

Abstract
Fibrosis, the hallmark of scleroderma or systemic sclerosis (SSc), is a complex, dynamic and generally irreversible pathophysiological process that leads to tissue disruption, and lacks effective therapy. While early-stage fibrosis resembles normal wound healing, in SSc fibrosis fails to resolve. Innate immune signaling via toll-like receptors (TLRs) has recently emerged as a key driver of persistent fibrotic response in SSc. Recurrent injury in genetically predisposed individual causes generation of “damage-associated molecular patterns” (DAMPs) such as fibronectin-EDA and tenascin-C. Sensing of these danger signals by TLR4 on resident cells elicits potent stimulatory effects on fibrotic gene expression and myofibroblast differentiation, and appears to sensitize fibroblasts to the profibrotic stimulatory effect of TGF-β. Thus, DAMPs induce TLR4-mediated innate immune signaling on resident mesenchymal cells which drives the emergence and persistence of fibrotic cells in tissues, and underlies the switch from a self-limited repair response to non-resolving pathological fibrosis characteristic of SSc. In this review, we present current views of the DAMP-TLR4 axis in driving sustained fibroblasts activation and its pathogenic roles in fibrosis progression in SSc, and potential anti-fibrotic approaches for selective therapeutic targeting of TLR4 signaling.
Keywords: Fibrosis, scleroderma, systemic sclerosis, fibroblast, innate immunity, damage-associated molecular patterns (DAMP), toll-like receptor (TLR), fibronectin-EDA, tenascin-C, A20, TNFAIP3/A20
INTRODUCTION
Systemic sclerosis (SSc) involves a complex interplay between autoimmunity, vasculopathy, and fibrosis in the skin and multiple internal organs [1]. The pathogenesis remains poorly understood; current therapies show only modest and variable efficacy and fail to alter disease course and mortality. The fibrotic process in SSc is most prominent in the skin and lungs, however fibrosis can also occur in the myocardium, gastrointestinal tract, tendons and muscles, and renal interstitium and contributes to mortality [2]. Activated T and B cells, monocytes, dendritic cells and macrophages, are prominent in early-stage SSc biopsies [3]. However, later stages of SSc are dominated by bland tissue fibrosis, and inflammatory cell infiltration is rare. Understanding the pathogenic network of inflammatory, vascular, and fibrotic processes driving non-resolving tissue fibrosis in SSc remains a major challenge in the field. Genomic analysis of SSc skin biopsies has identified distinct gene expression subsets, including an inflammatory intrinsic subset that is highly enriched in pathways related to innate immune signaling [4, 5]. Genetic studies further show that virtually all SSc-associated risk loci are located in genes related to innate immune signaling including the toll-like receptor (TLR) system [6–8]. Of great interest in this regard, variants of A20 or TNFAIP3, a deubiquitinase that is a key negative regulator of TLR signaling, and its partner (TNFAIP3)-interacting protein 1 (TNIP1), both showed strong association with SSc [7, 8]. While the role of TLR signaling in classical (bone marrow-derived) immune cells in multiple inflammatory and autoimmune diseases is well established, little is currently known regarding TLR signaling in stromal/mesenchymal cells, or its roles in SSc.
It has become recognized that the expression of TLR4 as well as several endogenous ligand DAMPs is elevated in lesional tissue from patients with SSc. DAMP-induced TLR4 activation elicits potent stimulatory effects on fibrotic gene expression and myofibroblast transformation and survival. When these cellular responses become persistent, either due to constitutive TLR activation by endogenous DAMPs or downstream signaling mediators, or impaired termination of signaling by endogenous inhibitors, they contribute to failure of fibrosis resolution [9]. This review highlights critical events in deregulated innate immune signaling that contribute to non-resolving tissue fibrosis in SSc and pathological fibrosis. Dissecting the pathogenic networks that underlie self-sustaining DAMP-induced fibroblast activation, DAMP interaction with TLRs and other pattern recognition receptors, downstream cellular signaling pathways, and regulation and function of endogenous inhibitors of innate immunity, will form the foundation for innovative targeted therapies to block fibrosis [10].
Damage-associated endogenous ligands
As the first line of host defense, the innate immune system comprising a diverse set of pattern recognition receptors (PRRs) recognizes exogenous pathogen-associated molecular patterns (PAMPs), derived from microbial pathogens, and DAMPs generated as the consequence of tissue damage [11]. Currently, the best characterized groups of PRRs include the Toll-like receptors (TLRs), the nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs) [9, 12]. Upon sensing PAMP or DAMP, these receptors trigger NF-κB activation and interferon response factor (IRF)-dependent signaling cascades, leading to secretion of proinflammatory cytokines. In the case of TLRs, ligand binding to its extracellular leucine-rich repeats region causes TLRs to dimerize and undergo conformational changes of their intracellular TIR domains, which in turn recruits and activates adaptor molecules such as MyD88, and/or TRIF to transduce signals [9].
In response to recognizing PAMPs such as bacterial lipopolysaccharide (LPS), TLR4 forms a complex with its co-receptor MD2 on the cell surface. Structural studies showed that five of six LPS lipid chains bind to the hydrophobic pocket of MD2, and the remaining lipid chain associates with TLR4 [13]. Danger signals that are generated in response to tissue injury contribute to the pathogenesis progression of many autoimmune diseases via TLR4 activation. Well-studied danger signals include intracellular molecules such as high mobility group box 1 (HMGB1) and heat shock proteins (HSPs), self-DNA and RNA, serum amyloid A (SAA), S100 proteins, fragments of extracellular matrix (ECM) molecules, and several chaperone proteins such as gp96, ER-resident chaperone proteins; and alternatively-spliced “oncofetal” variants of normal ECM components such as EDA-containing fibronectin [9]. In contrast to LPS, DAMPs may not require the same co-receptors and accessory molecules to achieve signaling-competent TLR4 conformation. It is noteworthy that many DAMPs were originally implicated as putative endogenous TLR4 ligands based on co-immunoprecipitation or functional cell-based assays in vitro, or using mutant mice deficient in TLRs or their adaptor proteins in vivo. No crystal structures of DAMP-TLR4 complex have been reported so far to confirm direct interactions, or the requirements of specific co-receptors for the formation of active signaling complexes.
In addition to TLR4, TLR2 might also play a role in mediating DAMP dependent pathogenic responses. Genome-wide association studies (GWAS) in SSc reveal a rare functional polymorphism in the TLR2 gene (Pro631His) that showed robust association with both anti-topoisomerase antibody-positivity and pulmonary arterial hypertension [14]. Dendritic cells from patient carrying this rare TLR2 variant exhibited a marked increase in IL-6 production upon stimulation with TLR2 ligand Pam3Cys. Additional studies showed elevated expression of TLR2 in SSc fibroblasts [15]. Antibody blockade of TLR2 reduced serum-amyloid-A (SAA)-induced IL-6 production, suggesting that SAA served as an endogenous DAMP for TLR2 [10, 15]. It is noteworthy in this context that serum levels of SAA were shown to be elevated in a subset of SSc patients with early diffuse cutaneous disease and pulmonary involvement [16].
One of the best-studied DAMP receptors is RAGE. The extracellular region of RAGE is responsible for ligand interaction and a cytoplasmic domain for downstream intracellular signaling [17]. Due to alternate splicing or protease processing RAGE can exist in truncated forms. RAGE was shown to serve as a receptor for a number of potentially SSc-relevent DAMPs, including HMGB1, S100 proteins, and amyloid-β protein [18–20].
TLR4 is implicated in SSc: elevated levels in lesional skin and lung
Recent studies indicate that TLR4 and its co-receptors, MD2 and CD14, are elevated in lesional skin biopsies from patients with diffuse cutaneous SSc, and show significant correlation with disease progression [21]. In lesional biopsies, TLR4 co-localized with myofibroblasts, as well as infiltrating macrophages and vascular cells [22]. Interstitial lung disease is a frequent SSc complication that can show sustained stability or rapid progression [23]. Lung biopsies from SSc-ILD patients showed elevated intracellular TLR4 expression prominently in parenchymal fibroblasts and infiltrating cells located at, or adjacent to, fibrotic loci. Numerous TLR4-positive interstitial cells in the fibrotic stroma showed strong α-SMA staining [22]. In contrast, only scant TLR4 immunostaining was noted around vascular structures. A recent study by Christmann et al. used lung biopsies from SSc-related interstitial lung disease (ILD) and controls to study pathogenesis [24]. Many SSc lung biopsies showed up-regulation of genes related to TLR and TGF-ß signaling, broadly consistent with gene activation clusters also seen in lesional skin [9]. Such widespread concordance in gene expression patterns across skin and lung implicates major immune-related processes as drivers of multi-organ pathology in SSc. However, the role of TLR4 in lung fibrosis appears to be complex, and the current evidence is contradictory. For instance, studies have shown that LPS- or bleomycin-induced pulmonary fibrosis was ameliorated in mice with TLR4 deleted using small hairpin RNA (shRNA), or in mice that are TLR4-null [25, 26]. In contrast, other studies showed worse bleomycin-induced lung inflammation and fibrosis and reduced survival after acute lung injury in TLR4 null mice, attributed to failure of alveolar epithelial cell regeneration in the absence of TLR4 [27, 28]. The findings suggest the possibility that TLR4 on epithelial cells might play a protective role during lung injury, and its absence aggravates the process. However, the conflicting observations in these studies remain difficult to reconcile at the moment.
TLR4 sensitizes fibroblasts to profibrotic stimulation by TGF-β
Presence of functionally intact TLR4 signaling axis in skin fibroblasts was confirmed by demonstrating stimulation of classic TLR4-dependent inflammatory genes and NF-κB-luc activity by LPS [22]. Treatment of skin fibroblasts by LPS, or by endogenous TLR4 ligands, elicited stimulation of a profibrotic gene expression program and transdifferentiation into α smooth muscle-actin-positive myofibroblasts. Moreover, TLR4 activation on fibroblasts dramatically enhanced its sensitivity to the profibrotic effect of TGF-β. Conversely, genetic targeting of TLR4, or of its endogenous DAMP ligands, or pharmacological disruption of signaling from TLR4 or its co-receptor MD2, ameliorated progressive tissue fibrosis in multiple disease models [22, 29, 30].
In skin fibroblasts, incubation with LPS elicited global changes in gene expression [22]. The TLR4-dependent LPS response was dominated by genes involved in ECM remodeling, tissue repair, and wound healing, while changes in inflammatory genes were relatively modest. These results suggest distinct functional roles for TLR4 in classical immune cells, where TLR4 serves to elicit a rapid and potent inflammatory response designed to deal with invading microbial pathogens, versus in tissue-resident stromal/parenchymal cells, where TLR4 might have evolved primarily to promote robust repair of injured tissue [9, 22]. Mechanistically, the TLR4-dependent profibrotic responses involved multiple pathways, including suppression of the endogenous TGF-β antagonist BAMBI (bone morphogenetic protein and activin membrane-bound inhibitor), and of miR-29, a microRNA known to function as a negative regulator of fibrosis [22, 31–33]. Additional transcriptional mechanisms and epigenetic mesenchymal cell reprograming underlying the persistent profibrotic effects of TLR4 are likely to be operative, and remain a vital area for investigation.
Sustained TLR4 activation by endogenous DAMPs underlies nonresolving tissue fibrosis in SSc
Sterile tissue injury results in the generation of DAMPs that enable cells to sense, and respond to, danger [11, 12]. Persistent DAMP exposure however contributes to chronic inflammatory and autoimmune diseases. To understand the role of DAMPs and TLRs in SSc, we performed an unbiased survey of lesional SSc skin for the expression of putative endogenous TLR4 ligands. By immunohistochemistry, we identified low molecular weight hyaluronic acid, HMGB1, alternatively-spliced isoform of fibronectin (FnEDA) and tenascin-C as most highly up-regulated DAMPs [34]. Alternatively spliced FnEDA and tenascin-C are normally detected in tissues during embryogenesis and then decline. The ‘embryonic’ splicing pattern is re-established transiently during tissue repair and angiogenesis; in contrast, persistent re-expression of these oncofetal isoforms in adults is a hallmark of cancer, and evidently also fibrosis [35, 36].
Alternatively spliced fibronectin EDA isoform drives intractable fibrotic response via TLR4
Fibronectin is one of the best known proteins generated by alternate splicing [35]. Fibronectin occurs in two main forms: dimeric soluble plasma fibronectin (pFN) lacking EDA and EDB domain and multimeric cellular fibronectin (cFN) including EDA or EDB which is deposited in the matrix. Isoforms of fibronectin that include or exclude the EDA and EDB exons arise due to alternative splicing from a single fibronectin pre-mRNA. During cutaneous wound healing, the inclusion of EDA and EDB domains is increased at the wound base [35]. The presence of EDA defines the ability of fibronectin to activate TLR4; recombinant EDA but not EDB can induce TLR4-dependent NF-kB signaling and cytokine synthesis [37–39].
Our studies showed that levels of FnEDA are significantly elevated in SSc skin biopsies and in circulation compared to healthy controls [29]. Ex vivo, treatment of normal skin fibroblasts or reconstituted 3D human skin equivalents with TGF-β induced isoform-specific preferential up-regulation of FnEDA [29]. These studies used antibodies that specifically detect EDA isoform. Serving as a bona fide endogenous TLR4 ligand, FnEDA elicited potent profibrotic responses, with enhanced synthesis of collagen and expression of the myofibroblast marker α-smooth muscle actin. Moreover, mice with genetic deletion of FnEDA were largely resistant to experimentally-induced skin and lung fibrosis [29, 40].
Additional evidence for a TLR4-dependent signaling mechanism of fibronectin was recently provided by Kelsh et al [41, 42]. Using Inflammatory Cytokine microarrays, these authors found that FnEDA, and its partially unfolded type III domain (FnIII-1c), induced TLR4-dependent inflammatory signaling in fibroblasts. This domain of fibronectin is exposed via tensional forces generated within rigid fibrotic microenvironments [41, 42]. These observations have clear implications for fibrosis, since fibrotic tissue is characterized by increased matrix stiffness [43]. We therefore speculate that in SSc, tensional forces within the stiff matrix of fibrotic skin and lungs drive exposure of the EDA and FnIII-1c domains of fibronectin, which, combined with increased generation of the EDA isoform via alternative splicing in resident fibroblasts, results in increased FnEDA bioavailability and profibrotic activity as an endogenous TLR4 ligands [29].
On the basis of these observations, we propose a working model for persistent cutaneous fibrosis where resident fibroblasts are chronically exposed to either fibronectin domains acting as endogenous TLR4 ligands within the rigid fibrotic microenvironment. In response, these fibroblasts undergo TLR4-mediated activation and reprogramming, resulting in unopposed TGF-ß signaling, enhanced profibrotic responses, myofibroblast phenoconversion and matrix remodeling. Moreover, TLR4-dependent profibrotic responses in these cells include preferential production of the EDA isoform of fibronectin, along with other endogenous TLR4 ligands, which in turn drive further TLR4 activation, generating a cell-autonomous fibrosis amplification loop underlying persistent tissue fibrosis.
Tenascin-C induced TLR4-dependent fibrotic responses
One of the best studied endogenous TLR4 ligands linked to SSc and fibrosis is the large modular glycoprotein tenascin-C [36]. The human tenascin-C protein comprises four domains: a tenascin assembly (TA) domain, epidermal growth factor-like (EGF-L) repeats, up to 17 FNIII-like repeats, and a fibrinogen-like globe (FBG) domain. Eight of the FNIII repeats are constitutively expressed (FNIII 1–8), and nine FNIII repeats can be alternatively spliced (FNIIIA1-D). Tenascin-C is widely expressed during embryogenesis but is highly restricted in healthy adult tissues. Expression reappears in wound healing and tissue regeneration; and also seen in several autoimmune diseases [36]. Moreover, elevated tenascin C deposition ia a hallmark of lung fibrosis in both IPF and lung cancer [44]Our unbiased survey for DAMPs associated with SSc identified tenascin-C as one of the most highly up-regulated ECM proteins in SSc skin and lung biopsies as well as in circulation [30, 34]. Furthermore, elevated serum levels of tenascin-C were correlated with the Modified Rodnan skin score, a measure of skin fibrosis [30]. The antibodies used in these studies specifically detected the FNIII-B and FNIII-C epitopes of the large tenascin-C (~250 kDa) isoforms. Treatment of normal fibroblasts with TGF-β or PDGF preferentially induced synthesis of the large tenascin-C variant, while SSc fibroblasts produce the large tenascin-C isoform constitutively. Meta-analysis using publicly available transcriptome data (GSE56038 and GSE59785) demonstrated significantly elevated tenascin-C mRNA levels in SSc skin biopsies mapping to the inflammatory intrinsic subset compared with healthy controls (P<0.0001) [30]. Interestingly, tenascin-C in these biopsies showed strong correlation with TLR4, as well as IL-6, a proinflammatory profibrotic cytokine and direct target of TLR4 that is implicated in SSc [30].
Skin fibroblasts explanted from SSc patients show constitutive production of tenascin-C ex vivo, indicating that its increased accumulation in SSc might at least in part result from its cell-autonomous overproduction [30]. Treatment of skin fibroblasts with tenascin-C elicited TLR4-dependent profibrotic responses, including up-regulation of IL-6, and of TLR4 itself [30]. Mice lacking tenascin-C were protected from skin and lung fibrosis. Moreover, loss of tenascin-C was associated with reduced hypodermal fibrosis in the Tsk/+ mouse, a spontaneous fibrosis model. Importantly, bleomycin-induced skin fibrosis showed accelerated resolution in mice lacking tenascin-C. While the EDA isoform of fibronectin is a potent TLR4 agonist [29], it might be insufficient to compensate for genetic loss of tenascin-C in null mice, since its expression is reduced in lesional skin. We propose that reduced TLR4 signaling in mice lacking tenascin-C accounts for attenuated skin and lung fibrosis and accelerated resolution. Pathological tissue fibrosis in SSc might be similarly perpetuated via a TLR4-dependent fibrosis amplification loop driven by endogenous DAMPs that accumulate and persist within injured microenvironments [30]. This notion is consistent with previous studies showing that tenascin-C mediates persistence of synovial inflammation and tissue destruction in arthritis [45] and induces inflammatory mediators in cardiac myofibroblasts [46, 47]. An intriguing recent study sought to determine whether the TLR4-dependent signaling pathways and biological readouts elicited by the tenascin-C FBG domain as an endogenous TLR4 ligand were similar to those elicited by the classic exogenous TLR4 ligand LPS [48]. Pursuing a comparative analysis of signaling pathways and biological outcomes in macrophages, the authors found that TLR4 activation elicited by FBG and LPS generated two distinct macrophage phenotypes, with only partially-overlapping sets of activation markers, secreted effector molecules, and phosphoproteomic profiles [48]. Remarkably, while LPS promoted a macrophage phenotype with matrix-degrading capacity, FBG promoted a TLR4-dependent “profibrotic” macrophage phenotype. These observations indicate that different microenvironmental cues can elicit distinct macrophage responses via the same receptor and confirm the profibrotic activity of tenascin-C. Whether TLR4 activated by LPS versus DAMPs will generate similarly divergent responses in fibroblasts, remains an important unanswered question with relevance to fibrosis.
A recent study demonstrated that, tenascin-C induced arterial constriction via its EGF-like (EGFL) domain and the EGF receptor (EGFR) [49]. Previous studies have already implicated EGFR activation in TLR4-mediated innate immune responses [50–53]. Importantly, the EGFR inhibitor erlotinib blocks TLR4-mediated NFκB activation and protects mice from LPS-induced lethality, indicating that EGFR kinase activity is necessary for TLR4 signaling [50, 53]. These observations involving TLR4 and EGFR gain significance in view of recent clinical observations linking EGFR signaling to fibrosis and SSc. A multicohort analysis of SSc skin transcriptomes identified a 415-gene SSc signature with transcriptional profiles of 314 ligand stimulations across different cell lines showed positive correlation with multiple EGFR ligands [54]. This SSc-specific 415 gene expression signature was independent of disease subtype, duration, and skin score [54]. These intriguing results collectively suggest a novel pathogenic role for EGFR signaling in SSc, possibly mediated via the EGFL domain of tenascin-C binding to EGFR and cross-talk with TLR4. Comparison of the SSc disease signature with transcriptional profiles of 314 ligand stimulations showed correlation with multiple EGFR ligands [54]. Targeting the EGFR pathways therefore may be a potential therapeutic approach for SSc.
The physiological and pathological regulation of tenascin-C splicing has received increasing attention in the past few years. In particular, the serine/arginine-rich splicing factor 6 (SRSF6) was shown to be an essential regulator of tenascin-C alternative splicing in melanoma [55]. Remarkably, transgenic mice overexpressing SRSF6 in collagen-producing cells were shown to spontaneously develop scleroderma-like skin hyperplasia, accompanied by aberrant tenascin-C splicing and accumulation of the large tenascin-C isoform [55]. By controlling tenascin-C alternative splicing, SRSF6 thus appear to exert powerful effect on skin homeostasis.
Significantly, we found that SRSF6 expression was highly elevated in SSc skin biopsies (Bhattacharyya S, Varga J; unpublished). Moreover, SRSF6 levels within the lesional dermis showed significant correlation with tenascin-C levels, and RNA-seq analysis indicated increased abundance of alternatively-spliced tenascin-C mRNA isoforms (Bhattacharyya S, Roberson E and Varga J; unpublished). Interestingly, SRSF6 was previously shown to be elevated in SSc skin biopsies, and implicated in driving pathogenic accumulation of alternatively spliced inhibitory vascular endothelial growth factor (VEGF) isoform [56]. While these intriguing observations implicate SRSF6 and aberrant regulation of alternative tenascin-C splicing in skin fibrosis, little is currently known about tenascin-C splicing in inflammation and tissue remodeling. It will be of great interest to determine if alternatively spliced tenascin-C isoforms, or its FBG or EGFL domains, are necessary and sufficient to elicit TLR4-dependent fibrotic signaling, and whether they can be targeted for anti-fibrotic therapy.
The endogenous TLR4 ligand HMGB1, a DAMP, is elevated in SSc skin
High mobility group box 1 (HMGB1) is a highly conserved nuclear DNA-binding protein and was the first DAMP recognized as a endogenous TLR ligand. HMGB1 is expressed as a single polypeptide chain of 215 amino acids. HMGB1 is composed of three distinct structural domains: A-box (amino acids 1–79), B-box (89–162) which is responsible for DNA binding and the acidic C tail (186–215) enriched with negatively-charged aspartic acid and glutamic acid that controls transcriptional stimulation [57]. Injury, infection or cellular stress induce posttranslational modification (acetylation or methylation) of HMGB1, triggering cytoplasmic translocation from nucleus, and facilitating subsequent release into the extracellular milieu [58–60].
One of the most extensively studied roles of HMGB1 is as an endogenous TLR ligand DAMP [58]. Extracellular HMGB1 induces cellular responses by diverse pattern recognition receptors including TLR2, TLR4, TLR9 and RAGE. While HMGB1 physically interacts with RAGE, interaction with TLR4 appears to be required for cytokine release [59]. The disulfide bonds between Cysteine 23 and C45 and Cysteine 106 thiol group of HMGB1 are required for its recognition by the TLR4/MD2 complex [59]. In order to identify specific inhibitors of HMGB1-TLR4 signaling, peptide libraries were screened by Yang et al [61]. These studies identified a tetramer (P5779) as a specific antagonist that selectively prevented HMGB1 interaction with MD2 and subsequent TLR4 signaling [61]. Since P5779 does not interfere with PAMP-induced TLR4-dependent cytokine/chemokine production, anti-microbial protective TLR4-MD2 responses are preserved. In preclinical studies, P5779 was shown to protect mice against hepatic ischemia/reperfusion injury, chemical toxicity, and sepsis. These findings reveal an exciting innovative strategy for selectively targeting DAMP-mediated harmful inflammation while preserving antimicrobial immune responsiveness, and therefore have clinical relevance.
Studies have implicated HMGB1-dependent TLR4 signaling in myocardial, pulmonary, renal and liver fibrosis via TLR4 signaling [62–68]. We therefore sought to characterize the expression of HMGB1 in SSc. Immunohistochemistry of skin biopsies from nine patients with early dcSSc and five healthy controls matched for sex and age studied in parallel demonstrated that, in contrast to healthy control biopsies where HMGB1 was localized primarily in the nucleus, in SSc biopsies a substantial increase in HMGB1 translocated from nucleus to cytoplasm was seen (Fig. 1), consistent with aberrant HMGB1 processing or secretion. It will therefore be of great interest to explore the implication of elevated HMGB1 in SSc.
Figure 1. Elevated HMGB1 expression and cytosolic localization in SSc skin biopsies.
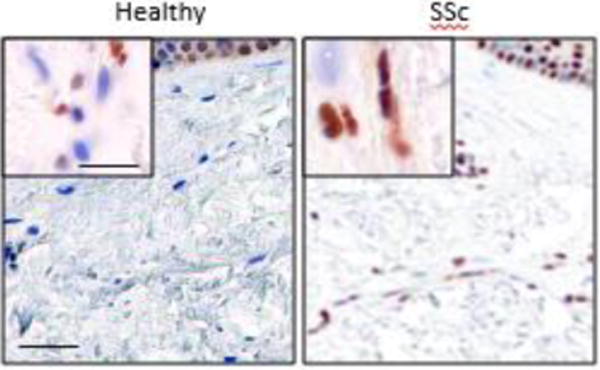
Immunohistochemistry of lesional skin biopsies from SSc patients with early-stage disease (n=6) and site-matched biopsies from age-matched healthy controls (n=4) using anti-HMGB1 antibodies; representative images. Note increased dermal HMGB1 expression (brown) and cytosolic localization in SSc biopsies. Nuclei are blue. Scale bar, 50 μm; inset, 10 μm.
S100 proteins as endogenous TLR4 ligands in SSc
S100 belongs to a superfamily of calcium (Ca2+)-binding proteins. S100 is composed of two distinct helix-loop-helix structural domain (EF-hands) connected by a central hinge region [69, 70]. Despite their small molecular size and conserved functional domains, S100 proteins have a plethora of tissue-specific intra- and extracellular functions and are implicated in diverse diseases such as cancer, cardiomyopathies, neurodegenerative, inflammatory and autoimmune diseases [71].
The S100 family members S100A8 and S100A9, also known as calgranulins A and B or myeloid-related proteins (MRP) 8 and 14 can form homo- and heterodimers as well as heterotetramers. While S100A8 and S100A9 can spontaneously form heterodimers in the absence of metal ions, tetramer formation of S100A8/S100A9 is strictly dependent on the presence of Zn2+ and Ca2+, which appears to be indispensable for the intracellular functions of S100A8/S100A9 [72, 73]. As endogenous TLR4 ligands, extracellular S100A8/A9 are involved in the pathogenesis of autoimmune diseases and cancer. Studies in SSc showed that circulating S100A8/A9 levels were significantly elevated and associated with lung fibrosis [74]. In addition, the S100A4 variant showed elevated tissue expression in SSc skin biopsies, and its production was stimulated by TGF-β in SSc skin fibroblasts [75]. Conversely, S100A4 knockout mice were protected from bleomycin-induced skin fibrosis. In light of these observation it will be interesting to explore the pathogenic roles of S100 family proteins as TLR4 ligand DAMPs in SSc.
DAMPs involved in regulation of TLR folding and trafficking
Appropriate protein folding is required for TLRs to traffic to their final cellular destination. Some TLRs, such as TLR4, traffic to the cell surface, while nucleic acid-sensing TLRs exit the endoplasmic reticulum (ER) for the endosomal system to interact with their ligands [76]. Folding of TLRs depends on the chaperone proteins gp96, CNPY3 and CNPY4 (also known as PRAT4A and PRAT4B). In particular gp96 enhanced TLR responses elicited by LPS (TLR4 agonist) or Pam3CSK4 (TLR2 agonist) [77]. On the other hand, deletion of gp96 in macrophages compromised signaling through both surface (TLR2 and TLR4) and endosomal (TLR7 and TLR9) receptors, presumably due to impaired trafficking of TLRs to their corresponding cellular compartments [78]. Rheumatoid arthritis (RA) synovial fluid-induced macrophage activation was suppressed by neutralizing antibodies to anti-gp96, demonstrating the role of gp96 as clinically relevant endogenous TLR ligand [79]. CNPY3 and CNPY4 belong to a family of ER-resident chaperone proteins also implicated in TLR trafficking and surface expression [80, 81]. Intriguingly, TLR4 is the only TLR only shown to be capable of interacting with endogenous CNPY3 [82]. Future studies are warranted to determine the regulation and pathogenic role of gp96, CNPY3 and CNPY4 as TLR chaperones in SSc.
Limiting the TLR response: negative TLR regulation in SSc
Limiting the duration and amplitude of TLR signaling is essential for preventing unchecked inflammation. Impaired regulation of negative regulators of TLR signaling might contribute to chronic inflammatory and fibrotic diseases. Mechanism for the negative regulation of TLR signaling include alternative splicing of TLR adaptors (e.g. MyD88s); the cell surface molecule radioprotective 105 (RP105); intracellular ubiquitin editing enzymes such as A20 (TNFAIP3) that modulate the activity of key TLR signaling intermediates; transcriptional regulators; and microRNAs [9, 83]. Although extensive discussion of negative-feedback mechanisms controlling TLR activation is beyond the scope of the current review, altered expression or function of the regulators appear to play important roles in a variety of TLR-dependent disease process.
Targeting TLR4 in SSc: potential therapeutic approaches
Current treatment options for patients with SSc are limited and associated morbidity and mortality remain substantial. Clinical heterogeneity is a major factor confounding understanding of SSc pathogenesis. Genome-wide profiling approaches have identified four “intrinsic” gene expression subsets among patients with SSc [4, 84]. Cluster analysis of skin biopsy transcriptomes reveals distinct gene subsets provisionally labeled fibroproliferative, inflammatory, limited, and normal-like [4, 5, 85, 86]. In contrast to other subsets, the inflammatory intrinsic subset showed robust upregulation of genes associated with innate immunity [4, 85]. However, paucity of immune cells within SSc biopsies suggests that the inflammatory gene signatures might originate primarily from resident stromal cells rather than infiltrating hematopoietic cells. To explore this hypothesis, we generated an experimentally-derived “fibroblast TLR4-regulated gene signature” using normal skin fibroblasts transfected with TLR4. Comparing our experimentally-generated TLR4 gene signatures to those in primary human monocytes showed partial overlap of differentially-expressed genes between the two cell types (hypergeometric test; p=0.02). Remarkably, only the fibroblast but not the monocytes, TLR4 gene signature showed enrichment with pathways related to wound healing, matrix organization and TGF-ß signaling, revealing important cell type-specific differences in how bone marrow-derived inflammatory cells versus stromal cells respond to TLR4 stimulation [9].
To determine if SSc is associated with altered TLR4 pathway activity, we compared the strength of TLR4 pathway activation in SSc and control skin biopsies using experimentally-derived TLR4 gene signatures. This analysis revealed significant TLR4 pathway activation in inflammatory intrinsic subset SSc biopsies. The same biopsies also showed a strong TGF-ß gene signature, and association with SSc-ILD [87] (Bhattacharyya S. and Varga J., unpublished). These findings suggest that elevated inflammatory gene expression in SSc skin biopsies might originate primarily from activated tissue-resident fibroblasts. Furthermore, this fibroblast TLR4 signature in skin biopsies might represent a predictive biomarker for identifying SSc patients with ongoing fibroblast activity who might be optimal responders to therapies that block TLR4 signaling.
Pharmacological strategies to block pathogenic TLR4 signaling
As TLR4 dysregulation is associated with a myriad of diseases, and may play a significant role in their pathogenesis or persistence, several groups have attempted to develop potent inhibitors of TLR4 activity. The two most clinically advanced TLR4 inhibitors to date are Eritoran, a lipid-A mimetic (Eisai Co., Ltd., Japan) and TAK-242 (Takeda Pharmaceutical Company Limited, Japan), a TLR4-binding small molecule antagonist. Both drugs reached Phase III clinical trials for sepsis but failed to demonstrate efficacy [9, 88, 89]. Therefore, developing and validating a TLR4 inhibitor using novel strategies remains a critical goal for effective anti-fibrotic therapy.
The studies highlighted above implicate TLR4 and its co-receptor MD2 in SSc, focusing attention of TLR4/MD2 as a novel therapeutic target in this disease [9]. Several endogenous TLR4 ligand DAMPs require MD2 as a co-receptor for signaling and initiation of fibrotic responses [11, 38]. Hence, disrupting ligand-MD2-TLR4 complex formation represents a logical anti-fibrotic strategy. Yin and colleagues undertook extensive structure-activity-relationship studies in order to identify drug candidates that selectively disrupt TLR4-MD2 interactions. The most efficient compound in this series, T5342126, competes with MD2 for binding to TLR4, and shows potent anti-fibrotic effects. In normal fibroblasts stimulated by microbial LPS or by endogenous DAMPs fibronectin EDA or tenascin-C, T5342126 prevented stimulation of collagen synthesis and myofibroblast differentiation (Fig. 2), and attenuated the constitutively activated phenotype of SSc fibroblasts (Bhattacharyya S and Varga J; unpublished). In vivo, T5342126 treatment was able to both prevent, and reverse, organ fibrosis in multiple disease models (Bhattacharyya S, Yin H and Varga J unpublished). An alternative therapeutic strategy for disrupting MD2-TLR4 complex formation using antibodies directed against TLR4 or the TLR4-MD-2 complex showed promising results in pre-clinical studies [90]. lnterleukin-1 Receptor Associated Kinases (IRAK) are a family of molecules that can also be therapeutically targeted to block TLR4-dependent responses. IRAK4 is the best characterized member of the family [91–93]. Pfizer recently patented an IRAK4 inhibitor (PF 06650833) for the treatment of autoimmune and inflammatory diseases.
Figure 2. The endogenous TLR4 ligand FnEDA induces TLR4-dependent myofibroblasts differentiation via TLR4.
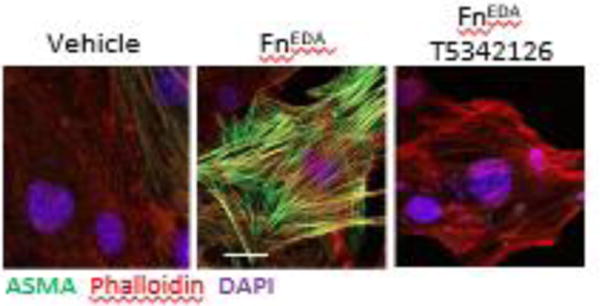
Skin fibroblasts were incubated with fnEDA for 72 h in absence or presence of the novel MD2-TLR4 inhibitor T5342126. Immunofluorescence using antibodies to ASMA demonstrating increased myofibroblasts differentiation and stress fiber formation which was completely abrogated by T5342126 treatment. Representative images. Scale bar, 10 μm.
An alternative approach to therapeutic TLR4 blockade involves selectively preventing TLR4 activation by disease-associated DAMPs. Carefully choosing a target unique to the tissue damage and distinct from pathogen-mediated activation of the immune response, may provide the advantage of preserving an intact host response to infection. Viable approaches include selective targeting of alternatively-spliced domains of fibronectin and tenascin-C using specific antibodies. For example, the F16 antibody targets the A1 domain of tenascin-C, whereas the 81C6 antibody recognizes the D domain; the EDA domain of fibronectin is the target of F8 antibody [94]. Of particular interest is a strategy to selectively target the tenascin-C FBG domain, which is responsible for TLR4 signaling [95]. In the same way, blocking S100A8/A9 and HMGB1 interaction with their cognate TLRs using small molecule inhibitors could prove to be useful therapeutic approaches for SSc [61, 73]. Small molecule inhibitors of S100A8/A9 and HMGB1 have also shown promises in animal models and could prove useful therapeutic approach for in SSc. Further preclinical studies and clinical data examining the therapeutic potential of targeting of the TLR4-DAMPs are clearly warranted. At the same time, comparative analysis of co-receptors, downstream kinases, and transcription factors involved in DAMP versus PAMP-induced signaling may identify key differences that could lead to novel therapeutic approaches by selectively silencing DAMP-TLR4 signaling while preserving intact PAMP-induced anti-microbial host responses. Blocking TLR4 may also lead to inappropriate immune responses in specific cell types or in response to certain injuries. Thus, the risks and benefits of manipulation of TLR-mediated immune responses need to be carefully balanced.
Summary and future perspective
Results from a growing body of in vitro experiments, animal models and clinical observations highlight a previously unsuspected fundamental pathogenic role of DAMP-TLR4 signaling in fibrosis development, progression and persistence in SSc (Fig. 3). However, the identity of pathogenic DAMPs, the nature of their cell-type-specific signaling, and their precise contribution to driving tissue pathology in SSc remain unclear. It will be essential to elucidate if targeting selective TLR4 activation by a particular DAMP is effective in SSc, or if it is more appropriate to target shared downstream signaling pathways common to many DAMPs and pattern recognition receptors. It is notable that anti-sepsis drugs targeting the TLR4 axis, including eritoran and TAK-242, failed to achieve primary endpoint goals in clinical trials, emphasizing the challenge for improving the design of clinical studies or optimizing new treatment strategy. Recent studies from our group and others highlight the association of aberrant DAMP-dependent TLR4 signaling with fibrosis in SSc, suggesting that subsets of SSc patients might be identified as the optimal responders to therapy targeting TLR4. Intriguingly, in initial studies the TLR4 inhibitor T5342126 displayed potent anti-fibrotic activity in vitro and in animal models, encouraging further optimization. Alternatively, selectively ablating the expression or function of individual pathogenic DAMPS, or restoring or boosting the expression of endogenous TLR inhibitors such as A20 or RP105 by pharmacologic agents, might hold promise for anti-fibrotic therapies. Results from both preclinical and clinical studies provide robust support for our premise that selective pharmacological targeting of the TLR4-DAMP pathway might have therapeutic potential to manage intractable fibrosis. In summary, this review highlighted an emerging paradigm implicating TLR4-DAMP signaling in persistent fibroblast activation as a key pathogenic mechanism in SSc, appealing opportunities for targeted therapeutic intervention and novel approaches for defining molecular classifiers to identify SSc patients who might be optimal responders to TLR4 inhibition.
Figure 3. DAMP-driven self-amplifying cycle of fibrosis in systemic sclerosis: a proposed model.
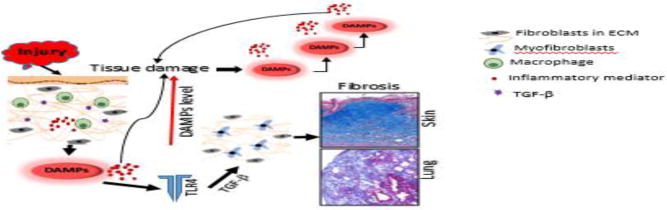
Tissue damage from sustained injury activates inflammatory cells to secrete cytokines and growth factors which in turn generate damage-associated endogenous TLR ligands (DAMPs) and pro-inflammatory mediators. Proinflammatory mediators and DAMPs trigger tissue damage leading to further increasing DAMPs levels. At the same time DAMPs activate innate immune signaling pathway in resident mesenchymal cells that transdifferentiate to myofibroblasts, enhancing fibrogenic responses and converting self-limited tissue repair into intractable scarring.
Highlights.
Triggering the innate immune system in resident stromal cells elicits fibrotic responses
In the fibrotic microenvironment, “damage-associated molecular patterns” act as endogenous ligands for pattern recognition receptors such as TLRs expressed in myofibroblasts
Persistent DAMP activation of myofibroblasts, coupled with impaired downregulation of innate immune signaling, underlies constitutive myofibroblast activation and failure of fibrosis resolution in SSc
The novel paradigm of fibrosis persistence driven by sustained innate immune activation by DAMPs presents translational and clinical opportunities for the development of fibrosis biomarkers and treatments.
Acknowledgments
Supported by grants from the NIH (AR42309 and AR064925 to JV; AR065800 to SB) and the Scleroderma Foundation (to SB). We are grateful to Kim S. Midwood and Hang Yin for helpful insights.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nature reviews Rheumatology. 2011;8(1):42–54. doi: 10.1038/nrrheum.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. The New England journal of medicine. 2009;360(19):1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 3.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117(3):557–567. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, Whitfield ML. Molecular subsets in the gene expression signatures of scleroderma skin. PloS one. 2008;3(7):e2696. doi: 10.1371/journal.pone.0002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson ME, Mahoney JM, Taroni J, Sargent JL, Marmarelis E, Wu MR, Varga J, Hinchcliff ME, Whitfield ML. Experimentally-derived fibroblast gene signatures identify molecular pathways associated with distinct subsets of systemic sclerosis patients in three independent cohorts. PloS one. 2015;10(1):e0114017. doi: 10.1371/journal.pone.0114017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin JE, Assassi S, Diaz-Gallo LM, Broen JC, Simeon CP, Castellvi I, Vicente-Rabaneda E, Fonollosa V, Ortego-Centeno N, Gonzalez-Gay MA, et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Human molecular genetics. 2013;22(19):4021–4029. doi: 10.1093/hmg/ddt248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korman BD, Criswell LA. Recent advances in the genetics of systemic sclerosis: toward biological and clinical significance. Current rheumatology reports. 2015;17(3):21. doi: 10.1007/s11926-014-0484-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koumakis E, Giraud M, Dieude P, Cohignac V, Cuomo G, Airo P, Hachulla E, Matucci-Cerinic M, Diot E, Caramaschi P, et al. Brief report: candidate gene study in systemic sclerosis identifies a rare and functional variant of the TNFAIP3 locus as a risk factor for polyautoimmunity. Arthritis and rheumatism. 2012;64(8):2746–2752. doi: 10.1002/art.34490. [DOI] [PubMed] [Google Scholar]
- 9.Bhattacharyya SMK, Yin H, Varga J. Toll Like Receptor-4 Signaling Drives Persistent Fibroblast Activation and Prevents Fibrosis Resolution in Scleroderma. Advances in wound care. 2017 doi: 10.1089/wound.2017.0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fullard N, O’Reilly S. Role of innate immune system in systemic sclerosis. Seminars in immunopathology. 2015;37(5):511–517. doi: 10.1007/s00281-015-0503-7. [DOI] [PubMed] [Google Scholar]
- 11.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators of inflammation 2010. 2010 doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryant CE, Gay NJ, Heymans S, Sacre S, Schaefer L, Midwood KS. Advances in Toll-like receptor biology: Modes of activation by diverse stimuli. Crit Rev Biochem Mol. 2015;50(5):359–379. doi: 10.3109/10409238.2015.1033511. [DOI] [PubMed] [Google Scholar]
- 13.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458(7242):1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 14.Broen JC, Bossini-Castillo L, van Bon L, Vonk MC, Knaapen H, Beretta L, Rueda B, Hesselstrand R, Herrick A, Worthington J, et al. A rare polymorphism in the gene for Toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis and rheumatism. 2012;64(1):264–271. doi: 10.1002/art.33325. [DOI] [PubMed] [Google Scholar]
- 15.O’Reilly S, Cant R, Ciechomska M, Finnigan J, Oakley F, Hambleton S, van Laar JM. Serum amyloid A induces interleukin-6 in dermal fibroblasts via Toll-like receptor 2, interleukin-1 receptor-associated kinase 4 and nuclear factor-kappaB. Immunology. 2014;143(3):331–340. doi: 10.1111/imm.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lakota K, Carns M, Podlusky S, Mrak-Poljsak K, Hinchcliff M, Lee J, Tomsic M, Sodin-Semrl S, Varga J. Serum amyloid A is a marker for pulmonary involvement in systemic sclerosis. PloS one. 2015;10(1):e0110820. doi: 10.1371/journal.pone.0110820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee EJ, Park JH. Receptor for Advanced Glycation Endproducts (RAGE), Its Ligands, and Soluble RAGE: Potential Biomarkers for Diagnosis and Therapeutic Targets for Human Renal Diseases. Genomics & informatics. 2013;11(4):224–229. doi: 10.5808/GI.2013.11.4.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Muller S, Iannacone M, Traversari C, et al. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO reports. 2004;5(8):825–830. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rani SG, Sepuru KM, Yu C. Interaction of S100A13 with C2 domain of receptor for advanced glycation end products (RAGE) Biochimica et biophysica acta. 2014;1844(9):1718–1728. doi: 10.1016/j.bbapap.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 20.Chaney MO, Stine WB, Kokjohn TA, Kuo YM, Esh C, Rahman A, Luehrs DC, Schmidt AM, Stern D, Yan SD, et al. RAGE and amyloid beta interactions: atomic force microscopy and molecular modeling. Biochimica et biophysica acta. 2005;1741(1–2):199–205. doi: 10.1016/j.bbadis.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 21.Stifano G, Affandi AJ, Mathes AL, Rice LM, Nakerakanti S, Nazari B, Lee J, Christmann RB, Lafyatis R. Chronic Toll-like receptor 4 stimulation in skin induces inflammation, macrophage activation, transforming growth factor beta signature gene expression, and fibrosis. Arthritis research & therapy. 2014;16(4):R136. doi: 10.1186/ar4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhattacharyya S, Kelley K, Melichian DS, Tamaki Z, Fang F, Su Y, Feng G, Pope RM, Budinger GR, Mutlu GM, et al. Toll-like receptor 4 signaling augments transforming growth factor-beta responses: a novel mechanism for maintaining and amplifying fibrosis in scleroderma. The American journal of pathology. 2013;182(1):192–205. doi: 10.1016/j.ajpath.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, Varga J. Systemic sclerosis. Nature reviews Disease primers. 2015;1:15002. doi: 10.1038/nrdp.2015.2. [DOI] [PubMed] [Google Scholar]
- 24.Christmann RB, Hayes E, Pendergrass S, Padilla C, Farina G, Affandi AJ, Whitfield ML, Farber HW, Lafyatis R. Interferon and alternative activation of monocyte/macrophages in systemic sclerosis-associated pulmonary arterial hypertension. Arthritis and rheumatism. 2011;63(6):1718–1728. doi: 10.1002/art.30318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He Z, Zhu Y, Jiang H. Inhibiting toll-like receptor 4 signaling ameliorates pulmonary fibrosis during acute lung injury induced by lipopolysaccharide: an experimental study. Respiratory research. 2009;10:126. doi: 10.1186/1465-9921-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi T, Asano Y, Ichimura Y, Toyama T, Taniguchi T, Noda S, Akamata K, Tada Y, Sugaya M, Kadono T, et al. Amelioration of tissue fibrosis by toll-like receptor 4 knockout in murine models of systemic sclerosis. Arthritis & rheumatology. 2015;67(1):254–265. doi: 10.1002/art.38901. [DOI] [PubMed] [Google Scholar]
- 27.Liang J, Zhang Y, Xie T, Liu N, Chen H, Geng Y, Kurkciyan A, Mena JM, Stripp BR, Jiang D, et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nature medicine. 2016;22(11):1285–1293. doi: 10.1038/nm.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, Yan J, Li Z, Liu H, Hua F, et al. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. The American journal of pathology. 2012;180(1):275–292. doi: 10.1016/j.ajpath.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, White ES, Varga J. FibronectinEDA promotes chronic cutaneous fibrosis through Toll-like receptor signaling. Science translational medicine. 2014;6(232):232ra250. doi: 10.1126/scitranslmed.3008264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, Lafyatis R, Lee J, Hinchcliff M, Feghali-Bostwick C, et al. Tenascin-C drives persistence of organ fibrosis. Nature communications. 2016;7:11703. doi: 10.1038/ncomms11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nature medicine. 2007;13(11):1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 32.Maurer B, Stanczyk J, Jungel A, Akhmetshina A, Trenkmann M, Brock M, Kowal-Bielecka O, Gay RE, Michel BA, Distler JH, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis and rheumatism. 2010;62(6):1733–1743. doi: 10.1002/art.27443. [DOI] [PubMed] [Google Scholar]
- 33.O’Reilly S. MicroRNAs in fibrosis: opportunities and challenges. Arthritis research & therapy. 2016;18:11. doi: 10.1186/s13075-016-0929-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhattacharyya S, Varga J. Emerging roles of innate immune signaling and toll-like receptors in fibrosis and systemic sclerosis. Current rheumatology reports. 2015;17(1):474. doi: 10.1007/s11926-014-0474-z. [DOI] [PubMed] [Google Scholar]
- 35.White ES, Muro AF. Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB life. 2011;63(7):538–546. doi: 10.1002/iub.493. [DOI] [PubMed] [Google Scholar]
- 36.Giblin SP, Midwood KS. Tenascin-C: Form versus function. Cell adhesion & migration. 2015;9(1–2):48–82. doi: 10.4161/19336918.2014.987587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gondokaryono SP, Ushio H, Niyonsaba F, Hara M, Takenaka H, Jayawardana ST, Ikeda S, Okumura K, Ogawa H. The extra domain A of fibronectin stimulates murine mast cells via toll-like receptor 4. Journal of leukocyte biology. 2007;82(3):657–665. doi: 10.1189/jlb.1206730. [DOI] [PubMed] [Google Scholar]
- 38.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., 3rd The extra domain A of fibronectin activates Toll-like receptor 4. The Journal of biological chemistry. 2001;276(13):10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 39.Lefebvre JS, Levesque T, Picard S, Pare G, Gravel A, Flamand L, Borgeat P. Extra domain A of fibronectin primes leukotriene biosynthesis and stimulates neutrophil migration through activation of Toll-like receptor 4. Arthritis and rheumatism. 2011;63(6):1527–1533. doi: 10.1002/art.30308. [DOI] [PubMed] [Google Scholar]
- 40.Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, Flaherty KR, Martinez FJ, Tsui JL, Sheppard D, et al. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. American journal of respiratory and critical care medicine. 2008;177(6):638–645. doi: 10.1164/rccm.200708-1291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kelsh R, You R, Horzempa C, Zheng M, McKeown-Longo PJ. Regulation of the innate immune response by fibronectin: synergism between the III-1 and EDA domains. PloS one. 2014;9(7):e102974. doi: 10.1371/journal.pone.0102974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.You R, Zheng M, McKeown-Longo PJ. The first type III repeat in fibronectin activates an inflammatory pathway in dermal fibroblasts. The Journal of biological chemistry. 2010;285(47):36255–36259. doi: 10.1074/jbc.C110.176990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. The Journal of pathology. 2013;229(2):298–309. doi: 10.1002/path.4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gocheva V, Naba A, Bhutkar A, Guardia T, Miller KM, Li CM, Dayton TL, Sanchez-Rivera FJ, Kim-Kiselak C, Jailkhani N, et al. Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(28):E5625–E5634. doi: 10.1073/pnas.1707054114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nature medicine. 2009;15(7):774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 46.Patel L, Sun W, Glasson SS, Morris EA, Flannery CR, Chockalingam PS. Tenascin-C induces inflammatory mediators and matrix degradation in osteoarthritic cartilage. BMC musculoskeletal disorders. 2011;12:164. doi: 10.1186/1471-2474-12-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maqbool A, Spary EJ, Manfield IW, Ruhmann M, Zuliani-Alvarez L, Gamboa-Esteves FO, Porter KE, Drinkhill MJ, Midwood KS, Turner NA. Tenascin C upregulates interleukin-6 expression in human cardiac myofibroblasts via toll-like receptor 4. World journal of cardiology. 2016;8(5):340–350. doi: 10.4330/wjc.v8.i5.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piccinini AM, Zuliani-Alvarez L, Lim JM, Midwood KS. Distinct microenvironmental cues stimulate divergent TLR4-mediated signaling pathways in macrophages. Science signaling. 2016;9(443):ra86. doi: 10.1126/scisignal.aaf3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujimoto M, Shiba M, Kawakita F, Liu L, Nakasaki A, Shimojo N, Imanaka-Yoshida K, Yoshida T, Suzuki H. Epidermal growth factor-like repeats of tenascin-C-induced constriction of cerebral arteries via activation of epidermal growth factor receptors in rats. Brain research. 2016;1642:436–444. doi: 10.1016/j.brainres.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 50.Chattopadhyay S, Veleeparambil M, Poddar D, Abdulkhalek S, Bandyopadhyay SK, Fensterl V, Sen GC. EGFR kinase activity is required for TLR4 signaling and the septic shock response. EMBO reports. 2015;16(11):1535–1547. doi: 10.15252/embr.201540337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hsu D, Fukata M, Hernandez YG, Sotolongo JP, Goo T, Maki J, Hayes LA, Ungaro RC, Chen A, Breglio KJ, et al. Toll-like receptor 4 differentially regulates epidermal growth factor-related growth factors in response to intestinal mucosal injury. Laboratory investigation; a journal of technical methods and pathology. 2010;90(9):1295–1305. doi: 10.1038/labinvest.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trussoni CE, Tabibian JH, Splinter PL, O’Hara SP. Lipopolysaccharide (LPS)-Induced Biliary Epithelial Cell NRas Activation Requires Epidermal Growth Factor Receptor (EGFR) PloS one. 2015;10(4):e0125793. doi: 10.1371/journal.pone.0125793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De S, Zhou H, DeSantis D, Croniger CM, Li X, Stark GR. Erlotinib protects against LPS-induced endotoxicity because TLR4 needs EGFR to signal. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(31):9680–9685. doi: 10.1073/pnas.1511794112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lofgren S, Hinchcliff M, Carns M, Wood T, Aren K, Arroyo E, Cheung P, Kuo A, Valenzuela A, Haemel A, et al. Integrated, multicohort analysis of systemic sclerosis identifies robust transcriptional signature of disease severity. JCI insight. 2016;1(21):e89073. doi: 10.1172/jci.insight.89073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jensen MA, Wilkinson JE, Krainer AR. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nature structural & molecular biology. 2014;21(2):189–197. doi: 10.1038/nsmb.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manetti M, Guiducci S, Romano E, Ceccarelli C, Bellando-Randone S, Conforti ML, Ibba-Manneschi L, Matucci-Cerinic M. Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circulation research. 2011;109(3):e14–26. doi: 10.1161/CIRCRESAHA.111.242057. [DOI] [PubMed] [Google Scholar]
- 57.Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. Journal of leukocyte biology. 2013;93(6):865–873. doi: 10.1189/jlb.1212662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488(7413):670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annual review of immunology. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang Y, Zhao X, Antoine D, Xiao X, Wang H, Andersson U, Billiar TR, Tracey KJ, Lu B. Regulation of Posttranslational Modifications of HMGB1 During Immune Responses. Antioxidants & redox signaling. 2016;24(12):620–634. doi: 10.1089/ars.2015.6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang H, Wang H, Ju Z, Ragab AA, Lundback P, Long W, Valdes-Ferrer SI, He M, Pribis JP, Li J, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. The Journal of experimental medicine. 2015;212(1):5–14. doi: 10.1084/jem.20141318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang W, Lavine KJ, Epelman S, Evans SA, Weinheimer CJ, Barger PM, Mann DL. Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. Journal of the American Heart Association. 2015;4(6):e001993. doi: 10.1161/JAHA.115.001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ebina M, Taniguchi H, Miyasho T, Yamada S, Shibata N, Ohta H, Hisata S, Ohkouchi S, Tamada T, Nishimura H, et al. Gradual increase of high mobility group protein b1 in the lungs after the onset of acute exacerbation of idiopathic pulmonary fibrosis. Pulmonary medicine. 2011;2011:916486. doi: 10.1155/2011/916486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hamada N, Maeyama T, Kawaguchi T, Yoshimi M, Fukumoto J, Yamada M, Yamada S, Kuwano K, Nakanishi Y. The role of high mobility group box1 in pulmonary fibrosis. American journal of respiratory cell and molecular biology. 2008;39(4):440–447. doi: 10.1165/rcmb.2007-0330OC. [DOI] [PubMed] [Google Scholar]
- 65.Li LC, Li DL, Xu L, Mo XT, Cui WH, Zhao P, Zhou WC, Gao J, Li J. High-Mobility Group Box 1 Mediates Epithelial-to-Mesenchymal Transition in Pulmonary Fibrosis Involving Transforming Growth Factor-beta1/Smad2/3 Signaling. The Journal of pharmacology and experimental therapeutics. 2015;354(3):302–309. doi: 10.1124/jpet.114.222372. [DOI] [PubMed] [Google Scholar]
- 66.Wang Q, Wang J, Wang J, Hong S, Han F, Chen J, Chen G. HMGB1 induces lung fibroblast to myofibroblast differentiation through NFkappaBmediated TGFbeta1 release. Molecular medicine reports. 2017;15(5):3062–3068. doi: 10.3892/mmr.2017.6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang M, Guo Y, Fu H, Hu S, Pan J, Wang Y, Cheng J, Song J, Yu Q, Zhang S, et al. Chop deficiency prevents UUO-induced renal fibrosis by attenuating fibrotic signals originated from Hmgb1/TLR4/NFkappaB/IL-1beta signaling. Cell death & disease. 2015;6:e1847. doi: 10.1038/cddis.2015.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang FP, Li L, Li J, Wang JY, Wang LY, Jiang W. High mobility group box-1 promotes the proliferation and migration of hepatic stellate cells via TLR4-dependent signal pathways of PI3K/Akt and JNK. PloS one. 2013;8(5):e64373. doi: 10.1371/journal.pone.0064373. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 69.Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. Journal of leukocyte biology. 2007;81(1):28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 70.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature) Biochemical and biophysical research communications. 2004;322(4):1111–1122. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 71.Turner NA. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs) Journal of molecular and cellular cardiology. 2016;94:189–200. doi: 10.1016/j.yjmcc.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 72.Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators of inflammation. 2013;2013:828354. doi: 10.1155/2013/828354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pruenster M, Vogl T, Roth J, Sperandio M. S100A8/A9: From basic science to clinical application. Pharmacology & therapeutics. 2016;167:120–131. doi: 10.1016/j.pharmthera.2016.07.015. [DOI] [PubMed] [Google Scholar]
- 74.van Bon L, Cossu M, Loof A, Gohar F, Wittkowski H, Vonk M, Roth J, van den Berg W, van Heerde W, Broen JC, et al. Proteomic analysis of plasma identifies the Toll-like receptor agonists S100A8/A9 as a novel possible marker for systemic sclerosis phenotype. Annals of the rheumatic diseases. 2014;73(8):1585–1589. doi: 10.1136/annrheumdis-2013-205013. [DOI] [PubMed] [Google Scholar]
- 75.Tomcik M, Palumbo-Zerr K, Zerr P, Avouac J, Dees C, Sumova B, Distler A, Beyer C, Cerezo LA, Becvar R, et al. S100A4 amplifies TGF-beta-induced fibroblast activation in systemic sclerosis. Annals of the rheumatic diseases. 2015;74(9):1748–1755. doi: 10.1136/annrheumdis-2013-204516. [DOI] [PubMed] [Google Scholar]
- 76.Leifer CA, Medvedev AE. Molecular mechanisms of regulation of Toll-like receptor signaling. Journal of leukocyte biology. 2016;100(5):927–941. doi: 10.1189/jlb.2MR0316-117RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Warger T, Hilf N, Rechtsteiner G, Haselmayer P, Carrick DM, Jonuleit H, von Landenberg P, Rammensee HG, Nicchitta CV, Radsak MP, et al. Interaction of TLR2 and TLR4 ligands with the N-terminal domain of Gp96 amplifies innate and adaptive immune responses. The Journal of biological chemistry. 2006;281(32):22545–22553. doi: 10.1074/jbc.M502900200. [DOI] [PubMed] [Google Scholar]
- 78.Yang Y, Liu B, Dai J, Srivastava PK, Zammit DJ, Lefrancois L, Li Z. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26(2):215–226. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang QQ, Pope RM. The role of glycoprotein 96 in the persistent inflammation of rheumatoid arthritis. Archives of biochemistry and biophysics. 2013;530(1):1–6. doi: 10.1016/j.abb.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shibata T, Takemura N, Motoi Y, Goto Y, Karuppuchamy T, Izawa K, Li X, Akashi-Takamura S, Tanimura N, Kunisawa J, et al. PRAT4A-dependent expression of cell surface TLR5 on neutrophils, classical monocytes and dendritic cells. International immunology. 2012;24(10):613–623. doi: 10.1093/intimm/dxs068. [DOI] [PubMed] [Google Scholar]
- 81.Kiyokawa T, Akashi-Takamura S, Shibata T, Matsumoto F, Nishitani C, Kuroki Y, Seto Y, Miyake K. A single base mutation in the PRAT4A gene reveals differential interaction of PRAT4A with Toll-like receptors. International immunology. 2008;20(11):1407–1415. doi: 10.1093/intimm/dxn098. [DOI] [PubMed] [Google Scholar]
- 82.Wakabayashi Y, Kobayashi M, Akashi-Takamura S, Tanimura N, Konno K, Takahashi K, Ishii T, Mizutani T, Iba H, Kouro T, et al. A protein associated with toll-like receptor 4 (PRAT4A) regulates cell surface expression of TLR4. Journal of immunology. 2006;177(3):1772–1779. doi: 10.4049/jimmunol.177.3.1772. [DOI] [PubMed] [Google Scholar]
- 83.Kondo T, Kawai T, Akira S. Dissecting negative regulation of Toll-like receptor signaling. Trends in immunology. 2012;33(9):449–458. doi: 10.1016/j.it.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 84.Sargent JL, Milano A, Connolly MK, Whitfield ML. Scleroderma gene expression and pathway signatures. Current rheumatology reports. 2008;10(3):205–211. doi: 10.1007/s11926-008-0034-5. [DOI] [PubMed] [Google Scholar]
- 85.Pendergrass SA, Lemaire R, Francis IP, Mahoney JM, Lafyatis R, Whitfield ML. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. The Journal of investigative dermatology. 2012;132(5):1363–1373. doi: 10.1038/jid.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hinchcliff M, Huang CC, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, Tamaki Z, Lee J, Carns M, Podlusky S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. The Journal of investigative dermatology. 2013;133(8):1979–1989. doi: 10.1038/jid.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sargent JL, Milano A, Bhattacharyya S, Varga J, Connolly MK, Chang HY, Whitfield ML. A TGFbeta-responsive gene signature is associated with a subset of diffuse scleroderma with increased disease severity. The Journal of investigative dermatology. 2010;130(3):694–705. doi: 10.1038/jid.2009.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Critical care medicine. 2010;38(8):1685–1694. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 89.Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA : the journal of the American Medical Association. 2013;309(11):1154–1162. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 90.Savva A, Roger T. Targeting toll-like receptors: promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Frontiers in immunology. 2013;4:387. doi: 10.3389/fimmu.2013.00387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Buckley GM, Gowers L, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, et al. IRAK-4 inhibitors. Part 1: a series of amides. Bioorganic & medicinal chemistry letters. 2008;18(11):3211–3214. doi: 10.1016/j.bmcl.2008.04.058. [DOI] [PubMed] [Google Scholar]
- 92.Buckley GM, Ceska TA, Fraser JL, Gowers L, Groom CR, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, et al. IRAK-4 inhibitors. Part II: a structure-based assessment of imidazo[1,2-a]pyridine binding. Bioorganic & medicinal chemistry letters. 2008;18(11):3291–3295. doi: 10.1016/j.bmcl.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 93.Buckley GM, Fosbeary R, Fraser JL, Gowers L, Higueruelo AP, James LA, Jenkins K, Mack SR, Morgan T, Parry DM, et al. IRAK-4 inhibitors. Part III: a series of imidazo[1,2-a]pyridines. Bioorganic & medicinal chemistry letters. 2008;18(12):3656–3660. doi: 10.1016/j.bmcl.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 94.Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nature reviews Drug discovery. 2011;10(10):767–777. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 95.Zuliani-Alvarez L, Midwood KS. Fibrinogen-Related Proteins in Tissue Repair: How a Unique Domain with a Common Structure Controls Diverse Aspects of Wound Healing. Advances in wound care. 2015;4(5):273–285. doi: 10.1089/wound.2014.0599. [DOI] [PMC free article] [PubMed] [Google Scholar]