann US, Schutz AK, Shirani H, et al. Structure-based drug design identifies polythiophenes as antiprion compounds. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.aab1923. [DOI] [PubMed] [Google Scholar]
- 46.Psonka-Antonczyk KM, Hammarstrom P, Johansson LB, Lindgren M, Stokke BT, Nilsson KP, Nystrom S. Nanoscale Structure and Spectroscopic Probing of Abeta1–40 Fibril Bundle Formation. Front Chem. 2016;4:44. doi: 10.3389/fchem.2016.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lendel C, Bjerring M, Dubnovitsky A, et al. A Hexameric Peptide Barrel as Building Block of Amyloid-beta Protofibrils. Angew Chem Int Edit. 2014;53:12756–60. doi: 10.1002/anie.201406357. [DOI] [PubMed] [Google Scholar]
- 48.Nystrom S, Psonka-Antonczyk KM, Ellingsen PG, et al. Evidence for age-dependent in vivo conformational rearrangement within Abeta amyloid deposits. ACS Chem Biol. 2013;8:1128–33. doi: 10.1021/cb4000376. [DOI] [PubMed] [Google Scholar]
- 49.Rasmussen J, Mahler J, Beschorner N, et al. Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1713215114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye L, Rasmussen J, Kaeser SA, et al. Abeta seeding potency peaks in the early stages of cerebral beta-amyloidosis. EMBO Rep. 2017;18:1536–44. doi: 10.15252/embr.201744067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caesar I, Jonson M, Nilsson KP, Thor S, Hammarstrom P. Curcumin promotes A-beta fibrillation and reduces neurotoxicity in transgenic Drosophila. PLoS One. 2012;7:e31424. doi: 10.1371/journal.pone.0031424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crowther DC, Kinghorn KJ, Miranda E, et al. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience. 2005;132:123–35. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 53.Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:6623–8. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jonson M, Pokrzywa M, Starkenberg A, Hammarstrom P, Thor S. Systematic Abeta Analysis in Drosophila Reveals High Toxicity for the 1–42, 3–42 and 11–42 Peptides, and Emphasizes N- and C-Terminal Residues. PLoS One. 2015;10:e0133272. doi: 10.1371/journal.pone.0133272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goedert M, Spillantini MG. Propagation of Tau aggregates. Mol Brain. 2017;10:18. doi: 10.1186/s13041-017-0298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tjernberg LO, Rising A, Johansson J, Jaudzems K, Westermark P. Transmissible amyloid. J Intern Med. 2016;280:153–63. doi: 10.1111/joim.12499. [DOI] [PubMed] [Google Scholar]
- 57.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 58.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 59.Eisele YS. From soluble abeta to progressive abeta aggregation: could prion-like templated misfolding play a role? Brain Pathol. 2013;23:333–41. doi: 10.1111/bpa.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heilbronner G, Eisele YS, Langer F, et al. Seeded strain-like transmission of beta-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013;14:1017–22. doi: 10.1038/embor.2013.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiang W, Yau WM, Lu JX, Collinge J, Tycko R. Structural variation in amyloid-beta fibrils from Alzheimer’s disease clinical subtypes. Nature. 2017;541:217–+. doi: 10.1038/nature20814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colvin MT, Silvers R, Ni QZ, et al. Atomic Resolution Structure of Monomorphic A beta(42) Amyloid Fibrils. J Am Chem Soc. 2016;138:9663–74. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmidt M, Rohou A, Lasker K, Yadav JK, Schiene-Fischer C, Fandrich M, Grigorieff N. Peptide dimer structure in an A beta(1–42) fibril visualized with cryo-EM. P Natl Acad Sci USA. 2015;112:11858–63. doi: 10.1073/pnas.1503455112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schmidt M, Sachse C, Richter W, Xu C, Fandrich M, Grigorieff N. Comparison of Alzheimer A beta(1–40) and A beta(1–42) amyloid fibrils reveals similar protofilament structures. P Natl Acad Sci USA. 2009;106:19813–8. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walti MA, Ravotti F, Arai H, et al. Atomic-resolution structure of a disease-relevant A beta(1–42) amyloid fibril. P Natl Acad Sci USA. 2016;113:E4976–E84. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schutz AK, Vagt T, Huber M, et al. Atomic-resolution three-dimensional structure of amyloid beta fibrils bearing the Osaka mutation. Angew Chem Int Ed Engl. 2015;54:331–5. doi: 10.1002/anie.201408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. P Natl Acad Sci USA. 2008;105:18349–54. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. P Natl Acad Sci USA. 2002;99:16742–7. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell. 2013;154:1257–68. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bousset L, Pieri L, Ruiz-Arlandis G, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gath J, Bousset L, Habenstein B, Melki R, Bockmann A, Meier BH. Unlike twins: an NMR comparison of two alpha-synuclein polymorphs featuring different toxicity. PLoS One. 2014;9:e90659. doi: 10.1371/journal.pone.0090659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Peelaerts W, Bousset L, Van der Perren A, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–4. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- 73.Tuttle MD, Comellas G, Nieuwkoop AJ, et al. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol. 2016;23:409–15. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Comellas G, Lemkau LR, Nieuwkoop AJ, et al. Structured Regions of alpha-Synuclein Fibrils Include the Early-Onset Parkinson’s Disease Mutation Sites. Journal of Molecular Biology. 2011;411:881–95. doi: 10.1016/j.jmb.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gath J, Bousset L, Habenstein B, Melki R, Bockmann A, Meier BH. Unlike Twins: An NMR Comparison of Two alpha-Synuclein Polymorphs Featuring Different Toxicity. Plos One. 2014;9 doi: 10.1371/journal.pone.0090659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gath J, Habenstein B, Bousset L, Melki R, Meier BH, Bockmann A. Solid-state NMR sequential assignments of alpha-synuclein. Biomol Nmr Assign. 2012;6:51–5. doi: 10.1007/s12104-011-9324-3. [DOI] [PubMed] [Google Scholar]
- 77.Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. P Natl Acad Sci USA. 2005;102:15871–6. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lv GH, Kumar A, Giller K, et al. Structural Comparison of Mouse and Human alpha-Synuclein Amyloid Fibrils by Solid-State NMR. Journal of Molecular Biology. 2012;420:99–111. doi: 10.1016/j.jmb.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 79.Verasdonck J, Bousset L, Gath J, Melki R, Bockmann A, Meier BH. Further exploration of the conformational space of alpha-synuclein fibrils: solid-state NMR assignment of a high-pH polymorph. Biomol Nmr Assign. 2016;10:5–12. doi: 10.1007/s12104-015-9628-9. [DOI] [PubMed] [Google Scholar]
- 80.Goedert M, Jakes R, Spillantini MG. The Synucleinopathies: Twenty Years On. J Parkinsons Dis. 2017;7:S53–S71. doi: 10.3233/JPD-179005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cohen FE, Prusiner SB. Pathologic conformations of prion proteins. Annu Rev Biochem. 1998;67:793–819. doi: 10.1146/annurev.biochem.67.1.793. [DOI] [PubMed] [Google Scholar]
- 82.Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol. 2011;18:504–6. doi: 10.1038/nsmb.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Magnusson K, Simon R, Sjolander D, Sigurdson CJ, Hammarstrom P, Nilsson KPR. Multimodal fluorescence microscopy of prion strain specific PrP deposits stained by thiophene-based amyloid ligands. Prion. 2014;8:319–29. doi: 10.4161/pri.29239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bett C, Joshi-Barr S, Lucero M, et al. Biochemical properties of highly neuroinvasive prion strains. PLoS Pathog. 2012;8:e1002522. doi: 10.1371/journal.ppat.1002522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bett C, Kurt TD, Lucero M, et al. Defining the conformational features of anchorless, poorly neuroinvasive prions. PLoS Pathog. 2013;9:e1003280. doi: 10.1371/journal.ppat.1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Legname G, Nguyen HO, Baskakov IV, Cohen FE, Dearmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci U S A. 2005;102:2168–73. doi: 10.1073/pnas.0409079102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Margalith I, Suter C, Ballmer B, et al. Polythiophenes inhibit prion propagation by stabilizing prion protein (PrP) aggregates. J Biol Chem. 2012;287:18872–87. doi: 10.1074/jbc.M112.355958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Annamalai K, Guhrs KH, Koehler R, et al. Polymorphism of Amyloid Fibrils In Vivo. Angew Chem Int Ed Engl. 2016;55:4822–5. doi: 10.1002/anie.201511524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Suhr OB, Lundgren E, Westermark P. One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J Intern Med. 2017;281:337–47. doi: 10.1111/joim.12585. [DOI] [PubMed] [Google Scholar]
- 90.Sekijima Y, Campos RI, Hammarstrom P, et al. Pathological, biochemical, and biophysical characteristics of the transthyretin variant Y114H (p.Y134H) explain its very mild clinical phenotype. J Peripher Nerv Syst. 2015;20:372–9. doi: 10.1111/jns.12143. [DOI] [PubMed] [Google Scholar]
- 91.Saelices L, Johnson LM, Liang WY, et al. Uncovering the Mechanism of Aggregation of Human Transthyretin. J Biol Chem. 2015;290:28932–43. doi: 10.1074/jbc.M115.659912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lashuel HA, Wurth C, Woo L, Kelly JW. The most pathogenic transthyretin variant, L55P, forms amyloid fibrils under acidic conditions and protofilaments under physiological conditions. Biochemistry. 1999;38:13560–73. doi: 10.1021/bi991021c. [DOI] [PubMed] [Google Scholar]
- 93.Groenning M, Campos RI, Hirschberg D, Hammarstrom P, Vestergaard B. Considerably Unfolded Transthyretin Monomers Preceed and Exchange with Dynamically Structured Amyloid Protofibrils. Sci Rep. 2015;5:11443. doi: 10.1038/srep11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Campos RI, Wu X, Elgland M, Konradsson P, Hammarstrom P. Novel trans-Stilbene-based Fluorophores as Probes for Spectral Discrimination of Native and Protofibrillar Transthyretin. ACS Chem Neurosci. 2016;7:924–40. doi: 10.1021/acschemneuro.6b00062. [DOI] [PubMed] [Google Scholar]
- 95.Zhang J, Sandberg A, Wu XY, Nystrom S, Lindgren M, Konradsson P, Hammarstrom P. trans-Stilbenoids with Extended Fluorescence Lifetimes for the Characterization of Amyloid Fibrils. Acs Omega. 2017;2:4693–704. doi: 10.1021/acsomega.7b00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schonhoft JD, Monteiro C, Plate L, et al. Peptide probes detect misfolded transthyretin oligomers in plasma of hereditary amyloidosis patients. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aam7621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Blancas-Mejia LM, Ramirez-Alvarado M. Systemic amyloidoses. Annu Rev Biochem. 2013;82:745–74. doi: 10.1146/annurev-biochem-072611-130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bellotti V, Mangione P, Merlini G. Review: immunoglobulin light chain amyloidosis–the archetype of structural and pathogenic variability. J Struct Biol. 2000;130:280–9. doi: 10.1006/jsbi.2000.4248. [DOI] [PubMed] [Google Scholar]
- 99.Schmidt A, Annamalai K, Schmidt M, Grigorieff N, Fandrich M. Cryo-EM reveals the steric zipper structure of a light chain-derived amyloid fibril. P Natl Acad Sci USA. 2016;113:6200–5. doi: 10.1073/pnas.1522282113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lundmark K, Westermark GT, Nystrom S, Murphy CL, Solomon A, Westermark P. Transmissibility of systemic amyloidosis by a prion-like mechanism. P Natl Acad Sci USA. 2002;99:6979–84. doi: 10.1073/pnas.092205999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kisilevsky R, Gruys E, Shirahama T. Does Amyloid Enhancing Factor (Aef) Exist - Is Aef a Single Biological Entity. Amyloid-International Journal of Experimental and Clinical Investigation. 1995;2:128–33. [Google Scholar]
- 102.Kisilevsky R, Lemieux L, Boudreau L, Dun-Sheng Y, Fraser P. New clothes for amyloid enhancing factor (AEF): silk as AEF. Amyloid-International Journal of Experimental and Clinical Investigation. 1999;6:98–106. doi: 10.3109/13506129909007309. [DOI] [PubMed] [Google Scholar]
- 103.Nystrom S, Vahdat Shariat Panahi A, Nilsson KPR, Westermark P, Westermark GT, Hammarstrom P, Lundmark K. Seed-dependent templating of murine AA amyloidosis. Amyloid. 2017;24:140–1. doi: 10.1080/13506129.2017.1290599. [DOI] [PubMed] [Google Scholar]
- 104.Westermark GT, Fandrich M, Westermark P. AA Amyloidosis: Pathogenesis and Targeted Therapy. Annu Rev Pathol-Mech. 2015;10:321–44. doi: 10.1146/annurev-pathol-020712-163913. [DOI] [PubMed] [Google Scholar]
- 105.Schuetz AK, Hornemann S, Walti MA, et al. Binding of polythiophenes to amyloids: structural mapping of the pharmacophore. ACS Chem Neurosci. 2017 doi: 10.1021/acschemneuro.7b00397. [DOI] [PubMed] [Google Scholar]
- 106.Luckgei N, Schutz AK, Bousset L, et al. The conformation of the prion domain of Sup35p in isolation and in the full-length protein. Angew Chem Int Ed Engl. 2013;52:12741–4. doi: 10.1002/anie.201304699. [DOI] [PubMed] [Google Scholar]
- 107.Luckgei N, Schutz AK, Habenstein B, et al. Solid-state NMR sequential assignments of the amyloid core of Sup35pNM. Biomol Nmr Assign. 2014;8:365–70. doi: 10.1007/s12104-013-9518-y. [DOI] [PubMed] [Google Scholar]
- 108.Schutz AK, Habenstein B, Luckgei N, et al. Solid-state NMR sequential assignments of the amyloid core of full-length Sup35p. Biomol Nmr Assign. 2014;8:349–56. doi: 10.1007/s12104-013-9515-1. [DOI] [PubMed] [Google Scholar]
- 109.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–6. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 110.Lockhart A, Ye L, Judd DB, et al. Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer’s disease PET imaging agents on beta-amyloid peptide fibrils. Journal of Biological Chemistry. 2005;280:7677–84. doi: 10.1074/jbc.M412056200. [DOI] [PubMed] [Google Scholar]
- 111.LeVine H. Quantification of beta-sheet amyloid fibril structures with thioflavin T. Method Enzymol. 1999;309:274–84. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 112.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 113.Eckroat TJ, Mayhoub AS, Garneau-Tsodikova S. Amyloid-beta probes: Review of structure-activity and brain-kinetics relationships. Beilstein J Org Chem. 2013;9:1012–44. doi: 10.3762/bjoc.9.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lockhart A, Ye L, Judd DB, et al. Evidence for the presence of three distinct binding sites for the thioflavin T class of Alzheimer’s disease PET imaging agents on beta-amyloid peptide fibrils. J Biol Chem. 2005;280:7677–84. doi: 10.1074/jbc.M412056200. [DOI] [PubMed] [Google Scholar]
- 115.Ye L, Morgenstern JL, Gee AD, Hong G, Brown J, Lockhart A. Delineation of PET imaging agent binding sites on beta -amyloid peptide fibrils. J Biol Chem. 2005;280:23599–604. doi: 10.1074/jbc.M501285200. [DOI] [PubMed] [Google Scholar]
- 116.LeVine H., III Multiple ligand binding sites on A beta(1–40) fibrils. Amyloid. 2005;12:5–14. doi: 10.1080/13506120500032295. [DOI] [PubMed] [Google Scholar]
- 117.Matveev SV, Kwiatkowski S, Sviripa VM, Fazio RC, Watt DS, LeVine H., 3rd Tritium-labeled (E,E)-2,5-bis(4′-hydroxy-3′-carboxystyryl)benzene as a probe for beta-amyloid fibrils. Bioorg Med Chem Lett. 2014;24:5534–6. doi: 10.1016/j.bmcl.2014.09.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Klunk WE, Lopresti BJ, Ikonomovic MD, et al. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer’s disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rosen RF, Walker LC, LeVine H., 3rd PIB binding in aged primate brain: Enrichment of high-affinity sites in humans with Alzheimer’s disease. Neurobiol Aging. 2011;32:223–34. doi: 10.1016/j.neurobiolaging.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fast R, Rodell A, Gjedde A, et al. PiB Fails to Map Amyloid Deposits in Cerebral Cortex of Aged Dogs with Canine Cognitive Dysfunction. Front Aging Neurosci. 2013;5:99. doi: 10.3389/fnagi.2013.00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Svedberg MM, Hall H, Hellstrom-Lindahl E, Estrada S, Guan Z, Nordberg A, Langstrom B. [(11)C]PIB-amyloid binding and levels of Abeta40 and Abeta42 in postmortem brain tissue from Alzheimer patients. Neurochem Int. 2009;54:347–57. doi: 10.1016/j.neuint.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 122.Rosen RF, Ciliax BJ, Wingo TS, et al. Deficient high-affinity binding of Pittsburgh compound B in a case of Alzheimer’s disease. Acta Neuropathol. 2010;119:221–33. doi: 10.1007/s00401-009-0583-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ikonomovic MD, Abrahamson EE, Price JC, et al. Early AD pathology in a [C-11]PiB-negative case: a PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol. 2012;123:433–47. doi: 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Scholl M, Wall A, Thordardottir S, et al. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012;79:229–36. doi: 10.1212/WNL.0b013e31825fdf18. [DOI] [PubMed] [Google Scholar]
- 125.Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shirani H, Appelqvist H, Back M, Klingstedt T, Cairns NJ, Nilsson KPR. Synthesis of Thiophene-Based Optical Ligands That Selectively Detect Tau Pathology in Alzheimer’s Disease. Chem Eur J. 2017;23:17127–35. doi: 10.1002/chem.201703846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Back M, Appelqvist H, LeVine H, 3rd, Nilsson KP. Anionic Oligothiophenes Compete for Binding of X-34 but not PIB to Recombinant Abeta Amyloid Fibrils and Alzheimer’s Disease Brain-Derived Abeta. Chemistry. 2016;22:18335–8. doi: 10.1002/chem.201604583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Neal KL, Shakerdge NB, Hou SS, et al. Development and screening of contrast agents for in vivo imaging of Parkinson’s disease. Mol Imaging Biol. 2013;15:585–95. doi: 10.1007/s11307-013-0634-y. [DOI] [PubMed] [Google Scholar]
- 129.Mathis CA, Lopresti BJ, Ikonomovic MD, Klunk WE. Small-molecule PET Tracers for Imaging Proteinopathies. Semin Nucl Med. 2017;47:553–75. doi: 10.1053/j.semnuclmed.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bagchi DP, Yu L, Perlmutter JS, Xu J, Mach RH, Tu Z, Kotzbauer PT. Binding of the radioligand SIL23 to alpha-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PLoS One. 2013;8:e55031. doi: 10.1371/journal.pone.0055031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Goedert M, Masuda-Suzukake M, Falcon B. Like prions: the propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain. 2017;140:266–78. doi: 10.1093/brain/aww230. [DOI] [PubMed] [Google Scholar]
- 132.LeVine H, III, Nilsson KPR, Hammarström P. Chapter 7 Reporters of Amyloid Structural Polymorphism. In: Uversky VN, Lyubchenko YL, editors. Bio-nanoimaging Protein Misfolding and Aggregation. Waltham, MA: Elsevier/Academic Press; 2014. pp. 69–79. [Google Scholar]
- 133.Sigurdson CJ, Nilsson KP, Hornemann S, et al. Prion strain discrimination using luminescent conjugated polymers. Nature methods. 2007;4:1023–30. doi: 10.1038/nmeth1131. [DOI] [PubMed] [Google Scholar]
- 134.Cohen ML, Kim C, Haldiman T, et al. Rapidly progressive Alzheimer’s disease features distinct structures of amyloid-beta. Brain. 2015;138:1009–22. doi: 10.1093/brain/awv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fiandaca MS, Kapogiannis D, Mapstone M, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015;11:600–7. doi: 10.1016/j.jalz.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Saman S, Kim W, Raya M, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid (CSF) in early Alzheimer’s Disease. J Biol Chem. 2012;287:3842–9. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Coleman BM, Hill AF. Extracellular vesicles - Their role in the packaging and spread of misfolded proteins associated with neurodegenerative diseases. Seminars in cell & developmental biology. 2015 doi: 10.1016/j.semcdb.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 138.Choi DW, Armitage R, Brady LS, et al. Medicines for the mind: policy-based “pull” incentives for creating breakthrough CNS drugs. Neuron. 2014;84:554–63. doi: 10.1016/j.neuron.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 139.Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. P Natl Acad Sci USA. 2012;109:9629–34. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ankarcrona M, Winblad B, Monteiro C, et al. Current and future treatment of amyloid diseases. J Intern Med. 2016;280:177–202. doi: 10.1111/joim.12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET, Kelly JW. Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov. 2015;14:759–80. doi: 10.1038/nrd4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Habchi J, Chia S, Limbocker R, et al. Systematic development of small molecules to inhibit specific microscopic steps of Abeta42 aggregation in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2017;114:E200–E8. doi: 10.1073/pnas.1615613114. [DOI] [PMC free article] [PubMed] [Google Scholar]