

Abstract
Macromolecular interactions occur with widely varying affinities. Strong interactions form well defined interfaces but weak interactions are more dynamic and variable. Weak interactions can collectively lead to large structures such as microvilli via cooperativity and are often the precursors of much stronger interactions, e.g. the initial actin-myosin interaction during muscle contraction. Electron tomography combined with subvolume alignment and classification is an ideal method for the study of weak interactions because a 3-D image is obtained for the individual interactions, which subsequently are characterized collectively. Here we describe a method to characterize heterogeneous F-actin-aldolase interactions in 2-D rafts using electron tomography. By forming separate averages of the two constituents and fitting an atomic structure to each average, together with the alignment information which relate the raw motif to the average, an atomic model of each crosslink is determined and a frequency map of contact residues computed. The approach should be applicable to any large structure composed of constituents that interact weakly and heterogeneously.
Keywords: electron microscopy, electron tomography, image processing
INTRODUCTION
Compared with NMR, X-ray crystallography and single particle image reconstruction, cryoelectron tomography (cryoET) produces 3-D images of generally lower resolution but does so within a larger context. All the image data is collected over the same area while fractionating the allowable dose, which depends on the desired resolution, over all the images. Tilt angle limitations in the microscope lead to some views of the specimen being inaccessible, the so-called missing wedge. Consequently, the limited number of images, the electron dose limit and the missing wedge effect combine to limit the tomogram resolution (Lucic et al., 2005).
However, cryoET has compensating advantages. Compared with other methods of structure determination, cryoET does not require specimen homogeneity or even spatial order, can image fields of large multimolecular complexes or even thin margins of whole cells. This makes cryoET uniquely capable of revealing interactions between molecules in complex structures in situ and for studying dynamic structural changes trapped by rapid freezing (Beck et al., 2004; Deng et al., 2008).
Although uniquely adapted for in situ imaging, cryoET suffers from high stochastic noise imposed by the specimen’s radiation sensitivity and the low contrast of unstained, frozen-hydrated specimens. Sub-volume alignment and averaging are both necessary and capable of producing averages to represent the raw motifs of the tomogram and in many cases can eliminate the missing wedge effect (Taylor et al., 2006). For specimens with positional or conformational heterogeneity, such as rapidly frozen, actively contracting muscle (Wu et al., 2010; Wu et al., 2012), classification strategies can identify similar motifs scattered almost randomly throughout a structure while preserving the conformational heterogeneity that provides mechanistic details.
For complex structures with heterogeneous arrangements of constituents, an effective approach is to break the structure down into individual components that themselves can be considered homogeneous. For example, rafts of actin filaments (F-actin) crosslinked with the protein villin were solved by aligning F-actin segments, which are the motifs within the raft with the largest, most homogeneous structure, followed by classification using a mask that eliminated everything except the villin crosslink (Hampton et al., 2008). A separate average for villin was then computed into which an atomic model was built. Interactions were determined by juxtaposing the two atomic models. This approach, alignment on the largest homogeneous structure followed by classification on the heterogeneous feature, was further developed to decipher the much more heterogeneous interactions between myosin heads and thin filaments in active muscle (Wu et al., 2009).
Actin is one of the most conserved proteins in eukaryotic cells due to its interactions with many different proteins (Perrin and Ervasti, 2010). F-actin forms numerous subcellular structures. Among the best known are the I-bands of the striated muscle sarcomere (Sjoblom et al., 2008), microvilli (Sauvanet et al., 2015), stereocilia (Tilney et al., 1992), and filopodia of motile cells (Khurana and George, 2011). In each of these cases, one or more crosslinkers form bundles of either parallel or antiparallel actin filaments. In the sarcomere, α-actinin is the best-known crosslinker (Sjoblom et al., 2008). Because of α-actinin’s length, i.e. ~38 nm, the cross-linked filaments can be either close together, as they appear to be in the Z-disk of honey-bee flight muscle (Rusu et al., 2017), or much further apart as in α-actinin-F-actin rafts (Hampton et al., 2007). Fimbrin/plastin, espin, villin and fascin crosslink bundles of F-actin with much closer interfilament separation in microvilli, stereocilia and filopodia (Bartles, 2000; Khurana and George, 2011).
Actin crosslinkers that in cells form 3-D bundles also form 2-D bundles (rafts) in vitro in which case, the crosslinking interaction can be viewed in detail by electron microscopy (Hampton et al., 2007; Hampton et al., 2008; Hanein et al., 1998). Such bundles can be heterogeneous in F-actin orientation (parallel, antiparallel or mixed) or in crosslinker orientation relative to F-actin (Hampton et al., 2007; Hanein et al., 1998). In some instances, 2-D actin rafts can be quite well ordered over comparatively small domains but show long-range heterogeneity (Taylor and Taylor, 1999). In such cases, conventional methods of averaging over the homogeneous area can produce a single average structure for these domains suitable for interpretation with molecular modeling (Hampton et al., 2008; Volkmann et al., 2001). In other cases, greater heterogeneity prevents obtaining a representative single structure for detailed interpretation.
Some proteins with no obvious physiological crosslinking function, such as several glycolytic enzymes (Clarke and Morton, 1976) will also form actin bundles and 2-D rafts (Taylor and Taylor, 1994), presumably reflecting a tendency to concentrate them in the myofibrils (Maughan et al., 2005). Aldolase, which is best known for its role in glycolysis, is one such protein. Aldolase has more recently been studied for its role in the cytoskeleton of Apicomplexan parasites, which are known causative agents of several diseases, of which the best known is malaria (Sibley, 2010). Aldolase forms an essential link between surface adhesion (micronemal) proteins and F-actin (Soldati and Meissner, 2004); disruption of this link severely impairs host cell invasion (Starnes et al., 2009).
Using F-actin-aldolase rafts as the model system the current study addresses the problem of identifying intermolecular interfaces in paracrystalline arrays having good filament alignment but heterogeneous crosslinks. We treat crosslink heterogeneity by forming independent averages of the F-actin and aldolase densities and fitting atomic models to the separate averages. Using inverse transformations, an atomic model is produced for each aldolase crosslink motif and a statistical description of the interacting regions obtained using contact analysis. The procedure can be used to analyze the complexity of weak interactions that occur in any system within which separate averages of the constituents can be obtained with sufficient resolution to fit their atomic structures.
MATERIALS AND METHODS
Actin-aldolase raft preparation
Actin-aldolase rafts were produced on a positively charged lipid monolayer as previously described (Taylor and Taylor, 1994). Briefly, actin was prepared from rabbit muscle acetone powder and purified by size exclusion chromatography (Pardee and Spudich, 1982). Rabbit muscle aldolase was obtained commercially (Sigma Chemical Co., St. Louis, MO), dialyzed and diluted into buffer A consisting of 2 mM Tris-Cl, 0.2mM Na2ATP, 0.02%, β-mercaptoethanol, 0.2 mM CaCl2, 0.01% NaN3, pH 8.0 (at 25°C). Positively charged lipid monolayers were prepared on Telflon blocks using a mixture of dilauryl phosphatidylcholine (Avanti, Alabaster, AL) and didodecyldimethyl ammonium bromide (Kodak) (Taylor and Taylor, 1992). The F-actin-aldolase arrays were obtained by polymerizing G-actin in the presence of aldolase under the monolayer (Hampton et al., 2007; Hampton et al., 2008; Taylor and Taylor, 1994). Actin rafts were recovered from the monolayer surface using hydrophobic reticulated carbon grids (Kubalek et al., 1991; Taylor et al., 2007a). The carbon grids with actin-aldolase rafts were negative stained with 2% uranyl acetate solution and stabilized with freshly evaporated carbon prior to tilt series data collection.
Electron microscopy
The specimens were examined under the low-dose conditions using an FEI CM300-FEG electron microscope equipped with a TemCam F224 2K X 2K CCD camera (TVIPS, Gauting, Germany). The microscope was operated at 300 kV and a magnification of 33,000x. Images were binned to yield an effective pixel size of 0.668 nm. Three single axis tilt series were collected using the EMmenu software following a 3.0° Saxton scheme (Saxton et al., 1984). The tilt angles ranged from −70° to +70°. Defocus was set to 5–7 μm.
Tilt series alignment and CTF correction
The tilt series were aligned with a marker-free method using the PROTOMO package (Winkler and Taylor, 2006). PROTOMO uses Radermacher’s weighted back-projection method (Radermacher, 2006).
The contrast transfer function (CTF) was determined for each tilt series image by averaging the power spectra from patches 256 × 256 pixels in size with a 50 pixel overlap along x and y axis (Huang et al., 2003). The average defocus value of the projection image was determined using the SPIDER command tf ed applied to the corresponding averaged power spectrum (Frank et al., 1996). Finally, a phase-flip CTF correction was calculated with the SPIDER command tf ct using the 0.8 cut off estimation. The CTF was measurable in individual images to 1.5 nm resolution.
Subvolume processing
All subvolume manipulations were done using the PROTOMO and I3 software packages (Winkler and Taylor, 2006; Winkler et al., 2009). I3 uses the “alignment by classification” philosophy (Dube et al., 1993) in which the class averages constitute the multiple references and the best alignment transformation for a class average is applied to its class members. The class averages have higher signal-to-noise ratio than the raw subvolumes, and depending on the specimen, will have less missing wedge. I3 also uses constrained correlation in which the missing wedge effect is explicitly taken into account in computing the cross correlation function and computes class averages by weighted averaging of Fourier coefficients. Motifs were picked manually using the i3display program (Winkler et al., 2009). The center of each raw motif was defined as the actin filament crossover position. A total of 3,708 raw F-actin motifs were selected.
A global average was generated from the 3,708 raw F-actin motifs using the preliminary alignment parameters, which oriented the filaments along the z-axis and with the same polarity. This global average was used as a reference for one cycle of translational alignment (no angular alignment in this step) to improve the centering of the raw motifs. In the next alignment step, a cylindrical, Boolean mask of length of 56 pixels (37.4 nm) and diameter of 12 pixels (8.0 nm) was applied to remove everything but the actin filament and the masked motifs subjected to multivariate data analysis (MDA) followed by hierarchical ascendant classification (van Heel, 1989). A multireference angular alignment was done to improve the angular alignment of the filament axis, which we refer to as polar alignment, using all of the F-actin class averages as the multiple references (Winkler et al., 2009). Six cycles of alignment by classification were run to improve the angular alignment, at which point the actin alignment ceased to improve. During the subvolume processing, no raw F-actin subvolumes are intentionally discarded.
After the actin filament alignment, an ellipsoidal cylindrical, Boolean mask ellipsoidal mask with axes a=56 pixels (37.4 nm), b=9 pixels (6 nm), and c=15 pixels (10.0 nm) was positioned in the interfilament space to the right side of the left side F-actin (the one on which alignment was done) and used to locate the aldolase crosslinks using MDA. For this classification, the first ranked eigenvector, numbered 0, which represents the global average, was excluded and only eigenvectors 1–12 used for the classification. After the aldolase was revealed by MDA and classification, the centers of the aldolase densities within the class averages were determined manually using i3display. The relative shift values between the center of the aldolase and the center of the actin in the class average were treated as preliminary translation parameters for the aldolase class averages and the aldolase global average was generated by directly averaging the aldolase class averages. The atomic structure of aldolase, PDB - 2EPH (Bosch et al., 2007), was then fitted into the global average of the aldolase class averages using CHIMERA (Pettersen et al., 2004) to bring the atomic structure into rough alignment with the aldolase global average. A density map of the appropriate dimensions and pixel size was then generated from the roughly aligned atomic structure and used as the reference for further angular and translational alignment of the aldolase class averages. Aldolase raw subvolumes were never aligned to this reference.
Mapping back
The original tomogram is noisy due to the low-dose requirement of ET for biological samples. The relative placement of the constituents can be viewed with greater clarity using the mapback method (Liu et al., 2006; Taylor et al., 2007b) in which the density of the raw motif or element of the raw motif is replaced with a higher signal-to-noise ratio representation, either class average or density map computed from an atomic structure.
Atomic model building
Fittings of atomic structures to global and class averages were done using CHIMERA (Pettersen et al., 2004). Density maps obtained from atomic models were calculated using the pdb2mrc utility of EMAN (Ludtke, 2010). We built a 16 subunit F-actin atomic model from PDB - 2ZWH (Oda et al., 2009) by continuation of the 13/6 helical symmetry of the atomic structure. The F-actin atomic model was fitted into the global average of the actin filament using CHIMERA (Pettersen et al., 2004). A density map of the F-actin atomic model was generated and then used as the reference to align the F-actin class averages. We used the atomic structure of aldolase from Plasmodium falciparum, PDB - 2EPH (Bosch et al., 2007), to compute the reference to align the aldolase class averages. Although this atomic structure is a different isoform than the rabbit muscle aldolase used for the specimens, the two structures are very similar. The peptides have a sequence identity of 53%. When the structure (Cα’s only) of an individual subunit from rabbit muscle aldolase is compared to a subunit of P. falciparum aldolase using the rmsd utility of CHIMERA, the difference is 0.96 Å (Supplemental Movie 1); when the tetramers are compared the RMS deviation is 1.89 Å (Supplemental Movie 2). The identified contact residues might be more readily used in studies of the Plasmodium cytoskeleton.
Contact Region Analysis
We used the VMD “atomselect” command to identify potential residue contacts (Humphrey et al., 1996). For each atomic model of each crosslink motif, the distance between the aldolase residues and actin residues from both the left and right actin filaments were calculated. Each time, one subunit of actin and one subunit of aldolase were chosen as a pair and all possible pairs were examined consecutively. The “atomselect” command lists the residue numbers of all residues that have any atom within the 4.2 Å threshold for the chosen subunit pair. Although this process can score specific atom-atom interactions, the resolution is too low to do so reliably. Instead, we collated only the frequency of contact between one residue on actin and any residue on aldolase and vice versa. Then for each crosslinked motif atomic model, all the contact residues on each F-actin and aldolase subunit were screened out and summed up for F-actin and aldolase respectively to produce the contact frequency histograms, which were graphed using Microsoft Excel.
APPROACH
The general approach used for this study is similar to previous methods for analyzing paracrystalline assemblies but modified for the goal of identifying molecular interactions (Fig. 1). Once the tomograms are computed with CTF corrections, motifs are picked and class averages computed. The actin rafts in each tomogram are continuous across the field of view (Fig. 2) and the actin filaments in each tomogram lie in the same direction enabling initial assignment of the first two Euler angles (polar angles) for each motif. This assignment minimized the polar angle alignment search of each F-actin segment. The subvolume processing involves first aligning the F-actin motifs, after which the aldolase cross-links are identified using MDA. The aldolase density in the class averages is separately aligned to produce a global average of the aldolase.
Figure 1.

Flow chart for subvolume processing.
Figure 2.

Tomogram of an F-actin-aldolase raft. A section from the raw tomogram taken through the middle of the raft. The resolution clearly resolves the actin subunits and the crosslinks. In some motifs, F-actin pairs are linked by one aldolase (white arrows) or by two aldolase molecules (white arrowheads). Note that some stretches of F-actin display no cross-links (white asterisk). When no cross-links form the filaments appear closer together than when crosslinks are present. This same region is shown in Figure 6 with the white box marking the area shown in Figure 6b–d. The direction of view is such that the monolayer lies below the F-actin raft.
Aldolase identification and alignment
Since the raw motifs were well aligned on the left side F-actin, cross-links could be identified by MDA (Fig. 1, right side pathway). A total of 19 classes revealed aldolase density. After aldolase was revealed by the MDA analysis, the relative positions of the aldolase center and actin filament center were manually determined, the aldolase density was boxed out from each class average and a global average generated for the aldolase. The aldolase atomic structure, PDB-2EPH (Bosch et al., 2007), was then fitted into the aldolase global average and a density map calculated from the fitted aldolase atomic coordinates. This map is used as a reference to align the aldolase class averages. The transformation matrix from the F-actin alignment results combined with the transformation derived from the alignment of aldolase class averages relative to the aldolase atomic structure and also the relative position of aldolase center to actin filament center are all that is needed to bring the aldolase raw subvolumes to the position and orientation of the atomic structure. In effect each raw aldolase subvolume is aligned to the map calculated from the fitted aldolase atomic structure, but indirectly through the class averages, not through alignment of the raw motifs. The inverse of this alignment transformation can position the aldolase atomic structure into the context of the raw tomogram.
Atomic model building of crosslink motif
The goal of this process is determination of interacting surfaces in a heterogeneous mixture, which requires that an atomic model of each interaction be obtained (Fig. 3). The class average alignment process aligns each class average of F-actin to an atomic model of F-actin through alignment to a density map computed from the atomic structure. The matrix to bring the class member (raw motifs) to the class average is already known from the motif alignment described above. Combining these two matrices generates the transformation matrix to bring each raw F-actin motif to the position and orientation of the reference generated from the F-actin atomic model. Application of the inverse of this alignment matrix to the F-actin model generates an atomic model for each actin filament segment within the tomogram itself.
Figure 3.

Contact analysis flow chart.
The aldolase class average alignment parameters relate the aldolase density from the class averages to the position and orientation of the density generated from the atomic structure and in turn to the atomic structure itself. Likewise, the matrix obtained by the combination of the motif alignment transformation matrix with the transformation matrix for each class average relative to the aldolase atomic model in turn relates each class member to the position and orientation of the aldolase atomic structure. Application of the inverse alignment matrix of each class member to the aldolase atomic structure generates the atomic model for the aldolase in the raw tomogram. In this way, an atomic model is generated for every F-actin-aldolase crosslink.
Identification of the second crosslinked F-actin segment
Each F-actin-aldolase motif contains a pair of F-actin segments and one or more intervening aldolase crosslinks. F-actin is the largest component of the motif and is used as the basis of all alignments. Thus, the alignment process has brought every raw F-actin segment and aldolase crosslink into alignment relative to an atomic model of a 13/6 F-actin positioned on the left side of the motif. The right side F-actin is poorly represented in the motif class averages and thus must be determined by an independent method. Although the left side F-actin segment is known from the alignment process, the right side F-actin in each motif is not directly known but can be obtained from knowledge of where each F-actin segment and aldolase molecule are positioned within the raw tomogram.
After alignment of the aldolase class averages to the aldolase atomic model, the aldolase centroid is back transformed to the position within the raw tomogram of each aldolase class member. In like manner the center of each F-actin segment of each motif as aligned to the F-actin atomic model is transformed to its position within the raw tomogram. It is then a simple matter to compare the centers of each F-actin segment to each aldolase center to identify the two F-actin segments closest to it. One of these, the left side one, is already known; the right side one is new information. Once the second crosslinked F-actin segment is determined, the atomic model for each actin-aldolase-actin crosslink motif is obtained.
RESULTS
General appearance of F-actin-aldolase rafts
Aldolase and F-actin form rafts covering extensive areas (Fig. 2). The area reconstructed in the tomograms, 1.87 μm2, understates their size; rafts often cover multiple holes in the reticulated carbon support film as a coherent unit sometimes 5–6 μm on edge. The rafts are essentially flat, displaying a variation in Z-height of only a few pixels, much less than the diameter of an actin filament. Compared with actin-villin rafts (Hampton et al., 2008), which also grow to considerable size, actin-aldolase rafts have a more variable appearance across the area of the raft. F-actin and villin form crosslinks in the ratio of one villin per actin crossover period. F-actin and aldolase have either one or two crosslinks per actin crossover. A pattern is visible across the raft in which crosslinks form stripes oriented oblique to the filament axis. The aldolase crosslinks occur either singly or in pairs every crossover repeat.
Well-ordered F-actin-aldolase rafts form occasionally (Sukow and DeRosier, 2003; Taylor et al., 1997), but these are rare and may be atypical thereby providing a biased impression of the specificity of the actin-aldolase interaction. Published images of the rafts generally indicate monopolar rather than bipolar F-actin orientation with multiple actin filaments per unit cell. The F-actin order is often excellent showing actin subunits clearly in averages. However, the aldolase crosslinks are often irregularly arranged producing a blurred density in spatial averages. Thus, good F-actin order does not indicate good crosslink order.
Filament polarity determination
F-actin-aldolase rafts in this study are less ordered than the published examples (Sukow and DeRosier, 2003; Taylor et al., 1997) necessitating that we determine the relative orientation of the filaments in the tomograms. The inter-filament spacing is ~12 nm as determined from the raw tomogram (Fig. 4a). The diffraction spots in the Fourier transform of bipolar rafts with alternating F-actin orientations would fall on an orthogonal reciprocal lattice with strong diffraction on the off-equatorial layer lines at a spacing corresponding to twice the inter-filament distance while the spacing along the equator would correspond to the spacing between individual filaments (Sukow and DeRosier, 2003). The Fourier transforms of small, well-ordered patches from the tomograms have an oblique lattice with the spacing between points in the off equatorial layer lines the same as that in the equator (Fig. 4b). This analysis also performed on the 0° image of each tilt series gave the same result indicating that the rafts are uni-polar.
Figure 4.
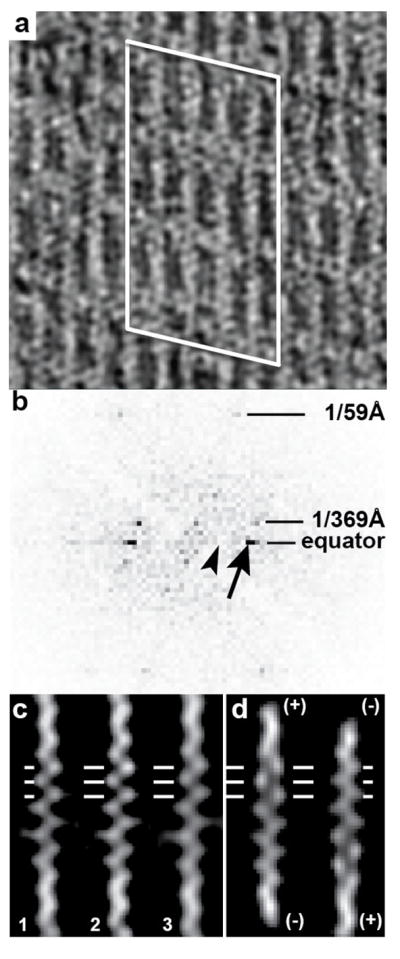
Polarity of F-actin-aldolase rafts. (a) Projection image of a region of the tomogram. Actin subunits are well resolved as are the crosslinks. The white lines outline the region from which the Fourier transform in (b) has been calculated. (b) Computed transform of the region shown in (a). The arrow points to the strong equatorial spot that arises from the interfilament spacing, 1/(12 nm). The arrowhead points to the expected location of a row line that would be present if the filaments were arranged antiparallel, 1/(24 nm). The lack of any off-equatorial diffraction in this location indicates that the filaments are parallel, with a small axial offset giving the lattice its oblique character. (c) Single filament averages of three adjacent filaments, labeled 1, 2 and 3. Horizontal lines denote the positions of actin subunits. At this location in this filament orientation, each actin subunit appears as a distinct round density. (d) Projections of the model F-actin aligned to the filaments in (c) in two different orientations. Note that the left-hand orientation in (d) is a much better fit than the right-hand orientation.
To further validate the uni-polar property of the actin-aldolase rafts, separate 2-D averages of individual actin filaments were computed from the 0° tilt projection using I3 (Winkler et al., 2009) and used to determine polarity by cross correlating against a reference generated from an atomic model of F-actin with 13/6 symmetry. Both parallel and antiparallel orientations were tested. In this way, the projections of the individual filament averages could be compared across the entire tomogram. Adjacent filaments in each tomogram were found to have the same polarity. The 2-D F-actin averages from the consecutive filaments are visually similar to only one orientation of the reference image computed from an F-actin atomic model (Fig. 4c, d) confirming the conclusion from the diffraction analysis: the actin-aldolase rafts are uni-polar.
Characterizing the heterogeneous crosslinks
Our subvolume alignment strategy was based on alignment of the left-side F-actin rather than crosslinked motifs (F-actin-aldolase-F-actin). Thus, the left-side F-actin was well aligned, but without regard to the position of aldolase. Details of the aldolase binding on actin are revealed through classification conducted using a mask ~30.0 nm in length placed to the right of the left-side F-actin centered at the crossover. Crosslinking heterogeneity is revealed more clearly in the class averages (Fig. 5). Crosslinks form on as many as five actin subunits along the F-actin long pitch helix. Crosslinks occur in pairs (Fig. 5a, b) and singly (Fig. 5c, d). The right-side F-actin segment of the motif generally shows poor actin subunit definition compared to the left-side F-actin. Thus, the axial offsets between crosslinked F-actins are variable confirming that alignment of complete motifs would generally fail to produce good averages.
Figure 5.

Class averages of F-actin-aldolase motifs generally arranged in order of the Y-position of the lower crosslink. (a) The central slices of those class averages with an apparent pair of aldolase crosslinks. In some class average, e.g. 5–7, both left and right actin filaments were well resolved. For other classes, the left actin filament subunits are clearly resolved but the density of the right side actin filaments are blurred and the subunits poorly resolved. (b) Surface view of the same class average as shown in (a). When both aldolase molecules function as cross links, both left and right actin filaments are clearly resolved. For the other classes, only one of the pair of aldolase molecules appears to function as a cross-link with the other molecules binding only one of the actin filaments. Consequently, the detail in the right actin filament is poor and subunits are not well resolved. (c) Ten class averages of single aldolase crosslinks. In this case, all the right-side F-actins are poorly ordered. The 19th class average (not shown) is very similar to the fourth from the left in rows (c) and (d). Note the progression up the filament of the cross-links even with the left side F-actin positioned identically in each image. (d) Surface view of the same class averages shown in (c).
Although the right side F-actin is poorly aligned relative to the left side within the motifs, all of the F-actin segments are aligned as a separate feature; it is only when the region being averaged shows both filaments that the disparity in the left-side-right side alignment becomes noticeable. The disparity in alignment is also more noticeable in the case of single cross-links (Fig. 5c, d). Thus, when paired cross-links are found and the right side F-actin shows poor alignment, a reasonable conclusion is that only one cross-link is present; the other aldolase molecule is bound to only one filament. Sometimes the lack of a second cross-link is apparent in isosurface views, but in other cases it is not (Fig. 5b). We cannot exclude the possibility that the long flexible C terminus might form a weak contact with the second actin. However, such a flexible link would not strongly influence the relative alignment between the cross-linked filaments.
Aldolase is well depicted in its class averages, but there are too many for useful interpretation. The aldolase class averages needed to be reduced to a single global average into which the aldolase crystal structure can be placed. To achieve this we computed a density map from an aldolase crystal structure, PDB - 2EPH, (Bosch et al., 2007) as a reference for alignment of the aldolase class averages. The aldolase class averages are contained within a cubic box 21 pixels on edge. The relative shifts observed when aligning the aldolase class averages to the density generated from the aldolase atomic model were within 3 pixels, and the cross correlation coefficient varied with the orientation/angle or separation from 0.71 to 0.79. After alignment, the transformation needed to bring each of the class members from the raw tomogram to the position and orientation of the aldolase map was determined. The inverse of this alignment transformation applied to both the aldolase map and atomic coordinates places them into the coordinate frame of the raw motifs at the location of every crosslink.
Reassembly of raw motifs for interpretation
The raw motifs have high stochastic noise. Consequently, atomic models built to fit them could be inaccurate. Our alignment procedures produce good alignment for F-actin and aldolase separately but to visualize the interaction between actin and aldolase in detail within the context of the rafts requires reassembly of the raw motifs from the F-actin and aldolase global averages or model densities. By virtue of the alignment of class averages to the density maps computed from atomic structures, every actin and aldolase subunit in the tomogram can be replaced by its corresponding atomic structure or a density map produced from it. This procedure can be done at the level of class averages or at the level of the raw motif and even for the raw tomogram itself. When done on the whole tomogram, the process is called “mapping back”. When done at the level of the raw motifs as extracted from the tomogram, the process is referred to as reassembly (Taylor et al., 2006). Comparison of Figure 2 with Figure 6a and the separate panels in Figures 6b–d shows nearly complete recovery of crosslinks by this procedure.
Figure 6.

Map backs. (a) Map back using actin and aldolase model densities of a complete tomogram. This is the same region of the tomogram shown in Figure 2 with the same points of interest identified. Arrows identify single cross-links, arrow heads identify double cross-links and the “*” identifies regions of a filament with no cross-linker. The square marks the region shown at higher magnification in panels (b–d). (b) A section taken from the right side of the raw tomogram shown in Figure 2. (c) Same area as (b) but a section from the map back using model densities. (d) The same as (c) but shown in surface view. Panels (b–d) all show the same area boxed in Figure 2.
The procedures for mapping back model density maps can also be applied to the atomic models of the constituents. An atomic model for the entire tomogram could be built, but this is impractical for deciphering intermolecular interactions. Consequently, intermolecular interactions were determined by reassembly of the individual raw crosslink motifs from atomic models of F-actin and aldolase. There are a total of 3,708 raw crosslink motifs in the dataset, which is far too many to examine individually. Moreover, up to five different actin subunits on each long-pitch helical strand interact with aldolase and up to three of the four aldolase subunits interact simultaneously with actin subunits. Identifying the contacting interfaces cannot be done individually but must be done statistically.
Analysis of the contact interfaces
Unlike the analysis of well-defined interactions, which can be done from one or a handful of atomic models, this many intermolecular interactions must be analyzed statistically for frequency. We used the VMD command “atomselect” (Humphrey et al., 1996) to analyze the residue contacts between every aldolase subunit and the actin subunit it contacted. The counts for each of the contact residues were then summed together for both actin and aldolase respectively. The plot of the counts versus the corresponding residue number reveals the surface residues responsible for the actin/aldolase crosslinking (Fig. 7a, b).
Figure 7.

Contact analysis for F-actin-aldolase interaction. (a) The collated analysis for actin shows that there are primarily four well-defined regions on the actin subunit where aldolase makes contact: residues 1–8, 20–30, 90–105, and 350–365. All of these are on subdomain 1. (b) The collated analysis for aldolase, indicates that the contacts are more complex and not as well defined. The identified residues, although widely separated in the sequence, are closely apposed in the 3D structure in two areas: 45–60, 310–330 & 200–210, 240–260. (c) A 3-D image of one motif shows how these disparate contacts can account for the complexity of a cross-link. Here we have use the B-factor representation in CHIMERA to illustrate the contract frequency. Gray (any shade) indicates no or very infrequent contact. For aldolase, the redder (hotter) the color the more frequent the contact; for actin, the lighter the shade of blue, the more frequent the contact.
The statistical analysis shows that the contact residues on the actin subunit are located in two groups on subdomain 1 of actin (Fig. 7a). One contact consists of residues 1–8, 20–30, 90–105 at the N terminus and the other contact residues 350–365 at the C-terminus. These areas are in close proximity on actin subdomain 1 (Fig. 7c). Contact analysis on aldolase identified two areas consisting of residues widely separated in the sequence (Fig. 7b), but closely positioned in the 3D structure (Fig. 7c, Supplemental Movie 3). One contact area is located at residues 45–60, and 310–330, and the other contact area at residues 200–210 and 240–260 (Fig. 4b, c). These two regions are quite distinct on the aldolase subunit.
DISCUSSION
Previous studies of the heterogeneous myosin-thin filament crosslinks built a single atomic model for the thin filament into the global average of the aligned motifs, and manually built atomic models for the actin-attached myosin heads observed in class averages (Wu et al., 2009). Myosin, unlike aldolase, has known points of flexibility in its structure facilitating modifications to fit the density. Aldolase cannot be so modified at this resolution and each crosslink must be examined individually due to the heterogeneity. The present procedure is designed to avoid entirely any manual model building, relying instead on the reassembly of raw motifs with atomic models built into the two constituents, F-actin and aldolase.
cryoET has become a widely used technique for structural studies of large complexes formed in vitro to much larger structures that can be found in sections (plastic or frozen hydrated) of tissues to the thin margins of cells and even whole bacteria. Successful efforts have reached subnanometer resolution in subvolume averages of relatively homogeneous structures (Schur et al., 2013). However, cryoET’s chief value may be in the imaging structures at 2–4 nm resolution with higher heterogeneity within a larger context. In cryoET, the heterogeneity can be examined in its original context within cells.
Analysis similar to that described here has been carried out on F-actin-villin rafts (Hampton et al., 2008). Sub-volume alignment on the left side F-actin and classification on just the villin position revealed not only the villin but also the second actin filament clearly because the axial offset between adjacent F-actins was well defined. Another study of well-ordered rafts of F-actin and fimbrin, observed two types of rafts requiring only a pair of atomic models to describe the interaction (Hanein et al., 1998). Here, the actin-aldolase-actin class averages did not show the second filament clearly requiring a modified approach.
The more complex the structure, the more difficult is the pattern recognition problem. Pattern recognition depends on an appropriate frame of reference. The complex pattern of actin-myosin interactions in contracting muscle is a case in point (Wu et al., 2009; Wu et al., 2010; Wu et al., 2012). The largest object within the repeating motifs that can be considered invariant was the thin filament, which comprises actin, tropomyosin and troponin (Gordon et al., 2000), making it the logical frame of reference. The highly heterogeneous myosin interactions could then be deciphered using a set of masks that looked for patterns along the surface of the thin filament at different locations. Such an approach is fundamentally segmentation (feature identification) by classification. No subsequent alignment of the different segmented features was attempted because they all differed.
One drawback of the above process is a lack of data reduction; at the end of the process there are as many reassembled motifs from atomic structures as there are raw motifs, substantially too many to be examined individually. Wu et al. solved this problem by determining the minimal number of reassembled motifs needed to represent all of the cross-bridge class averages at least once (Wu et al., 2009; Wu et al., 2010; Wu et al., 2012). Atomic models were then built directly into the reassembled motifs and the frequency of each cross-bridge was obtained from the number of class members. Our analysis of F-actin-aldolase rafts extends the previous effort by building indirectly an atomic model for each individual crosslink through alignment of class averages to a map generated from the appropriate atomic model. The reassembled atomic models retained the full range of heterogeneity present in the raw motifs.
Our method utilized alignment of class averages rather than alignment of raw subvolumes. The F-actin motifs are picked manually and aligned by classification; the aldolase crosslinks are not picked directly but are instead revealed by MDA and classification from the aligned F-actin segments. The F-actin and aldolase subunits are similar in size (42 kDa vrs 40 kDa). However, the F-actin subunits are much easier to identify and position because they are part of a larger and much more shapely helical segment of 16 subunits. The aldolase tetramer is 4x the size of an actin subunit, but still 1/4th the size of an F-actin segment. Its globular (rather than filamentous) shape, the close proximity of F-actin and the small size make the alignment prone to error if raw aldolase motifs were aligned. Given a set of well-aligned F-actin motifs, MDA and classification identified the aldolase crosslinks while at the same time grouping them according to their orientation and position on F-actin.
The aldolase class averages revealed variability of the aldolase position relative to the left-side F-actin frame of reference; aldolase subunits were found on up to five actin subunits on one side of F-actin. At the same time heterogeneity in the right-side F-actin position was so great that its average density failed to reveal actin subunits in averages of complete motifs. All the F-actin segments were well aligned in the F-actin global average; however, the crosslinked F-actin pairs that formed the motifs were not well aligned due to heterogeneity of the relative axial shift between the actin filament pairs caused by the heterogeneity of the aldolase crosslink. Nevertheless, an accurate atomic model for every raw crosslinked motif could be obtained by referring the F-actin and aldolase atomic models separately through the class averages to which they contributed. We built an atomic model for each of the individual raw crosslink motifs. Although not assembled into a single pdb file, in essence, an atomic model of the entire tomogram was produced. The quantity of intermolecular interfaces required an automated approach for identifying contacting residues.
Comparison with Previous Results
Our study finds that the contact residues on actin involved in aldolase binding are located in two areas. One area near the N-terminus includes residues 1–8, 20–30 and 90–105 (Fig. 7a); the other area near the C-terminus includes residues 350–365. Binding of aldolase to F-actin has been observed at two different sites, consistent with the two contact areas revealed here (Arnold and Pette, 1968; Arnold et al., 1971). A study comparing the binding of glycolytic enzymes to yeast and rabbit muscle F-actin showed that sequence differences at the actin N-terminus could be responsible for differences in aldolase binding affinity and identified actin residues 24–25 and 99–100 as important for aldolase binding (Gustafson, 1996). A Brownian dynamics simulation (Ouporov et al., 1999) demonstrated that mutating residues 1–4 of muscle actin to alanine either individually or in pairwise combinations reduced the estimated binding free energy consistent with participation of the actin N-terminus in aldolase binding. Their study suggested that residues 1, 2, 4, 25, 83, 99, 125, 359, 363 and 364 participate in aldolase binding (Ouporov et al., 1999). All of these actin residues fall into the two areas revealed by our study except residue 83, which was the least frequently identified residue (Ouporov et al., 1999). Residues 1, 2, and 4 had the highest number of occurrences with residue 363 having the second highest number. Our study also shows that the N-terminus has the highest number of occurrences and the C terminus the second highest (Fig. 7a). The actin N- and C-termini are positioned near each other within subdomain 1 of the actin subunit, which is important for the binding of other proteins. Thus, the contact residues revealed by the current study are consistent with previous reports.
Margi et al. (Magri et al., 1978) suggested that two different sites on the aldolase tetramer associate with G-actin. O’Reilly and Clarke (O’Reilly and Clarke, 1993) investigated the actin binding activity of isolated peptides of aldolase and found that a synthetic peptide corresponding to aldolase residues 32–52 competes with native aldolase for F-actin binding. Residues 40–52 of this peptide overlap with residues 40–60 revealed by the current study. Wang et al. (Wang et al., 1996) used site-directed mutagenesis to show that mutations R42A, K107A, and R148A resulted in minimal actin binding activity. Of these three residues, 42 falls into the segment 40–60 revealed by our study as well as the peptide study (O’Reilly and Clarke, 1993), but residues 107 and 148 do not. The reason for the discrepancy might be due to the type of experiment. In our work, wild type aldolase is used as a crosslinker between two actin filaments in a tight 2-D raft whereas Wang et al. used varying concentrations of aldolase and F-actin forming both tight 3-D bundles and crosslinked gels. Tight bundling/crosslink gels may facilitate some interactions not observed in 2-D rafts, which have different steric constraints.
Ouporov’s Brownian dynamics simulation suggested aldolase residues 67, 86, 139, 283, 293, 316–318, 321, 341 participate in actin binding (Ouporov et al., 1999). Of these, residues 283, 316–318 and 321 fall within the contact region revealed by our study. At the same time, our study revealed residues not identified by the simulation. The discrepancy could be due to the assumption of the predominance of Coulombic electrostatic forces while ignoring hydrophobic effects. The simulation involved one aldolase tetramer and one actin filament and thus did not fully mimic the actin-aldolase raft formation in the current study. In the rafts, aldolase crosslinks actin filament pairs, which may prevent some orientations suggested by the simulation of single actin filament single aldolase case while at the same time, cooperative effects that occur in F-actin 2-D bundles may facilitate others.
Potential Applications
Although the present study was conducted using specimens preserved in negative stain and image data collected on a CCD camera, a change to frozen-hydrated specimens, use of the newer direct electron detector cameras with capabilities for damage compensated motion correction, combined with radiation damage weighting during subvolume averaging could provide a considerable boost in resolution. The rafts have large numbers of motifs in a very thin arrangement requiring few tilt series images for a tomogram of even subnanometer resolution.
Apicomplexan parasites include over five thousand parasitic species that are responsible for severe diseases such as malaria, toxoplasmosis, and cryptosporidiosis (Sibley, 2010; Soldati and Meissner, 2004). Apicomplexans use a unique form of substrate-dependent motion called gliding motility to invade the target host cells (Morrissette and Sibley, 2002; Soldati and Meissner, 2004). Gliding motility requires an intact parasite actin cytoskeleton (Dobrowolski and Sibley, 1996). During gliding motility, adhesive proteins are translocated along the cell surface in a manner similar to moving cargo along a conveyor belt. The P. falciparum TRAP (thrombospondin-related anonymous protein) is an adhesive protein on the sporozoite surface that plays a central role in hepatocyte invasion. Between the actomyosin complex and the complexes of micronemal ligands (like TRAP) and host receptors, aldolase functions as a bridge (Jewett and Sibley, 2003). The interaction between aldolase and the actin filament is poorly understood even though different methods have been applied to study the interaction indirectly. Higher resolution analysis of the contact residues could reveal the nature of the actin/aldolase interaction and inform therapeutic development.
Supplementary Material
Supplemental Movie 1. Alignment and comparison of a single aldolase subunit from rabbit skeletal muscle (green), PDB – 1ADO, with the aldolase from P. falciparum (red), PDB - 2EPH. Comparison done using chain A of both structures.
Supplemental Movie 2. Alignment and comparison of the full aldolase tetramer from rabbit skeletal muscle (green), PDB – 1ADO, with the aldolase from P. falciparum (red), PDB - 2EPH.
Supplemental Movie 3. The movie shows the same atomic model that is shown in Figure 7c. The coloring scheme is identical to that of Figure 7. Note that the N-terminal extension of aldolase frequently contacts actin, as denoted by the yellow coloring, though its contact on actin is colored gray indicating infrequent contacts. The extension is stabilized by crystal contacts in the crystal structure so its structure may not accurately depict potential binding on actin. Nevertheless, the extension could very well stabilize the cross-links, but possibly not as depicted.
Acknowledgments
This work was supported by NIH Grants R01 GM30598 to KAT and U54 GM64346 to the Cell Migration Consortium. Data were collected at the Biological Science Imaging Resource at Florida State University. The Philips CM300-FEG was purchased with funds from NSF grant BIR-9512954, NIH grant S10 RR11357 and the Markey Charitable Trust.
Footnotes
ACCESSION NUMBERS
A representative subvolume class average has been deposited in the Electron Microscopy Data Bank under accession code, EMD-7064. The corresponding atomic model has been deposited in the Protein Data Bank under accession code PDB ID: 6B7X.
AUTHOR CONTRIBUTIONS
GH performed the data analysis, DWT prepared the 2-D rafts and the EM specimens, JL collected the tilt series, GH and KAT wrote the paper, KAT supervised the project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnold H, Pette D. Binding of glycolytic enzymes to structure proteins of the muscle. Eur J Biochem. 1968;6:163–171. doi: 10.1111/j.1432-1033.1968.tb00434.x. [DOI] [PubMed] [Google Scholar]
- Arnold H, Henning R, Pette D. Quantitative comparison of the binding of various glycolytic enzymes to F-actin and the interaction of aldolase with G-actin. Eur J Biochem. 1971;22:121–126. doi: 10.1111/j.1432-1033.1971.tb01522.x. [DOI] [PubMed] [Google Scholar]
- Bartles JR. Parallel actin bundles and their multiple actin-bundling proteins. Curr Opin Cell Biol. 2000;12:72–78. doi: 10.1016/s0955-0674(99)00059-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Forster F, Ecke M, Plitzko JM, Melchior F, Gerisch G, Baumeister W, Medalia O. Nuclear pore complex structure and dynamics revealed by cryoelectron tomography. Science. 2004;306:1387–1390. doi: 10.1126/science.1104808. [DOI] [PubMed] [Google Scholar]
- Bosch J, Buscaglia CA, Krumm B, Ingason BP, Lucas R, Roach C, Cardozo T, Nussenzweig V, Hol WG. Aldolase provides an unusual binding site for thrombospondin-related anonymous protein in the invasion machinery of the malaria parasite. Proc Natl Acad Sci U S A. 2007;104:7015–7020. doi: 10.1073/pnas.0605301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke FM, Morton DJ. Aldolase binding to actin-containing filaments. Formation of paracrystals. Biochem J. 1976;159:797–798. doi: 10.1042/bj1590797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng BB, O’Connor CM, Kedes DH, Zhou ZH. Cryo-electron tomography of Kaposi’s sarcoma-associated herpesvirus capsids reveals dynamic scaffolding structures essential to capsid assembly and maturation. J Struct Biol. 2008;161:419–427. doi: 10.1016/j.jsb.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski JM, Sibley LD. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell. 1996;84:933–939. doi: 10.1016/s0092-8674(00)81071-5. [DOI] [PubMed] [Google Scholar]
- Dube P, Tavares P, Lurz R, van Heel M. The portal protein of bacteriophage SPP1: a DNA pump with 13-fold symmetry. Embo J. 1993;12:1303–1309. doi: 10.1002/j.1460-2075.1993.tb05775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- Gustafson CD. Masters. University of North Dakota; Grand Forks, ND: 1996. Glycolytic enzyme interactions with wild type and mutant Saccharomyces cervisiae actin: comparison with skeletal muscle actin. [Google Scholar]
- Hampton CM, Taylor DW, Taylor KA. Novel structures for alpha-actinin:F-actin interactions and their implications for actin-membrane attachment and tension sensing in the cytoskeleton. J Mol Biol. 2007;368:92–104. doi: 10.1016/j.jmb.2007.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton CM, Liu J, Taylor DW, DeRosier DJ, Taylor KA. The 3D structure of villin as an unusual F-Actin crosslinker. Structure. 2008;16:1882–1891. doi: 10.1016/j.str.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanein D, Volkmann N, Goldsmith S, Michon AM, Lehman W, Craig R, DeRosier D, Almo S, Matsudaira P. An atomic model of fimbrin binding to F-actin and its implications for filament crosslinking and regulation. Nat Struct Biol. 1998;5:787–792. doi: 10.1038/1828. [DOI] [PubMed] [Google Scholar]
- Huang Z, Baldwin PR, Mullapudi S, Penczek PA. Automated determination of parameters describing power spectra of micrograph images in electron microscopy. J Struct Biol. 2003;144:79–94. doi: 10.1016/j.jsb.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph Model. 1996;14:33–38. 27–28. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jewett TJ, Sibley LD. Aldolase forms a bridge between cell surface adhesins and the actin cytoskeleton in apicomplexan parasites. Mol Cell. 2003;11:885–894. doi: 10.1016/s1097-2765(03)00113-8. [DOI] [PubMed] [Google Scholar]
- Khurana S, George SP. The role of actin bundling proteins in the assembly of filopodia in epithelial cells. Cell Adh Migr. 2011;5:409–420. doi: 10.4161/cam.5.5.17644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubalek EW, Kornberg RD, Darst SA. Improved transfer of two-dimensional crystals from the air/water interface to specimen support grids for high-resolution analysis by electron microscopy. Ultramicroscopy. 1991;35:295–304. doi: 10.1016/0304-3991(91)90082-h. [DOI] [PubMed] [Google Scholar]
- Liu J, Wu S, Reedy MC, Winkler H, Lucaveche C, Cheng Y, Reedy MK, Taylor KA. Electron tomography of swollen rigor fibers of insect flight muscle reveals a short and variably angled S2 domain. J Mol Biol. 2006;362:844–860. doi: 10.1016/j.jmb.2006.07.084. [DOI] [PubMed] [Google Scholar]
- Lucic V, Forster F, Baumeister W. Structural studies by electron tomography: from cells to molecules. Annu Rev Biochem. 2005;74:833–865. doi: 10.1146/annurev.biochem.73.011303.074112. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ. 3-D structures of macromolecules using single-particle analysis in EMAN. Methods Mol Biol. 2010;673:157–173. doi: 10.1007/978-1-60761-842-3_9. [DOI] [PubMed] [Google Scholar]
- Magri E, Zaccarini M, Grazi E. Versatility of G-actin as the building block of biological structures. FEBS Lett. 1978;89:276–278. doi: 10.1016/0014-5793(78)80235-x. [DOI] [PubMed] [Google Scholar]
- Maughan DW, Henkin JA, Vigoreaux JO. Concentrations of glycolytic enzymes and other cytosolic proteins in the diffusible fraction of a vertebrate muscle proteome. Mol Cell Proteomics. 2005;4:1541–1549. doi: 10.1074/mcp.M500053-MCP200. [DOI] [PubMed] [Google Scholar]
- Morrissette NS, Sibley LD. Cytoskeleton of apicomplexan parasites. Microbiol Mol Biol Rev. 2002;66:21–38. doi: 10.1128/MMBR.66.1.21-38.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly G, Clarke F. Identification of an actin binding region in aldolase. FEBS Lett. 1993;321:69–72. doi: 10.1016/0014-5793(93)80623-3. [DOI] [PubMed] [Google Scholar]
- Oda T, Iwasa M, Aihara T, Maeda Y, Narita A. The nature of the globular- to fibrous-actin transition. Nature. 2009;457:441–445. doi: 10.1038/nature07685. [DOI] [PubMed] [Google Scholar]
- Ouporov IV, Knull HR, Thomasson KA. Brownian dynamics simulations of interactions between aldolase and G- or F-actin. Biophys J. 1999;76:17–27. doi: 10.1016/S0006-3495(99)77174-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee JD, Spudich JA. Purification of muscle actin. Methods Enzymol. 1982;85(Pt B):164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- Perrin BJ, Ervasti JM. The actin gene family: function follows isoform. Cytoskeleton (Hoboken) 2010;67:630–634. doi: 10.1002/cm.20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Radermacher M. Weighted Back-projection Methods. In: Frank J, editor. Electron Tomography: Methods for Three-Dimensional Visualization of Structures in the Cell. Springer-Verlag; New York: 2006. pp. 245–273. [Google Scholar]
- Rusu M, Hu Z, Taylor KA, Trinick J. Structure of isolated Z-disks from honeybee flight muscle. J Muscle Res Cell Motil. 2017 doi: 10.1007/s10974-017-9477-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvanet C, Wayt J, Pelaseyed T, Bretscher A. Structure, regulation, and functional diversity of microvilli on the apical domain of epithelial cells. Annu Rev Cell Dev Biol. 2015;31:593–621. doi: 10.1146/annurev-cellbio-100814-125234. [DOI] [PubMed] [Google Scholar]
- Saxton WO, Baumeister W, Hahn M. Three-dimensional reconstruction of imperfect two-dimensional crystals. Ultramicroscopy. 1984;13:57–70. doi: 10.1016/0304-3991(84)90057-3. [DOI] [PubMed] [Google Scholar]
- Schur FK, Hagen WJ, de Marco A, Briggs JA. Determination of protein structure at 8.5A resolution using cryo-electron tomography and sub-tomogram averaging. J Struct Biol. 2013;184:394–400. doi: 10.1016/j.jsb.2013.10.015. [DOI] [PubMed] [Google Scholar]
- Sibley LD. How apicomplexan parasites move in and out of cells. Curr Opin Biotechnol. 2010;21:592–598. doi: 10.1016/j.copbio.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom B, Salmazo A, Djinovic-Carugo K. Alpha-actinin structure and regulation. Cell Mol Life Sci. 2008;65:2688–2701. doi: 10.1007/s00018-008-8080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldati D, Meissner M. Toxoplasma as a novel system for motility. Curr Opin Cell Biol. 2004;16:32–40. doi: 10.1016/j.ceb.2003.11.013. [DOI] [PubMed] [Google Scholar]
- Starnes GL, Coincon M, Sygusch J, Sibley LD. Aldolase is essential for energy production and bridging adhesin-actin cytoskeletal interactions during parasite invasion of host cells. Cell Host Microbe. 2009;5:353–364. doi: 10.1016/j.chom.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukow C, DeRosier DJ. Order, disorder, and perturbations in actin-aldolase rafts. Biophys J. 2003;85:525–536. doi: 10.1016/S0006-3495(03)74497-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DW, Kelly DF, Cheng A, Taylor KA. On the freezing and identification of lipid monolayer 2-D arrays for cryoelectron microscopy. J Struct Biol. 2007a;160:305–312. doi: 10.1016/j.jsb.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KA, Taylor DW. Formation of 2-D paracrystals of F-actin on phospholipid layers mixed with quaternary ammonium surfactants. J Struct Biol. 1992;108:140–147. doi: 10.1016/1047-8477(92)90013-z. [DOI] [PubMed] [Google Scholar]
- Taylor KA, Taylor DW. Formation of two-dimensional complexes of F-actin and crosslinking proteins on lipid monolayers: demonstration of unipolar alpha-actinin-F-actin crosslinking. Biophys J. 1994;67:1976–1983. doi: 10.1016/S0006-3495(94)80680-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KA, Taylor DW. Structural studies of cytoskeletal protein arrays formed on lipid monolayers. J Struct Biol. 1999;128:75–81. doi: 10.1006/jsbi.1999.4167. [DOI] [PubMed] [Google Scholar]
- Taylor KA, Liu J, Winkler H. Localization and classification of repetitive structures in electron tomograms of paracrystalline assemblies. In: Frank J, editor. Electron Tomography: Methods for Three-dimensional Visualization of Structures in the Cell. Springer-Verlag; 2006. pp. 417–439. [Google Scholar]
- Taylor KA, Tang J, Cheng Y, Winkler H. The use of electron tomography for structural analysis of disordered protein arrays. J Struct Biol. 1997;120:372–386. doi: 10.1006/jsbi.1997.3932. [DOI] [PubMed] [Google Scholar]
- Taylor KA, Wu S, Reedy MC, Reedy MK. Imaging actomyosin in situ. In: McIntosh JR, editor. Cellular Electron Microscopy. Academic Press; San Diego, CA: 2007b. pp. 321–368. [DOI] [PubMed] [Google Scholar]
- Tilney LG, Tilney MS, DeRosier DJ. Actin filaments, stereocilia, and hair cells: how cells count and measure. Annu Rev Cell Biol. 1992;8:257–274. doi: 10.1146/annurev.cb.08.110192.001353. [DOI] [PubMed] [Google Scholar]
- van Heel M. Classification of very large electron microscopical image data sets. Optik. 1989;82:114–126. [Google Scholar]
- Volkmann N, DeRosier D, Matsudaira P, Hanein D. An atomic model of actin filaments cross-linked by fimbrin and its implications for bundle assembly and function. J Cell Biol. 2001;153:947–956. doi: 10.1083/jcb.153.5.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Morris AJ, Tolan DR, Pagliaro L. The molecular nature of the F-actin binding activity of aldolase revealed with site-directed mutants. J Biol Chem. 1996;271:6861–6865. [PubMed] [Google Scholar]
- Winkler H, Taylor KA. Accurate marker-free alignment with simultaneous geometry determination and reconstruction of tilt series in electron tomography. Ultramicroscopy. 2006;106:240–254. doi: 10.1016/j.ultramic.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Winkler H, Zhu P, Liu J, Ye F, Roux KH, Taylor KA. Tomographic subvolume alignment and subvolume classification applied to myosin V and SIV envelope spikes. J Struct Biol. 2009;165:64–77. doi: 10.1016/j.jsb.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Liu J, Reedy MC, Winkler H, Reedy MK, Taylor KA. Methods for identifying and averaging variable molecular conformations in tomograms of actively contracting insect flight muscle. J Struct Biol. 2009;168:485–502. doi: 10.1016/j.jsb.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Liu J, Reedy MC, Tregear RT, Winkler H, Franzini-Armstrong C, Sasaki H, Lucaveche C, Goldman YE, Reedy MK, Taylor KA. Electron tomography of cryofixed, isometrically contracting insect flight muscle reveals novel actin-myosin interactions. PLoS ONE. 2010;5:e12643. doi: 10.1371/journal.pone.0012643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Liu J, Reedy MC, Perz-Edwards RJ, Tregear RT, Winkler H, Franzini-Armstrong C, Sasaki H, Lucaveche C, Goldman YE, Reedy MK, Taylor KA. Structural changes in isometrically contracting insect flight muscle trapped following a mechanical perturbation. PLoS ONE. 2012;7:e39422. doi: 10.1371/journal.pone.0039422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie 1. Alignment and comparison of a single aldolase subunit from rabbit skeletal muscle (green), PDB – 1ADO, with the aldolase from P. falciparum (red), PDB - 2EPH. Comparison done using chain A of both structures.
Supplemental Movie 2. Alignment and comparison of the full aldolase tetramer from rabbit skeletal muscle (green), PDB – 1ADO, with the aldolase from P. falciparum (red), PDB - 2EPH.
Supplemental Movie 3. The movie shows the same atomic model that is shown in Figure 7c. The coloring scheme is identical to that of Figure 7. Note that the N-terminal extension of aldolase frequently contacts actin, as denoted by the yellow coloring, though its contact on actin is colored gray indicating infrequent contacts. The extension is stabilized by crystal contacts in the crystal structure so its structure may not accurately depict potential binding on actin. Nevertheless, the extension could very well stabilize the cross-links, but possibly not as depicted.